

Abstract
The nuclear farnesoid X receptor (FXR) and the enzyme soluble epoxide hydrolase (sEH) are validated molecular targets to treat metabolic disorders such as non‐alcoholic steatohepatitis (NASH). Their simultaneous modulation in vivo has demonstrated a triad of anti‐NASH effects and thus may generate synergistic efficacy. Here we report dual FXR activators/sEH inhibitors derived from the anti‐asthma drug Zafirlukast. Systematic structural optimization of the scaffold has produced favorable dual potency on FXR and sEH while depleting the original cysteinyl leukotriene receptor antagonism of the lead drug. The resulting polypharmacological activity profile holds promise in the treatment of liver‐related metabolic diseases.
Keywords: Polypharmacology, non-alcoholic steatohepatitis, NASH, NAFLD, nuclear receptor
Changing targets: The anti‐asthma drug Zafirlukast was used as a lead to develop dual farnesoid X receptor activators/soluble epoxide hydrolase inhibitors. Systematic structural modification produced the desired polypharmacological profile and depleted the drug's original on‐target activity while conserving the favorable molecular scaffold. Optimized compounds engaged the desired targets in cellular settings. The designed multi‐target profile may generate synergies in treating liver‐related metabolic diseases.
Introduction
The nuclear farnesoid X receptor (FXR) is a key metabolic regulator governing the homeostasis of bile acids, lipids and glucose.1, 2 It has a profound protective role in liver and has been identified as a valuable target for the treatment of metabolism‐associated liver diseases, particularly non‐alcoholic fatty liver (NAFL) and non‐alcoholic steatohepatitis (NASH).3, 4, 5 This hepatic manifestation of the metabolic syndrome is considered as global health burden with alarming prevalence but there is no pharmacological treatment option to date.6, 7 The FXR agonist obeticholic acid (1)8 (Scheme 1) has shown great promise in NASH treatment in several clinical trials5, 9 and has validated FXR as a valuable drug target.
Scheme 1.
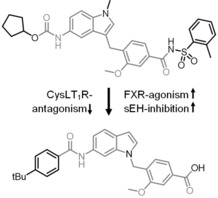
Clinically relevant FXR agonist obeticholic acid (1), dual FXR agonist/sEH inhibitor (FXRA/sEHi) 2 and lead compound Zafirlukast (3).
Soluble epoxide hydrolase (sEH) as a downstream enzyme in the CYP‐pathway of the arachidonic acid cascade converts epoxyeicosatrienoic acids (EETs) to the corresponding dihydroxyeicosatrienoic acids (DHETs).10 Since EETs are anti‐inflammatory lipid mediators, sEH inhibition constitutes an anti‐inflammatory strategy that was repeatedly shown to have considerable potential in NASH treatment.11, 12 FXR activation in NASH is mainly effective through metabolic effects and anti‐steatotic activity,5 sEH inhibition has anti‐inflammatory and anti‐fibrotic effects in liver.13 Thus, combination of FXR activation and sEH inhibition may produce additive therapeutic efficacy in NASH. Additionally, a recent report14 links FXR activation with induction of cytochrome P450 epoxygenases. This in turn increases sEH substrate concentrations which further illustrates potential synergies of FXR and sEH.
We have recently reported the development and characterization of the first‐in‐class dual FXR agonist/sEH inhibitor (FXRA/sEHi) 2.15 With its designed polypharmacological profile, this innovative multi‐target agent16 showed robust therapeutic efficacy in animal models of toxin‐ and diet‐induced NASH by simultaneously exhibiting anti‐steatotic, anti‐fibrotic and anti‐inflammatory activity.17 The dual mode of action was superior to pure FXR activation in these in vivo studies confirming the value of the multi‐target approach.
In designing a triple modulator of soluble epoxide hydrolase (sEH), peroxisome proliferator‐activated receptor γ (PPARγ) and cysteinyl leukotriene receptor 1 (CysLT1R) from zafirlukast (3),18 we also observed weak agonistic potency of 3 in a FXR‐Gal4 hybrid assay (4.8‐fold activation at 10 μM).18 Full dose‐response characterization of 3 in a more physiological full length FXR‐BSEP reporter gene assay confirmed this activity. Zafirlukast (3) possessed an EC50 value of 3.9 μM and 28 % relative activation compared to GW4064 (at 3 μM). This combined FXR agonism and sEH inhibition (IC50=2.0 μM) appeared as promising activity profile for the development of a new class of dual FXR/sEH modulators and prompted us to study the structure‐activity relationship (SAR) of 3 on both molecular targets. Based on the strong therapeutic efficacy of the first‐in‐class FXRA/sEHi 2 in rodent models of NASH,15, 17 we aimed to design a balanced activity profile of partial FXR agonism and sEH inhibition. In addition, the original antagonism of 3 on CysLT1R had to be diminished according to the concept of selective optimization of side‐activities.19 We succeeded in specifically optimizing 3 to the dual FXR/sEH modulators 13 and 16 which simultaneously display reduced activity on CysLT1R. These novel FXRA/sEHi valuably expand the collection of multi‐target agents for NASH and related diseases.
Results and Discussion
Synthesis
Zafirlukast descendants 4–16 were synthesized according to Schemes 2–6 in five‐ to ten‐step procedures that were partially adopted from published18, 20, 21, 22 approaches to 3 with suitable modifications. Analogues 4–6 and 9 were prepared in a six‐step procedure from 5‐nitroindole (17) which was N‐methylated with DMS to generate 18. Buildings blocks 19 and 20 were prepared by bromination of 21 and 22 under radical conditions. 18 was then coupled with 19 or 20 to 23 or 24 in a Friedel‐Crafts reaction using FeCl3 as catalyst. Reduction of 23 and 24 with H2 and Pd(C) afforded anilines 25 and 26. Urethane 27 was then available by reacting 25 with cyclopentyl chloroformate (28). Reaction of 25 with 4‐tert‐butylbenzoyl chloride (29) yielded ester 30, while 26 was coupled with 4‐tert‐butylbenzoic acid (31) using EDC*HCl and 4‐DMAP to form ester 32. Alkaline hydrolysis of 27, 30 and 32 then yielded 4, 5 and 9 (Scheme 2).18, 20 6 and 7 were prepared accordingly using isonicotinic acid (33) or GW4064‐derived building block 34 as N‐substituents. The required isoxazolecarboxylate 34 was prepared according to the published procedure21 from 2,6‐dichlorobenzaldehyde (35) which was transformed to aldoxime 36 using hydroxylamine hydrochloride followed by chlorination to 37 with NCS.
Scheme 2.
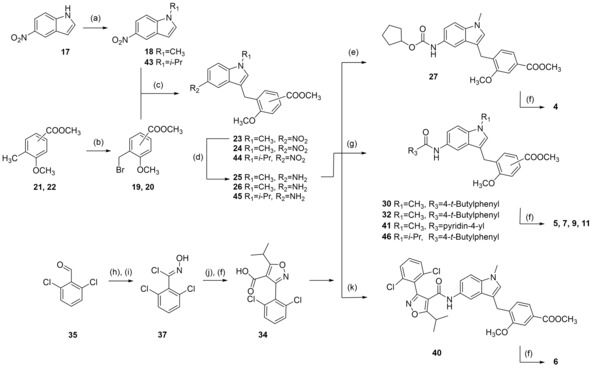
Reagents & conditions: (a) DMS or diisopropyl sulfate (42), dioxane, 40 °C, 2.5 h; (b) NBS, AIBN, CHCl3, reflux, 4 h; (c) FeCl3, dioxane, rt, 12 h; (d) H2, Pd(C), MeOH, rt, 12 h; (e) C5H9‐OCOCl (28), DIPEA, 4 °C to rt, 4 h; (f) LiOH, MeOH/H2O, rt, 16 h; (g) 4‐tert‐butylbenzoyl chloride (29), pyridine, DMF, CHCl3, reflux; or 4‐tert‐butylbenzoic acid (31) or isonicotinic acid (33), EDC*HCl, 4‐DMAP, CHCl3, reflux, 6 h; (h) H2N−OH*HCl, NaOH, EtOH, 90 °C, 24 h; (i) NCS, DMF, rt, 5 h; (j) methyl isobutyrylacetate (38), NaOMe, THF, rt, 16 h; (k) 34, EDC*HCl, 4‐DMAP, CHCl3, reflux, 6 h.
Scheme 6.
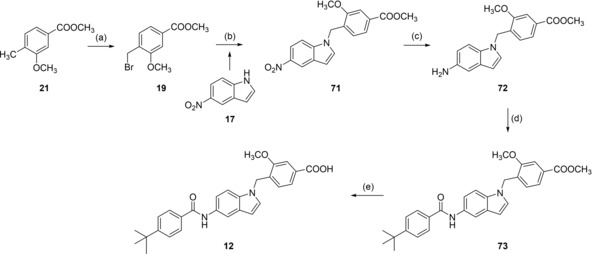
Reagents & conditions: (a) NBS, AIBN, CHCl3, reflux, 4 h; (b) K2CO3, DMF, reflux, 12 h; (c) H2, Pd(C), MeOH, rt, 12 h; (d) 4‐tert‐butylbenzoyl chloride (29), pyridine, DMF, CHCl3, reflux, 6 h; (e) LiOH, MeOH/H2O, rt, 16 h.
37 was cyclized with methyl isobutyrylacetate (38) to isoxazole 39 and alkaline hydrolysis of 39 yielded 34. Coupling of 25 with 33 or 34 using EDC*HCl and 4‐DMAP to esters 40 and 41 followed by alkaline hydrolysis yielded 6 and 7 (Scheme 2). For the synthesis of 11, 5‐nitroindole (17) was N‐alkylated with diisopropylsulfate (42) to generate 43 which was then transformed to 11 in four further steps according to the synthesis of 5.
8 and 10 were prepared from N‐methyl‐5‐nitroindole (18) which was coupled with methyl 4‐formylbenzoate (47) or methyl 3‐formylbenzoate (48) in presence of triethylsilane and trifluoroacetic acid22 to 49 and 50. 49 and 50 were then reduced with H2 and Pd(C) to anilines 51 and 52, and subsequently reacted with 4‐tert‐butylbenzoyl chloride (29) to generate 53 or 54 whose alkaline hydrolysis yielded 8 and 10 (Scheme 3).
Scheme 3.
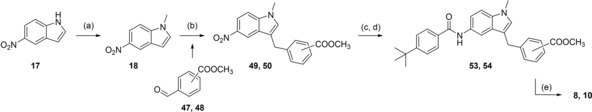
Reagents & conditions: (a) DMS, dioxane, 40 °C, 2.5 h; (b) methyl 4‐formylbenzoate (47) or methyl 3‐formylbenzoate (48), triethylsilane, trifluoroacetic acid, CH2Cl2, 0 °C (10 min) ‐> rt (12 h); (c) H2, Pd(C), MeOH, rt, 12 h; (d) 4‐tert‐butylbenzoyl chloride (29), pyridine, DMF, CHCl3, reflux, 6 h; (e) LiOH, MeOH/H2O, rt, 16 h.
Analogues 13, 15 and 16 with inverted indole orientation were prepared in a five‐step procedure according to Scheme 4. 6‐Nitroindole (55) was N‐alkylated with 19, 56 or 57 to generate 58, 59 and 60. After reduction with H2 and Pd(C) to anilines 61–63, coupling with 4‐tert‐butylbenzoic acid (31) in presence of EDC*HCl and 4‐DMAP formed 64–66 and alkaline hydrolysis yielded 13, 15 and 16 (Scheme 4).
Scheme 4.
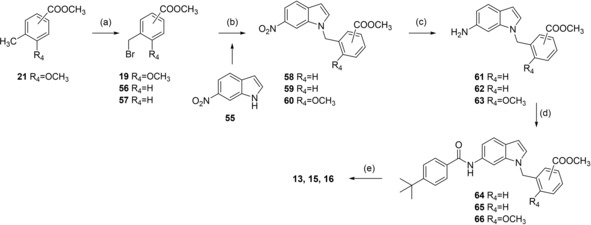
Reagents & conditions: (a) NBS, AIBN, CHCl3, reflux, 4 h; (b) K2CO3, DMF, reflux, 12 h; (c) H2, Pd(C), MeOH, rt, 12 h; (d) 4‐tert‐butylbenzoic acid (31), EDC*HCl, 4‐DMAP, CHCl3, reflux, 6 h; (e) LiOH, MeOH/H2O, rt, 16 h.
Benzimidazole analogue 14 was prepared from 6‐nitro‐1H‐benzimidazole (67) by N‐alkylation with 19 to 68 followed by reduction with H2 and Pd(C) to aniline 69, reaction with 4‐tert‐butylbenzoyl chloride (29) to 70 and alkaline hydrolysis to 14 (Scheme 5).
Scheme 5.
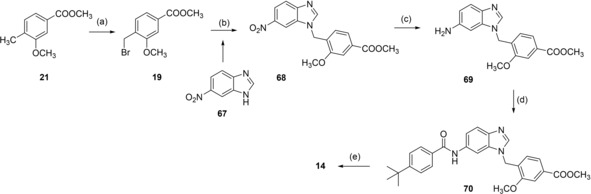
Reagents & conditions: (a) NBS, AIBN, CHCl3, reflux, 4 h; (b) K2CO3, DMF, reflux, 12 h; (c) H2, Pd(C), MeOH, rt, 12 h; (d) 4‐tert‐butylbenzoyl chloride (29), pyridine, DMF, CHCl3, reflux, 6 h; (e) LiOH, MeOH/H2O, rt, 16 h.
Preparation of 12 proceeded in a similar fashion as analogues 13, 15 and 16 starting with N‐alkylation of 5‐nitroindole (17) with 19 to 71, followed by reduction of 71 with H2 and Pd(C) to aniline 72. 72 was then reacted with 4‐tert‐butylbenzoyl chloride (29) to 73 and saponification of 73 yielded 12 (Scheme 6).
Biological Evaluation
4‐16 were characterized in a full‐length FXR reporter gene assay that relies on a firefly luciferase reporter under the control of the FXR response element from the promoter region of the bile salt export protein (BSEP) gene.25, 26 In this assay, HeLa cells are transiently transfected with the reporter construct, constitutive expression plasmids coding for human full‐length FXR and its heterodimer partner retinoid X receptor α (RXRα) as well as a constitutively expressed renilla luciferase as internal control for transfection efficiency and test compound toxicity. Inhibitory potency of 4–16 was determined in a sEH activity assay27, 28 using recombinant human enzyme and (3‐phenyloxiranyl)acetic acid cyano‐(6‐methoxynaphthalen‐2‐yl)methyl ester (PHOME) as fluorogenic substrate. CysLT1R antagonism was determined in a cell‐based Ca2+‐flux assay29 in competition with 0.1 nM leukotriene D4.
Structure‐Activity Relationship Analysis
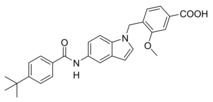
As first step in optimizing 3 towards dual FXR/sEH modulators (Table 1), we evaluated the effect of replacing the N‐acyl sulfonamide moiety by a carboxylic acid (4). According to SAR studies on 3 as CysLT1R antagonist, this structural change is accompanied by a remarkable, 500‐fold loss in CysLT1R antagonistic potency (Ki 0.3 nM vs. 157 nM)20 and, for the development of dual FXR/sEH modulators according to the SOSA19, 30 concept, reduction in activity on CysLT1R was desired. On sEH, 4 gained in inhibitory potency while the structural modification was not favored by FXR.
