

Abstract
Circulating tumor DNA (ctDNA) is assumed to reflect tumor burden and has been suggested as a tool for prognostication and follow‐up in patients with metastatic pancreatic ductal adenocarcinoma (mPDAC). However, the prognostic value of ctDNA and its relation with tumor burden has yet to be substantiated, especially in mPDAC. In this retrospective analysis of prospectively collected samples, cell‐free DNA from plasma samples of 58 treatment‐naive mPDAC patients was isolated and sequenced using a custom‐made pancreatobiliary NGS panel. Pathogenic mutations were detected in 26/58 (44.8%) samples. Cross‐check with droplet digital PCR showed good agreement in Bland–Altman analysis (p = 0.217, nonsignificance indicating good agreement). In patients with liver metastases, ctDNA was more frequently detected (24/37, p < 0.001). Tumor volume (3D reconstructions from imaging) and ctDNA variant allele frequency (VAF) were correlated (Spearman's ρ = 0.544, p < 0.001). Median overall survival (OS) was 3.2 (95% confidence interval [CI] 1.6–4.9) versus 8.4 (95% CI 1.6–15.1) months in patients with detectable versus undetectable ctDNA (p = 0.005). Both ctDNA VAF and tumor volume independently predicted OS after adjustment for carbohydrate antigen 19.9 and treatment regimen (hazard ratio [HR] 1.05, 95% CI 1.01–1.09, p = 0.005; HR 1.00, 95% CI 1.01–1.05, p = 0.003). In conclusion, our study showed that ctDNA detection rates are higher in patients with larger tumor volume and liver metastases. Nevertheless, measurements may diverge and, thus, can provide complementary information. Both ctDNA VAF and tumor volume were strong predictors of OS.
Keywords: pancreatic cancer, tumor volume, circulating tumor DNA, KRAS, prognosis
Short abstract
What's new?
Circulating tumor DNA (ctDNA) attracts much interest as a possible prognostic tool for cancer. Here, the authors showed that the quantity of ctDNA correlated strongly with tumor volume in metastatic pancreatic ductal adenocarcinoma (mPDAC). They conducted a retrospective analysis using samples collected from 58 untreated mPDAC patients. For this study, the authors designed a pancreatobiliary NGS panel, which they used to test the patients’ cell‐free DNA, along with droplet digital PCR. Both ctDNA variant allele frequency and tumor volume predicted overall survival, they found.
Abbreviations
- 95% CI
95% confidence interval
- CA19.9
carbohydrate antigen 19.9
- cfDNA
cell‐free DNA
- ctDNA
circulating tumor DNA
- ddPCR
droplet digital PCR
- HR
hazard ratio
- mPDAC
metastatic pancreatic ductal adenocarcinoma
- NGS
next‐generation sequencing
- OS
overall survival
- PDAC
pancreatic ductal adenocarcinoma
- VAF
variant allele fraction
Introduction
Pancreatic ductal adenocarcinoma (PDAC) is predicted to become the second leading cause of cancer‐related death by 2030.1 Around 40% of PDAC patients present with metastatic disease (mPDAC), which is associated with an extremely poor prognosis of two to three months median overall survival (OS).2 Tools to predict prognosis, personalize treatment and monitor treatment response are urgently needed.
Circulating tumor DNA (ctDNA) has recently gained attention as a promising minimally invasive tumor marker. Cells release short fragments of cell‐free DNA (cfDNA) in the circulation; fragments shed specifically by tumor cells are referred to as ctDNA. The somatic mutations present in ctDNA can be detected and quantified using several techniques, including next‐generation sequencing (NGS) and droplet digital PCR (ddPCR). Potential clinical applications of ctDNA analysis include diagnosis, molecular characterization, prognostication, detection of residual disease, monitoring of treatment response and assessment of clonal evolution.3, 4
As illustrated by the joint review of the American Society of Clinical Oncology and the College of American Pathologists, questions remain about the validity and reproducibility of ctDNA analysis, hampering clinical application.5 ctDNA detection rates vary widely depending on cancer type and analysis technique; for mPDAC, rates between 38.8% and 86.1% have been reported.6, 7, 8, 9 In addition, ctDNA detection appears to have a strong prognostic value in patients with PDAC, but validation of this finding is required.7, 10 Lastly, it has been suggested that ctDNA is a surrogate marker for tumor burden, but very few studies have formally assessed the relationship between ctDNA quantity and 3D tumor volumes, and never in PDAC.11, 12, 13 As PDAC is comprised of dense desmoplastic stroma with comparatively few tumor cells, the relationship between tumor volume and ctDNA quantity is difficult to predict.
In our study, we evaluated the capability of targeted sequencing using a custom‐made pancreatobiliary specific NGS panel and of ddPCR to detect ctDNA in mPDAC. We tested the relationship between 3D‐measured tumor volume and ctDNA quantity. Finally, the independent prognostic value of ctDNA detection and tumors volumes on OS was assessed.
Material and Methods
Study design
All patients were selected from the prospective biobanks of the Amsterdam UMC, location AMC (September 2016–December 2017) and University of Verona Hospital (September 2017–December 2017). Both are tertiary referral hospitals and the majority of patients only visited once for a second opinion. All consecutive treatment‐naive patients presenting with PDAC (any stage) in the institutions were asked to participate in the biobank, excluding patients <18 years, and patients diagnosed with hepatitis B, C or HIV/AIDS. Both biobanks were approved by the institution's ethics committees (AMC 2014_181 and Verona 1101cesc) and all patients provided written informed consent for participation after the nature of the study was explained.
All patients with pathologically confirmed mPDAC of whom at least 4 mL of plasma was available at baseline (i.e., before start of treatment) were selected for the current study. All patients had metastatic disease at time of blood draw. This included patients with distant lymph node metastases and patients with metastatic disease detected upon surgical exploration. Patients were treated according to standard clinical practice, and received palliative chemotherapy (FOLFIRINOX or gemcitabin‐nabpaclitaxel) or supportive care. If patients underwent palliative chemotherapy in the Amsterdam UMC or University of Verona Hospital, they were approached for follow‐up blood samples at the moment of response evaluation. This was after four cycles of FOLFIRINOX or after two cycles in the case of gemcitabin‐nabpaclitaxel. However, on clinical grounds response evaluation could be delayed or expedited. Patient characteristics (age, sex, ECOG performance score, CA19.9, vascular involvement primary tumor, localization of metastases, type of first line chemotherapy, OS) were retrieved from medical records by a trained MD. Vascular involvement was defined as involvement of the coeliac trunc, hepatic artery, superior mesenteric artery, portal vein, superior mesenteric vein or splenic vein. Administration of chemotherapy was defined as at least one cycle of chemotherapy. Primary outcome was OS. The sample size calculation is provided in Supplementary Material and Methods. Our study was reported according to the Reporting Recommendations for tumor Marker Prognostic Studies and Strengthening the Reporting of Observational studies in Epidemiology guidelines.14, 15
Reconstruction from CT and MRI
Tumor volumes at baseline and follow‐up were measured using Syngo.via (Siemens Healthcare, Forchheim, Germany). Contrast‐enhanced scans as performed during clinical practice in the university hospital or one of the referring hospitals were used. For CT scans a semiautomated software (MM Oncology workflow) with manual correction if needed was used when possible (depending on tumor morphology; Supplementary Fig. S2). If semiautomated volume measurement was not possible (e.g., in the case of poorly defined margins) a manual setting was used (MM reading workflow). For MRI scans, the workflow MR Liver Spleen was used. Assessment was done by a trained MD; each scan was finally approved by an expert abdominal radiologist blinded for outcome (KvL, 22 years of experience). Lymph nodes were included when the short axis was larger than 10 mm, according to the definitions for pathological lymph nodes reported in the RECIST 1.1 criteria.16 All primary tumors and organ metastases were measured regardless of size. Volumes were calculated in milliliters and analyzed as both a continious variable and as a binary variable after dichotomization by median.
