

Abstract
Gastric cancer (GC) is the third leading cause of cancer deaths and the fourth most prevalent malignancy worldwide. The high incidence and mortality rates of gastric cancer result from multiple factors such as ineffective screening, diagnosis, and limited treatment options. In our study, we sought to systematically identify predictive molecular networks and key regulators to elucidate complex interacting signaling pathways in GC. We performed an integrative network analysis of the transcriptomic data in The Cancer Genome Atlas (TCGA) gastric cancer cohort and then comprehensively characterized the predictive subnetworks and key regulators by the matched genetic and epigenetic data. We identified 221 gene subnetworks (modules) in GC. The most prognostic subnetworks captured multiple aspects of the tumor microenvironment in GC involving interactions among stromal, epithelial and immune cells. We revealed the genetic and epigenetic underpinnings of those subnetworks and their key transcriptional regulators. We computationally predicted and experimentally validated specific mechanisms of anticancer effects of GKN2 in gastric cancer proliferation and invasion in vitro. The network models and the key regulators of the tumor microenvironment in GC identified here pave a way for developing novel therapeutic strategies for GC.
Keywords: gastric cancer, tumor microenvironment, gene coexpression network, network module, key driver, DNA methylation, somatic mutations
Short abstract
What's new?
Gene signatures have been identified for diagnosis and classification of gastric cancer (GC) as well as prediction of therapeutic response. However, key molecular mechanisms underlying prognosis remain to be revealed. Our study systematically identifies and characterizes predictive molecular networks and key regulators. The most prognostic subnetworks capture multiple aspects of the tumor microenvironment in GC involving interactions among stromal, epithelial, and immune cells. The authors computationally predicted and experimentally validated specific mechanisms of anti‐cancer effects of GKN2 in GC proliferation and invasion in vitro. These network models and key regulators pave the way for developing novel therapeutic strategies for GC.
Abbreviations
- DMR
differentially methylated region
- EBV
Epstein–Barr virus
- EBV‐HEMG
EBV‐specific hyper‐methylation gene signature
- EBV‐HOMG
EBV‐specific hypo‐methylation gene signature
- eMSGs
expression associated methylation site genes
- EMT
epithelial‐mesenchymal transition
- FC
expression fold change
- FE
fold enrichment
- FET
Fisher's Exact Test
- GC
gastric cancer
- GEO
gene expression omnibus
- GIST
gastrointestinal stromal tumor
- GMB
gastric mucosal barrier
- IMAC
intermicrovillar adhesion complex
- IMB
intestinal mucosal barrier
- SMGs
somatic mutation associated genes
- TCGA
The Cancer Genome Atlas
- TCGA‐GCC
TCGA gastric cancer discovery cohort
- vTCGA‐GCC
TCGA gastric cancer validation cohort
Introduction
Gastric cancer (GC) is the third leading cause of cancer death and the fourth most prevalent malignancy worldwide, accounting for 8% of cancer incidence and 10% of cancer deaths, and approximately 21,320 cases of GC (13,020 men and 8,300 women) were diagnosed and 10,540 patients died from GC in 2012 in the United States.1 The molecular mechanisms driving tumorigenesis of GC include a number of biological and cellular processes activated in tumor pathogenesis such as proliferation, angiogenesis, the bypass of senescence and cell death pathways, evasion of tumor suppressing mechanisms, immortality, invasion and so on.2 leading inconsistent treatment responses and marginal improvements.3
High‐throughput molecular profiling data makes it possible to dissect the heterogeneity of GC in a comprehensive and unbiased manner.3 A number of gene signatures have been identified for diagnosis and classification of GC as well as prediction of therapeutic response.3 However, the reproducibility of such gene signature‐based models are usually poor due to multiple factors including limited cross validation of predictive gene lists per tumor type and outcome.4 Recently, The Cancer Genome Atlas (TCGA), a comprehensive multi‐Omics cohort for studying multiple cancers was developed. TCGA includes genomic, transcriptomic and epigenomic molecular data of primary gastric adenocarcinomas. Analysis of the TCGA gastric cancer data stratifies five distinct molecular subtypes GC, specifically, Epstein–Barr virus positive (EBV), microsatellite instability (MSI), genomically stable (GS) and chromosomal instability (CIN) and these subtypes complement histopathological classifications by their distinctive patterns of DNA methylation, somatic genomic alterations and gene/protein expressions.5
Network biology has been successfully established to systematically model molecular interactions underlying complex human diseases.6 Here, we employed an integrative multiscale gene network analysis framework to the gastric cancer data in The Cancer Genome Atlas (TCGA‐GCC)5 to reveal key molecular mechanisms underlying GC prognosis in a data‐driven manner. We postulate that these key pathways emerge as coherent modules and associate to respective key genetic and/or epigenetic alterations. Combination of these key molecular events will further generate hypothesis on multiple molecular “niche” exploited in GC etiology. Note that all the p‐values reported in the manuscript were corrected for multiple testing unless stated otherwise.