Table 1.
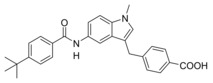
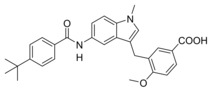
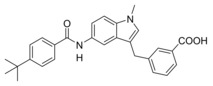
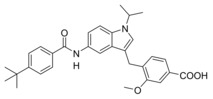
In vitro activity of Zafirlukast (3) and derivatives 4–16 in a full‐length FXR‐BSEP reporter gene assay[a] and an sEH activity assay on recombinant protein.[b]
|
ID |
Structure |
FXR EC50 (max. rel. act.)[a] |
sEH IC50 |
|---|---|---|---|
|
3 |
|
3.9 μM (28±1 %) |
2.0±0.3 μM |
|
4 |
|
<10 % act. (30 μM)[c] |
0.30±0.02 μM |
|
5 |
|
0.6±0.2 μM (29±1 %) |
>30 μM |
|
6 |
|
inactive |
3.3±0.6 μM |
|
7 |
|
0.91±0.06 μM (15±1 %) |
>100 μM |
|
8 |
|
>10 μM[c] |
12±1 μM |
|
9 |
|
inactive |
86±10 μM |
|
10 |
|
1.19±0.08 μM (18±1 %) |
22±3 μM |
|
11 |
|
2.3±0.1 μM (11±1 %) |
>100 μM |
|
12 |
|
9.2±0.8 μM (23±1 %) |
>100 μM |
|
13 |
|
0.26±0.05 μM (15±1 %) |
1.7±0.1 μM |
|
14 |
|
5.3±0.4 μM (26±1 %) |
18±5 μM |
|
15 |
|
inactive |
4.3±0.4 μM |
|
16 |
|
3.5±0.3 μM (24±1 %) |
2.9±0.1 μM |
[a] Maximum relative activation on FXR refers to the activity of GW4064 at 3 μM defined as 100 %. Inactive ‐ no significant activation up to 30 μM. Results are the mean±S.E.M.; n≥3. [b] Results are the mean±S.E.M.; n≥3. [c] Compounds 4 and 8 caused statistically significant FXR activation, but no dose‐response curve was recorded when activation efficacy was <10 % or EC50>10 μM.
Molecular docking of 4 (Figure 1) suggested a favorable binding mode to the sEH active site (PDB‐ID: 3OTQ23) with the urethane engaging polar contact with the catalytic Asp335 residue which is typical for sEH inhibitors. Additionally, the urethane carbonyl oxygen was bound to Tyr383 and Tyr466 and the carboxylate formed a hydrogen bond to Ser312. The cyclopentyl substituent though bound in a lipophilic environment seemed to offer optimization potential by enabling a stacking interaction with Trp366 with an aromatic replacement.
Figure 1.
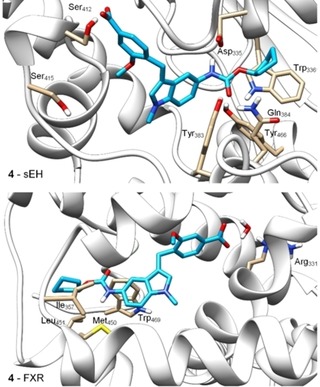
Molecular docking of 4 in the sEH active site (PDB‐ID: 3OTQ23) and the ligand binding site of FXR (PDB‐ID: 4QE824) particularly suggested potential for structural optimization in the terminal cyclopentylurethane moiety. Docking was performed in MOE and visualized using UCSF Chimera.
In the FXR ligand binding site (PDB‐ID: 4QE824), the carboxylate moiety of 4 formed a water‐mediated hydrogen bond to Arg331 as exclusive polar contact while the urethane structure appeared not favored in its lipophilic environment close to Ile357, Met450, Leu451, and Trp469 further suggesting that this terminal residue might hold potential to enhance activity on FXR. Thus, to compensate the loss in activity on FXR resulting from the modification of 3 to 4, we aimed to introduce known FXR ligand moieties replacing the cyclopentylurethane in 4.
A 4‐tert‐butylbenzamide (5) known from various FXR partial agonist series15, 31, 32 indeed strongly improved FXR agonistic potency but caused a loss in sEH inhibitory activity which is likely due to a weakening of the polar interaction with Asp335 when the urethane is replaced by an amide. The isoxazole “hammerhead” structure (6) derived from GW4064 and analogues,33, 34 in contrast, further diminished activity on FXR while it was tolerated by sEH. The inactivity of 6 on FXR may be due to the rigid amide linker between isoxazole and indole but this moiety could not be replaced since the amide is the key pharmacophore element for sEH inhibition.15, 35, 36 To mimic the isoxazole which forms key contacts with the FXR ligand binding site (His447 and Trp469 of FXR)2, 37 with a sterically less demanding scaffold, we replaced the “hammerhead” structure with an isonicotinic amide (7) which indeed regained activity on FXR but was inactive on sEH. Thus, the 4‐tert‐butylbenzamide in 5 turned out as the most favored moiety to replace the urethane and was conserved for further structural optimization.
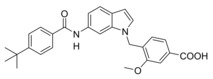
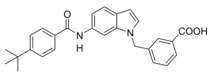
We then focused on the SAR of the methoxybenzoate in 6 and first removed the methoxy group (8) which promoted inhibitory potency on sEH but was not tolerated by FXR. Shifting the carboxylate residue from 4‐position (6) to the 3‐position (9) diminished activity on both targets. However, combining removal of the methoxy group and the 3‐benzoate geometry resulted in the first dual modulator 10 with low micromolar activity on FXR and sEH.
Next, we studied the SAR of the central indole scaffold of 6. Enlargement of the N‐methyl substituent to an isopropyl moiety (11) was not favored by either target. The same held true when the substitution geometry was modified by linking central indole and methoxybenzoate via the indole nitrogen (12). However, inverting the central indole while retaining the overall geometry in 13 strongly improved inhibitory potency on sEH and was also favored by FXR.
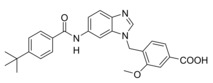
Compared to 3, 13 comprises a 15‐fold improved EC50 value on FXR and equal activity on sEH but is remarkably less active on CysLT1R (IC50: <0.001 μM29 (3) vs. ∼1 μM (13, as estimated from the compound′s activity at 1 μM) Figure 2a). When we replaced the indole scaffold of 13 by a benzimidazole (14), activity on both targets was diminished by approx. 10‐fold.
Figure 2.
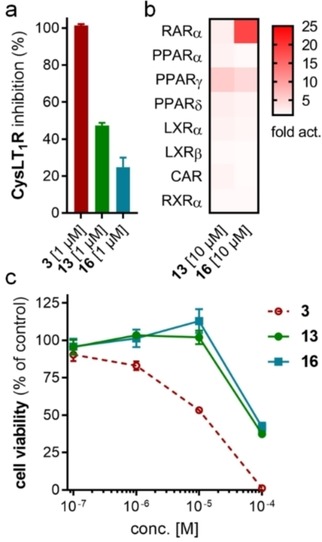
In vitro profiling of Zafirlukast derived FXRA/sEHi 13 and 16. (a) Both 13 and 16 are markedly less active on Zafirlukast's (3) molecular target CysLT1R (IC50: <0.001 μM29 (3), ∼1 μM (13), >1 μM (16) as estimated from their activity at 1 μM). Results are the mean±SD, n=2. (b) Apart from considerable RARα agonism of 16, the FXRA/sEHi 13 and 16 are selective over nuclear receptors related to FXR. Results are the mean, n=2. (c) Compared to 3, 13 and 16 comprise reduced cytotoxicity (WST‐1 assay in HepG2 cells). Results are the mean±S.E.M., n=4.
In an attempt to combine the favored structural variations on the indole and the benzoate moieties, we merged the modifications of 10 and 13 in 3‐benzoic acid derivative 15 which retained activity on sEH but was inactive on FXR. However, conserving the original 4‐benzoate geometry resulted in the well‐balanced dual modulator 16, which was even less active on the lead compound's original receptor target CysLT1R (IC50: <0.001 μM29 (3) vs. >1 μM (16, as estimated from the compound′s activity at 1 μM) Figure 2a).
Selectivity profiling of the most favorable Zafirlukast derived FXRA/sEHi 13 and 16 demonstrated high selectivity over nuclear receptors related to FXR apart from marked RARα activation by 16 (Figure 2b). Moreover, the optimized descendants 13 and 16 comprised strongly reduced toxicity compared to 3 (Figure 2c). While 3 caused dose‐dependent anti‐proliferative activity at concentrations of 1 μM and above, 13 and 16 had no toxic effect up to 10 μM.
13 and 16 were then assessed for FXR activation and sEH inhibition in a more physiological cellular setting in hepatocytes to probe target engagement in cells. Both compounds blocked EET conversion to DHETs in HepG2 cells demonstrating cellular sEH inhibition (Figure 4a). Moreover, 13 and 16 induced expression of FXR regulated genes organic solute transporter α (OSTα) and small heterodimer‐partner (SHP) while the expression of the indirectly FXR regulated cholesterol 7α hydroxylase (CYP7 A1) was reduced upon treatment with 13 or 16 (Figure 4b). Therein, both dual modulators behaved as partial FXR agonists and only caused a favorably modest effect on gene expression compared to the endogenous FXR activator CDCA.
Figure 4.
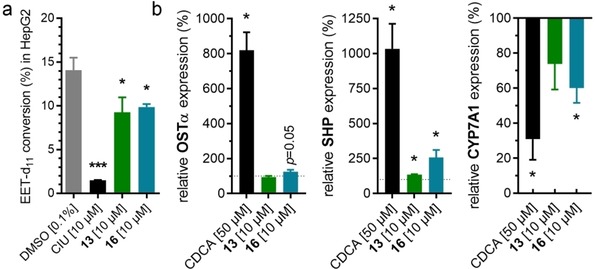
Biological activity of 13 and 16 in hepatocytes. (a) 13 and 16 blocked conversion of Deuterium‐labelled (±)‐14(15)‐EET to the corresponding diol in HepG2 homogenates demonstrating inhibition of sEH. sEH inhibitor CIU for comparison. Results are the mean±S.E.M.; n≥4. (b) 13 and 16 induced expression of FXR regulated genes organic solute transporter α (OSTα) and small heterodimer partner (SHP) while expression of the indirectly FXR regulated cholesterol 7α hydroxylase (CYP7 A1) was decreased. Results are the mean±S.E.M.; n=3. *p<0.05; ***p<0.001.
To computationally assess the interaction of the optimized dual modulators 13 and 16 with FXR and sEH, we predicted their binding modes to both proteins by molecular docking (Figure 4). The co‐crystal 3OTQ23 served as structural template for sEH, while binding to FXR was studied based on the X‐ray data of the FXR ligand binding domain in complex with a partial agonist (4QE824) owing to the partial agonistic profile of 13 and 16.
Figure 3.
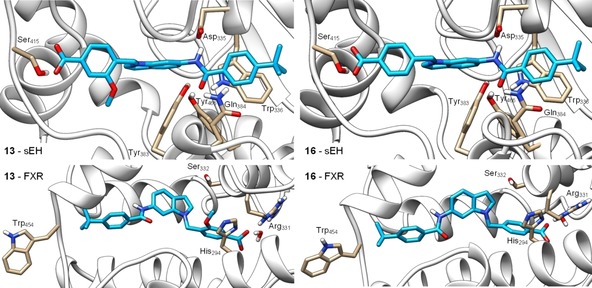
Molecular docking of 13 (left) and 16 (right) in the sEH active site (PDB‐ID: 3OTQ,23 upper images) and the ligand binding site of FXR in partially active conformation (PDB‐ID: 4QE8,24 lower images). 13 and 16 form similar predicted binding modes in the sEH active site with the amide nitrogen participating in the typical hydrogen bond to the catalytic Asp335 residue and the amide oxygen interacting with Tyr383 and Tyr466. In addition, the benzamide aromatic ring makes a stacking interaction with Trp336 and the carboxylate moiety engages a hydrogen bond with Ser415. In the FXR ligand binding site, 13 and 16 interact with Arg331 in water mediated hydrogen bonds as exclusive polar contact. The tert‐butylbenzamide moieties occupy the region of Trp454 which consequently obtains an outward conformation resulting in partial FXR activation.24 The methoxy group of 13 occupies a polar region between His294 and Ser332 which agrees with the higher potency of 13 on FXR. Docking was performed in MOE and visualized using UCSF Chimera.
Within the sEH active site, 13 and 16 formed similar favorable binding modes. As typical for sEH inhibitors, the amide nitrogen engaged a hydrogen bond with the catalytic Asp335 and the amide oxygen made polar contacts with Tyr383 and Tyr466. In addition, the carboxylic acid residues of 13 and 16 formed a hydrogen bond with Ser415 and the terminal benzamide aromatic ring made a stacking interaction with Trp336. No specific interactions were observed for the methoxy substituent of 13 which agrees with the almost identical sEH inhibitory potency of 13 and 16.
In the FXR ligand binding site, the predicted binding modes of 13 and 16 revealed water mediated hydrogen bonding of the carboxylate residues with Arg331 as exclusive polar contact. As observed for other partial agonists,24 the tert‐butylbenzamide moieties of 13 and 16 occupied the terminal region of the binding site where Trp454 is located leading to an outward conformation of this residue which results in partial FXR activation. The methoxy substituent of 13 favorably occupied a polar region of the binding site between His294 and Ser332 which agrees with the approx. 10‐fold higher potency of 13 on FXR compared to 16.
Conclusions
The metabolic syndrome with its multiple manifestations including insulin resistance, dyslipidemia and NAFLD is a major global health burden with continuously increasing prevalence.38 Despite a variety of approved drugs for several metabolic diseases, the therapy of this disease complex is not satisfying. For its multifactorial nature, multimodal treatment may be required to achieve improved clinical efficacy. In NASH, FXR activation demonstrated favorable anti‐steatotic effects and countered fibrosis5, 9 while sEH inhibition produced strong anti‐inflammatory activity in liver accompanied by anti‐steatotic and anti‐fibrotic action.11, 12 The combination of these two unrelated mechanisms has the potential to generate synergistic efficacy for NASH treatment.15, 17 Based on these considerations we have employed the CysLT1R antagonist Zafirlukast comprising side activities on FXR and sEH as lead compound and systematically optimized the scaffold to dual FXR activators/sEH inhibitors with markedly reduced activity on CysLT1R. The resulting Zafirlukast‐derived FXRA/sEHi 13 and 16 inhibited sEH and activated FXR in hepatocytes (HepG2) demonstrating target engagement in a native cellular setting. Thus, the designed multi‐target compounds are suitable for further evaluation as innovative agents to counter NASH and related metabolic diseases and may open new avenues to polypharmacological agents as superior future treatment options for the metabolic syndrome.
