

Abstract
Once‐weekly injection of 56.5‐μg teriparatide formulation is a potent therapeutic agent for osteoporosis treatment. However, this treatment has an issue of difficulty in continuing the treatment by its adverse side effects including nausea, vomiting, and headaches. To reduce these adverse side effects, we conducted a randomized, single‐blind, placebo‐controlled study to examine the pharmacokinetics, changes in bone turnover markers, and safety profiles of twice‐weekly 28.2‐μg teriparatide injections. Different dosing intervals of the twice‐weekly 28.2‐μg injections were also studied. A total of 100 healthy Japanese postmenopausal women were enrolled in this multiple‐dosing study. The systemic exposure of teriparatide acetate in the twice‐weekly 28.2‐μg injection was half that of the once‐weekly 56.5‐μg injection. Changes in bone turnover markers in the twice‐weekly 28.2‐μg injection were comparable to those in the once‐weekly 56.5‐μg injection. Incidences of adverse events including nausea, vomiting, and headaches were lower in the twice‐weekly 28.2‐μg injections than those in the once‐weekly 56.5‐μg injection. Findings were similar in the twice‐weekly 28.2‐μg injections regardless of the dosing interval. Thus, the new dosing regimen using twice‐weekly 28.2‐μg injections maintained comparable efficacy to the once‐weekly 56.5‐μg injections; however, it improved the safety profile and contributed to better continuity of care with teriparatide.
Keywords: bone turnover marker, pharmacokinetics, safety, teriparatide, twice‐weekly regimen
Teriparatide is an active polypeptide that consists of the 1–34 amino acid fragment of the parathyroid hormone and is used as a therapeutic agent for osteoporosis in a once‐weekly administered 56.5‐μg teriparatide injection as well as daily 20‐μg treatment.1 Once‐weekly teriparatide injections, approved in Japan and South Korea, are subcutaneously administered in an outpatient setting. Daily 20‐μg teriparatide injections are self‐administered and are approved in many countries including the United States, European Union, and Japan.
The pharmacokinetic properties of weekly teriparatide in humans have been widely reported in the literature. Imai et al2 reported that the elimination half‐life of weekly teriparatide was significantly prolonged in subjects with severe renal impairment (estimated glomerular filtration rate, 15.0–29.9 mL/min/1.73 m2). Similarly, population pharmacokinetic analysis of weekly teriparatide revealed that renal function significantly influenced apparent total body clearance.3 In a study giving 24 weeks’ treatment with once‐weekly 56.5‐μg teriparatide, maximum plasma concentration (Cmax) value and area under the concentration‐time curve from time zero to infinity (AUCinf) value were almost the same compared to those with single dose.4
In the TOWER trial, a phase 3 trial of once‐weekly 56.5‐μg teriparatide injections, the drug showed a high reduction of fracture incidence in patients with osteoporosis.1 However, there is difficulty associated with continuing such treatment owing to adverse side effects.5 In the TOWER trial, adverse events were observed in 271 of 290 (93.4%) subjects in the safety assessment population. Commonly reported adverse events were nausea (n = 59; 20.3%), vomiting (n = 33; 11.4%), headache (n = 39; 13.4%), malaise (n = 21; 7.2%), and abdominal discomfort (n = 22; 7.6%).1 In another phase 3 trial, the 24‐month open‐label teriparatide once‐weekly efficacy research trial, 181 of 189 subjects (95.8%) in the safety assessment population experienced adverse events, including nausea (n = 69; 36.5%), vomiting (n = 42; 22.2%), headache (n = 37; 19.6%), malaise (n = 33; 17.5%), and abdominal discomfort (n = 25; 13.2%).6 As most of the adverse events leading to the discontinuation of treatment during these phase 3 trials were nausea, vomiting, and headache; therefore, the continuation of teriparatide treatment might be improved by reducing the incidence of these adverse events.
Incidences of adverse events from once‐weekly teriparatide injections during a phase 2b trial were 19.2% (14 of 73 subjects) in the 14.1‐μg dosing group, 18.7% (14 of 75) in the 28.2‐μg dosing group, and 41.7% (30 of 72) in the 56.5‐μg dosing group, indicating similar incidences between the 28.2‐μg and 14.1‐μg dosing groups, but lower than the 56.5‐μg dosing group.7 The frequency of the most commonly reported adverse event (ie, nausea) showed a dose response (5.5% [4 of 73 patients] in the 14.1‐μg group, 9.3% [7 of 75] in the 28.2‐μg group, and 20.8% [15 of 72] in the 56.5‐μg group), although the response in the 28.2‐μg group decreased to <10%.7 In addition to weekly teriparatide, it was reported that incidences of adverse events after daily teriparatide dosing were also lowered by reducing the dose by one time.8, 9 Based on these findings, a ≤28.2‐μg dose of weekly teriparatide is recommended to decrease the incidence of adverse events. Conversely, once‐weekly 28.2‐μg injections were found to be less effective in improving bone mineral density (BMD) compared to once‐weekly 56.5‐μg injections during a phase 2b trial.7 However, given the same level of exposure per week, the effects on BMD and bone strength were found to be approximately equivalent regardless of the single dose in a nonclinical study.10 Based on the above, a new dosing treatment of twice‐weekly 28.2‐μg injections was expected to maintain the same efficacy as that obtained for the once‐weekly 56.5‐μg injection while improving the safety profile.