Materials and Methods
Bioinformatics analysis
Gene expression data processing
Ilumina HiSeq RNA Sequencing data, processed by Reads per kilo base per million (RPKM) method from TCGA, have been downloaded and comprehensive data quality control (QC) has been performed: log2(RPKM +1) transform, quantile‐normalization, correction for batch effects by center, platform and tissue source site (TSS) ids from TCGA sample barcodes, and correction for confounding factors including ethnic group, age and gender, resulting to 218 annotated primary tumor tissue samples across 26,539 genes.
Integrative network analyses of TCGA GC cohort
In order to handle multifaceted, large‐scale –omics data for TCGA GC cohort (TCGA‐GCC; see Supporting Information Table S1 for cohort description), we developed an integrative network analysis framework to prioritize altered pathways in GC etiology (illustrated in Supporting Information Fig. S1). The backbone of the framework work lies in constructing multiscale coexpression network (Supporting Information Fig. S1C) by Multiscale Embedded Gene Co‐expression Network Analysis (MEGENA).7 Briefly, MEGENA first selects gene pairs with significant correlations (FDR < 0.05) and then embeds them onto a three‐dimensional topological sphere, leading to planar filtered network (PFN). Multiscale clustering is performed on PFN to unveil gene modules varying compactness, leading to a hierarchy of parent and child modules. The gene modules are annotated by enrichments of known pathway and functions from MSigDB8 (Supporting Information Fig. S1d).
Then, we generated a compendium of gene signatures reflecting genomic and epigenomic alterations (Supporting Information Fig. S1b; see Supporting Materials and Methods in Supporting Material 1). These gene signatures were intersected with gene modules to test enrichments. For instance, mutation signature is identified as differentially expressed genes in somatic mutants of a gene through limma9 with FDR < 0.05 and fold change >1.2. Gene modules enriched for the mutation signatures are then associated to this somatic gene mutation.
The gene modules were prioritized by associations to overall and recurrence‐free survival across the whole TCGA‐GCC as well as within clinical subtypes (Supporting Information Fig. S2). For each group of patients, the association between a module and an outcome was tested by two approaches. First, the module eigen‐gene10 (i.e., first principal component of module gene expressions) was modeled by univariate Cox proportional hazard model.11 Second, median expression of the module eigen‐gene was used to stratify patients into low‐ and high‐expression groups, and these were tested for significant difference in outcomes by logrank test (see Supporting Materials and Methods in Supporting Material 1). Overall, the importance of each gene module is determined by a score MS m that summarizes the module's correlations to GC survival by
where m = mth module, T = number of tests, p t = Cox or logrank p‐values.
To validate the network interactions of the nominated drivers, differentially expressed genes by respective gene perturbation from in vivo/in vitro experiments were projected to the network neighbor genes to test for enrichments (Supporting Information Fig. S1f; see Supporting Materials 1).
Experimental procedures
Patients and specimens
One hundred and four patients who underwent gastric carcinoma resection between July 2012 and April 2015 were recruited in our study. The study protocol was approved by the Research Ethics Committee of the Fujian Provincial Cancer Hospital, and informed consent was obtained from all participants (namely, Fujian cohort; Approval number: SQ2015‐068‐01). There were 82 men and 22 women with a median age of 58.2 years (interquartile range, 22.0–82.0 years) (see Supporting Information Table S2). Histologic type was determined according to Lauren.12 None of the patients had received any chemotherapy prior to surgery. Fresh tumor tissues and adjacent nontumorous stomach tissues were obtained immediately after tumor resection. One part of the tissues was immediately snap‐frozen in liquid nitrogen and stored at −80°C, and the other part was fixed in 10% buffered formalin and embedded in paraffin.