Experimental Section
Chemistry
General: All chemicals and solvents were of reagent grade and used without further purification unless otherwise specified. All reactions were conducted in oven‐dried glassware under argon atmosphere and in absolute solvents. NMR spectra were recorded on a Bruker AV 500, Bruker AV 300 or a Bruker am250xp spectrometer (Bruker Corporation, Billerica, MA, USA). Chemical shifts (δ) are reported in ppm relative to tetramethylsilane (TMS) as reference. Multiplicity is reported: s, singlet; d, doublet; dd, doublet of doublets; t, triplet; m, multiplet. Approximate coupling constants (J) are given in hertz (Hz). Mass spectra were obtained on a VG Platform II (Thermo Fischer Scientific, Inc., Waltham, MA, USA) using electrospray ionization (ESI). High‐resolution mass spectra were recorded on a MALDI LTQ ORBITRAP XL instrument (Thermo Fisher Scientific). Compounds were purified by preparative HPLC using a Shimadzu preparative LC‐20 A Prominence (Shimadzu, Kyoto, Japan) with the following conditions: column, Luna (10 μ C18(2) 100 Å; 250×21.2 mm; Phenomenex, Torrance, CA, U.S.A.); mobile phase, isocratic 50 : 50 acetonitrile/H2O+0.1 formic acid for 30 min at a flow rate of 21 mL/min and UV‐detection at 245 nm and 280 nm. Compound purity was analyzed on a Waters 600 Controller HPLC (Waters, Milford, MA, U.S.A.) using a Waters 2487 Dual Absorbance Detector and a Waters 717 plus Autosampler or a VWR Chromaster (VWR, Radnor, PA, U.S.A.) with a 5160 pump system, using a DAD 5430 and 5260 Autosampler both equipped with a MultoHigh100 RP18‐5 μ 250×4 mm column (CS‐Chromatographie Service GmbH, Langerwehe, Germany) using a gradient (H2O+0.1 % formic acid/MeOH 80 : 20 isocratic for 5 min to MeOH after additional 45 min and MeOH for additional 10 min) at a flow rate of 1 mL/min or a gradient (H2O+0.1 % formic acid/MeOH 60 : 40 isocratic for 5 min to MeOH after additional 25 min and MeOH for additional 10 min) at a flow rate of 1 mL/min with UV‐detection at 245 nm and 280 nm. Compound 4 as well as its precursors 18, 19, 23 and 27 have been reported previously.18
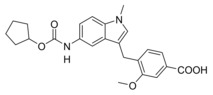
4‐((5‐(4‐(tert‐Butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoic acid (5): Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (30, 0.22 g, 0.45 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (15 mL), H2O (10 mL) and lithium hydroxide (33 mg, 1.4 mmol, 3.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 5 as colorless solid (0.13 g, 62 %). 1H NMR (250 MHz, DMSO) δ=12.79 (s, 1H), 9.99 (s, 1H), 7.89 (d, J=8.6, 3H), 7.56–7.41 (m, 5H), 7.35 (d, J=8.8, 1H), 7.13 (d, J=7.8, 1H), 7.08 (s, 1H), 4.00 (s, 2H), 3.92 (s, 3H), 3.73 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.98, 165.40, 162.36, 156.64, 154.29, 144.71, 139.89, 134.70, 133.70, 132.43, 129.36, 128.21, 127.49, 125.16, 121.74, 119.25, 115.74, 111.15, 102.04, 55.71, 42.94, 35.82, 34.70, 30.98. MS (ESI+): m/z 493.12 ([M+Na]+). HRMS (MALDI): m/z calculated for C29H30N2O4Na 493.20978, found 493.20917 [M+Na]+.
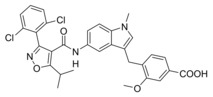
4‐((5‐(3‐(2,6‐Dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoic acid (6): Methyl 4‐((5‐(3‐(2,6‐dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (40, 10 mg, 0.017 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (10 mL), H2O (5 mL) and lithium hydroxide (24 mg, 0.10 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 6 as a yellow solid (1.2 mg, 10 %). 1H NMR (500 MHz, DMSO) δ=12.84 (s, 1H), 9.90 (s, 1H), 7.68 (s, 1H), 7.60 (d, J=7.6, 2H), 7.55–7.51 (m, 1H), 7.45 (d, J=1.3, 1H), 7.42 (dd, J=7.8, 1.4, 1H), 7.31 (d, J=8.8, 1H), 7.19 (d, J=8.4, 1H), 7.11 (d, J=7.8, 1H), 7.07 (s, 1H), 3.95 (s, 2H), 3.84 (s, 3H), 3.70 (s, 3H), 1.37 (d, J=6.7, 6H). 13C NMR (126 MHz, DMSO) δ=176.07, 167.29, 158.41, 157.95, 156.56, 134.64, 134.06, 132.20, 130.15, 129.78, 129.49, 128.70, 128.38, 127.18, 127.03, 121.57, 115.71, 113.36, 111.66, 110.70, 109.50, 100.31, 55.35, 32.37, 30.72, 27.00, 20.31. MS (ESI+): m/z 592.19 ([M+H]+). HRMS (MALDI): m/z calculated for C31H27Cl2N3O5 519.13223, found 519.13117 [M]*.
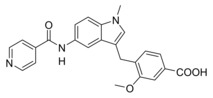
4‐((5‐(Isonicotinamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoic acid (7): Methyl 4‐((5‐(isonicotinamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (41, 0.14 g, 0.33 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (10 mL), H2O (5 mL) and lithium hydroxide (24 mg, 0.99 mmol, 3.0 eq) were added, and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 7 as a yellow solid (53 mg, 38 %). 1H NMR (500 MHz, DMSO) δ=8.51 (t, J=2.9 Hz, 1H), 8.34 (d, J=5.3 Hz, 2H), 8.07–7.87 (m, 5H), 7.81 (dd, J=9.6, 5.4 Hz, 1H), 7.66 (d, J=7.8 Hz, 1H), 7.53–7.49 (m, 1H), 4.55 (s, 2H), 4.44 (s, 3H), 4.25 (s, 3H). 13C NMR (126 MHz, DMSO) δ 177.31, 173.83, 167.75, 160.89, 153.33, 145.98, 145.30, 141.33, 140.14, 140.06, 139.22, 138.38, 132.45, 131.87, 126.38, 122.94, 121.49, 121.42, 119.80, 65.63, 42.49, 35.42. MS (ESI‐): m/z 414.17 ([M−H]−). HRMS (MALDI): m/z calculated for C24H22N3O4 416.16048, found 416.16027 [M+H]+.
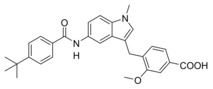
4‐((5‐(4‐(tert‐Butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoic acid (8): Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (53, 0.35 g, 0.77 mmol, 1.0 eq) was dissolved in EtOH (20 mL), H2O (10 mL) and lithium hydroxide (55 mg, 2.3 mmol, 3.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 8 as pale purple solid (0.19 g, 58 %). 1H NMR (500 MHz, DMSO) δ=10.01 (s, 1H), 7.89–7.84 (m, 5H), 7.52 (d, J=8.5, 2H), 7.48 (dd, J=8.8, 1.8, 1H), 7.38–7.35 (m, 3H), 7.15 (s, 1H), 4.08 (s, 2H), 3.74 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.31, 165.03, 154.00, 146.93, 133.98, 132.64, 131.08, 129.48, 129.44, 128.48, 127.40, 126.92, 125.07, 116.26, 112.16, 110.61, 109.44, 34.66, 32.40, 30.99. MS (ESI‐): m/z 439.23 ([M−H]−). HRMS (MALDI): m/z calculated for C28H29N2O3 441.21727, found 441.21604 [M+H]+.
3‐((5‐(4‐tert‐Butylbenzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐4‐methoxybenzoic acid (9): Methyl 3‐((5‐(4‐tert‐butylbenzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐4‐methoxybenzoate (32, 12 mg, 0.02 mmol, 1.0 eq) was dissolved in EtOH (20 mL), H2O (10 mL) and lithium hydroxide (2.4 mg, 0.1 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 9 as yellow solid (5 mg, 53 %). 1H NMR (500 MHz, DMSO) δ=10.00 (s, 1H), 7.93–7.87 (m, 3H), 7.82 (dd, J=8.6, 2.2, 1H), 7.64 (d, J=2.1, 1H), 7.52 (d, J=8.4, 2H), 7.47–7.43 (m, 1H), 7.36 (d, J=8.8, 1H), 7.13–7.06 (m, 2H), 3.98 (s, 2H), 3.74 (s, 3H), 3.73 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=168.04, 167.17, 159.19, 154.04, 133.32, 132.66, 131.73, 131.03, 130.32, 129.71, 129.26, 127.38, 126.80, 125.07, 121.48, 116.64, 112.12, 110.60, 109.40, 108.58, 55.86, 34.65, 32.39, 30.99, 25.11. MS (ESI+): m/z 493.25 ([M+Na]+). HRMS (MALDI): m/z calculated for C29H30N2O4 471.22783, found 471.22751 [M+H]+.
3‐((5‐(4‐(tert‐Butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoic acid (10): Methyl 3‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (54, 0.32 g, 0.70 mmol, 1.0 eq) was dissolved in EtOH (20 mL), H2O (10 mL) and lithium hydroxide (50 mg, 2.1 mmol, 3.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 10 as pale purple solid (0.26 g, 87 %). 1H NMR (500 MHz, DMSO) δ=10.01 (s, 1H), 7.89–7.86 (m, 3H), 7.81 (s, 1H), 7.74 (d, J=7.7, 1H), 7.55–7.47 (m, 4H), 7.42–7.35 (m, 2H), 7.15 (s, 1H), 4.08 (s, 2H), 3.75 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.44, 164.99, 153.98, 142.05, 133.98, 132.79, 132.63, 131.07, 129.07, 128.50, 127.39, 126.91, 126.85, 125.06, 116.26, 112.50, 110.57, 109.43, 34.65, 32.39, 30.98, 30.52. MS (ESI‐): m/z 439.22 ([M−H]−). HRMS (MALDI): m/z calculated for C28H29N2O3 441.21727, found 441.21588 [M+H]+.
4‐((5‐(4‐(tert‐Butyl)benzamido)‐1‐isopropyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoic acid (11): Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐isopropyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (46, 82 mg, 0.16 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (10 mL), H2O (5 mL) and lithium hydroxide (12 mg, 0.48 mmol, 3.0 eq) were added, and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 11 as a colorless solid (0.03 g, 38 %). 1H NMR (500 MHz, DMSO) δ=9.99 (s, 1H), 7.91–7.85 (m, 3H), 7.50 (dd, J=15.6, 4.9 Hz, 3H), 7.46–7.40 (m, 3H), 7.29 (s, 1H), 7.10 (d, J=7.8 Hz, 1H), 4.73–4.65 (m, 1H), 4.01 (s, 2H), 3.93 (s, 3H), 1.44 (d, J=6.6 Hz, 6H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.30, 164.95, 156.65, 153.96, 134.78, 132.70, 132.64, 131.00, 129.75, 129.19, 127.39, 127.23, 125.04, 123.73, 121.59, 115.95, 111.59, 110.75, 110.69, 109.56, 55.45, 46.47, 34.64, 30.98, 24.85, 22.52. MS (ESI‐): m/z 497.13 ([M−H]−). HRMS (MALDI): m/z calculated for C31H35N2O4 499.25913, found 499.25707 [M+H]+.
4‐((5‐(4‐(tert‐Butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoic acid (12): Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (73, 89 mg, 0.19 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (15 mL), H2O (10 mL) and lithium hydroxide (23 mg, 0.95 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 12 as white solid (0.08 g, 93 %). 1H NMR (500 MHz, DMSO) δ=10.03 (s, 1H), 8.02 (d, J=1.7, 1H), 7.89 (d, J=8.4, 2H), 7.55–7.51 (m, 3H), 7.46–7.37 (m, 3H), 7.34 (dd, J=8.8, 3.3, 1H), 6.69 (d, J=7.7, 1H), 6.48 (d, J=2.5, 1H), 5.41 (s, 2H), 3.94 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.03, 165.13, 156.34, 154.05, 133.01, 132.69, 131.69, 131.50, 131.30, 131.14, 127.95, 127.46, 127.42, 125.11, 121.67, 116.30, 112.50, 110.97, 109.72, 101.21, 55.65, 52.25, 34.67, 31.00. MS (ESI+): m/z 479.23 ([M+Na]+). HRMS (MALDI): m/z calculated for C28H29N2O4 457.21218, found 457.21028 [M+H]+.
4‐((6‐(4‐(tert‐Butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoic acid (13): Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (66, 42 mg, 0.09 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (15 mL), H2O (10 mL) and lithium hydroxide (11 mg, 0.45 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 13 as yellow solid (40 mg, 98 %). 1H NMR (500 MHz, DMSO) δ=10.09 (s, 1H), 7.97 (s, 1H), 7.87 (d, J=8.5, 2H), 7.53 (dd, J=9.3, 4.9, 4H), 7.44–7.40 (m, 2H), 7.36 (dd, J=8.5, 1.7, 1H), 6.63 (d, J=7.9, 1H), 6.49–6.46 (m, 1H), 5.38 (s, 2H), 3.97 (s, 3H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.03, 165.19, 156.25, 154.12, 135.78, 133.88, 132.58, 131.31, 131.00, 129.40, 127.43, 127.14, 125.09, 124.69, 121.72, 120.27, 113.77, 110.90, 101.59, 101.14, 55.66, 44.29, 34.67, 30.97. MS (ESI+): m/z 479.24 ([M+Na]+). HRMS (MALDI): m/z calculated for C28H29N2O4 457.21218, found 457. 20796 [M+H]+.
4‐((6‐(4‐(tert‐Butyl)benzamido)‐1H‐benzoimidazol‐1‐yl)methyl)‐3‐methoxybenzoic acid (14): Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐benzoimidazol‐1‐yl)methyl)‐3‐methoxybenzoate (70, 0.41 g, 0.87 mmol, 1.0 eq) was dissolved in THF (10 mL) and EtOH (15 mL), H2O (10 mL) and lithium hydroxide (0.10 g, 4.4 mmol, 5.0 eq) were added, and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 14 as colorless solid (0.22 g, 54 %). 1H NMR (500 MHz, DMSO) δ=8.28 (s, 1H), 8.11 (d, J=1.6 Hz, 1H), 7.95 (s, 1H), 7.87 (d, J=8.5 Hz, 2H), 7.61 (d, J=8.7 Hz, 1H), 7.54–7.52 (m, 3H), 7.49–7.46 (m, 2H), 7.01 (d, J=7.9 Hz, 1H), 5.46 (s, 2H), 3.95 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.98, 165.40, 162.36, 156.64, 154.29, 144.71, 139.89, 134.70, 133.70, 132.43, 129.36, 128.21, 127.49, 125.16, 121.74, 119.25, 115.74, 111.15, 102.04, 55.71, 42.94, 34.70, 30.98. MS (ESI+): m/z 458.13 ([M+H]+). HRMS (MALDI): m/z calculated for C27H28N3O4 458.20743, found 458. 20778 [M+H]+.