Taking into consideration the patients’ burden of hospital visits, a twice‐weekly treatment should preferably be by self‐medication so that regular hospital visits become unnecessary. However, depending on the circumstances of the patient, keeping consistent dosing intervals when administering self‐medication twice a week can be difficult. Therefore, it is necessary to clarify the influence of different dosing intervals on the efficacy and safety under a twice‐weekly regimen.
Thus, we examined the following in healthy postmenopausal Japanese women in the present study:
Changes in pharmacokinetics, bone turnover markers, and safety profiles for twice‐weekly 28.2‐μg teriparatide injections over 6 weeks compared with once‐weekly 56.5‐μg teriparatide injections.
The influence of different dosing intervals on the pharmacokinetics, changes in bone turnover markers, and safety profiles of twice‐weekly 28.2‐μg teriparatide injections over 6 weeks.
Methods
The present study was conducted at MEDICAL Co. LTA Hakata Clinic, MEDICAL Co. LTA Sumida Hospital, and MEDICAL Co. LTA Nishi‐Kumamoto Hospital. The protocol was reviewed by the central Institutional Review Board at MEDICAL Co. LTA Hakata Clinic. Written informed consent was obtained from all subjects before enrollment in the study. The present study was conducted in compliance with the World Medical Association Declaration of Helsinki—Ethical Principles for Medical Research Involving Human Subjects and Good Clinical Practice.
Subjects
Recruitment was undertaken in August 2014, targeting 100 postmenopausal women aged 45 years or older and confirmed healthy at the time of screening. Menopause was defined as 2 or more years since the last menstruation. Key exclusion criteria were as follows:
-
1.
History of radiotherapy potentially influential on the bone
-
2.
Systolic blood pressure <90 mm Hg
-
3.
Serum calcium level >10.4 mg/dL
-
4.
History of gastrectomy
-
5.
History of hyperthyroidism, Cushing's syndrome, hyperparathyroidism, systemic steroid therapy, or urinary calculi
-
6.
Administration of drug(s) affecting bone metabolism within 8 weeks before administration of the study drug
-
7.
History of treatment with bisphosphonate(s)
-
8.
History of treatment with antireceptor activator of nuclear factor kappa‐B ligand (RANKL) antibody
-
9.
History of treatment with teriparatide agent(s)
-
10.
Receiving investigational drug(s) for the indication of osteoporosis within 1 year before administration of the study drug
-
11.
History of fracture(s) within 12 weeks before administration of the study drug
Study Design and Treatment
The study comprised 3 cohorts, all conducted as a randomized, single‐blind, placebo‐controlled, parallel group and twice‐weekly dosing design. Subject randomization for the assignment of the study drug was performed using a key code table, and saline was used for the placebo. Cohort 1 consisted of 3 groups; twice‐weekly 28.2‐μg teriparatide injection group (dosing intervals of 3 and 4 days), once‐weekly 56.5‐μg teriparatide injection group (an active control) and placebo group. Additionally, the injection was administered to 2 sites (abdomen and upper arm) simultaneously in Cohort 1. Different dosing intervals of the twice‐weekly 28.2‐μg injection regimen were examined in Cohort 2 (dosing intervals of 2 and 5 days) and Cohort 3 (1 and 6 days). Tables 1 and S1 show the method of administration for subjects in each treatment group in each cohort. The study treatment was given for 6 weeks.
Table 1.
Method of Administration in Each Treatment Group
| Cohort | Group | Injection Site | Dosing Interval | Study Drug |
|---|---|---|---|---|
| 1 | BIW | Abdomen | 3, 4 days | Teriparatide 28.2 μg |
| Upper arm | Week | Saline | ||
| QW | Abdomen | 3, 4 days | Saline | |
| Upper arm | Week | Teriparatide 56.5 μg | ||
| P | Abdomen | 3, 4 days | Saline | |
| Upper arm | Week | Saline | ||
| 2 | BIW | Abdomen | 2, 5 days | Teriparatide 28.2 μg |
| P | Abdomen | 2, 5 days | Saline | |
| 3 | BIW | Abdomen | 1, 6 days | Teriparatide 28.2 μg |
| P | Abdomen | 1, 6 days | Saline |
BIW, twice‐weekly 28.2‐μg injection group; QW, once‐weekly 56.5‐μg injection group; P, placebo group.
The purpose of Cohort 1 was to compare the changes in trough values in bone turnover markers, pharmacokinetics, and safety profile between the once‐weekly 56.5‐μg injection regime and the twice‐weekly 28.2‐μg injection regime (dosing intervals of 3 and 4 days). Cohort 2 (dosing intervals of 2 and 5 days) and Cohort 3 (1 and 6 days) aimed to compare changes in trough values in bone turnover markers, pharmacokinetics, and safety profile with other dosing intervals that were different from the twice‐weekly 28.2‐μg injection regime applied in Cohort 1. We defined bone turnover marker values at baseline (before treatment) of each injection on days 1 (defined as the day of the initial dosing), 8, 15, 22, 29, and 36, and at the corresponding time on day 43 as the trough values.
Vial formulations consisted of synthetic human parathyroid hormone 1‐34 and were used for the 28.2–μg teriparatide dosing and the active control of 56.5‐μg teriparatide dosing. In each cohort, the study drugs were administered using the administration method shown in Tables 1 and S1. When administration of 28.2‐μg teriparatide was given in the abdomen, 1 vial was dissolved with 0.45 mL saline at the time of usage, half of which was subcutaneously administered. When 56.5‐μg teriparatide was administered to the upper arm, 1 vial was dissolved in 1 mL saline at usage and the entire amount was given subcutaneously. Subjects stayed in the clinical site on each administration day. The study drug was prepared at time of use, followed by administration to each subject by the medical doctor.