Cell lines
Human gastric cancer cell line (AGS ‐ RRID:CVCL_0139) was purchased from the Cell Banks, the Chinese Academy of Sciences (Shanghai, China). AGS cell line was authenticated by DNA finger printing analysis. AGS was grown in RPMI1640 (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA), supplemented with 10% fetal bovine serum (FBS) (Gibco; Thermo Fisher Scientific, Inc.). AGS was cultured in a 5% CO2 incubator at 37°C. When the cells reached the logarithmic growth phase, succeeding experiments were performed. All experiments were performed with mycoplasma‐free cells.
Ethics approval and consent to participate
The study protocol for Fujian cohort was approved by the Research Ethics Committee of the Fujian Provincial Cancer Hospital, and informed consent was obtained from all participants (approval number: SQ2015‐068‐01).
Data availability
The original gene expression data, analyzed as “TCGA‐GCC” and “vTCGA‐GCC,” that support findings of our study are available from TCGA data portal (https://portal.gdc.cancer.gov/). Respective patient barcodes for TCGA‐GCC and vTCGA‐GCC are provided in Supporting Material 2C within this manuscript. Another independent validation data, GSE84437, that support findings our study, are available from Gene Expression Omnibus (GEO) with the accession code “GSE84437.”
The primary software MEGENA (version 1.3.7) for network analysis is publicly available as R package in The Comprehensive R Archive Network (CRAN) (https://cran.r-project.org/web/packages/MEGENA/index.html). The development version of MEGENA is available from GitHub repository (https://github.com/songw01/MEGENA).
Results
Multiscale gene coexpression network of gastric cancer
We developed an integrative network analysis framework to analyze multi‐Omics data in the TCGA gastric cancer cohort (denoted TCGA‐GCC; see Materials and Methods), using Multiscale Embedded Gene co‐Expression Network Analysis (MEGENA)7 (Fig. 1 a). Two hundred and twenty‐one modules pertaining a parent–child hierarchy were identified and were associated with known pathways (e.g., cell cycles, extra‐cellular matrix, focal adhesions, immune system process, DNA metabolic process, proteasome, biological oxidations) or reflected some unknown biological processes (see Supporting Material 2A). These modules were prioritized by their prognostic power of survival in the whole TCGA‐GCC population as well as various subtypes via Cox proportional hazard model (Fig. 1 b; Supporting Information Fig. S2; see Materials and Methods). Among 221 modules, top five modules were further interrogated (M666, M434, M28, M226 and M121). Taking account of module overlaps by the hierarchy, the biology of top five modules was effectively captured within M121/M666 (associated with intestinal brush border), M434 (associated with digestion) and M28 (associated with epithelial cell differentiation) and respective module hub genes were nominated as key drivers (Fig. 1 c).
Figure 1.