3‐((6‐(4‐(tert‐Butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoic acid (15): Methyl 3‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoate (65, 0.41 g, 0.93 mmol, 1.0 eq) was dissolved in EtOH (30 mL), H2O (10 mL) and lithium hydroxide (0.11 g, 4.7 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 1) as mobile phase. 15 was obtained as a colorless solid (0.28 g, 70 %). 1H NMR (500 MHz, DMSO) δ=12.98 (s, 1H), 10.10 (s, 1H), 7.98 (s, 1H), 7.89–7.84 (m, 2H), 7.82 (d, J=7.7, 1H), 7.69 (s, 1H), 7.52 (d, J=8.5, 3H), 7.49–7.43 (m, 2H), 7.39 (d, J=7.7, 1H), 7.35 (dd, J=8.5, 1.7, 1H), 6.48 (dd, J=3.1, 0.6, 1H), 5.47 (s, 2H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.09, 165.20, 154.11, 138.96, 135.62, 133.85, 132.56, 131.03, 129.26, 128.90, 128.26, 127.42, 127.30, 125.09, 124.85, 120.30, 113.92, 101.71, 101.21, 48.67, 34.67, 30.97. MS (ESI‐): m/z 425.27 ([M−H]−). HRMS (MALDI): m/z calculated for C27H27N2O3 427.20162, found 427.20184 [M+H]+.
4‐((6‐(4‐(tert‐Butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoic acid (16): Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoate (64, 0.53 g, 1.2 mmol, 1.0 eq) was dissolved in EtOH (30 mL), H2O (10 mL) and lithium hydroxide (0.14 g, 6.0 mmol, 5.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 1) as mobile phase. 16 was obtained as a yellow solid (0.41 g, 80 %). 1H NMR (500 MHz, DMSO) δ=12.88 (s, 1H), 10.09 (s, 1H), 7.96 (s, 1H), 7.90–7.85 (m, 4H), 7.52 (d, J=8.5, 4H), 7.48 (d, J=3.1, 1H), 7.35 (dd, J=8.5, 1.7, 1H), 7.22 (d, J=8.3, 1H), 6.48 (d, J=2.6, 1H), 5.48 (s, 2H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.02, 165.20, 154.11, 143.37, 135.64, 133.85, 132.55, 129.67, 129.20, 127.42, 126.67, 125.40, 125.08, 124.85, 120.29, 113.92, 101 .75, 101.23, 48.82, 34.66, 30.96. MS (ESI‐): m/z 425.28 ([M−H]−). HRMS (MALDI): m/z calculated for C27H27N2O3 427.20162, found 427.20128 [M+H]+.
1‐Methyl‐5‐nitro‐1H‐indole (18): 5‐Nitro‐1H‐indole (17, 0.48 g, 3.0 mmol, 1.0 eq) was dissolved in DMF (abs., 20 mL), NaOH (0.25 g, 6.3 mmol, 2.1 eq) was added and the mixture was stirred for 10 min at 40 °C. Dimethyl sulfate (0.34 mL, 3.6 mmol, 1.2 eq) was carefully added and the mixture was stirred for another 2 h at 40 °C. Then, H2O was added to precipitate the title compound as a yellow solid (0.47 g, 91 %). 1H NMR (250 MHz, DMSO) δ=8.55 (d, J=2.2, 1H), 8.02 (dd, J=9.1, 2.3, 1H), 7.62 (d, J=9.1, 1H), 7.58 (d, J=3.2, 1H), 6.73 (dd, J=3.2, 0.6, 1H), 3.86 (s, 3H). 13C NMR (126 MHz, DMSO) δ=141.10, 139.68, 133.99, 127.67, 117.93, 116.77, 110.73, 103.81, 33.45. MS (ESI+): no molecular ion.
Methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19): Methyl 3‐methoxy‐4‐methylbenzoate (21, 4.26 g, 23.7 mmol, 1.00 eq) was dissolved in CHCl3 (abs., 150 mL). N‐Bromosuccinimide (5.10 g, 28.4 mmol, 1.20 eq) and azobisisobutyronitrile (0.38 g, 2.3 mmol, 0.10 eq) were added and the mixture was refluxed for 12 h. After cooling to room temperature, the precipitated title compound was filtered off as a colorless solid (4.48 g, 73 %). 1H NMR (250 MHz, DMSO) δ=7.59 (dd, J=7.8, 2.3, 3H), 4.72 (s, 2H), 3.98 (s, 3H), 3.92 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.69, 156.16, 136.77, 129.36, 127.02, 121.90, 110.33, 55.80, 52.55, 28.96. MS (ESI+): no molecular ion.
Methyl 3‐(bromomethyl)‐4‐methoxybenzoate (20): Methyl 4‐methoxy‐3‐methylbenzoate (22, 0.48 g, 2.7 mmol, 1.0 eq) was dissolved in CHCl3 (abs., 30 mL). N‐Bromosuccinimide (0.57 g, 3.2 mmol, 1.2 eq) and azobisisobutyronitrile (44 mg, 0.27 mmol, 0.10 eq) were added and the mixture was refluxed for 12 h. After cooling to room temperature, the precipitated title compound was filtered off as a colorless solid (0.61 g, 88 %). 1H NMR (500 MHz, DMSO) δ=8.03 (d, J=2.2, 1H), 7.95 (dd, J=8.7, 2.3, 1H), 7.16 (d, J=8.7, 1H), 4.70 (s, 2H), 3.94 (s, 3H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.59, 161.01, 131.99, 127.75, 126.11, 121.70, 111.48, 56.20, 51.96, 24.45. MS (ESI+): no molecular ion.
Methyl 3‐methoxy‐4‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (23): 1‐Methyl‐5‐nitro‐1H‐indole (18, 699 mg, 4.00 mmol, 1.00 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19, 1.00 g, 4.00 mmol, 1.00 eq) were dissolved in 1,4‐dioxane (abs., 50 mL), FeCl3 (1.94 g, 12.0 mmol, 3.00 eq) was carefully added, and the mixture was stirred at room temperature for 12 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 5) as mobile phase to obtain 23 as yellow solid (0.53 g, 38 %). 1H NMR (250 MHz, DMSO) δ=8.50 (d, J=2.2, 1H), 8.02 (dd, J=9.1, 2.3, 1H), 7.59 (d, J=9.1, 1H), 7.53–7.45 (m, 2H), 7.37 (s, 1H), 7.28 (d, J=8.3, 1H), 4.13 (s, 2H), 3.92 (s, 3H), 3.83 (s, 3H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.58, 157.16, 140.76, 139.91, 135.11, 132.10, 130.39, 129.41, 126.94, 122.08, 116.88, 116.43, 115.56, 111.20, 110.84, 56.02, 52.59, 33.31, 24.94. MS (ESI+): m/z 377.05 ([M+Na]+).
Methyl 4‐methoxy‐3‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (24): 1‐Methyl‐5‐nitro‐1H‐indole (18, 0.15 g, 0.85 mmol, 1.0 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (20, 0.22 g, 0.85 mmol, 1.0 eq) were dissolved in 1,4‐dioxane (abs., 20 mL), FeCl3 (0.97 g, 6.0 mmol, 7.0 eq) was carefully added, and the mixture was stirred at room temperature for 12 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 1) as mobile phase to obtain the title compound as yellow solid (0.14 g, 47 %). 1H NMR (500 MHz, DMSO) δ=8.53 (d, J=2.2, 1H), 8.02 (dd, J=9.1, 2.3, 1H), 7.84 (dd, J=8.6, 2.2, 1H), 7.78 (d, J=2.2, 1H), 7.59 (d, J=9.1, 1H), 7.38 (s, 1H), 7.13 (d, J=8.7, 1H), 4.10 (s, 2H), 3.94 (s, 3H), 3.82 (s, 3H), 3.75 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.97, 160.66, 140.27, 139.45, 131.52, 130.80, 129.56, 129.19, 126.45, 121.58, 116.40, 116.01, 115.62, 110.80, 110.38, 55.85, 51.78, 32.85, 24.16. MS (ESI+): m/z 377.10 ([M+Na]+).
Methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (25): Methyl 3‐methoxy‐4‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (23, 0.17 g, 0.50 mmol, 1.0 eq) was dissolved in MeOH (10 mL) and Pd(C) (loading 10 % w/w, 53 mg, 0.05 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.15 g, 94 %). 1H NMR (250 MHz, DMSO) δ=7.53–7.41 (m, 2H), 7.20–6.83 (m, 4H), 6.52 (d, J=7.3, 3H), 3.91 (s, 3H), 3.90 (s, 2H), 3.83 (s, 3H), 3.62 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.30, 156.77, 141.25, 135.80, 133.52, 129.74, 128.40, 127.95, 123.16, 121.40, 111.80, 111.75, 110.76, 110.42, 110.24, 55.55, 52.12, 32.27, 24.98. MS (ESI+): m/z 325.19 ([M+H]+).
Methyl 3‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐4‐methoxybenzoate (26): Methyl 4‐methoxy‐3‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (24, 0.14 g, 0.40 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 43 mg, 0.040 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (95 mg, 73 %). 1H NMR (500 MHz, DMSO) δ=7.81 (dd, J=8.6, 2.3, 1H), 7.59 (d, J=2.2, 1H), 7.12–7.04 (m, 2H), 6.89 (s, 1H), 6.53–6.51 (m, 2H), 3.93 (s, 3H), 3.86 (s, 2H), 3.72 (s, 3H), 3.63 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.12, 160.77, 141.04, 130.90, 130.22, 129.92, 129.11, 128.32, 127.64, 121.35, 111.80, 110.39, 109.78, 109.61, 101.78, 55.82, 51.73, 32.26, 24.40. MS (ESI+): m/z 325.17 ([M+Na]+).
Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (30): Methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (25, 0.24 g, 0.74 mmol, 1.0 eq) and 4‐tert‐butylbenzoyl chloride (29, 0.19 g, 0.96 mmol, 1.3 eq) were dissolved in THF (abs., 10 mL), DMF (abs., 2 mL) and pyridine (0.18 mL, 2.2 mmol, 3.0 eq). The mixture was stirred at room temperature for 12 h. After acidification with aqueous hydrochloric acid (2 N, 15 mL), the mixture was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 3) as mobile phase. 30 was obtained as a yellow solid (0.22 g, 61 %). 1H NMR (500 MHz, DMSO) δ=10.00 (s, 1H), 7.90–7.88 (m, 3H), 7.53–7.45 (m, 5H), 7.35 (d, J=8.8, 1H), 7.15 (d, J=7.8, 1H), 7.09 (s, 1H), 4.01 (s, 2H), 3.93 (s, 3H), 3.82 (s, 3H), 3.73 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.20, 164.97, 156.75, 153.98, 135.26, 133.87, 132.64, 131.05, 129.49, 128.64, 128.59, 127.38, 127.09, 125.06, 121.48, 116.10, 111.19, 110.60, 110.47, 109.39, 55.55, 52.08, 34.65, 32.38, 30.98, 24.72. MS (ESI+): m/z 507.22 ([M+Na]+).
Methyl 3‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐4‐methoxybenzoate (32): 4‐tert‐Butylbenzoic acid (31, 62 mg, 0.35 mmol, 1.2 eq) was dissolved in CHCl3 (abs., 10 mL). EDC⋅HCl (67 mg, 0.35 mmol, 1.2 eq), 4‐DMAP (54 mg, 0.44 mmol, 1.5 eq) and methyl 3‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐4‐methoxybenzoate (26, 95 mg, 0.29 mmol, 1.0 eq) were added. The reaction mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (10 %, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×10 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 3) as mobile phase to obtain 32 as a yellow solid (12 mg, 8 %). 1H NMR (500 MHz, DMSO) δ=10.01 (s, 1H), 7.92–7.88 (m, 3H), 7.82 (dd, J=8.6, 2.2, 2H), 7.63 (d, J=2.2, 1H), 7.52 (d, J=8.5, 2H), 7.44 (d, J=1.8, 1H), 7.37 (s, 1H), 7.12 (d, J=8.7, 1H), 3.98 (s, 2H), 3.95 (s, 3H), 3.74 (s, 3H), 3.73 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.10, 165.00, 160.78, 154.02, 133.93, 132.66, 131.02, 130.33, 129.73, 129.28, 128.59, 127.40, 127.10, 125.09, 121.42, 116.15, 111.66, 110.62, 110.51, 109.42, 55.87, 51.74, 34.67, 32.41, 31.00, 24.32. MS (ESI+): m/z 507.23 ([M+Na]+).
3‐(2,6‐Dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxylic acid (34): Methyl 3‐(2,6‐dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxylate (39, 0.75 g, 2.4 mmol, 1.0 eq) was dissolved in EtOH (30 mL), H2O (10 mL) and lithium hydroxide (0.31 g, 7.2 mmol, 3.0 eq) were added and the mixture was stirred at room temperature for 16 h. Aqueous hydrochloric acid (2 N, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (5 : 1) as mobile phase to obtain the title compound as colorless solid (0.35 g, 49 %). 1H NMR (500 MHz, DMSO) δ=7.62 (d, J=1.4, 1H), 7.60 (s, 1H), 7.62–7.60 (m, 1H), 3.89–3.79 (m, 1H), 1.35 (d, J=7.0, 6H). 13C NMR (126 MHz, DMSO) δ=182.39, 161.85, 158.56, 134.28, 132.10, 128.14, 127.89, 27.06, 20.05. MS (ESI+): no molecular ion.
2,6‐Dichlorobenzaldehyde oxime (36): 2,6‐Dichlorobenzaldehyde (35, 4.99 g, 28.5 mmol, 1.00 eq) was dissolved in EtOH (40 mL) and added to a solution of hydroxylamine hydrochloride (2.28 g, 32.8 mmol, 1.15 eq) and sodium hydroxide (1.31 g, 32.8 mmol, 1.15 eq) in H2O (20 mL). The mixture was stirred at 90 °C for 24 h. The volume of the reaction mixture was then reduced in vacuum to induce precipitation of the product which was filtered off, washed with cold H2O and dried in vacuum to yield 36 as a colorless solid (3.2 g, 60 %). 36 was used without further purification. 1H NMR (250 MHz, DMSO) δ=11.80 (s, 1H), 8.22 (s, 1H), 7.61–7.50 (m, 2H), 7.47–7.37 (m, 1H). 13C NMR (126 MHz, DMSO) δ=143.84, 133.93, 131.05, 129.42, 128.96. MS (ESI+): no molecular ion.
2,6‐Dichloro‐N‐hydroxybenzimidoyl chloride (37): N‐chlorosuccinimide (0.502 g, 3.76 mmol, 1.00 eq) was slowly added at room temperature to a solution of 2,6‐dichlorobenzaldehyde oxime (36, 0.714 g, 3.76 mmol, 1.00 eq) in DMF (abs., 8 mL). The reaction mixture was stirred for 5 h at room temperature. H2O (20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. 37 was obtained as a colorless solid (0.836, 99 %) and was used without further purification. 1H NMR (250 MHz, DMSO) δ=12.71 (s, 1H), 7.67–7.51 (m, 3H). 13C NMR (126 MHz, DMSO) δ=134.36, 132.83, 131.82, 128.70, 128.04. MS (ESI+): no molecular ion.