Study End Points
Pharmacokinetics
Plasma samples were collected before dosing and at 0.25, 0.5, 0.75, 1, 2, 3, 4, and 6 hours after dosing on days 1 and 36. Plasma teriparatide acetate concentrations were determined by an immunoradiometric assay, as previously described.2 All samples were frozen and analyzed centrally (Sekisui Medical Corporation, Tokyo, Japan). The lower limit of quantification of teriparatide acetate in plasma was 21.46 pg/mL.
Bone Turnover Markers
Serum bone formation markers, type 1 procollagen‐N‐propeptide (P1NP) and intact osteocalcin (OC), were measured at the baseline (pretreatment) of each injection on days 1, 8, 15, 22, 29, and 36, and at the corresponding time on day 43. P1NP and OC were determined by electrochemiluminescence immunoassay11 and immunoradiometric assay,12 respectively. Serum bone resorption markers, crosslinked N‐telopeptide of type I collagen (NTX), and C‐terminal telopeptide type I collagen (CTX), were measured at the baseline (before treatment) of each injection on days 1, 8, 15 (NTX only), 22 (NTX only), 29, and 36 (NTX only) and at the corresponding time on day 43. NTX, a urinary bone resorption marker, was measured at the baseline (before treatment) of each injection on days 1, 8, 15, 22, 29, and 36, and at the corresponding time on day 43. NTX and CTX were determined by enzyme‐linked immunosorbent assay13 and electrochemiluminescence immunoassay,14 respectively. Serum was frozen, while urine was stored under refrigeration until analysis. All samples were analyzed at a central laboratory (LSI Medience Corporation, Tokyo, Japan).
Safety
Safety assessments included adverse events, vital signs, 12‐lead electrocardiogram, and clinical laboratory tests (hematology tests, a biochemical blood test, and urine analysis). Subjects were assessed for adverse events through self‐reporting, medical interviews, or other tests; and the adverse event name, date of onset, intervention, outcome, seriousness, severity, and relationship with the study drug were recorded. The adverse events reported were then classified and tabulated using the Medical Dictionary for Regulatory Activities (Version 17.1) preferred terms.
Analysis Methods
Summary statistics for plasma teriparatide acetate concentrations were calculated, as were pharmacokinetic parameters using noncompartmental analysis (WinNonlin, Version 6.1; Pharsight Corporation, Mountainview, California).
The geometric mean ratio of log‐transformed trough levels to baseline levels of each bone turnover marker (P1NP, OC, serum NTX, CTX, and urine NTX) and the 95% confidence intervals were calculated using analysis of variance in each dosing group. For the calculation, a linear mixed‐effects model was used with time points as the fixed effect and subjects as the random effect. A pooled placebo group was also analyzed in the same manner. The SAS Release 9.2 software program (SAS Institute Japan Ltd, Tokyo, Japan) was used for the analysis.
Results
Subjects
In the present study, 100 subjects were assigned to receive the study treatment. Figure 1 shows the participant flow. Of the 100 subjects receiving the study treatment, 97 subjects completed the trial and 3 subjects discontinued. The reasons behind the discontinuation of the study by these 3 subjects were adverse events in 2 subjects and the subject's wishes in the third subject. Table 2 shows the background safety population per cohort. There were no differences recognized in each item, except for the 25‐hydroxyvitamin D3 values, among each cohort and group.
Figure 1.
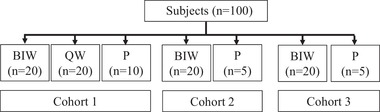
Participant flow. BIW, twice‐weekly 28.2‐μg injection group; P, placebo group; QW, once‐weekly 56.5‐μg injection group.
Table 2.
Background of Safety Population per Cohort
| Cohort 1 | Cohort 2 | Cohort 3 | Pooled | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Measure | BIW | QW | P | BIW | P | BIW | P | P | |
| Age | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (y) | Mean | 61.3 | 58.9 | 58.3 | 56.4 | 57.8 | 58.7 | 63.8 | 59.6 |
| SD | 5.8 | 5.5 | 5.1 | 4.3 | 8.2 | 5.5 | 6.3 | 6.4 | |
| Height | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (cm) | Mean | 154.7 | 154.9 | 157.3 | 156.1 | 153.6 | 155.4 | 154.7 | 155.7 |
| SD | 5.1 | 5.6 | 3.5 | 5.8 | 6.4 | 5.7 | 3.8 | 4.5 | |
| Body weight | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (kg) | Mean | 55.1 | 52.9 | 58.9 | 51.6 | 51.2 | 55.2 | 51.6 | 55.2 |
| SD | 6.5 | 6.4 | 4.9 | 7.6 | 5.2 | 7.7 | 7.0 | 6.5 | |
| BMI | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (kg/m2) | Mean | 23.0 | 22.0 | 23.8 | 21.2 | 21.7 | 22.9 | 21.6 | 22.7 |
| SD | 2.5 | 2.4 | 2.4 | 2.5 | 1.4 | 3.1 | 3.1 | 2.5 | |
| 25‐Hydroxyvitamin D3 | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (ng/mL) | Mean | 16.4 | 18.6 | 17.5 | 23.5 | 23.9 | 21.0 | 19.2 | 19.5 |
| SD | 3.8 | 4.3 | 4.7 | 4.0 | 6.0 | 5.2 | 3.6 | 5.3 | |
| Serum albumin‐corrected calcium | n | 20 | 20 | 10 | 20 | 5 | 20 | 5 | 20 |
| (mg/dL) | Mean | 9.01 | 9.06 | 8.99 | 9.17 | 9.32 | 8.95 | 8.82 | 9.03 |
| SD | 0.17 | 0.22 | 0.20 | 0.20 | 0.45 | 0.25 | 0.25 | 0.33 | |
BIW, twice‐weekly 28.2‐μg injection group; BMI, body mass index; P, placebo group; pooled P, pooled placebo group; QW, once‐weekly 56.5‐μg injection group.