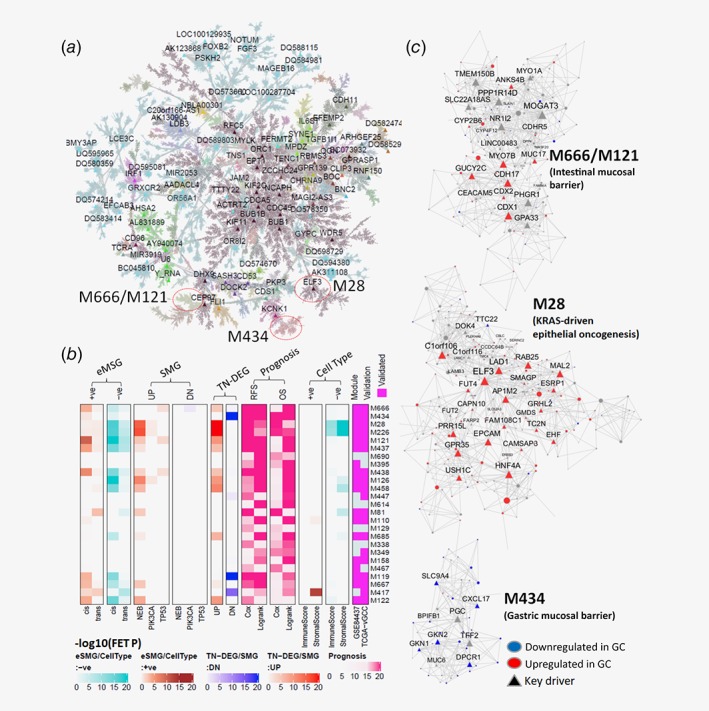
Multiscale coexpression network of TCGA‐GCC illustrates GC microenvironment. (a) TCGA‐GCC coexpression network: A global Planar Filtered Network (PFN) of the primary tumor gene expression data from TCGA gastric cancer. Nodes are colored by MEGENA modules identified at the default compactness scale α = 1. Global hub genes are labeled. (b) Heatmap for enrichments (i.e., ‐log10(FET p‐value)) of key molecular features of the top 25 gene modules most predictive of survival. The color bars are shown below. The columns are organized into several different categories including (right to left): (i) Module conservation (columns 1,2) in GSE84437 and vTCGA‐GCC. (ii) Cell type specificity (tracks 3–6): modules enriched for genes correlated to immune and/or stromal scores inferred by ESTIMATE. Negative (−ve) and positive (+ve) correlation signatures are in columns 3–4 and 5–6. (iii) Prognosis power of a module defined as ∑ − log10(Cox/logrank p‐value) based on overall survival in the whole cohort and in subtypes (column 7–8) (track 8), and based on recurrence‐free survival in the whole cohort and subtypes (columns 9–10); (iv) Enrichments of tumor vs normal DEGs, down‐/up‐regulated in tumor (column 11/12) (vi) Enrichments of key gene SMGs: module enrichment for the SMGs down‐/up‐regulated (DN: columns 13‐15/UP: columns 16–18) by the mutations in NEB, PIK3CA or TP53; (vii) eMSG: module enrichments of negatively (columns 19–20) or positively (columns 21–22) correlated cis‐eMSGs and trans‐eMSGs. (c) Subnetworks of top modules enriched in down‐/up‐regulated (blue/red) DEGs in tumor, compared to normal tissue. Network key drivers are labeled as triangles. Module names are respective locations in the global networks are labeled. [Color figure can be viewed at http://wileyonlinelibrary.com]
Reproducibility of the TCGA‐GCC modules was assessed using two additional independent cohorts. As TCGA‐GCC has expanded with more samples during our study, we collected the RNA‐seq data from an additional 135 primary gastric tumor samples (termed vTCGA‐GCC) to verify the findings from the original TCGA‐GCC study. Also, we collected a microarray based gene expression study of 433 gastric cancer patients (Illumina HumanHT‐12 V3.0 expression beadchip) from GEO with an accession number GSE84437 (denoted as GSE84437). The module preservation analysis13 showed that 76.0% (168) and 94.6% (209) of the 221 TCGA‐GCC modules were significantly preserved in GSE84437 and vTCGA‐GCC, respectively (Fig. 1 b and Supporting Material 2B).
The most prognostic modules capture multiple facets of GC tumor microenvironment
To characterize the GC network modules, we identified genetic, epigenetic and cell type gene signatures associated with GC. We systematically identified gene signatures as potential functional manifestation of nonsynonymous somatic mutations as differentially expressed genes between the mutants and wildtypes (Somatic Mutation associated Genes (SMGs); see the section Identification of Somatic Mutation Associated Gene Signatures in Supporting Material 1). Also, gene signatures correlated to cis−/trans‐methylation sites were identified (expression associated methylation site gene (eMSGs); see Extraction of cis−/trans‐methylation significantly correlated gene signatures in Supporting Material 1). These signatures were then used to characterize genetic and/or epigenetic alterations in the previously identified gene modules and key regulators using Fisher's Exact Test (FET) (Fig. 1 b; SMGs: Supporting Material 2B, eMSGs: Supporting Material 7). We further inferred abundances of stromal and immune cell components in the primary tumors by using ESTIMATE14 and CIBERSORT,15 respectively. Gene signatures significantly correlated with cell type compositions were identified and then projected onto gene modules to determine their cell type specificity (Fig. 1 b; see Supporting Material 4).