Methyl 3‐(2,6‐dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxylate (39): A stirred solution of methyl isobutyrylacetate (38, 0.544 g, 3.77 mmol, 1.00 eq) in THF (abs., 50 mL) was treated with a solution of sodium methoxide (0.76 mL, 0.50 M in MeOH) followed by a solution of 2,6‐dichloro‐N‐hydroxybenzimidoyl chloride (37, 0.848 g, 3.77 mmol, 1.00 eq) in THF (abs., 20 mL). After stirring at room temperature for 16 h, H2O (20 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 5) as mobile phase to obtain the title compound as colorless solid (0.75 g, 63 %). 1H NMR (500 MHz, DMSO) δ=7.65 (d, J=1.6, 1H), 7.63 (d, J=0.5, 1H), 7.60–7.56 (m, 1H), 3.85–3.77 (m, 1H), 3.68 (s, 3H), 1.36 (d, J=7.0, 6H). 13C NMR (126 MHz, DMSO) δ=182.71, 167.99, 160.72, 158.28, 134.22, 132.38, 128.25, 107.15, 51.79, 33.24, 20.00. MS (ESI+): no molecular ion.
Methyl 4‐((5‐(3‐(2,6‐dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (40): 3‐(2,6‐Dichlorophenyl)‐5‐isopropylisoxazole‐4‐carboxylic acid (34, 0.18 g, 0.50 mmol, 1.0 eq) was dissolved in CHCl3 (abs., 20 mL). EDC⋅HCl (0.12 g, 0.60 mmol, 1.2 eq), 4‐DMAP (92 mg, 0.75 mmol, 1.5 eq) and methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (25, 0.19 g, 0.60 mmol, 1.2 eq) were added. The mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by preparative HPLC to obtain 40 as a yellow solid (50 mg, 14 %). 1H NMR (500 MHz, DMSO) δ=9.89 (s, 1H), 7.68 (s, 1H), 7.62–7.58 (m, 2H), 7.55–7.52 (m, 1H), 7.45–7.43 (m, 2H), 7.31 (d, J=8.8, 1H), 7.18 (d, J=8.1, 1H), 7.13 (d, J=7.7, 1H), 7.07 (s, 1H), 3.96 (s, 2H), 3.84 (s, 3H), 3.83 (s, 3H), 3.70 (s, 3H), 1.37 (d, J=6.6, 6H). 13C NMR (126 MHz, DMSO) δ=176.07, 166.20, 157.95, 156.67, 135.21, 134.63, 134.06, 132.21, 130.17, 129.67, 128.74, 128.60, 128.38, 127.18, 127.02, 121.46, 115.72, 113.35, 111.51, 110.49, 109.53, 55.44, 52.11, 32.38, 27.00, 24.60, 20.31. MS (ESI+): m/z 606.25 ([M+H]+).
Methyl 4‐((5‐(isonicotinamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (41): Isonicotinic acid (33, 0.12 g, 1.1 mmol, 1.0 eq) was dissolved in CHCl3 (abs., 20 mL). EDC⋅HCl (0.46 g, 2.4 mmol, 2.4 eq), 4‐DMAP (0.46 g, 3.0 mmol, 3.0 eq) and methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (25, 0.32 g, 1.0 mmol, 1.0 eq) were added. The reaction mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (2 N, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 5) as mobile phase to obtain 41 as a yellow solid (0.14 g, 33 %). 1H NMR (500 MHz, DMSO) δ=8.77 (d, J=4.5, 2H), 7.87 (dd, J=4.5, 1.5, 3H), 7.50–7.43 (m, 4H), 7.40 (d, J=8.7, 2H), 7.14 (s, 1H), 7.12 (s, 1H), 3.99 (s, 2H), 3.92 (s, 3H), 3.75 (s, 3H). 13C NMR (126 MHz, DMSO) δ=167.33, 167.30, 156.66, 150.20, 142.31, 134.65, 134.13, 130.35, 129.79, 129.32, 128.16, 127.09, 126.98, 121.61, 115.98, 113.14, 111.48, 110.89, 110.70, 109.56, 55.48, 35.55, 32.40. MS (ESI+): no molecular ion.
1‐Isopropyl‐5‐nitro‐1H‐indole (43): 5‐Nitro‐1H‐indole (17, 2.0 g, 12 mmol, 1.0 eq) was dissolved in DMF (abs., 20 mL), NaOH (1.0 g, 25 mmol, 2.1 eq) was added and the mixture was stirred for 10 min at 40 °C. Diisopropyl sulfate (2.5 g, 14 mmol, 1.2 eq) was carefully added and the mixture was stirred for another 2 h at 40 °C. Then, H2O was added to precipitate the title compound as a yellow solid (0.5 g, 20 %). 1H NMR (500 MHz, DMSO) δ=8.57–8.56 (m, 1H), 8.03–7.97 (m, 1H), 7.78 (d, J=3.3, 1H), 7.73 (d, J=9.2, 1H), 6.78 (d, J=3.3, 1H), 4.91–4.83 (m, 1H), 1.47 (d, J=6.7, 6H). 13C NMR (126 MHz, DMSO) δ=140.60, 138.05, 128.75, 127.26, 117.61, 116.21, 110.33, 103.99, 47.40, 22.42. MS (ESI+): no molecular ion.
Methyl 4‐((1‐isopropyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (44): 1‐Isopropyl‐5‐nitro‐1H‐indole (43, 0.50 g, 2.5 mmol, 1.0 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19, 0.65 g, 2.5 mmol, 1.0 eq) were dissolved in 1,4‐dioxane (abs., 30 mL), FeCl3 (1.2 g, 7.5 mmol, 3.0 eq) was carefully added, and the mixture was stirred at room temperature for 12 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 5) as mobile phase to obtain the title compound as yellow solid (0.15 g, 16 %). 1H NMR (500 MHz, DMSO) δ=8.46 (d, J=2.3, 1H), 7.99 (dd, J=9.1, 2.3, 1H), 7.69 (d, J=9.1, 1H), 7.62 (s, 1H), 7.51–7.45 (m, 1H), 7.26 (d, J=7.7, 2H), 4.87–4.73 (m, 1H), 4.12 (s, 2H), 3.94 (s, 3H), 3.82 (s, 3H), 1.45 (d, J=6.7, 6H). 13C NMR (126 MHz, DMSO) δ=166.13, 156.70, 140.25, 138.30, 134.73, 129.85, 128.93, 126.91, 126.53, 121.67, 116.29, 116.08, 115.48, 110.78, 110.42, 55.55, 52.14, 47.30, 24.67, 22.41. MS (ESI+): m/z 405.18 ([M+Na]+).
Methyl 4‐((5‐amino‐1‐isopropyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (45): Methyl 4‐((1‐isopropyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (44, 0.15 g, 0.39 mmol, 1.0 eq) was dissolved in MeOH (15 mL) and Pd(C) (loading 10 % w/w, 43 mg, 0.04 mmol, 0.1 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.12 g, 86 %). 1H NMR (500 MHz, DMSO) δ=7.49–7.40 (m, 2H), 7.36 (d, J=8.5, 1H), 7.24 (s, 1H), 7.21–7.10 (m, 2H), 7.08 (d, J=7.1, 1H), 4.65 (m, 1H), 3.92 (s, 3H), 3.90 (s, 2H), 3.82 (s, 3H), 1.38 (d, J=6.7, 6H). 13C NMR (126 MHz, DMSO) δ=133.15, 130.07, 128.42, 128.39, 125.73, 124.17, 123.42, 121.44, 111.60, 111.24, 110.56, 110.41, 109.51, 109.02, 55.51, 52.08, 46.17, 28.16, 20.06. MS (ESI+): m/z 353.22 ([M+H]+).
Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐isopropyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (46): Methyl 4‐((5‐amino‐1‐isopropyl‐1H‐indol‐3‐yl)methyl)‐3‐methoxybenzoate (45, 0.10 g, 0.28 mmol, 1.0 eq) and 4‐tert‐butylbenzoyl chloride (29, 71 mg, 0.36 mmol, 1.3 eq) were dissolved in THF (abs., 20 mL), DMF (abs., 2 mL) and pyridine (0.07 mL, 0.84 mmol, 3.0 eq). The mixture was stirred at room temperature for 12 h. After acidification with aqueous hydrochloric acid (2 N, 15 mL), the mixture was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 3) as mobile phase. 46 was obtained as a yellow solid (0.08 g, 57 %). MS (ESI+): m/z 513.26 ([M+H]+). 1H NMR (250 MHz, DMSO) δ=10.06 (s, 1H), 7.64–7.38 (m, 7H), 7.29–7.04 (m, 4H), 4.03 (s, 2H), 3.93 (s, 3H), 3.82 (s, 3H), 1.44–1.37 (m, 15H). 13C NMR (126 MHz, DMSO) δ=167.45, 166.81, 155.48, 152.83, 136.12, 133.83, 131.64, 129.65, 128.85, 127.57, 127.12, 124.93, 121.98, 121.60, 117.34, 111.04, 110.63, 110.27, 108.68, 55.45, 52.32, 46.56, 34.52, 31.65, 23.96, 21.13. MS (ESI+): m/z 513.26 ([M+H]+).
Methyl 4‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (49): A solution of 1‐methyl‐5‐nitro‐1H‐indole (18, 0.36 g, 2.0 mmol, 1.0 eq) and methyl 4‐formylbenzoate (47, 0.33 g, 2.0 mmol, 1.0 eq) in CH2Cl2 (abs., 30 mL) was cooled to 0 °C, and triethylsilane (0.96 mL, 6.0 mmol, 3.0 eq) and trifluoroacetic acid (0.31 mL, 4.0 mmol, 2.0 eq) were added. After 10 min at 0 °C, the ice bath was removed and the mixture was stirred for 5 h. Aqueous NaHCO3 (saturated, 30 mL) was added, phases were separated, and the aqueous layer was extracted with CH2Cl2 (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (10 : 1) as mobile phase to yield 49 as a yellow solid (0.29 g, 45 %). 1H NMR (250 MHz, DMSO) δ=8.03–7.90 (m, 3H), 7.61 (d, J=8.8, 1H), 7.29–7.15 (m, 4H), 3.84 (s, 5H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.23, 148.35, 146.97, 137.37, 129.39, 129.36, 129.05, 128.91, 128.22, 126.32, 125.33, 112.02, 110.45, 52.01, 35.77, 30.75. MS (ESI+): m/z 347.12 ([M+Na]+).
Methyl 3‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (50): A solution of 1‐methyl‐5‐nitro‐1H‐indole (18, 0.36 g, 2.0 mmol, 1.0 eq) and methyl 3‐formylbenzoate (48, 0.33 g, 2.0 mmol, 1.0 eq) in CH2Cl2 (abs., 30 mL) was cooled to 0 °C, and triethylsilane (0.96 mL, 6.0 mmol, 3.0 eq) and trifluoroacetic acid (0.31 mL, 4.0 mmol, 2.0 eq) were added. After 10 min at 0 °C, the ice bath was removed and the mixture was stirred for 5 h. Aqueous NaHCO3 (saturated, 30 mL) was added, phases were separated, and the aqueous layer was extracted with CH2Cl2 (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (10 : 1) as mobile phase to yield 50 as a yellow solid (0.31 g, 48 %). 1H NMR (500 MHz, DMSO) δ=8.02 (dd, J=9.1, 2.3, 1H), 7.89 (s, 1H), 7.79 (d, J=7.7, 1H), 7.49–7.42 (m, 5H), 4.21 (s, 2H), 3.83 (s, 3H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.45, 143.37, 133.53, 131.68, 131.27, 129.11, 128.99, 128.63, 127.56, 127.01, 116.64, 115.96, 112.10, 110.52, 52.19, 32.93, 29.99. MS (ESI+): m/z 347.13 ([M+Na]+).
Methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (51): Methyl 4‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (49, 0.29 g, 0.89 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 96 mg, 0.089 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.22 g, 85 %). 1H NMR (500 MHz, DMSO) δ=7.90 (d, J=8.2, 1H), 7.87–7.85 (m, 3H), 7.43 (d, J=8.3, 1H), 7.39–7.36 (m, 3H), 6.84–6.80 (m, 2H), 3.82 (s, 5H), 3.71 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.21, 147.48, 133.77, 129.27, 129.21, 128.67, 127.25, 125.13, 112.31, 111.36, 110.46, 110.18, 52.02, 35.80, 30.79. MS (ESI+): m/z 295.19 ([M+H]+).
Methyl 3‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (52): Methyl 3‐((1‐methyl‐5‐nitro‐1H‐indol‐3‐yl)methyl)benzoate (50, 0.31 g, 0.95 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 0.11 g, 0.10 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.25 g, 89 %). 1H NMR (500 MHz, DMSO) δ=7.83 (d, J=7.7, 2H), 7.75 (d, J=7.9, 2H), 7.57 (d, J=7.6, 2H), 7.47 (t, J=7.6, 2H), 7.44–7.39 (m, 2H), 4.56 (s, 2H), 3.85 (s, 3H), 3.84 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.45, 143.37, 138.29, 134.03, 131.28, 129.60, 128.64, 127.56, 127.01, 126.39, 123.36, 111.97, 110.72, 109.90, 108.99, 52.17, 32.32, 30.79. MS (ESI+): m/z 294.95 ([M+H]+).
Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (53): Methyl 4‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (51, 0.30 g, 1.0 mmol, 1.0 eq) and 4‐tert‐butylbenzoyl chloride (29, 0.25 g, 1.3 mmol, 1.3 eq) were dissolved in THF (abs., 30 mL), DMF (abs., 5 mL) and pyridine (0.40 mL, 5.0 mmol, 5.0 eq). The mixture was stirred at room temperature for 12 h. After acidification with aqueous hydrochloric acid (2 N, 30 mL), the mixture was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 1) as mobile phase. 53 was obtained as a pale purple solid (0.35 g, 65 %). 1H NMR (250 MHz, DMSO) δ=10.01 (s, 1H), 7.90 (s, 1H), 7.87 (s, 2H), 7.81 (s, 1H), 7.74 (d, J=7.5, 1H), 7.56–7.46 (m, 4H), 7.37 (dd, J=8.2, 4.6, 2H), 7.14 (s, 1H), 4.08 (s, 2H), 3.75 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.21, 165.00, 153.99, 147.45, 133.98, 132.62, 131.09, 128.67, 127.60, 127.39, 127.23, 127.12, 126.90, 125.06, 116.27, 112.00, 110.59, 109.44, 51.99, 34.65, 32.39, 30.98. MS (ESI+): m/z 477.19 ([M+Na]+).