DNA isolation techniques
Blood was collected before the start of therapy (baseline) and, if possible, during follow‐up at the time of response evaluation. For patients included at the Amsterdam UMC, one 5 mL serum tube and two 10 mL EDTA tubes were collected. For plasma separation, the tubes were centrifuged twice within 1 hr of blood collection, first at 1,300g for 10 min followed by transfer of the plasma into 1.5 mL Eppendorf tubes, then centrifuged at 20,000g for 10 min. Plasma was stored at −80°C until cfDNA isolation. For patients included in the University of Verona Hospital, blood was collected in cfDNA BCT tube (Streck Inc., La Vista, NE) and sent to the Amsterdam UMC. The tubes were handled and centrifuged within 14 days according to the manufacturer's instructions (double spin at 1,600g and 16,000g). Previous research has shown that the cfDNA quantity from BTC and EDTA tubes was highly comparable, with cfDNA quantity from the BTC tubes remaining stable over time.17 cfDNA was isolated from 4 mL of plasma using the QIAamp Circulating Nucleic Acid Kit (Qiagen, Venlo, the Netherlands) according to the manufacturer's instructions. When available, matching tumor tissue was sequenced for comparison (Supplementary Material and Methods).
Next‐generation sequencing
A custom targeted NGS amplicon panel was designed specifically for pancreatobiliary adenocarcinoma for the preparation of libraries from cfDNA and tumor tissue DNA. Hotspot locations included in this panel were based on large whole‐genome sequencing studies (Bailey 2016, Witkiewicz 2015, TCGA data set) and the COSMIC database.18, 19, 20, 21 KRAS, TP53, SMAD4, CDKN2A, PIK3CA, GNAS, BRAF and NRAS were included in the panel, based on a combination of frequency of occurrence of mutations (all mutations reported more than once per dataset in these genes were included) and the potential to cover the relevant parts of the gene in a small, cost‐effective panel of 34 amplicons (Table S3).
The DNA libraries were produced using the custom Ion AmpliSeq Panel (Life Technologies, Bleiswijk, the Netherlands) according to the manufacturer's instructions. Libraries were barcoded (Ion Xpress Barcodes adapters kit, Life Technologies) and quantified using a Qubit dsDNA HS assay kit (Life Technologies). Tumor DNA libraries were sequenced on a 316 chip in the Personal Genome Machine system (Ion Torrent, Life Technologies). Torrent suite software v5.8.0 was used for signal processing, run quality reports and to generate BAM files. Sequences were analyzed using SeqNext software v4.1.2 (JSI Medical Systems GmbH, Ettenheim, Germany). The target sequencing depth was 5,000× for cfDNA and 1,500× for tissue DNA. For mutation calling in cfDNA, a variant allele fraction (VAF) cutoff value of 1% was used. The minimum threshold for successful sequencing was set at a minimum of 1,000 reads for both KRAS amplicons, which covered codon 12, 13 and 61. For analyses, the highest VAF of all detected mutant alleles in the cfDNA of that particular patient was used.
Droplet digital PCR (ddPCR)
NGS results were cross checked with ddPCR using the QX200™ Droplet Digital™ PCR System from Bio‐Rad (Bio‐Rad Laboratories, Veenendaal, the Netherlands). KRAS somatic alterations were detected using a commercial KRAS Screening Multiplex Kit by Bio‐Rad, which screens for seven mutations in KRAS codons 12 and 13 (exon 2); G12A (c.35G>C), G12C (c.34G>T), G12D (c.35G>A), G12 V (c.35G>T), G12R (c.34G>C), G12S (c.34G>A), G13D (c.38G>A). The QuantaSoft Software version 1.7.4 (Bio‐Rad Laboratories) was used for data analysis, including the calculation of the fractional abundance. The fractional abundance quantifies the abundance of mutant DNA alleles in the wild‐type background. In ddPCR analysis, it is not possible to determine the change in base pair; therefore, the type of mutation was not reported. To accommodate for the possibility of false positive droplets, the threshold for mutation calling was set at ≥5 positive droplets. The reaction was considered acceptable if >200 droplets containing DNA were generated.
Serum antigen CA19.9 analysis
For patients included at the Amsterdam UMC, carbohydrate antigen 19.9 (CA19.9) levels were measured using 50 μL of serum in one batch with an immunochemical assay on the Roche e602 (Roche Diagnostics, Almere, the Netherlands) integrated in a Cobas c8000 system (Roche Diagnostics). For patients included in the University of Verona Hospital, measurements available from the electronic patient file were used if the measurement was performed less than 14 days before/after blood draw for cfDNA and before the start of systemic treatment.
Statistical analysis
Characteristics of patients with and without detectable ctDNA were compared using a Student's t‐test (normally distributed continuous variables), Mann–Whitney U‐test (nonnormally distributed continuous variables) or χ 2‐test/Fisher exact test (categorical variables). Continuous variables were not categorized except for tumor volume. A Bland–Altman plot was used to assess agreement between the VAF of KRAS as detected using NGS and ddPCR (only for the mutations covered by ddPCR analysis).22 Correlation between VAF and tumor volumes and between VAF and CA19.9 was visualized using a scatterplot and statistically tested using Pearson's (normally distributed) or Spearman's (nonnormally distributed) correlation test. OS was calculated from the date of baseline blood sample drawn until death of any cause. OS was visualized using the Kaplan–Meier method and reported as median with 95% confidence interval (95% CI). OS between groups was compared using a logrank test.
For regression analyses, missing data were handled using multiple imputation (Predictive Mean Matching) with the construction of 10 databases.23, 24 Hazard RAtios (HR) and 95% CI for ctDNA VAF and tumor volumes were calculated using Cox regression analysis, and adjusted for known prognostic factors or that were of (borderline) significance (p < 0.10) in univariable analysis. In all analyses two‐sided tests were used and a p‐value below 0.05 was considered statistically significant. Data were analyzed using IBM SPSS Statistics for Windows version 24.0 (IBM Corp., Armonk, NY).