Pharmacokinetics
Plasma concentration plots are provided in Figure 2 for teriparatide acetate on days 1 and 36 when teriparatide was administered once weekly at 56.5 μg or twice weekly at 28.2 μg. One subject in Cohort 2 on day 1 was excluded from the pharmacokinetic analysis because all plasma concentrations in this subject showed obvious abnormal values (below the limit of quantification), despite having received 28.2 μg of teriparatide. Plasma concentrations of teriparatide acetate on day 36 were similar to that on day 1 in all 3 twice‐weekly 28.2‐μg groups and once‐weekly 56.5‐μg groups. Pharmacokinetic parameters are shown in Table 3. Mean AUCinf and Cmax values of the 3 twice‐weekly 28.2‐μg groups on day 1 were 576.1 to 636.8 pg • hr/mL and 270.1 to 305.1 pg/mL, respectively. Mean AUCinf and Cmax values of the once‐weekly 56.5‐μg group on day 1 were 1214.5 pg • hr/mL and 461.0 pg/mL, respectively. In the twice‐weekly 28.2‐μg groups, AUCinf and Cmax were approximately half that of the once‐weekly 56.5‐μg group.
Figure 2.
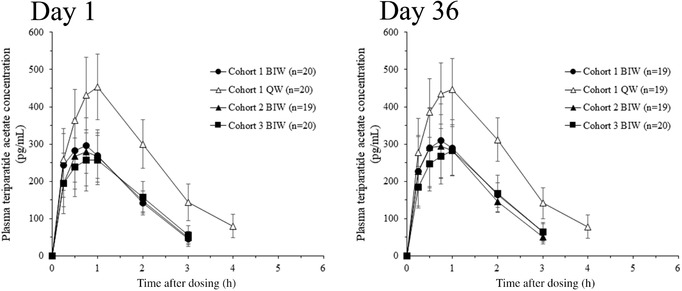
Mean and standard deviation of plasma teriparatide acetate concentration over time on days 1 and 36. Each value below the limit of quantification was set at zero in the calculation of mean values. Mean values at 6 hours after injection of teriparatide QW, and at 4 and 6 hours after injection of teriparatide BIW were below the limit of quantification. BIW, twice‐weekly 28.2‐μg injection group; QW, once‐weekly 56.5‐μg injection group.
Table 3.
Pharmacokinetic Parameters of Twice‐Weekly 28.2‐μg and Once‐Weekly 56.5‐μg Teriparatide
| Day 1 | Day 36 | |||||||
|---|---|---|---|---|---|---|---|---|
| Cohort 1 BIW | Cohort 1 QW | Cohort 2 BIW | Cohort 3 BIW | Cohort 1 BIW | Cohort 1 QW | Cohort 2 BIW | Cohort 3 BIW | |
| Cmax (pg/mL) | 305.1 ± 73.1, n = 20 | 461.0 ± 93.3, n = 20 | 288.3 ± 89.7, n = 19 | 270.1 ± 76.1, n = 20 | 317.1 ± 106.5, n = 19 | 457.0 ± 83.0, n = 19 | 314.4 ± 91.8, n = 19 | 285.1 ± 68.3, n = 20 |
| AUCinf (pg • h/mL) | 576.1 ± 91.7, n = 17 | 1214.5 ± 220.3, n = 20 | 583.6 ± 119.2, n = 16 | 636.8 ± 101.4, n = 13 | 679.8 ± 91.3, n = 16 | 1196.9 ± 170.2, n = 19 | 612.6 ± 111.3, n = 19 | 648.8 ± 128.4, n = 15 |
| t1/2 (h) | 0.738 ± 0.102, n = 17 | 1.325 ± 0.467, n = 20 | 0.822 ± 0.196, n = 16 | 0.804 ± 0.261, n = 13 | 0.747 ± 0.137, n = 16 | 1.180 ± 0.463, n = 19 | 0.789 ± 0.131, n = 19 | 0.861 ± 0.158, n = 15 |
AUCinf, area under the concentration‐time curve from time zero to infinity; BIW, twice‐weekly 28.2‐μg injection group; Cmax, maximum plasma concentration; t1/2, terminal elimination half‐life; QW, once‐weekly 56.5‐μg injection group.
Data are expressed as arithmetic mean ± standard deviation.
Bone Turnover Markers
For bone formation markers and bone resorption markers when teriparatide was administered by once‐weekly 56.5‐μg injection or twice‐weekly 28.2‐μg injections, geometric mean ratios (GMRs) of log‐transformed trough levels to baseline levels, hereinafter referred to as the GMRs (trough to baseline), were calculated using analysis of variance in each dosing group. The estimated GMRs (trough to baseline) and 95% confidence intervals are shown in Figures 3 and 4. GMRs (trough to baseline) of serum P1NP and serum OC were similarly increased to day 29 or day 36 in all 3 twice‐weekly 28.2‐μg groups and in the once‐weekly 56.5‐μg group, and then were steady until day 43 (Figure 3). The GMRs (trough to baseline) in the 3 twice‐weekly 28.2‐μg groups were comparable regardless of dosing intervals and did not change compared to the once‐weekly 56.5‐μg group. Conversely, the GMRs (trough to baseline) of serum NTX, serum CTX, and urine NTX did not increase after the 6‐week treatments in all twice‐weekly 28.2‐μg groups and in the once‐weekly 56.5‐μg group (Figure 4).