Interestingly, the top modules of TCGA‐GCC network suggest strong cross‐talks among different cell types in GC tumor microenvironment (Fig. 5). As summarized in Figure 5a, our data reveal these modules are regulated by somatic mutations in NEB and PIK3CA, epigenetic alterations (methylation changes by EBV infection) and loss of GKN1/2 and TFF1/2 expressions. The complex signaling network across stromal, epithelial and immune cells demonstrates activated or suppressed key oncogenic/tumor suppressive pathways in GC such as epithelial‐mesenchymal (EMT), gastrointestinal mucosal barrier and cytotoxic CD8+ T‐cell/Natural Killer (NK) cell activation (Fig. 5a, Table 1). Furthermore, eigen‐genes of these modules (i.e., first principal components of gene modules)10 are significantly correlated with each other, suggesting cross‐talks (Fig. 5b).
Table 1.
Summaries of top modules of TCGA‐GCC and their functions in GC microenvironment
| Key module | Associated pathways and functions |
|---|---|
| M28: GC carcinogenesis, intestinal fibrosis |
|
| M666/M121: intestinal mucosa homeostasis |
|
| M102: NK‐/T‐cell cytotoxic pathway |
|
| M434: gastric mucosal protection, GKN1/2‐TFF1/2 axis |
|
In the rest of the article, we will focus on the modules shown in Figure 1 c and comprehensively investigate their network structures and biological implications as well as their interactions in the context of GC tumor microenvironment.
Deactivation of gastric mucosal barrier drives GC proliferation and invasion
The second ranked module, M434, represents downregulation of tumor suppressors such as TFF2, GKN1 and GKN2 in gastric mucosal barrier (GMB) homeostasis in GC (Fig. 1 c). M434 was highly enriched for the genes downregulated in tumor compared to adjacent normal tissue (Bonferroni corrected FET p = 1.04E‐25, 9.25 fold enrichment (FE)) and highly reproduced in vTCGA‐GCC and GSE84437 with high overlaps (% overlap with vTCGA‐GCC = 43%, GSE84437 = 48%).
Suppression of the genes in M434 is associated with poor overall survival in GC and increased tumor sizes. For instance, lower expression levels of the key drivers GKN1 and GKN2 are associated with poor survival in node positive cases in TCGA‐GCC (Fig. 2 a). Within node positive patients, tumor weights in the group with median‐low GKN1 or GKN2 expressions were significantly higher than those in the group with median‐high GKN1 expression (Supporting Information Fig. S3b ), but are independent of pathological T stage (Supporting Information Fig. S3c ). We confirmed prognostic significance of GKN1/2 in an independent km‐plotter cohort17 (Supporting Information Fig. S3e ).
Figure 2.
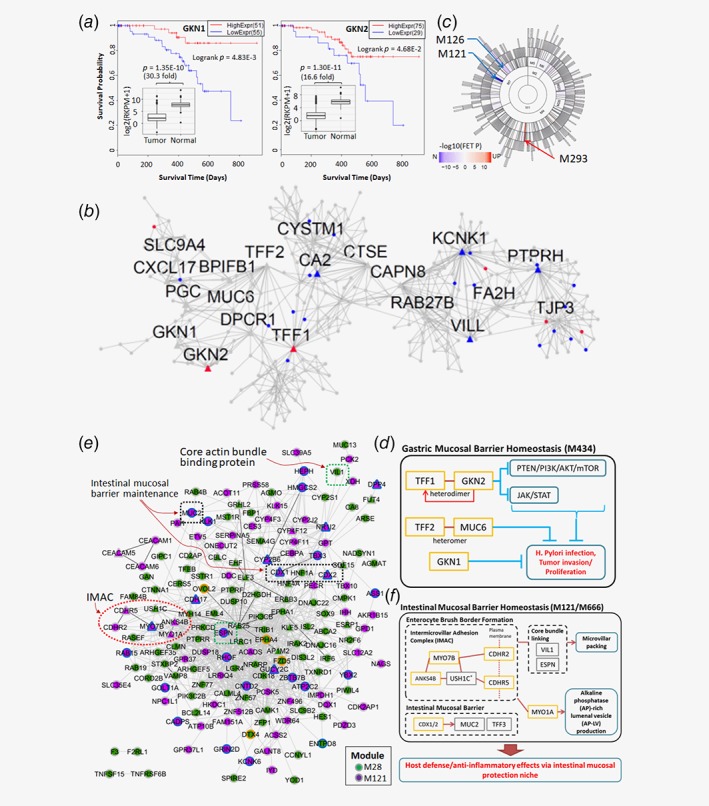
(a‐c) Gastric Mucosal Barrier (GMB) module, M434, is suppressed in primary GC. (a) Prognostic significance of M434 key drivers, GKN1/2, in TCGA‐GCC. Kaplan–Meier plots of node positive patients in TCGA‐GCC by GKN1/2 expressions. Significant suppressions of GKN1/2 in primary compared to adjacent normal tissues are shown as embedded mini‐plot with relevant t‐test statistics on the top. (b) 5‐layer neighborhood network of GKN2 in TCGA‐GCC MEGENA. Up‐/down‐regulated genes in GKN2 overexpressed AGS cells shown in red/blue. (c) Significant enrichment of up‐/down‐regulated