Results
Patient inclusion
We included 60 treatment‐naive patients with pathologically confirmed mPDAC (Amsterdam UMC n = 46; University of Verona Hospital n = 14). For all patient samples, cfDNA was isolated successfully with a median concentration of 3.2 ng/μL (range 0.58–23 ng/μL). Two patients (cfDNA concentrations 2.2 and 9.3 ng/μL) were excluded as both NGS and ddPCR results were lower than the predefined sequence quality after two attempts, leaving 58 patients for further analysis. Of the 39 patients receiving palliative chemotherapy, 15 patients discontinued before first evaluation and a follow‐up sample could not be obtained. In total, 19 follow‐up samples from 10 individual patients were available and sequenced successfully. The number of follow‐up samples varied between one and six per patient.
Cohort description
Thirty‐seven (64%) patients presented with liver metastases, 11 (19%) with metastases to other organs and 2 (3%) with only distant lymph node metastases. Baseline CA19.9 values were available for 49 patients (median value 786 kU/L, interquartile range 142–5,910). In eight patients, metastases were detected during surgical exploration. Fourteen patients received no chemotherapy treatment and 49 completed at least one cycle of chemotherapy (FOLFIRINOX or gemcitabin‐nabpaclitaxel). Data on chemotherapy could not be retrieved for five patients (baseline characteristics in Table 1). Median OS of the total cohort after 50 events was 4.9 months (95% CI 2.3–7.4). Median follow‐up of the eight censored patients was 12.3 months (range 2.3–27.7). The cohort from the Amsterdam UMC and Verona did not show significant differences regarding the parameters presented in Table 1, with the exception of first line chemotherapy. Most patients from the Amsterdam UMC received FOLFIRINOX, whereas the majority of Verona patients received gemcitabin‐nabpaclitaxel.
Table 1.
Baseline characteristics of patients with and without detectable circulating tumor DNA using next generation sequencing
| All (n = 58) | ctDNA detected (n = 26) | No ctDNA detected (n = 32) | p | |
|---|---|---|---|---|
| Age | 67 (58–73) | 68 (59–74) | 65 (53–72) | 0.719 |
| Male sex | 31 (53.4) | 17 (65.4) | 14 (43.8) | 0.100 |
| ECOG performance status | ||||
| 0–1 | 39 (69.6) | 19 (76.0) | 20 (64.5) | 0.353 |
| 2–3 | 17 (30.4) | 6 (24.0) | 11 (35.5) | |
| Unknown | 2 | 1 | 1 | |
| CA19.9 (kU/L) | 797 (138–6,063) | 3,941 (102–78,342) | 449 (143–3,261) | 0.338 |
| Maximum diameter primary tumor (mm) | 36 (28–54) | 36 (27–58) | 36 (29–50) | 0.727 |
| Vascular involvement primary tumor1 | ||||
| No | 41 (71.9) | 7 (26.9) | 9 (29.0) | 0.860 |
| Yes | 16 (28.1) | 19 (73.1) | 22 (71.0) | |
| Unknown | 1 | 0 | 1 | |
| Metastases on baseline imaging1 | ||||
| No or distant lymph nodes only | 10 (17.5) | 0 | 10 (32.3) | <0.001 |
| Liver metastases2 | 37 (64.9) | 24 (92.3) | 13 (41.9) | |
| Metastases other than liver | 10 (17.5) | 2 (7.7) | 8 (25.8) | |
| Unknown | 1 | 0 | 1 | |
| Total tumor volume3 | 47.9 (22.7–88.9) | 70.0 (41.9–264.0) | 35.4 (18.7–65.6) | 0.004 |
| Tumor volume primary tumor3 | 25.1 (13.7–50.1) | 27.0 (18.4–69.0) | 21.5 (10.8–38.2) | 0.149 |
| Tumor volume metastases3 | 12.6 (1.7–48.9) | 36.2 (12.0–153.4) | 4.6 (0–22.2) | 0.001 |
| First line chemotherapy | ||||
| No chemotherapy | 14 (26.4) | 8 (36.4) | 6 (19.4) | 0.642 |
| FOLFIRINOX4 | 23 (39.6) | 8 (36.4) | 15 (46.9) | |
| Gemcitabin‐nabpaclitaxel4 | 16 (27.6) | 6 (27.3) | 10 (31.3) | |
| Unknown | 5 | 4 | 1 |
Continuous variables were presented as median with Interquartile ranges (IQR); Categorical variables were presented as counts with percentages.
On baseline imaging, baseline imaging data were missing for one patient.
With or without metastases to other organs.
Available for 47 cases.
At least one completed cycle of chemotherapy.
Baseline imaging
Baseline imaging of 51 patients was available (48 CT scans, 3 MRI scans). Lesions of four patients with massive liver metastases (n = 1), diffuse lung metastases (n = 1) or omental cake (n = 2) could not be reliably quantified. However, the volumes of the measurable lesions in these patients were higher than the median value used for dichotomization, hence categorization was possible. The median tumor volume was 47.9 mL (interquartile range 22.7–88.9) for all patients. For patients without and patients with detectable ctDNA. median tumor volume was 35.4 mL (interquartile range 18.7–65.6) and 70.0 mL (interquartile range 41.9–264.0), respectively (p = 0.004; Table 1).
Results of cfDNA detection using NGS
In 26/58 (45%) individual patients, a pathogenic mutation was detected at baseline using the targeted sequencing approach (Table 2). This was 24/37 (65%) in the subgroup with liver metastases at baseline imaging. Detection rates were highest in the subgroup of patients with liver metastases at baseline and high tumor volume (15/20, 75%). No ctDNA was detected in patients with lymph node metastases only or metastases detected during surgical exploration. The median total coverage of all cfDNA samples was 5,142 reads per amplicon (range 1,237–13,487 reads). The median coverage of KRAS was 3,573 reads (range 1,058–12,985 reads).
Table 2.