Methyl 3‐((5‐(4‐(tert‐butyl)benzamido)‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (54): Methyl 3‐((5‐amino‐1‐methyl‐1H‐indol‐3‐yl)methyl)benzoate (52, 0.30 g, 1.0 mmol, 1.0 eq) and 4‐tert‐butylbenzoyl chloride (29, 0.25 g, 1.3 mmol, 1.3 eq) were dissolved in THF (abs., 30 mL), DMF (abs., 5 mL) and pyridine (0.40 mL, 5.0 mmol, 5.0 eq). The mixture was stirred at room temperature for 12 h. After acidification with aqueous hydrochloric acid (2 N, 30 mL), the mixture was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 1) as mobile phase to obtain 54 as a pale purple solid (0.32 g, 59 %). 1H NMR (500 MHz, DMSO) δ=8.12 (s, 1H), 7.74 (d, J=7.5, 1H), 7.38–7.30 (d, J=8.5, 2H), 7.38–7.30 (m, 6H), 7.14 (s, 1H), 6.97 (dd, J=8.5, 2.0, 1H), 5.40 (s, 1H), 4.07 (s, 2H), 3.80 (s, 3H), 3.79 (s, 3H), 1.30 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.65, 166.60, 155.36, 135.77, 134.26, 133.66, 133.56, 132.27, 131.54, 129.45, 129.22, 128.28, 125.64, 125.58, 125.34, 125.22, 118.75, 113.73, 110.93, 109.71, 52.33, 35.00, 34.93, 32.60, 31.17. MS (ESI+): m/z 477.22 ([M+Na]+).
Methyl 4‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (58): 6‐Nitro‐1H‐indole (55, 0.30 g, 1.9 mmol, 1.0 eq), potassium carbonate (0.52 g, 3.8 mmol, 2.0 eq) and methyl 4‐(bromomethyl)benzoate (56, 0.43 g, 1.9 mmol, 1.0 eq) were dissolved in DMF (abs., 20 mL). The mixture was stirred under reflux for 12 h. After cooling to room temperature, aqueous hydrochloric acid (5 %, 30 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (30 : 1) as mobile phase. 58 was obtained as a yellow solid (0.57 g, 97 %). 1H NMR (500 MHz, DMSO) δ=8.48 (d, J=1.9, 1H), 7.96 (d, J=3.1, 1H), 7.92 (d, J=8.2, 3H), 7.78 (d, J=8.8, 1H), 7.31 (d, J=8.4, 2H), 6.75 (dd, J=3.0, 0.7, 1H), 5.73 (s, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.91, 143.16, 142.26, 135.93, 134.36, 133.28, 129.64, 128.90, 127.13, 120.99, 114.53, 107.18, 102.68, 52.17. MS (ESI+): m/z 333.04 ([M+Na]+).
Methyl 3‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (59): 6‐Nitro‐1H‐indole (55, 0.30 g, 1.9 mmol, 1.0 eq), potassium carbonate (0.52 g, 3.8 mmol, 2.0 eq) and methyl 3‐(bromomethyl)benzoate (57, 0.43 g, 1.9 mmol, 1.0 eq) were dissolved in DMF (abs., 20 mL). The mixture was stirred under reflux for 12 h. After cooling to room temperature, aqueous hydrochloric acid (5 %, 30 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (10 : 1) as mobile phase. 59 was obtained as a yellow solid (0.48 g, 81 %). 1H NMR (500 MHz, DMSO) δ=8.53 (d, J=2.0, 1H), 7.98 (d, J=3.1, 1H), 7.91 (dd, J=8.8, 2.1, 1H), 7.87–7.84 (m, 2H), 7.77 (d, J=8.8, 1H), 7.51–7.47 (m, 2H), 6.75 (dd, J=3.0, 0.6, 1H), 5.72 (s, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.96, 142.24, 138.51, 135.84, 134.28, 131.86, 130.03, 129.29, 128.92, 128.43, 128.23, 120.96, 114.51, 107.21, 102.64, 52.25. MS (ESI+): m/z 333.05 ([M+Na]+).
Methyl 3‐methoxy‐4‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (60): 6‐Nitro‐1H‐indole (55, 0.16 g, 1.0 mmol, 1.0 eq), potassium carbonate (0.25 g, 2.0 mmol, 2.0 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19, 0.26 g, 1.0 mmol, 1.0 eq) were dissolved in DMF (abs., 20 mL). The mixture was stirred under reflux for 12 h. After cooling to room temperature, aqueous hydrochloric acid (5 %, 30 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (30 : 1) as mobile phase to obtain 60 as a yellow solid (0.13 g, 38 %). 1H NMR (500 MHz, DMSO) δ=8.50 (d, J=1.9, 1H), 7.93–7.87 (m, 2H), 7.76 (d, J=8.8, 1H), 7.53 (d, J=1.4, 1H), 7.48 (dd, J=7.8, 1.5, 1H), 6.96 (d, J=7.9, 1H), 6.72 (dd, J=3.1, 0.7, 1H), 5.60 (s, 2H), 3.94 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.85, 156.59, 142.15, 135.97, 134.43, 133.10, 130.75, 130.51, 128.29, 121.69, 120.86, 114.37, 111.00, 107.27, 102.47, 55.74, 52.28, 44.69. MS (ESI+): m/z 363.14 ([M+Na]+).
Methyl 4‐((6‐amino‐1H‐indol‐1‐yl)methyl)benzoate (61): Methyl 4‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (58, 0.57 g, 1.8 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 0.19 g, 0.18 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.32 g, 64 %). 1H NMR (500 MHz, DMSO) δ=7.89 (d, J=8.3, 2H), 7.23–7.18 (m, 3H), 7.13 (d, J=3.1, 1H), 6.43–6.38 (m, 2H), 6.27 (d, J=3.1, 1H), 5.33 (s, 2H), 4.74 (s, 2H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.02, 144.30, 144.20, 137.38, 129.46, 128.49, 126.85, 125.99, 120.77, 119.96, 110.01, 101.17, 93.50, 52.13. MS (ESI+): m/z 281.21 ([M+H]+).
Methyl 3‐((6‐amino‐1H‐indol‐1‐yl)methyl)benzoate (62): Methyl 3‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (59, 0.49 g, 1.6 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 0.17 g, 0.16 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.44 g, 98 %). 1H NMR (500 MHz, DMSO) δ=7.83 (d, J=7.8, 1H), 7.72 (s, 1H), 7.46 (d, J=7.7, 1H), 7.37 (d, J=7.7, 1H), 7.21 (d, J=8.3, 1H), 7.16 (d, J=3.1, 1H), 6.45–6.39 (m, 2H), 6.28 (dd, J=3.1, 0.7, 1H), 5.32 (s, 2H), 3.81 (s, 3H). 13C NMR (126 MHz, DMSO) δ=166.06, 143.90, 139.54, 137.29, 131.53, 129.88, 129.03, 127.98, 127.18, 126.02, 120.76, 120.08, 110.03, 101.12, 93.62, 52.19, 48.49. MS (ESI+): m/z 281.22 ([M+H]+).
Methyl 4‐((6‐amino‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (63): Methyl 3‐methoxy‐4‐((6‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (60, 0.13 g, 0.38 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 43 mg, 0.038 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.07 g, 59 %). 1H NMR (250 MHz, DMSO) δ=7.53 (d, J=1.4, 1H), 7.43 (dd, J=7.9, 1.4, 1H), 7.21 (d, J=8.2, 1H), 7.08 (d, J=3.1, 1H), 6.57 (d, J=7.9, 1H), 6.43–6.38 (m, 1.6, 2H), 6.27 (d, J=3.2, 1H), 5.23 (s, 2H), 4.73 (s, 2H), 3.96 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.98, 156.24, 144.23, 137.47, 131.92, 129.81, 127.07, 126.10, 121.55, 120.74, 119.80, 110.59, 109.94, 101.08, 93.35, 55.71, 52.22, 44.08. (ESI+): m/z 311.53 ([M+H]+).
Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoate (64): 4‐tert‐Butylbenzoic acid (31, 0.24 g, 1.4 mmol, 1.2 eq) was dissolved in CHCl3 (abs., 20 mL). EDC⋅HCl (0.26 g, 1.4 mmol, 1.2 eq), 4‐DMAP (0.20 g, 1.7 mmol, 1.5 eq) and methyl 4‐((6‐amino‐1H‐indol‐1‐yl)methyl)benzoate (61, 0.32 g, 1.1 mmol, 1.0 eq) were added. The reaction mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (10 %, 10 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×10 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using toluene/acetone (10 : 1) as mobile phase to obtain 64 as pale purple solid (0.47 g, 97 %). 1H NMR (500 MHz, DMSO) δ=10.09 (s, 1H), 7.96 (s, 1H), 7.93–7.83 (m, 4H), 7.52 (d, J=8.3, 3H), 7.48 (d, J=3.1, 1H), 7.34 (dd, J=8.5, 1.7, 1H), 7.24 (d, J=8.4, 2H), 6.50–6.47 (m, 1H), 5.49 (s, 2H), 3.81 (s, 3H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=165.96, 165.19, 154.12, 143.88, 135.64, 133.87, 132.54, 129.54, 129.28, 128.63, 127.42, 126.84, 125.09, 124.85, 120.30, 113.95, 101.75, 101.27, 52.12, 48.80, 34.67, 30.96. MS (ESI+): m/z 463.19 ([M+Na]+).
Methyl 3‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)benzoate (65): 4‐tert‐Butylbenzoic acid (31, 0.34 g, 1.9 mmol, 1.2 eq) was dissolved in CHCl3 (abs., 20 mL). EDC⋅HCl (0.36 g, 1.9 mmol, 1.2 eq), 4‐DMAP (0.29 g, 2.4 mmol, 1.5 eq) and methyl 3‐((6‐amino‐1H‐indol‐1‐yl)methyl)benzoate (62, 0.45 g, 1.6 mmol, 1.0 eq) were added. The reaction mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (10 %, 20 mL) was then added, phases were separated, and the aqueous layer was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using toluene/acetone (10 : 1) as mobile phase. 65 was obtained as a yellow solid (0.41 g, 59 %). 1H NMR (500 MHz, DMSO) δ=10.10 (s, 1H), 7.98 (s, 1H), 7.88–7.82 (m, 3H), 7.75 (s, 1H), 7.54–7.46 (m, 3H), 7.41 (d, J=7.8, 1H), 7.34 (dd, J=8.5, 1.7, 1H), 7.27–7.23 (m, 2H), 6.49 (dd, J=3.1, 0.6, 1H), 5.48 (s, 2H), 3.81 (s, 3H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=166.02, 165.19, 154.12, 139.21, 137.36, 135.59, 133.84, 132.55, 131.51, 129.96, 128.92, 128.22, 127.41, 127.20, 125.33, 125.09, 120.30, 113.95, 101.75, 101.25, 52.20, 48.63, 34.66, 30.96. MS (ESI+): m/z 463.19 ([M+Na]+).
Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (66): 4‐tert‐Butylbenzoic acid (31, 50 mg, 0.28 mmol, 1.2 eq) was dissolved in CHCl3 (abs., 10 mL). EDC⋅HCl (54 mg, 0.28 mmol, 1.2 eq), 4‐DMAP (43 mg, 0.35 mmol, 1.5 eq) and methyl 4‐((6‐amino‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (63, 70 mg, 0.23 mmol, 1.0 eq) were added. The reaction mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (10 %, 10 mL) was then added, phases were separated, and the product was extracted with EtOAc (3×10 mL). The combined organic layers were dried over MgSO4, the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 3) as mobile phase to yield 66 as a yellow solid (42 mg, 42 %). 1H NMR (500 MHz, DMSO) δ=10.09 (s, 1H), 7.96 (s, 1H), 7.86 (d, J=8.4, 2H), 7.54–7.51 (m, 3H), 7.46–7.41 (m, 3H), 7.35 (dd, J=8.5, 1.4, 1H), 6.65 (d, J=7.9, 1H), 6.48 (d, J=2.9, 1H), 5.38 (s, 2H), 3.98 (s, 3H), 3.82 (s, 3H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=165.96, 165.19, 156.34, 154.13, 135.78, 133.90, 132.56, 131.56, 130.05, 129.41, 129.21, 127.43, 127.29, 125.39, 125.10, 124.70, 120.29, 113.79, 110.71, 101.58, 101.20, 55.76, 52.25, 44.29, 34.68, 30.98. MS (ESI+): m/z 493.24 ([M+Na]+).
Methyl 3‐methoxy‐4‐((6‐nitro‐1H‐benzimidazol‐1‐yl)methyl)benzoate (68): 6‐Nitro‐1H‐benzimidazole (67, 0.33 g, 2.0 mmol, 1.0 eq), potassium carbonate (0.52 g, 2.0 mmol, 2.0 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19, 0.52 g, 2.0 mmol, 1.0 eq) were dissolved in DMF (abs., 20 mL). The mixture was stirred under reflux for 12 h. After cooling to room temperature, aqueous hydrochloric acid (5 %, 30 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (10 : 1) as mobile phase. 68 was obtained as a yellow solid (0.45 g, 66 %). 1H NMR (500 MHz, DMSO) δ=8.57 (d, J=2.1, 1H), 7.95 (s, 1H), 7.90 (d, J=8.9, 1H), 7.81 (d, J=9.0, 1H), 7.53–7.50 (m, 2H), 7.31 (d, J=7.8, 1H), 5.63 (s, 2H), 3.90 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.79, 157.01, 149.32, 148.62, 143.13, 137.87, 131.07, 129.46, 129.28, 121.63, 119.30, 118.53, 111.20, 111.14, 55.78, 52.34, 44.01. MS (ESI+): m/z 342.14 ([M+H]+).
Methyl 4‐((6‐amino‐1H‐benzimidazol‐1‐yl)methyl)‐3‐methoxybenzoate (69): Methyl 3‐methoxy‐4‐((6‐nitro‐1H‐benzmidazol‐1‐yl)methyl)benzoate (68, 0.68 g, 2.0 mmol, 1.0 eq) was dissolved in MeOH (20 mL) and Pd(C) (loading 10 % w/w, 0.21 g, 0.20 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.50 g, 80 %). 1H NMR (500 MHz, DMSO) δ=7.55–7.46 (m, 2H), 7.34–7.26 (m, 1H), 7.12–7.03 (m, 1H), 6.91 (d, J=7.9, 1H), 6.51 (dd, J=8.5, 2.0, 1H), 6.45 (d, J=1.9, 1H), 5.31 (s, 2H), 3.94 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.98, 156.68, 145.18, 141.89, 135.48, 135.05, 130.43, 130.31, 128.10, 121.64, 119.63, 111.39, 110.93, 93.40, 55.82, 52.34, 42.71. MS (ESI+): m/z 311.94 ([M+H]+).
Methyl 4‐((6‐(4‐(tert‐butyl)benzamido)‐1H‐benzoimidazol‐1‐yl)methyl)‐3‐methoxybenzoate (70): Methyl 4‐((6‐amino‐1H‐benzoimidazol‐1‐yl)methyl)‐3‐methoxybenzoate (69, 0.50 g, 1.6 mmol, 1.0 eq) and 4‐tert‐butylbenzoyl chloride (29, 0.41 g, 2.1 mmol, 1.3 eq) were dissolved in THF (abs., 50 mL), DMF (abs., 10 mL) and pyridine (0.39 mL, 4.8 mmol, 3.0 eq). The mixture was stirred at room temperature for 12 h. After acidification with aqueous hydrochloric acid (2 N, 30 mL), the mixture was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 5) as mobile phase to obtain 70 as a yellow solid (0.41 g, 55 %). 1H NMR (500 MHz, DMSO) δ=7.92–7.88 (m, 7H), 7.56–7.53 (m, 4H), 5.68 (s, 2H), 3.94 (s, 3H), 3.83 (s, 3H), 1.31 (s, 9H). 13C NMR (126 MHz, DMSO) δ=167.26, 167.00, 158.15, 156.06, 145.36, 137.41, 132.37, 132.32, 132.23, 131.10, 128.33, 127.48, 126.28, 122.49, 120.12, 117.02, 112.09, 56.65, 53.34, 49.60, 35.19, 31.82. MS (ESI+): m/z 472.16 ([M+H]+).