genes in GKN2 overexpression in TCGA‐GCC MEGENA modules. Hierarchical organization of MEGENA modules are illustrated in the sunburst plot, fill colors representing –log10(FET p‐value) of up−/down‐regulated signatures in blue/red, respectively. Most significantly enriched modules are labeled by arrows, namely M121, M294 and M126. (d) GMB signaling pathways, captured by key drivers of M434. (e) F: Intestinal Mucosal Barrier (IMB) modules, M121/M666, are suppressed through hyper‐methylation by EBV infection. Experimentally validated protein–protein interactions from the String database16 are intersected with M28 and M121. Node colors show module membership (M28: green, M121: purple), border colors show hyper‐/hypo‐methylation (hyper: blue, hypo: orange), and dotted box shows specific pathways/complexes in IMB niche. Network structure of M666, a daughter module of M121. The red and blue nodes are the genes up‐ and down‐regulated in EBV subtype compared to other molecular subtypes, respectively. (f) IMB pathways captured by M121/M666. Some components of IMB are from GC epithelial module, M28 (USH1C and VIL1). [Color figure can be viewed at http://wileyonlinelibrary.com]
Novel downstream functions of overexpressing GKN2 in GC
M434 captures a tumor suppressive axis TFF1‐TFF2‐GKN1‐GKN2 (Fig. 1 c), where TFF2, GKN1/2 are found in M434, and TFF1 is found at a neighboring module directly connected to M434 (Supporting Information Fig. S3f ). Within the TFF1‐TFF2‐GKN1‐GKN2 axis, we investigated functional impacts of overexpressing GKN2 in GC, as detailed downstream mechanisms of GKN2 are largely unknown, compared to other genes in the axis. We first comprehensively validated clinic‐pathological features of GKN2 expression in an independent cohort of 104 patients who underwent gastric carcinoma resection in the Fujian Provincial Cancer Hospital (Fujian cohort; see Patients and Specimens in Materials and Methods, Supporting Material 1). The correlations between GKN2 gene expression in cancer tissues and pathological features are shown in Supporting Information Table S2. GKN2 mRNA expression was significantly correlated with tumor size and location with p = 0.023 and 0.006, respectively. The average mRNA level of GKN2 in the group with large tumors was 5.2‐fold lower than the group smaller tumors while the average expression in the patients with non‐EGJ (esophagogastric junction) was 3.8‐fold higher than EGJ.
Next, we transfected human gastric cancer cell line AGS, with lenti‐virus containing GKN2 (GKN2‐LV), and performed RNA‐sequencing of GKN2‐LV cells with the control (ctrl‐LV) (see Supporting Materials and Methods, Supporting Material 1; Supporting Information Fig. S6). We confirmed GKN2 over‐expression confers antitumoral effect and inhibiting invasion in AGS by inhibiting PTEN/PI3K/AKT/mTOR and JAK/STAT axes (see Supporting Results, Supporting Material 1; Supporting Information Figs. S7, S8, S9 and S10). Using edgeR 18 (see Supporting Materials and Methods, Supporting Material 1), we identified 253 upregulated and 401 downregulated genes (FDR < 0.05 and fold change >1.2 or < 0.833) that represent the down‐stream pathways of GKN2 over‐expression in GC (Supporting Information Fig. S3a ; Supporting Material 4; Fig. S7).