Detected mutations in cfDNA of patients with metastatic pancreatic ductal adenocarcinoma using next‐generation sequencing (IonTorrent) and ddPCR. [Color table can be viewed at https://wileyonlinelibrary.com]
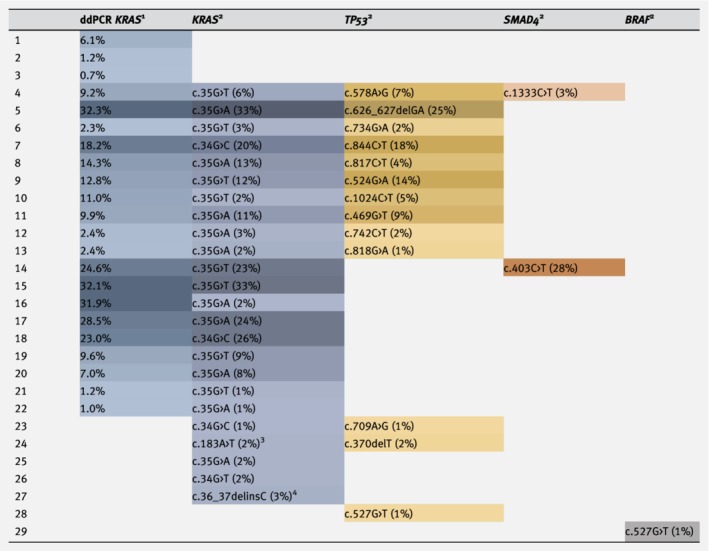
|
The darker the color, the higher the FA or VAF.
Fractional abundance (FA).
Variant allele frequency (VAF).
Exon 3 mutation, not covered by ddPCR.
Deletion–insertion, not covered by ddPCR.
In the baseline samples, 40 pathogenic mutations (22 unique mutations) were identified in KRAS (n = 24), TP53 (n = 13), SMAD4 (n = 2) and BRAF (n = 1). Notably, one patient showed a deletion–insertion in KRAS codon 13 (c.36_37delinsC). Thirteen patients showed multiple mutations including KRAS and 11 patients showed only a KRAS mutation. In two patients without KRAS mutations, BRAF V600E or a TP53 mutation was detected. The median VAF of KRAS in baseline samples with detectable ctDNA was 4% (range 1–40%). Mutations were detected in 6/19 (32%) follow‐up samples (3/10 individual patients) in KRAS, GNAS, TP53 and SMAD4. In one patient without any mutations at baseline, the follow‐up samples showed a mutation in KRAS c.34G>T and GNAS c.559C>T. The sequencing results of tissue and the follow‐up samples are stated in Tables S1 and S2, respectively.
Agreement between ddPCR and NGS
For 77 samples (baseline and follow‐up), results of seven KRAS hotspot loci were available for both NGS and ddPCR, demonstrating a good correlation (Pearson's ρ = 0.96, p < 0.001; Table 2; Fig. S1 a). Bland–Altman analysis showed good agreement (p = 0.217; nonsignificance indicating good agreement), with a mean difference of −0.56 (95% CI −1.45 to 0.33) and limits of agreement of −8.26 and 7.14 (mean difference ±1.96*SD of mean difference; Fig. S1 b). In three samples, values were outside the limits of agreement. In 24 samples, a KRAS mutation was detected by both NGS and ddPCR. The ddPCR analysis detected a mutation in six (7.8%) samples for which NGS did not detect a mutation (Table 2). NGS identified four (5.2%) KRAS exon 2 mutations which were not detected by ddPCR, including the sample with the deletion–insertion in KRAS (c.36_37delinsC). Three other samples only showed mutations in loci not covered by the KRAS Screening Multiplex Kit used for ddPCR (KRAS codon 61, BRAF and TP53).
Correlations between ctDNA, tumor volumes and CA19.9
Tumor volumes and the maximum VAF of ctDNA were correlated in all patients (Spearman's ρ = 0.544, p < 0.001) and in the subgroup of patients with detectable ctDNA (Spearman's ρ = 0.781, p < 0.001; Fig. 1 a). There was no statistically significant correlation between baseline serum CA19.9 (n = 49) and quantity of ctDNA (Spearman's ρ = 0.199, p = 0.17; Fig. 1 b).
Figure 1.
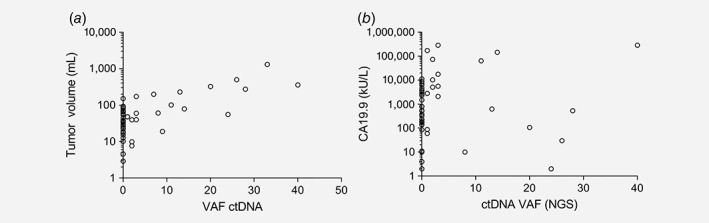
Correlation between quantity of circulating tumor DNA and other measurements. (a) Correlation between quantity of circulating tumor DNA and 3D measured tumor volume (n = 47). Total group: Spearman's ρ = 0.544, p < 0.001. Subgroup with detectable ctDNA: Spearman's ρ = 0.781, p < 0.001. (b) Correlation between quantity of circulating tumor DNA and CA19.9 (n = 49). Total group: Spearman's ρ = 0.199, p = 0.170. Subgroup with detectable ctDNA: Spearman's ρ = −1.20, p = 0.616.
Prognostic value of ctDNA and tumor volumes
Median OS was 3.2 (95% CI 1.6–4.9) versus 8.4 (95% CI 1.6–15.1) months in patients with detectable or undetectable ctDNA, respectively (p = 0.005, Fig. 2). For high versus low tumor volume, this was 3.2 (95% CI 2.9–3.5) or 10.8 (95% CI 7.0–14.5) months (p = 0.001). It was not possible to construct a multivariable Cox model including both ctDNA VAF and tumor volume, due to multicollinearity (variance inflation factor >3). Therefore, separate multivariable models were constructed (Table 3). Both ctDNA VAF and tumor volume predicted OS after adjustment for CA19.9 and type of treatment (HR 1.05, 95% CI 1.01–1.09, p = 0.005; HR 1.00, 95% CI 1.01–1.05, p = 0.003, respectively).
Figure 2.
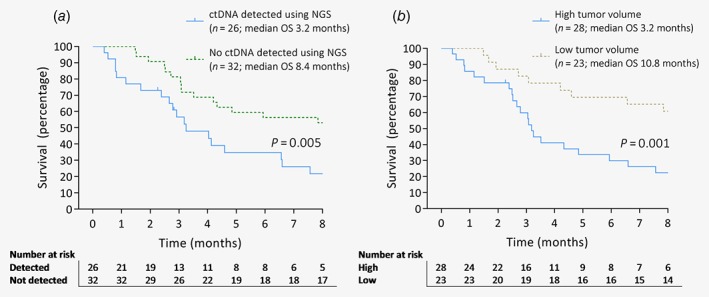
Overall survival of patients with metastatic pancreatic ductal adenocarcinoma. (a) Patients with and without detectable ctDNA. (b) Patients with high and low tumor volumes dichotomized by median volume (47.9 mL).
Table 3.