Methyl 3‐methoxy‐4‐((5‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (71): 5‐Nitro‐1H‐indole (17, 0.16 g, 1.0 mmol, 1.0 eq), potassium carbonate (0.25 g, 2.0 mmol, 2.0 eq) and methyl 4‐(bromomethyl)‐3‐methoxybenzoate (19, 0.26 g, 1.0 mmol, 1.0 eq) were dissolved in DMF (abs., 20 mL). The mixture was stirred under reflux for 12 h. After cooling to room temperature, aqueous hydrochloric acid (5 %, 30 mL) was added, phases were separated, and the aqueous layer was extracted with EtOAc (3×30 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. The crude product was purified by column chromatography using toluene/acetone (10 : 1) as mobile phase. 71 was obtained as a yellow solid (0.16 g, 47 %). 1H NMR (500 MHz, DMSO) δ=8.50 (d, J=1.9, 1H), 7.91 (dd, J=8.8, 2.1, 1H), 7.89 (d, J=3.1, 1H), 7.76 (d, J=8.8, 1H), 7.53 (d, J=1.4, 1H), 7.48 (dd, J=7.8, 1.5, 1H), 6.96 (d, J=7.9, 1H), 6.72 (dd, J=3.1, 0.7, 1H), 5.60 (s, 2H), 3.94 (s, 3H), 3.83 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.85, 156.59, 142.15, 135.97, 134.43, 133.10, 130.75, 130.51, 128.29, 121.69, 120.86, 114.37, 111.00, 107.27, 102.47, 55.74, 52.28, 44.69. MS (ESI+): m/z 363.08 ([M+Na]+).
Methyl 4‐((5‐amino‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (72): Methyl 3‐methoxy‐4‐((5‐nitro‐1H‐indol‐1‐yl)methyl)benzoate (71, 0.16 g, 0.47 mmol, 1.0 eq) was dissolved in MeOH (15 mL) and Pd(C) (loading 10 % w/w, 53 mg, 0.05 mmol, 0.10 eq) was added. The suspension was stirred at room temperature under H2 atmosphere for 6 h. The mixture was then filtered through celite, the filtrate was dried over MgSO4, and the solvent was evaporated in vacuum to obtain the title compound as pale purple solid (0.13 g, 90 %). 1H NMR (500 MHz, DMSO) δ=7.51 (d, J=1.4, 1H), 7.42 (dd, J=7.8, 1.5, 1H), 7.32 (d, J=3.1, 1H), 7.14 (d, J=8.6, 1H), 6.92 (d, J=1.9, 1H), 6.67 (d, J=7.9, 1H), 6.62 (dd, J=8.6, 2.0, 1H), 6.29 (dd, J=3.0, 0.5, 1H), 5.33 (s, 2H), 3.94 (s, 3H), 3.82 (s, 3H). 13C NMR (126 MHz, DMSO) δ=165.96, 156.36, 137.05, 131.87, 131.12, 129.98, 129.56, 128.93, 127.55, 121.52, 113.04, 110.72, 110.29, 106.38, 100.10, 55.71, 52.24, 44.29. MS (ESI+): m/z 311.21 ([M+H]+).
Methyl 4‐((5‐(4‐(tert‐butyl)benzamido)‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (73): 4‐tert‐Butylbenzoic acid (31, 0.16 g, 0.89 mmol, 1.2 eq) was dissolved in CHCl3 (abs., 20 mL). EDC⋅HCl (0.17 g, 0.89 mmol, 1.2 eq), 4‐DMAP (0.29 g, 1.1 mmol, 1.5 eq) and methyl 4‐((5‐amino‐1H‐indol‐1‐yl)methyl)‐3‐methoxybenzoate (72, 0.23 g, 0.74 mmol, 1.0 eq) were added. The mixture was stirred at 50 °C for 12 h. Aqueous hydrochloric acid (10 %, 20 mL) was then added, phases were separated, and the product was extracted with EtOAc (3×20 mL). The combined organic layers were dried over MgSO4, and the solvents were evaporated in vacuum. Further purification was performed by column chromatography using EtOAc/hexane (1 : 3) as mobile phase. 73 was obtained as a yellow solid (0.1 g, 29 %). 1H NMR (500 MHz, DMSO) δ=10.03 (s, 1H), 8.02 (d, J=1.7, 1H), 7.89 (d, J=8.5, 2H), 7.56–7.51 (m, 3H), 7.45–7.43 (m, 2H), 7.39 (dd, J=8.8, 1.8, 1H), 7.33 (d, J=8.8, 1H), 6.70 (d, J=7.9, 1H), 6.49 (d, J=2.9, 1H), 5.42 (s, 2H), 3.95 (s, 3H), 3.83 (s, 3H), 1.32 (s, 9H). 13C NMR (126 MHz, DMSO) δ=165.94, 165.11, 156.40, 154.03, 132.99, 132.68, 131.68, 131.52, 130.05, 129.98, 127.94, 127.56, 127.41, 125.10, 121.56, 116.30, 112.49, 110.76, 109.69, 101.25, 55.73, 52.24, 44.38, 34.66, 30.98. MS (ESI+): m/z 493.24 ([M+Na]+).
In vitro Pharmacology
FXR transactivation assay: Plasmids: pcDNA3‐hFXR39 contains the sequence of human FXR. pSG5‐hRXR40 contains the sequence of human RXRα. pGL3basic (Promega Corporation, Fitchburg, WI, USA) was used as a reporter plasmid and contains a shortened construct of the promotor of the bile salt export protein (BSEP) cloned into the SacI/NheI cleavage site in front of the luciferase gene.41 pRL‐SV40 (Promega) served as a control for normalization of transfection efficiency and cell growth. Assay procedure: HeLa cells were grown in DMEM high glucose supplemented with 10 % fetal calf serum (FCS), sodium pyruvate (1 mM), penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C and 5 % CO2. 24 h before transfection, HeLa cells were seeded in 96‐well plates with a density of 8000 cells per well. 3.5 h before transfection, medium was changed to DMEM high glucose, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL) and 0.5 % charcoal‐stripped FCS. Transient transfection of HeLa cells with BSEP‐pGL3, pRL‐SV40 and the expression plasmids pcDNA3‐hFXR and pSG5‐hRXR was carried out using calcium phosphate transfection method. 16 h after transfection, medium was changed to DMEM high glucose, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL) and 0.5 % charcoal‐stripped FCS. 24 h after transfection, medium was changed to DMEM without phenol red, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL), L‐glutamine (2 mM) and 0.5 % charcoal‐stripped FCS, now additionally containing 0.1 % DMSO and the respective test compound or 0.1 % DMSO alone as untreated control. Each concentration was tested in triplicate wells and each experiment was repeated independently at least three times. Following 24 h incubation with the test compounds, cells were assayed for luciferase activity using Dual‐Glo™ Luciferase Assay System (Promega) according to the manufacturer's protocol. Luminescence was measured with a Tecan Infinite M200 luminometer (Tecan Deutschland GmbH, Crailsheim, Germany) or a Tecan Spark luminometer (Tecan). Normalization of transfection efficiency and cell growth was done by division of firefly luciferase data by renilla luciferase data multiplied by 1000 resulting in relative light units (RLU). Fold activation was obtained by dividing the mean RLU of the tested compound at a respective concentration by the mean RLU of untreated control. Relative activation was obtained by dividing the fold activation of the tested compound at a respective concentration by the fold activation of FXR full agonist GW4064 at 3 μM. EC50 and standard deviation values were calculated with the mean relative activation values of at least three independent experiments set up in triplicates by SigmaPlot 12.5 (Systat Software GmbH, Erkrath, Germany) using a four‐parameter logistic regression. The assay was validated with FXR agonists OCA (EC50=0.16±0.02 μM, 87±3% rel. max. act.) and GW4064 (EC50=0.51±0.16 μM, 3 μM defined as 100 %).32, 42
Hybrid reporter gene assays for nuclear receptors: Plasmids: The Gal4‐fusion receptor plasmids pFA‐CMV‐hPPARα‐LBD,43 pFA‐CMV‐hPPARγ‐LBD,43 pFA‐CMV‐hPPARδ‐LBD,43 pFA‐CMV‐hLXRα‐LBD,44 pFA‐CMV‐hLXRβ‐LBD,44 pFA‐CMV‐hRXRα‐LBD,26 pFA‐CMV‐hRARα‐LBD26 and pFA‐CMV‐hCAR‐LBD26 coding for the hinge region and ligand binding domain (LBD) of the canonical isoform of the respective nuclear receptor have been reported previously. pFR‐Luc (Stratagene, Agilent Technologies, Santa Clara, California, USA) was used as reporter plasmid and pRL‐SV40 (Promega) for normalization of transfection efficiency and cell growth. Assay procedure: HEK293T cells were grown in DMEM high glucose, supplemented with 10 % FCS, sodium pyruvate (1 mM), penicillin (100 U/mL) and streptomycin (100 μg/mL) at 37 °C and 5 % CO2. The day before transfection, HEK293T cells were seeded in 96‐well plates (3 ⋅ 104 cells/well). Before transfection, medium was changed to Opti‐MEM without supplements. Transient transfection was carried out using Lipofectamine LTX reagent (Invitrogen, Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer's protocol with pFR‐Luc (Stratagene), pRL‐SV40 (Promega) and the corresponding Gal4‐fusion nuclear receptor plasmid. 5 h after transfection, medium was changed to Opti‐MEM supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL), now additionally containing 0.1 % DMSO and the respective test compound or 0.1 % DMSO alone as untreated control. Each concentration was tested in duplicates and each experiment was repeated independently at least three times. Following overnight (14–16 h) incubation with the test compounds, cells were assayed for luciferase activity using Dual‐Glo™ Luciferase Assay System (Promega) according to the manufacturer's protocol. Luminescence was measured with a Tecan Spark luminometer (Tecan). Normalization of transfection efficiency and cell growth was done by division of firefly luciferase data by renilla luciferase data and multiplying the value by 1000 resulting in relative light units (RLU). Fold activation was obtained by dividing the mean RLU of a test compound at a respective concentration by the mean RLU of the untreated control. Relative activation was obtained by dividing the fold activation of a test compound at a respective concentration by the fold activation of a respective reference agonist at 1 μM (PPARα: GW7647; PPARγ: pioglitazone; PPARδ: L165,041; LXRα/β: T0901317; RXRα: bexarotene; RARα: tretinoin; CAR: CITCO). All hybrid assays were validated with the above‐mentioned reference agonists, which yielded EC50 values in agreement with literature.
sEH activity assay on recombinant protein: sEH inhibitory potency was determined in a fluorescence‐based 96‐well sEH activity assay using recombinant human enzyme.27, 28 Non‐fluorescent PHOME ((3‐phenyloxiranyl)aetic acid cyano‐(6‐methoxynapthalen‐2‐yl)methyl ester) was used as substrate which is hydrolyzed by sEH to fluorescent 6‐methoxynaphthaldehyde. Recombinant human sEH (in BisTris buffer, pH 7, with 0.1 mg/mL BSA containing a final concentration of 0.01 % Triton‐X 100) was preincubated with test compounds (in DMSO, final DMSO concentration: 1 %) for 30 min at room temperature. Then, substrate was added (final concentration 50 μM) and hydrolysis of the substrate was determined by measuring fluorescent product formation on a Tecan Infinite F200 Pro (λem=330 nm, λex=465 nm) for 30 min (one point per minute). A blank control (no protein and no compound) as well as a positive control (no compound) were executed. All experiments were conducted in triplicate and repeated in at least three independent experiments. For IC50 calculation, dose‐response curves of increasing compound concentrations were recorded.
CysLT 1 R antagonism assay: CysLT1R antagonism of 3, 13 and 16 at a concentration of 1 μM was determined in a cell‐based (CHO−K1 cells) Ca2+‐flux assay on human CysLT1R in competition with 0.1 nM leukotriene D4. The experiments were conducted in two independent repeats. The assay was performed by Cerep (Celle L′Evescault, France; assay reference number 1607) on a fee‐for‐service basis.
Evaluation of FXR regulated gene expression: Cell culture and treatment: HepG2 cells were grown in DMEM high glucose, supplemented with 10 % FCS, 1 mM sodium pyruvate, penicillin (100 U/mL), and streptomycin (100 μg/mL) at 37 °C and 5 % CO2 in 6‐well plates (2×106 per well). 24 h after seeding, medium was changed to MEM supplemented with 1 % charcoal stripped FCS, penicillin (100 U/mL), streptomycin (100 μg/mL), and 2 mM L‐glutamate. After an additional 24 h, medium was again changed to MEM now additionally containing the test compounds (10 μM 13; 10 μM 16, 50 μM CDCA) solubilized with 0.1 % DMSO or DMSO alone as untreated control. After 12 h incubation, the cells were harvested, washed with cold phosphate‐buffered saline (PBS), and then directly used for RNA extraction. Quantitative polymerase chain reaction (qPCR): 2 μg of total RNA were extracted from HepG2 cells by the Total RNA mini Kit (R6834‐02, Omega Bio‐Tek, Inc., Norcross, GA). RNA was reverse‐transcribed into cDNA using the High‐Capacity cDNA Reverse Transcription Kit (Thermo Fischer Scientific, Inc.) according to the manufacturer's protocol. FXR regulated gene expression was evaluated by quantitative real‐time PCR analysis with a StepOnePlus System (Life Technologies, Carlsbad, CA) using Power SYBR Green (Life Technologies; 12.5 μL/well). Each sample was set up in duplicates and repeated in at least three independent experiments. Expression was quantified by the comparative 2−ΔΔCt method and glyceraldehyde 3‐phosphate dehydrogenase (GAPDH) served as the reference gene. The following PCR primers were used: GAPDH: 5′‐ATA TGA TTC CAC CCA TGG CA‐3′ (fwd) and 5′‐GAT GAT GAC CCT TTT GGC TC‐3′ (rev); SHP: 5′‐GCT GTC TGG AGT CCT TCT GG‐3′ (fwd) and 5′‐CCA ATG ATA GGG CGA AAG AAG AG‐3′ (rev); CYP7A1: 5′‐CAC CTT GAG GAC GGT TCC TA‐3′ (fwd) and 5′‐CGA TCC AAA GGG CAT GTA GT‐3′ (rev); OSTα: 5′‐TGC TGC TCA CCA GGA AGA AG‐3′ (fwd) and 5′‐ATA GAG CTG TGC TCC CCT CA‐3′ (rev).