The altered genes by GKN2‐LV were captured in GKN2's neighborhood in the TCGA‐GCC network. The downregulated genes were significantly enriched in GKN2's network neighborhood (Fig. 2 b). Interestingly, TFF1 was upregulated by GKN2 overexpression (Fig. 2 a,b), known synergistic heterodimeric interaction partner of GKN2 conferring antiproliferative and proapoptotic effects on gastric cancer cells.19 This suggests GKN2 alone is capable of conferring antitumor effect.
We systematically identified de novo downstream functions of GKN2 as enriched gene modules by the GKN2 overexpression signatures (Fig. 2 c). M121 and M126 were most significantly downregulated whereas M293 was most significantly upregulated. M121 represents intestinal niche/fibrosis in GC as discussed in later sections, and M293 is enriched for cholesterol biosynthesis pathway.
GC epithelium interacts with stromal cells via an actin‐binding protein, Nebulin
The third ranked module, M28 (Fig. 1 c) contains several GC carcinogenic pathways activated in GC epithelium. M28 is upregulated in tumor compared to adjacent normal tissue (Boferroni FET p = 4.43E‐29, 4.72 FE), and its gene interactions conserved in independent cohorts, vTCGA‐GCC and GSE84437 (Fig. 1 b; Supporting Material 2B, C).
M28 contains several oncogenic pathways. For instance, M28 significantly intersects with cell viability signatures from KRAS mutant epithelial cells20 (Bonferroni corrected FET p‐value = 5.421E‐2, 21.8 FE). Comparing by KRAS mutation status, KRAS mutant viability signatures genes in M28, MST1R and SH2D3A, were consistently upregulated in nonsynonymous KRAS mutants in TCGA‐GCC and vTCGA‐GCC. Furthermore, M28 is epithelial tumor specific. Several key drivers of M28 such as EPCAM 14 and ESRP1 21 are epithelial tumor cell specific markers (Fig. 1 c). Expression of the genes in M28 is negatively correlated with ESTIMATE inferred stromal score (Fig. 3 a).
Figure 3.
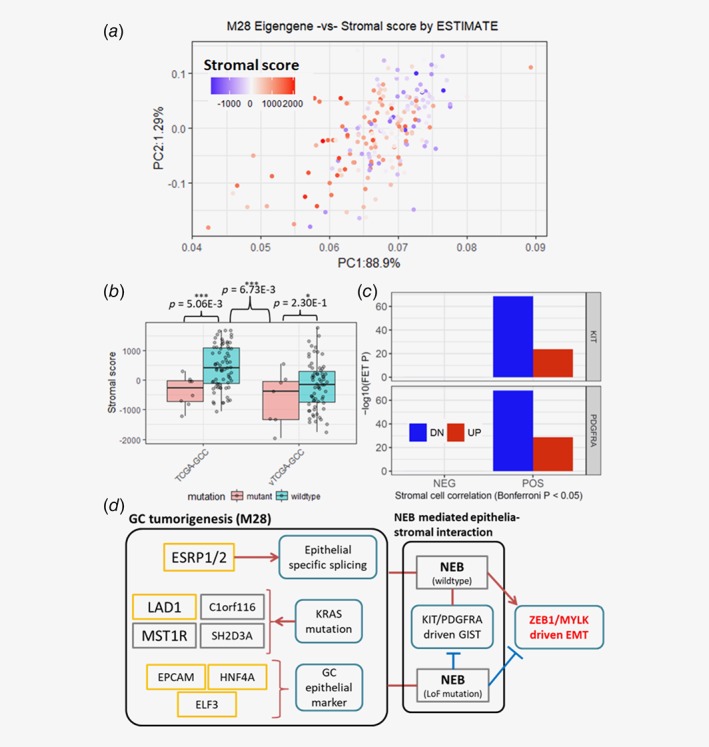
GC epithelial/tumorigenic module, M28, is upregulated in primary GC. (a) Correlation between M28 eigen‐gene and TCGA‐GCC stromal score inferred by ESTIMATE algorithm. The x‐ and y‐ axes are the first and second PC of M28, respectively. (b) ESTIMATE inferred stromal score distributions by NEB mutation status, in TCGA‐GCC (left) and vTCGA‐GCC (right). (c) Enrichment of oncogenic KIT/PDGFRA mutation signatures from gastrointestinal stromal tumors (GISTs) in stromal score correlated gene signature in TCGA‐GCC. (d) A GC tumorigenesis pathway captured by M28, interacts with GC stromal cells via NEB to promote epithelial‐mesenchymal transition (EMT) in GC. [Color figure can be viewed at http://wileyonlinelibrary.com]