Univariable and multivariable Cox regression analyses predicting overall survival in 58 patients with metastatic pancreatic ductal adenocarcinoma
| Univariable | Multivariable VAF ctDNA | Multivariable tumor volume | ||||
|---|---|---|---|---|---|---|
| HR (95% CI) | p | HR (95% CI) | p | HR (95% CI) | p | |
| Age (per year increase) | 1.02 (0.99–1.05) | 0.202 | – | – | – | – |
| ECOG performance score1 | ||||||
| 0–1 | 1 | – | – | – | ||
| 2–3 | 1.37 (0.74–2.54) | 0.319 | ||||
| Increasing CA19.9 (per 1,000 kU/L increase) | 1.00 (1.00–1.01) | 0.073 | 1.00 (1.00–1.01) | 0.147 | 1.00 (1.00–1.01) | 0.039 |
| Vascular involvement primary tumor2 , 3 | ||||||
| No | 1 | – | – | – | – | |
| Yes | 0.94 (0.50–1.81) | 0.856 | ||||
| Localization metastases on baseline imaging3 | ||||||
| No or distant lymph nodes only | 1 | |||||
| Liver metastases4 | 1.59 (0.77–3.29) | 0.210 | – | – | – | – |
| Other metastases | 1.15 (0.46–2.89) | 0.763 | ||||
| Palliative chemotherapy | ||||||
| No | 1 | 1 | 1 | |||
| First line FOLFIRINOX5 | 0.37 (0.19–0.75) | 0.005 | 0.34 (0.16–0.71) | 0.004 | 0.29 (0.14–0.61) | 0.001 |
| First line gem‐nab5 | 0.43 (0.20–1.92) | 0.030 | 0.48 (0.22–1.06) | 0.070 | 0.43 (0.19–0.95) | 0.024 |
| Increasing cfDNA concentration | 1.02 (0.96–1.08) | 0.559 | – | – | – | – |
| NGS ctDNA detected | ||||||
| No | 1 | 0.009 | – | – | – | – |
| Yes | 2.16 (1.21–3.85) | |||||
| VAF ctDNA (per 1% increase) | 1.06 (1.03–1.09) | 0.001 | 1.05 (1.01–1.09) | 0.005 | 6 | |
| Total tumor volume (per 10 mL increase) | 1.03 (1.01–1.04) | 0.001 | 6 | 1.00 (1.01–1.05) | 0.003 | |
| Primary tumor volume (per 10 mL increase) | 1.03 (1.01–1.05) | 0.003 | 6 | 6 | ||
| Metastases volume (per 10 mL increase) | 1.06 (1.01–1.11) | 0.014 | 6 | 6 | ||
When analyzing ECOG performance status using each score as a separate category (0, 1, 2, 3; 0 = ref) only ECOG 3 (n = 3) was a significant prognostic factor (HR 8.6, p = 0.002); VAF, variant allele frequency (highest VAF of detected mutations per patients used for analyses).
Vascular involvement was defined as involvement of the coeliac trunc, hepatic artery, superior mesenteric artery, portal vene, superior mesenteric vene or splenic vene.
On baseline imaging.
With or without metastases to other organs.
At least one completed cycle of chemotherapy.
Not included to avoid multicollinearity.
Circulating tumor DNA dynamics
As ctDNA could possibly be used to monitor treatment response and clonal evolution, follow‐up samples were collected. For four patients with follow‐up serum samples, relations among ctDNA VAF, tumor volumes and CA19.9 are illustrated in Figures 3 a–3 d. Dynamics in tumor volume and ctDNA seemed similar in most patients, but showed inversed changes in one of the patients: ctDNA VAF decreased, while tumor volume and CA19.9 increased (Fig. 3 b). In one patient (Fig. 3 c), very high ctDNA VAF (exceeding 50%) was found at the moment of progression after nine cycles of chemotherapy, while absolute tumor volume was lower compared to baseline.
Figure 3.
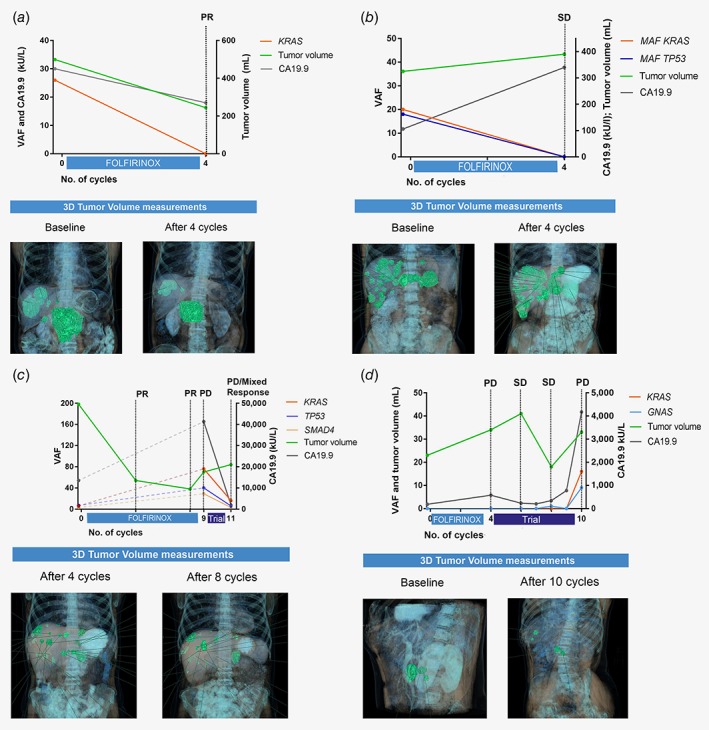
ctDNA dynamics in four individual patients during follow‐up. (a) Tumor volumes, CA19.9 and ctDNA VAF all showed similar dynamics. (b) CA19.9 and ctDNA VAF showed opposite responses in relatively stable tumor volume. (c) While VAF and CA19.9 showed similar trends in this patient, tumor volume differed. NB: One follow‐up measurement showed a KRAS VAF of 76%, indicating amplification of KRAS. The measurement was confirmed with ddPCR and observed again in the consecutive sample collected 2 weeks later (no treatment was started in the interval). (d) ctDNA and CA19.9 showed similar trends. Abbreviations: PD, progressive disease; PR, partial response; SD, stable disease; all according to RECIST 1.1. In the lower parts of the figures (illustrations), 3D tumor volumes are shown in green. The left and right Y‐axes represent different parameters in per subfigure, as the absolute values of the three parameter (ctDNA, CA19.9, tumor volumes) differed widely.
Discussion
To the best of our knowledge, this international study is the first to show a correlation between ctDNA and tumor volumes in PDAC. After adjustment for CA19.9 and type of treatment, both ctDNA and tumor volume predicted OS.