Figure 3.
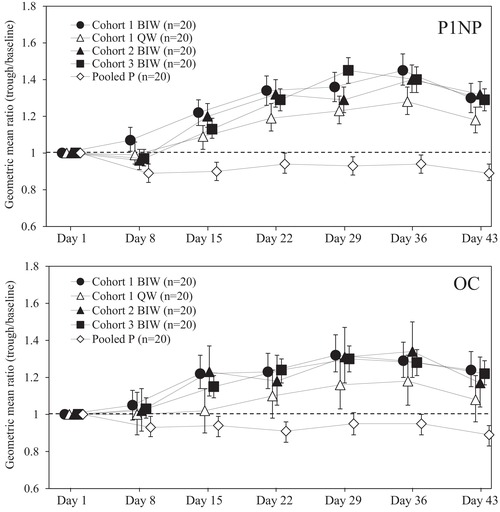
Estimated geometric mean ratios and 95% confidence intervals of trough levels to baseline levels in serum bone formation markers. Each point represents the estimated geometric mean ratio and its 95% confidence interval. The dashed line indicates the estimated geometric mean ratio as 1. BIW, twice‐weekly 28.2‐μg injection group; OC, intact osteocalcin; P1NP, type 1 procollagen‐N‐propeptide; pooled P, pooled placebo group; QW, once‐weekly 56.5‐μg injection group.
Figure 4.
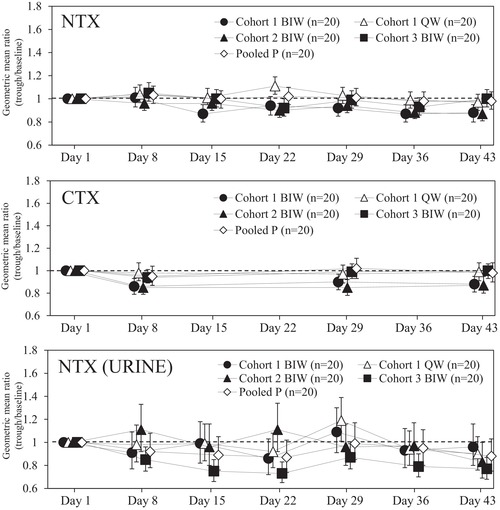
Estimated geometric mean ratios and 95% confidence intervals of trough levels to baseline levels in serum and urinary bone resorption markers. Each point represents the estimated geometric mean ratio and its 95% confidence interval. The dashed line indicates the estimated geometric mean ratio as 1. BIW, twice‐weekly 28.2‐μg injection group; CTX, C‐terminal telopeptide type I collagen; NTX, crosslinked N‐telopeptide of type I collagen; pooled P, pooled placebo group; QW, once‐weekly 56.5‐μg injection group
Safety
The incidences of adverse events in the once‐weekly 56.5‐μg injection regimen and twice‐weekly 28.2‐μg injection regimen are shown in Table 4. Most of the events, including nausea, vomiting, and headache were lower in the twice‐weekly 28.2‐μg injection treatment compared to the once‐weekly 56.5‐μg injection. Incidences of adverse events observed in the other dosing intervals of twice‐weekly 28.2‐μg treatment, ie, Cohort 2 (dosing intervals of 2 and 5 days) and Cohort 3 (1 and 6 days) were similar to that in Cohort 1 (3 and 4 days).
Table 4.
Summary of Adverse Events
| Cohort 1 | Cohort 2 | Cohort 3 | Pooled | |||||
|---|---|---|---|---|---|---|---|---|
| BIW | QW | P | BIW | P | BIW | P | P | |
| Adverse Event | n = 20 | n = 20 | n = 10 | n = 20 | n = 5 | n = 20 | n = 5 | n = 20 |
| Total | 12 (60.0) | 18 (90.0) | 0 (0.0) | 12 (60.0) | 0 (0.0) | 16 (80.0) | 0 (0.0) | 0 (0.0) |
| Nasopharyngitis | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dizziness | 1 (5.0) | 3 (15.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Headache | 5 (25.0) | 15 (75.0) | 0 (0.0) | 4 (20.0) | 0 (0.0) | 6 (30.0) | 0 (0.0) | 0 (0.0) |
| Somnolence | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Abdominal pain | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Constipation | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Dyspepsia | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Nausea | 9 (45.0) | 18 (90.0) | 0 (0.0) | 10 (50.0) | 0 (0.0) | 10 (50.0) | 0 (0.0) | 0 (0.0) |
| Vomiting | 4 (20.0) | 14 (70.0) | 0 (0.0) | 5 (25.0) | 0 (0.0) | 6 (30.0) | 0 (0.0) | 0 (0.0) |
| Pruritus | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) |
| Rash | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) |
| Arthralgia | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Chest discomfort | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Feeling abnormal | 0 (0.0) | 0 (0.0) | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Injection site erythema | 2 (10.0) | 2 (10.0) | 0 (0.0) | 3 (15.0) | 0 (0.0) | 13 (65.0) | 0 (0.0) | 0 (0.0) |
| Malaise | 2 (10.0) | 4 (20.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Alanine aminotransferase increased | 0 (0.0) | 2 (10.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Aspartate aminotransferase increased | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Blood pressure decreased | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Gamma‐glutamyltransferase increased | 0 (0.0) | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Fractured coccyx | 1 (5.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
BIW, twice‐weekly 28.2‐μg injection group; P, placebo group; Pooled P, pooled placebo group; QW, once‐weekly 56.5‐μg injection group.