Cellular sEH Assay: Quantification of cellular sEH metabolic activity was performed as described by Zha et al.45 Cellular assay: 10 μg of total cell homogenate from HepG2 cells (diluted in 100 μL of PBS containing 0.1 mg/mL BSA) was incubated with the test compounds (10 μM 13; 10 μM 16), N‐cyclohexyl‐N‐(4‐iodophenyl)urea (CIU, 10 μM) as positive control, or vehicle (DMSO in a final concentration of 1 %) as negative control for 15 min at 37 °C. An amount of 25 ng of (±)14(15)‐EET‐d11 (Cayman Chemical, Ann Arbor, USA) was added, and incubation was continued for additional 10 min at 37 °C. A blank test was performed with PBS (containing 0.1 mg/mL BSA) treated likewise. The reactions were stopped by adding 100 μL of ice‐cold methanol. After centrifugation (2000 rpm, 4 °C, 5 min), the amounts of (±)14(15)‐EET‐d11 and the corresponding (±)14(15)‐DHET‐d11 were determined from the supernatants by UPLC‐MS. Each experiment was independently repeated four times. Sample preparation for UPLC‐MS: The samples received from the assay were extracted twice with 700 μL of ethyl acetate. The organic phase was removed at a temperature of 45 °C under a gentle stream of nitrogen. The residues were dissolved in 100 μL of methanol/water (50 : 50, v/v), and centrifuged for 15 minutes at 13000 rpm. The supernatants were used for UPLC‐MS analysis. UPLC‐MS analysis: The UPLC‐MS analyses were carried out on an ACQUITY UPLC‐MS system (Waters, Eschborn) with TUV detector (Waters) coupled to a single quadrupole mass detector (QDa, Waters). 10 μL per sample were injected. Separation was achieved on an ACQUITY UPLC® HSS T3 (1.8 μm, 2.1×100 mm) reverse phase column with column temperature of 45 °C. The mobile phases A water, B acetonitrile and D water/0.1 % formic acid were used as eluents. The gradient range started with B 5 % to 95 % whereas D remained at 5 % within four minutes at a flow rate of 0.5 mL/min. ( )14(15)‐EET‐d11 (m/z 332.2) and ( )14(15)‐DHET‐d11 (m/z 350.2) were analyzed in positive single ion mode [M+H]+ using Selected Ion Monitoring (SIM). The obtained ion chromatograms were integrated with Epower 3 software and the peak areas were compared to estimate sEH inhibition.
WST‐1 assay (Roche Diagnostics International AG, Rotkreuz, Switzerland) was performed according to manufacturer's protocol. In brief, HepG2 cells were seeded in DMEM high glucose, supplemented with sodium pyruvate (1 mM), penicillin (100 U/mL), streptomycin (100 μg/mL) and 10 % FCS in 96 well plates (3 ⋅ 104 cells/well). After 24 h, medium was changed to DMEM high glucose, supplemented with penicillin (100 U/mL), streptomycin (100 μg/mL) and 1 % charcoal stripped FCS additionally containing 0.1 % DMSO and the test compounds (final concentrations 0.1 μM, 1 μM, 10 μM, and 100 μM), or Revlotron as positive control, or 0.1 % DMSO alone as negative control. After 24 h, WST reagent (Roche Diagnostics International AG) was added to each well according to manufacturer's instructions. After 45 min. incubation, absorption (450 nm/reference: 620 nm) was determined with a Tecan Spark (Tecan). Each experiment was performed in triplicates in four independent repeats.
Molecular Docking
The X‐ray structures 3OTQ23 (sEH) and 4QE824 (FXR) were selected for molecular docking for the high structural similarity of the co‐crystallized ligands to 4, 13 and 16. The structures were prepared for docking using the QuickPrep routine of the MOE software suite (v2018.01, Chemical Computing Group, Montreal, Canada). In the sEH co‐crystal structure water molecule HOH577 in a hydrophobic sub‐pocket was removed in order to leave more space for the bulky lipophilic cyclopentylurethane or tert‐butyl group. Subsequently, molecular docking was performed using the MOE induced fit protocol. For initial placement, the Template substructure (N‐phenyl amide substructure) option was used, followed by refinement using the GBVI/WSA dG scoring function. The 5 top scored binding poses were inspected visually with particular emphasis on saturated hydrogen bonding. The most probable binding mode based on these considerations was subjected to global energy minimization with Amber10:EHT forcefield using default settings. Results were visualized with UCSF Chimera.46
Acknowledgements
This research was financially supported by the LOEWE center “Translational Medicine and Pharmacology (TMP)” Frankfurt, Germany and by the Else‐Kroener‐Fresenius‐Foundation funding the graduate school “Translational Research Innovation – Pharma” (TRIP). A.P. and E.P. thank the German Research Foundation (DFG) for financial support (SFB 1039 TP A07 and Heisenberg‐Professur PR1405/4‐1). Molecular graphics and analyses were performed with UCSF Chimera, developed by the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco, with support from NIH P41‐GM103311.
S. Schierle, M. Helmstädter, J. Schmidt, M. Hartmann, M. Horz, A. Kaiser, L. Weizel, P. Heitel, A. Proschak, V. Hernandez-Olmos, E. Proschak, D. Merk, ChemMedChem 2020, 15, 50.
References
- 1. Fiorucci S., Mencarelli A., Distrutti E., Zampella A., Future Med. Chem. 2012, 4, 877–891. [DOI] [PubMed] [Google Scholar]
- 2. Merk D., Steinhilber D., Schubert-Zsilavecz M., Future Med. Chem. 2012, 4, 1015–1036. [DOI] [PubMed] [Google Scholar]
- 3. Gadaleta R. M., Cariello M., Sabbà C., Moschetta A., Biochim. Biophys. Acta 2014, 1851, 30–39. [DOI] [PubMed] [Google Scholar]
- 4. Oseini A. M., Sanyal A. J., Liver Int. 2017, 37, 97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Neuschwander-Tetri B. A., Loomba R., Sanyal A. J., Lavine J. E., Van Natta M. L., Abdelmalek M. F., Chalasani N., Dasarathy S., Diehl A. M., Hameed B., Lancet 2014, 385, 956–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Younossi Z., Tacke F., Arrese M., Chander Sharma B., Mostafa I., Bugianesi E., Wai-Sun Wong V., Yilmaz Y., George J., Fan J., Hepatology 2019, 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- 7. Younossi Z. M., Koenig A. B., Abdelatif D., Fazel Y., Henry L., Wymer M., Hepatology 2016, 64, 73–84. [DOI] [PubMed] [Google Scholar]
- 8. Pellicciari R., Fiorucci S., Camaioni E., Clerici C., Costantino G., Maloney P. R., Morelli A., Parks D. J., Willson T. M., J. Med. Chem. 2002, 45, 3569–3572. [DOI] [PubMed] [Google Scholar]
- 9. Mudaliar S., Henry R. R., Sanyal A. J., Morrow L., Marschall H. U., Kipnes M., Adorini L., Sciacca C. I., Clopton P., Castelloe E., Gastroenterology 2013, 145, 574–582.e1. [DOI] [PubMed] [Google Scholar]
- 10. Shen H. C., Hammock B. D., J. Med. Chem. 2012, 55, 1789–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Liu Y., Dang H., Li D., Pang W., Hammock B. D., Zhu Y., PLoS One 2012, 7, e39165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schuck R. N., Zha W., Edin M. L., Gruzdev A., Vendrov K. C., Miller T. M., Xu Z., Lih F. B., DeGraff L. M., Tomer K. B., PLoS One 2014, 9, e110162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. López-Vicario C., Alcaraz-Quiles J., García-Alonso V., Rius B., Hwang S. H., Titos E., Lopategi A., Hammock B. D., Arroyo V., Clària J., Proc. Mont. Acad. Sci. 2015, 112, 536–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gai Z., Visentin M., Gui T., Zhao L., Thasler W. E., Häusler S., Hartling I., Cremonesi A., Hiller C., Kullak-Ublick G. A., Mol. Pharmacol. 2018, 94, 802–811. [DOI] [PubMed] [Google Scholar]
- 15. Schmidt J., Rotter M., Weiser T., Wittmann S., Weizel L., Kaiser A., Heering J., Goebel T., Angioni C., Wurglics M., J. Med. Chem. 2017, 60, 7703–7724. [DOI] [PubMed] [Google Scholar]
- 16. Proschak E., Stark H., Merk D., J. Med. Chem. 2019, 62, 420–444. [DOI] [PubMed] [Google Scholar]
- 17. Hye Khan M. A., Schmidt J., Stavniichuk A., Imig J. D., Merk D., Biochem. Pharmacol. 2019, 166, 212–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Schierle S., Flauaus C., Heitel P., Willems S., Schmidt J., Kaiser A., Weizel L., Goebel T., Kahnt A. S., Geisslinger G., J. Med. Chem. 2018, 61, 5758–5764. [DOI] [PubMed] [Google Scholar]
- 19. Wermuth C. G., J. Med. Chem. 2004, 47, 1303–14. [DOI] [PubMed] [Google Scholar]
- 20. Matassa V. G., Maduskuie T. P., Shapiro H. S., Hesp B., Snyder D. W., Aharony D., Krell R. D., Keith R. A., J. Med. Chem. 1990, 33, 1781–90. [DOI] [PubMed] [Google Scholar]
- 21. Boggs S., Collins J., Hyatt S., Maloney P., Farnesoid X Receptor Agonists, 2003, US7319109B2. [Google Scholar]
- 22. Mahadevan A., Sard H., Gonzalez M., McKew J. C., Tetrahedron Lett. 2003, 44, 4589–4591. [Google Scholar]
- 23. Lo H. Y., Man C. C., Fleck R. W., Farrow N. A., Ingraham R. H., Kukulka A., Proudfoot J. R., Betageri R., Kirrane T., Patel U., Bioorg. Med. Chem. Lett. 2010, 20, 6379–6383. [DOI] [PubMed] [Google Scholar]
- 24. Merk D., Sreeramulu S., Kudlinzki D., Saxena K., Linhard V., Gande S. L., Hiller F., Lamers C., Nilsson E., Aagaard A., Nat. Commun. 2019, 10, 2915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gabler M., Kramer J., Schmidt J., Pollinger J., Weber J., Kaiser A., Löhr F., Proschak E., Schubert-Zsilavecz M., Merk D., Sci. Rep. 2018, 8, 6846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Flesch D., Cheung S.-Y., Schmidt J., Gabler M., Heitel P., Kramer J. S., Kaiser A., Hartmann M., Lindner M., Lüddens-Dämgen K., J. Med. Chem. 2017, 60, 7199–7205. [DOI] [PubMed] [Google Scholar]
- 27. Klingler F.-M., Wolf M., Wittmann S., Gribbon P., Proschak E., J. Biomol. Screen. 2016, 21, 689–694. [DOI] [PubMed] [Google Scholar]
- 28. Blöcher R., Lamers C., Wittmann S. K., Merk D., Hartmann M., Weizel L., Diehl O., Brüggerhoff A., Boß M., Kaiser A., J. Med. Chem. 2016, 59, 61–81. [DOI] [PubMed] [Google Scholar]
- 29. Sarau H. M., Ames R. S., Chambers J., Ellis C., Elshourbagy N., Foley J. J., Schmidt D. B., Muccitelli R. M., Jenkins O., Murdock P. R., Mol. Pharmacol. 1999, 56, 657–663. [DOI] [PubMed] [Google Scholar]
- 30. Langer T., Wermuth C. G., in Polypharmacology Drug Discov., John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012, pp. 227–243. [Google Scholar]
- 31. Schierle S., Schmidt J., Kaiser A., Merk D., ChemMedChem 2018, 13, 2530–2545. [DOI] [PubMed] [Google Scholar]
- 32. Merk D., Lamers C., Ahmad K., Carrasco Gomez R., Schneider G., Steinhilber D., Schubert-Zsilavecz M., J. Med. Chem. 2014, 57, 8035–8055. [DOI] [PubMed] [Google Scholar]
- 33. Maloney P. R., Parks D. J., Haffner C. D., Fivush A. M., Chandra G., Plunket K. D., Creech K. L., Moore L. B., Wilson J. G., Lewis M. C., J. Med. Chem. 2000, 43, 2971–2974. [DOI] [PubMed] [Google Scholar]
- 34. Bass J. Y., Caravella J. A., Chen L., Creech K. L., Deaton D. N., Madauss K. P., Marr H. B., McFadyen R. B., Miller A. B., Mills W. Y., Bioorg. Med. Chem. Lett. 2011, 21, 1206–1213. [DOI] [PubMed] [Google Scholar]
- 35. Pecic S., Pakhomova S., Newcomer M. E., Morisseau C., Hammock B. D., Zhu Z., Rinderspacher A., Deng S.-X., Bioorg. Med. Chem. Lett. 2013, 23, 417–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pecic S., Zeki A. A., Xu X., Jin G. Y., Zhang S., Kodani S., Halim M., Morisseau C., Hammock B. D., Deng S.-X., Prostaglandins Other Lipid Mediators 2018, 136, 90–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Akwabi-Ameyaw A., Bass J. Y., Caldwell R. D., Caravella J. A., Chen L., Creech K. L., Deaton D. N., Jones S. A., Kaldor I., Liu Y., Bioorg. Med. Chem. Lett. 2008, 18, 4339–4343. [DOI] [PubMed] [Google Scholar]
- 38. Gellrich L., Merk D., Nucl. Recept. Res. 2017, 4, 101310. [Google Scholar]
- 39. Steri R., Achenbach J., Steinhilber D., Schubert-Zsilavecz M., Proschak E., Biochem. Pharmacol. 2012, 83, 1674–1681. [DOI] [PubMed] [Google Scholar]
- 40. Seuter S., Väisänen S., Rådmark O., Carlberg C., Steinhilber D., Biochim. Biophys. Acta 2007, 1771, 864–872. [DOI] [PubMed] [Google Scholar]
- 41. Ananthanarayanan M., Balasubramanian N., Makishima M., Mangelsdorf D. J., Suchy F. J., J. Biol. Chem. 2001, 276, 28857–28865. [DOI] [PubMed] [Google Scholar]
- 42. Flesch D., Gabler M., Lill A., Gomez R. C., Steri R., Schneider G., Stark H., Schubert-Zsilavecz M., Merk D., Bioorg. Med. Chem. 2015, 3, 1–9. [DOI] [PubMed] [Google Scholar]
- 43. Rau O., Wurglics M., Paulke A., Zitzkowski J., Meindl N., Bock A., Dingermann T., Abdel-Tawab M., Schubert-Zsilavecz M., Planta Med. 2006, 72, 881–887. [DOI] [PubMed] [Google Scholar]
- 44. Heitel P., Achenbach J., Moser D., Proschak E., Merk D., Bioorg. Med. Chem. Lett. 2017, 27, 1193–1198. [DOI] [PubMed] [Google Scholar]
- 45. Zha W., Edin M. L., Vendrov K. C., Schuck R. N., Lih F. B., Jat J. L., Bradbury J. A., DeGraff L. M., Hua K., Tomer K. B., J. Lipid Res. 2014, 55, 2124–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Pettersen E. F., Goddard T. D., Huang C. C., Couch G. S., Greenblatt D. M., Meng E. C., Ferrin T. E., J. Comput. Chem. 2004, 25, 1605–12. [DOI] [PubMed] [Google Scholar]