The ctDNA detection rate of 46% in the total cohort and 75% in patients with high tumor volume and liver metastases are within the ranges reported in literature. Studies reporting exclusively on mPDAC showed detection rates of 29.3–33%.7, 25 When various tumor stages were studied, a sensitivity between 38.8% and 86.1% was determined for those patients with metastatic disease.6, 8, 9, 26 This wide range may be explained by the various methods used. In the current study as well as in literature, it has been shown that detection rates of ctDNA are dependent on many factors, related to both tumor characteristics and technical aspects (volume of plasma input, method of cfDNA isolation, sequencing techniques and number of interrogated hotspots/genes).5, 27 In our cohort, the full spectrum of metastatic disease was included, varying from patients with small metastases, undetectable on standard imaging, to those with only distant lymph node or extensive organ metastases. This provides a good representation of the patient population but might have resulted in lower detection rates. In most other studies with mPDAC patients, patient flow and selection methods were unclear, hampering comparisons.6, 7, 9, 25, 26 Our finding that higher ctDNA detection rates occurred in patients with liver metastases of PDAC has also been reported for colorectal cancer.28 We hypothesize that this may be explained by the relatively large size of liver metastases and the high vascularity of the organ.
Although some previous studies have correlated ctDNA to imaging findings (e.g., RECIST criteria or sum of target lesions), exact quantification of total 3D tumor volumes based on imaging has never been performed for PDAC.11, 29, 30 For other cancer types, a very small number of studies have correlated exact 3D‐measured tumor volume to ctDNA VAF (ovarian carcinoma, n = 40; colorectal cancer, n = 45).31, 32 The focus of these studies was on the correlation between variations in tumor volumes and in ctDNA between serial measurements. A significant correlation between differences in VAF and tumor volume was reported.31, 32 Although the VAF shows a strong correlation with tumor volume in our study, it should be noted that there are still patients with high tumor volumes in whom no ctDNA was detected and vice versa. Additionally, in two of our follow‐up samples the ctDNA VAF did not parallel changes in tumor volume. In one of these patients, follow‐up samples showed an increasing VAF of KRAS, while tumor volume and CA19.9 decreased. The fact that VAF exceeded 50% indicates amplification of the mutated KRAS gene, and likely explains part of the extreme increase in ctDNA. Notably, two other articles have shown incongruence between ctDNA quantity and imaging‐based tumor response (non 3D‐measured) in a minority of patients.12, 33
These findings illustrate that more factors than tumor volume contribute to the detectability of ctDNA, underscoring that these two measurements provide complementary information. Whether one measurement confers more accurate prognostic information than the other requires further investigation. The prognostic value of ctDNA for OS (p = 0.005) is in line with the results of a recent meta‐analysis of ctDNA in PDAC.7 In our study, ctDNA was an predictor of OS after adjustment for CA19.9 and treatment regimen, while CA19.9 was not an independent predictor after adjustment for ctDNA, an observation also reported by others.25, 34, 35 At baseline, there was no statistically significant relationship between CA19.9 and ctDNA (n = 47; p = 0.170). Other studies have shown a correlation between CA19.9 and ctDNA, although the cohorts were of limited size.25, 36 Surprisingly, we did not find ECOG status to be a significant prognosticator in the regression analysis. This seems to be due to the fact that only ECOG performance status 3 had prognostic strength, but that there were only three patients with ECOG 3. Therefore, we had to combine ECOG status into two categories (ECOG 0–1 and 1–2). Another explanation could be the relatively small sample size of our study.
In relation to technical aspects, previous studies have interrogated regions ranging from a single locus of KRAS to comprehensive cancer panels covering >20 genes.9, 30, 35, 37 PDAC shows a very homogeneous mutational landscape with KRAS mutations in approximately 90% of cases, barring the need for extensive coverage and patient‐tailored primers.18, 38, 39 This implies that ddPCR, which has a lower limit of detection than NGS, would be eminently suited to PDAC. On the other hand, ddPCR is limited to a small number of loci, which limit future clinical applications and lower sensitivity. In one patient, the deletion–insertion in KRAS exon 2 likely prohibited binding of all ddPCR primers on the mutated allele, resulting in the generation of only wild‐type droplets. This sample and three other samples only showed mutations in loci not covered by the KRAS Screening Multiplex Kit used for ddPCR (KRAS codon 61, BRAF and TP53). Nonetheless, we observed good agreement between ddPCR and NGS, which was also reported in other studies.40, 41, 42
Our primary limitation was the relatively small sample size, which increases the risk of a type 1 error. The limited number of follow‐up samples prevented further insight into ctDNA dynamics. Moreover, pancreatic tumors are difficult to measure because of their often poorly defined margins and irregular growth patterns; errors in measurements may have occurred but seem inevitable in PDAC. The most important strength of our study is the multidisciplinary approach of the assessment of ctDNA and the precise assessment of tumor volumes, which enabled us to assess to what extent ctDNA VAF are reflective of tumor volume within the same stage (IV) of disease. Also, the use of a concise custom‐made NGS panel allowed us to interrogate frequently mutated loci other than KRAS at relatively limited costs; specific genes of interest can easily be added to this kind of custom‐made panels.
In order to determine whether clinical implementation of ctDNA is useful, future studies need to determine whether serial testing of ctDNA quantity in PDAC can provide additional information in the assessment of response to therapy. After administration of chemotherapy, interpretation of imaging can be difficult, as imaging cannot distinguish between viable carcinoma and fibrosis.43 In patients who show mixed response or progression on CT but with a clinical response, ctDNA may be able to help classify tumor response. Moreover, serial testing can reveal changes in the mutational profile under pressure of chemotherapy. Future studies may also be able to use the further advanced techniques that have been introduced since the start of our study, including unique molecular identifiers and selection based on differences in fragmentation patterns between cfDNA derived from normal cells and ctDNA.44, 45 When using unique molecular identifiers, all input DNA‐molecules are indexed with an identifier before PCR. This can decrease the limit of detection, as true mutations can be distinguished from PCR errors or sequencing errors. When using optimal technical conditions for NGS, including unique molecular identifiers, detection rates of 0.01–0.0015% have been documented for both IonTorrent NGS and ddPCR.46, 47 Finally, it seems sensible to combine ctDNA and other biomarkers in one test, attempting to increase sensitivity and specificity. An interesting illustration of this approach in the diagnostic setting is the CancerSEEK test, which used a combination of ctDNA and established serum makers to detect early stage cancers, showing a sensitivity of 69–98% for the detection of five cancer types, including pancreatic cancer.48
Supporting information
Appendix S1: Supplementary Information
Fig. S1 Comparison of NGS and ddPCR results in detection of circulating tumor DNA (n = 77).
A: Plot showing the correlation between the fractional abundance (FA) of KRAS exon 2 mutations in ddPCR and the variant allele frequency (VAF) of KRAS exon 2 in NGS.
B: Bland–Altman plot assessing agreement between ctDNA quantity measured using NGS (VAF) and ddPCR (FA). The mean difference of −0.56 (95% CI ‐1.47‐0.31) was small, with most measurements within the limits of agreement (P = 0.217; non‐significance indicating good agreement).
Fig. S2 Illustrations of 3D tumor volume measurements.
A: Semi‐automated measurement (MM oncology workflow)
B: Manual measurement (MM reading workflow)
C: 3D‐reconstruction showing several semi‐automated measurements
Table S1 Results tissue comparison.