Data are expressed as number of subjects (%).
Discussion
Osteoporosis is a condition characterized by impaired bone strength and increased risk of fracture.15, 16 Patients with osteoporosis‐associated fractures have shown impaired quality of life due to pain and decreased motor function, as well as aggravated life prognosis.17, 18, 19, 20 Once‐weekly 56.5‐μg teriparatide can reduce the incidence of fractures and is useful for treating patients with high risk of fractures.1 However, the drug has an issue in continuing treatment owing to adverse side effects.5 We focused on the adverse events (nausea, vomiting, and headache) caused by once‐weekly 56.5‐μg teriparatide. The purpose of the present study was to confirm whether a twice‐weekly teriparatide treatment regimen had a better safety profile compared to the once‐weekly dosing regimen; thus, we evaluated the pharmacokinetics, changes in bone turnover markers, and safety profiles of twice‐weekly teriparatide treatment using half the dose compared to the weekly full‐dose regimen. In addition, to evaluate the influence of dosing intervals on efficacy and safety, a 3‐cohort design was employed with different dosing intervals to compare pharmacokinetics, changes in bone turnover markers, and safety profiles.
The pharmacokinetic profile after dosing with once‐weekly 56.5‐μg teriparatide in the present study (Figure 2 and Table 3) was similar in comparison with previous reports: (1) AUCinf and Cmax values in the present study were almost the same compared to subjects with normal renal function and mild impairment2; and (2) no accumulation of teriparatide acetate in plasma through multiple dosing of once‐weekly 56.5‐μg teriparatide was observed in the present study as well as a study giving 24 weeks’ treatment with once‐weekly 56.5‐μg teriparatide.4
The AUCinf and Cmax were approximately half the values in the twice‐weekly 28.2‐μg injection regime than in the once‐weekly 56.5‐μg injection regime. No influence of dosing intervals on AUCinf or Cmax was observed. Therefore, the weekly AUC of the twice‐weekly 28.2‐μg injection was the same as that of the once‐weekly 56.5‐μg injection regardless of dosing intervals. Additionally, accumulation in the plasma from the twice‐weekly 28.2‐μg injection treatment was not observed regardless of dosing intervals, which is reasonable, as the pharmacokinetics of the twice‐weekly 28.2‐μg injection treatment demonstrated its plasma peak concentration of teriparatide within 1 hour followed by quick elimination.
Changes in bone turnover markers in the twice‐weekly 28.2‐μg injection treatment (dosing interval: 3 and 4 days) were similar to those in the once‐weekly 56.5‐μg injection (Figures 3 and 4). Therefore, the increase in bone formation markers was comparable between the 2 regimens, and the decrease in the bone resorption markers was also comparable even with the increased frequency of administration. Moreover, changes in the bone turnover markers in the twice‐weekly 28.2‐μg injection treatment showed the same trend as that in the once‐weekly 56.5‐μg injection treatment regardless of dosing intervals (Figures 3 and 4). In general, the efficacy of therapeutic agents for osteoporosis can be predicted from bone turnover markers.21, 22, 23, 24 Thus, a twice‐weekly 28.2‐μg injection is expected to have comparable efficacy to that of the once‐weekly 56.5‐μg injection, with the dosing interval being irrelevant to this efficacy.
Subject demographics were similar among each cohort and group; however, the values of 25‐hydroxyvitamin D3 at baseline in subjects in Cohort 1 were relatively low in comparison with subjects in other cohorts (Table 2). Dawson‐Hughes et al25 concluded that the response to teriparatide did not differ significantly in women even with baseline 25‐hydroxyvitamin D3 insufficiency. Therefore, it is reasonable to think that this unbalance of 25‐hydroxyvitamin D3 at baseline did not have a large effect on the exploratory evaluation of efficacy.
Changes in the bone turnover markers were similar between once‐weekly 56.5‐μg injections and twice‐weekly 28.2‐μg injections, with those in the twice‐weekly 28.2‐μg injections comparable regardless of dosing intervals. Takakura et al10 showed that an increase in the administration frequency of teriparatide increased the level of urinary CTX and the development of cortical porosity in ovariectomized rats. It was also reported in rabbits that the once‐daily administration of teriparatide induced a marked increase in the cortical porosity, which was not observed following the once‐weekly administration of equivalent total weekly doses.26, 27 Based on these previous studies, the shortening of dosing intervals to more than twice‐weekly 28.2‐μg injections in humans might induce the elevation of bone resorption markers, resulting in cortical porosity.