Corresponding tumor material was available in 17 patients (eight cytology and nine histology). Unfortunately, sequencing of cytology material failed in seven of the eight patients due to low DNA yield). Most of the tissue samples were derived from patients who underwent exploration. Nine of the 10 successfully sequenced tissue samples showed a mutation in KRAS exon 2. Five samples showed an additional mutation in TP53 and/or SMAD4 and/or CDKN2A. Using NGS, in only one of these 10 patients (KRAS wildtype) we were able to detect a mutation in cfDNA; this BRAF V600E mutation was confirmed in tissue. Using ddPCR we detected a KRAS mutation in cfDNA of one other patient.
Table S2 NGS results follow‐up samples.
Table S3 Amplicons in the custom‐made pancreatobiliary NGS panel.
For each amplicon the start and end position are presented in reference genome hg 19. Mean amplicon length was 87 base pair (range 67–137).
Acknowledgements
The authors acknowledge Paul Eijk for helping set up droplet digital PCR at the VUmc Cancer Center Amsterdam and serving as the point of contact for researchers using the platform. This work was supported by the Dutch Cancer Society (KWF) grant to M.J.V., O.R.B. and H.W.L. (UVA 2014‐6803).
Conflict of interests: M.S. has received a travel grant sponsored by IPSEN. S.S. has received a travel grant (travel expenses and accommodation) from Roche. K.P.L. is a consultant for AngioDynamics and Cook. M.J.V. has received research funding from Seno Medical Instruments, is involved in Agendia and travel/accommodation expenses were paid by Hoffmann‐La Roche. J.W.W. has received research funding from Servier, Halozyme, Novartis, Celgene, Astra Zenica, Pfizer, Roche and Merck. B.Y. received a travel grant from Servier, and disclosed that one of the employees on the payroll of his group is paid by GenM. M.F.B. has received research funding from Celgene and acted as a consultant to Servier. H.W.L. has acted as a consultant for Celgene, Eli Lilly and Company, Nordic Pharma Group and Philips, has received research grants from, Amgen, Bayer Schering Pharma AG, Celgene, Eli Lilly and Company, GlaxoSmithKline Pharmaceuticals, Nordic Pharma Group, Philips, Roche Pharmaceuticals.
Data availability
The data that support the findings of our study are available from the corresponding author upon reasonable request.
References
- 1. Rahib L, Smith BD, Aizenberg R, et al. Projecting cancer incidence and deaths to 2030: the unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Res 2014;74:2913–21. [DOI] [PubMed] [Google Scholar]
- 2. van der Geest LGM, Haj Mohammad N, Besselink MGH, et al. Nationwide trends in chemotherapy use and survival of elderly patients with metastatic pancreatic cancer. Cancer Med 2017;6:2840–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Corcoran RB, Chabner BA. Application of cell‐free DNA analysis to cancer treatment. N Engl J Med 2018;379:1754–65. [DOI] [PubMed] [Google Scholar]
- 4. Wan JCMM, Massie C, Garcia‐Corbacho J, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 5. Merker JD, Oxnard GR, Compton C, et al. Circulating tumor DNA analysis in patients with cancer: American Society of Clinical Oncology and College of American Pathologists joint review. J Clin Oncol 2018;36:1631–41. [DOI] [PubMed] [Google Scholar]
- 6. Kim MK, Woo SM, Park B, et al. Prognostic implications of multiplex detection of KRAS mutations in cell‐free DNA from patients with pancreatic ductal adenocarcinoma. Clin Chem 2018;64:726–34. [DOI] [PubMed] [Google Scholar]
- 7. Chen L, Zhang Y, Cheng Y, et al. Prognostic value of circulating cell‐free DNA in patients with pancreatic cancer: a systemic review and meta‐analysis. Gene 2018;679:328–34. [DOI] [PubMed] [Google Scholar]
- 8. Singh N, Gupta S, Pandey RM, et al. High levels of cell‐free circulating nucleic acids in pancreatic cancer are associated with vascular encasement, metastasis and poor survival. Cancer Invest 2015;33:78–85. [DOI] [PubMed] [Google Scholar]
- 9. Pietrasz D, Pécuchet N, Garlan F, et al. Plasma circulating tumor DNA in pancreatic cancer patients is a prognostic marker. Clin Cancer Res 2017;23:116–23. [DOI] [PubMed] [Google Scholar]
- 10. Creemers A, Krausz S, Strijker M, et al. Clinical value of ctDNA in upper‐GI cancers: a systematic review and meta‐analysis. Biochim Biophys Acta Rev Cancer 2017;1868:394–403. [DOI] [PubMed] [Google Scholar]
- 11. Tjensvoll K, Lapin M, Buhl T, et al. Clinical relevance of circulating KRAS mutated DNA in plasma from patients with advanced pancreatic cancer. Mol Oncol 2016;10:635–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. García‐Saenz JA, Ayllón P, Laig M, et al. Tumor burden monitoring using cell‐free tumor DNA could be limited by tumor heterogeneity in advanced breast cancer and should be evaluated together with radiographic imaging. BMC Cancer 2017;17:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tie J, Kinde I, Wang Y, et al. Circulating tumor DNA as an early marker of therapeutic response in patients with metastatic colorectal cancer. Ann Oncol 2015;26:1715–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. McShane LM, Altman DG, Sauerbrei W, et al. Reporting recommendations for tumor marker prognostic studies (REMARK). JNCI J Natl Cancer Inst 2005;97:1180–4. [DOI] [PubMed] [Google Scholar]
- 15. von Elm E, Altman DG, Egger M, et al. The Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) statement: guidelines for reporting observational studies. Lancet 2007;370:1453–7. [DOI] [PubMed] [Google Scholar]
- 16. Schwartz LH, Litière S, de Vries E, et al. RECIST 1.1—update and clarification: from the RECIST committee. Eur J Cancer 2016;62:132–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parpart‐Li S, Bartlett B, Popoli M, et al. The effect of preservative and temperature on the analysis of circulating tumor DNA. Clin Cancer Res 2017;23:2471–7. [DOI] [PubMed] [Google Scholar]
- 18. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature 2016;531:47–52. [DOI] [PubMed] [Google Scholar]
- 19. Witkiewicz AK, McMillan EA, Balaji U, et al. Whole‐exome sequencing of pancreatic cancer defines genetic diversity and therapeutic targets. Nat Commun 2015;6:6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tate JG, Bamford S, Jubb HC, et al. COSMIC: the catalogue of somatic mutations in cancer. Nucleic Acids Res 2019;47(D1):D941–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Raphael BJ, Hruban RH, Aguirre AJ, et al. Integrated genomic characterization of pancreatic ductal adenocarcinoma. Cancer Cell 2017;32:185–203.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bland JM, Altman DG. Statistical methods for assessing agreement between two methods of clinical measurement. Lancet 1986;1:307–10. [PubMed] [Google Scholar]
- 23. He Y. Missing data analysis using multiple imputation: getting to the heart of the matter. Circ Cardiovasc Qual Outcomes 2010;3:98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tie J, Wang Y, Tomasetti C, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med 2016;8:346ra92–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Perets R, Greenberg O, Shentzer T, et al. Mutant KRAS circulating tumor DNA is an accurate tool for pancreatic cancer monitoring. Oncologist 2018;23:566–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hadano N, Murakami Y, Uemura K, et al. Prognostic value of circulating tumour DNA in patients undergoing curative resection for pancreatic cancer. Br J Cancer 2016;115:59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bettegowda C, Sausen M, Leary RJ, et al. Detection of circulating tumor DNA in early‐ and late‐stage human malignancies. Sci Transl Med 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bachet JB, Bouché O, Taieb J, et al. RAS mutation analysis in circulating tumor DNA from patients with metastatic colorectal cancer: the AGEO RASANC prospective multicenter study. Ann Oncol 2018;29:1211–9. [DOI] [PubMed] [Google Scholar]
- 29. Sausen M, Phallen J, Adleff V, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun 2015;6:7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheng H, Liu C, Jiang J, et al. Analysis of ctDNA to predict prognosis and monitor treatment responses in metastatic pancreatic cancer patients. Int J Cancer 2017;140:2344–50. [DOI] [PubMed] [Google Scholar]
- 31. Schøler LV, Reinert T, Ørntoft M‐BW, et al. Clinical implications of monitoring circulating tumor DNA in patients with colorectal cancer. Clin Cancer Res 2017;23:5437–45. [DOI] [PubMed] [Google Scholar]
- 32. Parkinson CA, Gale D, Piskorz AM, et al. Exploratory analysis of TP53 mutations in circulating tumour DNA as biomarkers of treatment response for patients with relapsed high‐grade serous ovarian carcinoma: a retrospective study. PLoS Med 2016;13:e1002198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nygaard AD, Holdgaard PC, Spindler K‐LG, et al. The correlation between cell‐free DNA and tumour burden was estimated by PET/CT in patients with advanced NSCLC. Br J Cancer 2014;110:363–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Chen H, Tu H, Meng ZQ, et al. K‐ras mutational status predicts poor prognosis in unresectable pancreatic cancer. Eur J Surg Oncol 2010;36:657–62. [DOI] [PubMed] [Google Scholar]
- 35. Kinugasa H, Nouso K, Miyahara K, et al. Detection of K‐ras gene mutation by liquid biopsy in patients with pancreatic cancer. Cancer 2015;121:2271–80. [DOI] [PubMed] [Google Scholar]
- 36. Nordgård O, Tjensvoll K, Gilje B, et al. Circulating tumour cells and DNA as liquid biopsies in gastrointestinal cancer. Br J Surg 2018;105:e110–20. [DOI] [PubMed] [Google Scholar]
- 37. Lin M, Alnaggar M, Liang S, et al. Circulating tumor DNA as a sensitive marker in patients undergoing irreversible electroporation for pancreatic cancer. Cell Physiol Biochem 2018;47:1556–64. [DOI] [PubMed] [Google Scholar]
- 38. Waddell N, Pajic M, Patch A‐M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Biankin AV, Waddell N, Kassahn KS, et al. Pancreatic cancer genomes reveal aberrations in axon guidance pathway genes. Nature 2012;491:399–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Demuth C, Spindler KLG, Johansen JS, et al. Measuring KRAS mutations in circulating tumor DNA by droplet digital PCR and next‐generation sequencing. Transl Oncol 2018;11:1220–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sato KA, Hachiya T, Iwaya T, et al. Individualized mutation detection in circulating tumor DNA for monitoring colorectal tumor burden using a cancer‐associated gene sequencing panel. PLoS One 2016;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Garcia J, Forestier J, Dusserre E, et al. Cross‐platform comparison for the detection of RAS mutations in cfDNA (ddPCR biorad detection assay, BEAMing assay, and NGS strategy). Oncotarget 2018;9:21122–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ferrone CR, Marchegiani G, Hong TS, et al. Radiological and surgical implications of neoadjuvant treatment with FOLFIRINOX for locally advanced and borderline resectable pancreatic cancer. Ann Surg 2015;261:12–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kou R, Lam H, Duan H, et al. Benefits and challenges with applying unique molecular identifiers in next generation sequencing to detect low frequency mutations. PLoS One 2016;11:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Mouliere F, Chandrananda D, Piskorz AM, et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci Transl Med 2018;4921:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stasik S, Schuster C, Ortlepp C, et al. An optimized targeted next‐generation sequencing approach for sensitive detection of single nucleotide variants. Biomol Detect Quantif 2018;15:6–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Milbury CA, Zhong Q, Lin J, et al. Determining lower limits of detection of digital PCR assays for cancer‐related gene mutations. Biomol Detect Quantif 2014;1:8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Cohen JD, Li L, Wang Y, et al. Detection and localization of surgically resectable cancers with a multi‐analyte blood test. Science 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supplementary Information
Fig. S1 Comparison of NGS and ddPCR results in detection of circulating tumor DNA (n = 77).
A: Plot showing the correlation between the fractional abundance (FA) of KRAS exon 2 mutations in ddPCR and the variant allele frequency (VAF) of KRAS exon 2 in NGS.
B: Bland–Altman plot assessing agreement between ctDNA quantity measured using NGS (VAF) and ddPCR (FA). The mean difference of −0.56 (95% CI ‐1.47‐0.31) was small, with most measurements within the limits of agreement (P = 0.217; non‐significance indicating good agreement).
Fig. S2 Illustrations of 3D tumor volume measurements.
A: Semi‐automated measurement (MM oncology workflow)
B: Manual measurement (MM reading workflow)
C: 3D‐reconstruction showing several semi‐automated measurements
Table S1 Results tissue comparison.
Corresponding tumor material was available in 17 patients (eight cytology and nine histology). Unfortunately, sequencing of cytology material failed in seven of the eight patients due to low DNA yield). Most of the tissue samples were derived from patients who underwent exploration. Nine of the 10 successfully sequenced tissue samples showed a mutation in KRAS exon 2. Five samples showed an additional mutation in TP53 and/or SMAD4 and/or CDKN2A. Using NGS, in only one of these 10 patients (KRAS wildtype) we were able to detect a mutation in cfDNA; this BRAF V600E mutation was confirmed in tissue. Using ddPCR we detected a KRAS mutation in cfDNA of one other patient.
Table S2 NGS results follow‐up samples.
Table S3 Amplicons in the custom‐made pancreatobiliary NGS panel.
For each amplicon the start and end position are presented in reference genome hg 19. Mean amplicon length was 87 base pair (range 67–137).
Data Availability Statement
The data that support the findings of our study are available from the corresponding author upon reasonable request.