Weekly AUC in the twice‐weekly 28.2‐μg injection was the same as that in the once‐weekly 56.5‐μg injection treatment regardless of dosing intervals. Additionally, changes in the bone turnover markers in the twice‐weekly 28.2‐μg injection treatment did not differ with once‐weekly 56.5‐μg injection regardless of the dosing intervals. Therefore, the pharmacokinetic parameter related to the efficacy of teriparatide in the once‐weekly 56.5‐μg and twice‐weekly 28.2‐μg injection treatments was weekly AUC, not Cmax. This is consistent with a previous study.3
Commonly reported adverse events in the once‐weekly 56.5‐μg injections were nausea, vomiting, and headache.1 Incidences of these adverse events in the twice‐weekly 28.2‐μg injection group were lower than those in the once‐weekly 56.5‐μg injection group regardless of dosing intervals (Table 4). Therefore, reduction in treatment discontinuation owing to adverse events can be expected in the new dosing regimen (twice‐weekly 28.2‐μg injections), suggesting its contribution to improving the rate of continuous treatment with teriparatide. In the pharmacokinetic profile observed in our study, Cmax of plasma teriparatide acetate concentrations was approximately halved. This might cause adverse events such as nausea to be reduced, even though the dosing time of twice‐weekly 28.2‐μg injection is twice that of the once‐weekly 56.5‐μg injection. Consequently, Cmax, not weekly AUC, is suggested as being an important indicator to control the development of adverse events.
Thus, the new dosing regimen of twice‐weekly 28.2‐μg teriparatide injections maintained comparable efficacy to once‐weekly 56.5‐μg teriparatide injections and demonstrated a more favorable safety profile. However, the evaluation of the efficacy in this study was exploratory while bone turnover markers were selected as indicators of efficacy, BMD was not. Furthermore, subjects in the study were healthy postmenopausal women and the study was conducted at a relatively small‐scale setting. Therefore, it is necessary to examine the BMD and safety profiles whether similar discussions are feasible in other clinical studies enrolling more patients with osteoporosis.
Conclusions
The injections of 28.2‐μg teriparatide twice‐weekly showed roughly halved Cmax in comparison with once‐weekly 56.5‐μg injections. Weekly AUC and change of trough values in bone turnover markers were similar in both twice‐weekly 28.2‐μg injection groups and once‐weekly 56.5‐μg injection group. The incidence of adverse events were lower in the twice‐weekly 28.2‐μg injection group compared with the once‐weekly 56.5‐μg injection group. These findings were similar to those found in the twice‐weekly 28.2‐μg injection groups regardless of the dosing interval. Therefore, the new dosing regimen of twice‐weekly 28.2‐μg teriparatide injections maintained comparable efficacy to once‐weekly 56.5‐μg teriparatide injections and improved the safety profile, suggesting its contribution to improving the rate of continuous treatment with teriparatide.
Declaration of Conflicting Interests
YK received consulting fees from Asahi Kasei Pharma. AO and KT are employees of Asahi Kasei Pharma. TS received research grants from Astellas Pharma, Eisai, Daiichi‐Sankyo, Chugai Pharmaceutical, and Eli Lilly Japan as well as consulting and/or lecture fees from Asahi Kasei Pharma and Daiichi‐Sankyo.
Funding
This work was funded by Asahi Kasei Pharma Corporation.
Supporting information
Supplemental Table 1 Method of administration in each treatment group
Acknowledgments
The authors acknowledge the subjects for their participation in this study. The authors thank the investigators and the staff at MEDICAL Co. LTA Hakata Clinic, MEDICAL Co. LTA Sumida Hospital, and MEDICAL Co. LTA Nishi‐Kumamoto Hospital for their support in performing this study. The authors also thank Drs. Toshitaka Nakamura and Masataka Shiraki for their valuable expertise during this study.
References
- 1. Nakamura T, Sugimoto T, Nakano T, et al. Randomized Teriparatide [human parathyroid hormone (PTH) 1–34] Once‐Weekly Efficacy Research (TOWER) trial for examining the reduction in new vertebral fractures in subjects with primary osteoporosis and high fracture risk. J Clin Endocrinol Metab. 2012;97(9):3097‐3106. [DOI] [PubMed] [Google Scholar]
- 2. Imai H, Watanabe M, Fujita T, Watanabe H, Harada K, Moritoyo T; ANTCliPh (Academic Network for Trials in Clinical Pharmacology) Trial 04 Study Group . Pharmacokinetics of teriparatide after subcutaneous administration to volunteer with renal failure: a pilot study. Int J Clin Pharmacol Ther. 2014;52(2):166‐174. [DOI] [PubMed] [Google Scholar]
- 3. Ose A, Serada M, Yamashita K, Tsurui K, Tanigawara Y. Population pharmacokinetic and exposure‐response analysis of weekly teriparatide in osteoporosis patients. J Clin Pharmacol. 2017;57(12):1545‐1553. [DOI] [PubMed] [Google Scholar]
- 4. Sugimoto T, Nakamura T, Nakamura Y, Isogai Y, Shiraki M. Profile of changes in bone turnover markers during once‐weekly teriparatide administration for 24 weeks in postmenopausal women with osteoporosis. Osteoporos Int. 2014;25(3):1173‐1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Usui T, Funagoshi M, Seto K, Ide K, Tanaka S, Kawakami K. Persistence of and switches from teriparatide treatment among women and men with osteoporosis in the real world: a claims database analysis. Arch Osteoporos. 2018;13(1):54. [DOI] [PubMed] [Google Scholar]
- 6. Sugimoto T, Shiraki M, Fukunaga M, et al. 24‐month open‐label teriparatide once‐weekly efficacy research trial examining bone mineral density in subjects with primary osteoporosis and high fracture risk. Adv Ther. 2017;34(7):1727‐1740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fujita T, Inoue T, Morii H, et al. Effect of an intermittent weekly dose of human parathyroid hormone (1‐34) on osteoporosis: a randomized double‐masked prospective study using three dose levels. Osteoporos Int. 1999;9(4):296‐306. [DOI] [PubMed] [Google Scholar]
- 8. Neer RM, Arnaud CD, Zanchetta JR, et al. Effect of parathyroid hormone (1‐34) on fractures and bone mineral density in postmenopausal women with osteoporosis. N Engl J Med. 2001;344(19):1434‐1441. [DOI] [PubMed] [Google Scholar]
- 9. Miyauchi A, Matsumoto T, Shigeta H, Tsujimoto M, Thiebaud D, Nakamura T. Effect of teriparatide on bone mineral density and biochemical markers in Japanese women with postmenopausal osteoporosis: a 6‐month dose‐response study. J Bone Miner Metab. 2008;26(6):624‐634. [DOI] [PubMed] [Google Scholar]
- 10. Takakura A, Lee JW, Hirano K, et al. Administration frequency as well as dosage of PTH are associated with development of cortical porosity in ovariectomized rats. Bone Res. 2017;5:17002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lüftner D, Jozereau D, Schildhauer S, et al. PINP as serum marker of metastatic spread to the bone in breast cancer patients. Anticancer Res. 2005;25(3A):1491‐1499. [PubMed] [Google Scholar]
- 12. Price PA, Nishimoto SK. Radioimmunoassay for the vitamin K–dependent protein of bone and its discovery in plasma. Proc Natl Acad Sci U S A. 1980;77(4):2234‐2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hanson DA, Weis MA, Bollen AM, Maslan SL, Singer FR, Eyre DR. A specific immunoassay for monitoring human bone resorption: quantitation of type I collagen cross‐linked N‐telopeptides in urine. J Bone Miner Res. 1992;7(11):1251‐1258. [DOI] [PubMed] [Google Scholar]
- 14. Okabe R, Nakatsuka K, Inaba M, et al. Clinical evaluation of the Elecsys beta‐CrossLaps serum assay, a new assay for degradation products of type I collagen C‐telopeptides. Clin Chem. 2001;47(8):1410‐1414. [PubMed] [Google Scholar]
- 15. Assessment of fracture risk and its application to screening for postmenopausal osteoporosis. Report of a WHO study group. WHO technical report series 1994;843. [PubMed] [Google Scholar]
- 16. NIH Consensus Development Panel on Osteoporosis Prevention, Diagnosis, and Therapy. Osteoporosis prevention, diagnosis, and therapy. JAMA. 2001;285(6):785‐795. [DOI] [PubMed] [Google Scholar]
- 17. Bliuc D, Nguyen ND, Milch VE, Nguyen TV, Eisman JA, Center JR. Mortality risk associated with low‐trauma osteoporotic fracture and subsequent fracture in men and women. JAMA. 2009;301(5):513‐521. [DOI] [PubMed] [Google Scholar]
- 18. Johnell O, Kanis JA, Odén A, et al. Mortality after osteoporotic fractures. Osteoporos Int. 2004;15(1):38‐42. [DOI] [PubMed] [Google Scholar]
- 19. Hasserius R, Karlsson MK, Nilsson BE, Redlund‐Johnell I, Johnell O; European Vertebral Osteoporosis Study . Prevalent vertebral deformities predict increased mortality and increased fracture rate in both men and women: a 10‐year population‐based study of 598 individuals from the Swedish cohort in the European Vertebral Osteoporosis Study. Osteoporos Int. 2003;14(1):61‐68. [DOI] [PubMed] [Google Scholar]
- 20. Chrischilles EA, Butler CD, Davis CS, Wallace RB. A model of lifetime osteoporosis impact. Arch Intern Med. 1991;151(10):2026‐2032. [PubMed] [Google Scholar]
- 21. Chen P, Satterwhite JH, Licata AA, et al. Early changes in biochemical markers of bone formation predict BMD response to teriparatide in postmenopausal women with osteoporosis. J Bone Miner Res. 2005;20(6):962‐970. [DOI] [PubMed] [Google Scholar]
- 22. Diez‐Perez A, Naylor KE, Abrahamsen B, et al. Adherence Working Group of the International Osteoporosis Foundation and the European Calcified Tissue Society. International Osteoporosis Foundation and European Calcified Tissue Society Working Group. Recommendations for the screening of adherence to oral bisphosphonates. Osteoporos Int. 2017;28(3):767‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tsujimoto M, Chen P, Miyauchi A, Sowa H, Krege JH. PINP as an aid for monitoring patients treated with teriparatide. Bone. 2011;48(4):798‐803. [DOI] [PubMed] [Google Scholar]
- 24. Mori Y, Kasai H, Ose A, et al. Modeling and simulation of bone mineral density in Japanese osteoporosis patients treated with zoledronic acid using tartrate‐resistant acid phosphatase 5b, a bone resorption marker. Osteoporos Int. 2018;29(5):1155‐1163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Dawson‐Hughes B, Chen P, Krege JH. Response to teriparatide in patients with baseline 25‐hydroxyvitamin D insufficiency or sufficiency. J Clin Endocrinol Metab. 2007;92(12):4630‐4636. [DOI] [PubMed] [Google Scholar]
- 26. Yamane H, Takakura A, Shimadzu Y, et al. Acute development of cortical porosity and endosteal naive bone formation from the daily but not weekly short‐term administration of PTH in rabbit. PLoS One 2017;12:e0175329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zebaze R, Takao‐Kawabata R, Peng Y, et al. Increased cortical porosity is associated with daily, not weekly, administration of equivalent doses of teriparatide. Bone. 2017;99:80‐84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1 Method of administration in each treatment group