To some extent, the association between M28 and stromal score can be explained by nonsynonymous somatic mutations in an actin‐binding protein, Nebulin (NEB). NEB somatic nonsynonymous mutations were present in 26 (~11.6%) of the 224 samples, and associated to key stromal phenotypes such as lower stromal scores (Fig. 3 b) and less occurrence of N3 patients (Supporting Information Fig. S4c ) in NEB mutants. On the contrary, the downregulated NEB SMGs are enriched in an epithelial‐mesenchymal transition (EMT) module, M109 (Supporting Information Fig. S4a,B ), a key pathway mediating epithelial‐stromal interaction within tumor microenvironment.22
The epithelial‐stromal interaction may involve key tumorigenic pathways in gastrointestinal stromal tumor (GIST). KIT/PDGFRA mutations serve as characteristic oncogenic mutations, present in ~98% of GISTs.23 The genes downregulated in active KIT/PDGFRA mutations from GISTs, curated from the CREED database (initial source data: GSE17743),24 are significantly enriched with the stromal score correlated gene signature (Fig. 3 c).
Taken together, these results suggest NEB may serve as a gateway for epithelial‐stromal interactions in advanced GC microenvironment (Fig. 3 d), mediating the cross‐talk between tumorigenic pathways of epithelial and stromal tumor cells, and activating (NEB wildtype) or suppressing (NEB mutant) EMT, a critical pathway for cancerous cell dissemination.
GC epithelium leverages intestinal mucosal barrier niche against EBV infection
M28 strongly interacts with the intestinal epithelium specific/goblet cell‐like modules, M666 and its parent module, M121 (Fig. 1 c). Eigen‐gene of M666 strongly correlates to M28 eigen‐gene (Fig. 5b), and M121/M666 contains genes in several key intestinal mucosal barrier (IMB) functions including the members of the intermicrovillar adhesion complex (IMAC) such as MYO7B, ANKS4B, CDHR2 and CDHR5, which are critical for enterocyte brush border formation25 ESPN involved in actin bundling26 mucins (TFF3 and MUC2) secreted by intestinal goblet cells27 (Fig. 2 e,f). M121/M666 are also conserved in vTCGA‐GCC and GSE84437 (Fig. 1 b; Supporting Material 2B, C).
M121/M666 is specifically silenced in the EBV subtype via hypermethylation. By comparing the EBV subtype and the rest of the samples, we identified the genes downregulated in the EBV subtype (using the threshold of FDR < 0.05 and fold change >1.2) and the cis‐hyper‐methylated genes in the EBV subtype (FDR < 0.05) and then intersected them to derive an EBV‐specific hypermethylation gene signature (EBV‐HEMG; see Supporting Material 6A, B). These EBV‐HEMGs are significantly enriched in M121 (Bonferroni corrected FET p < 2.63E‐22, 6.49 FE). Projection of M121 and M28 onto the protein–protein interaction network from the String database16 (Fig. 2 e) revealed that many key features of IMB maintenance involve interactions with GC epithelium specific pathways (M28), and are suppressed in the EBV subtype. Furthermore, suppression of key genes in M121/M666 including MYO7B and PPP1R14D, were associated with poor recurrence‐free survival (see Supporting Information Fig. S5a,b ).
PIK3CA mutations and EBV infection aberrantly upregulate T‐cell receptors in CD8+ T‐cell cytotoxic pathways
In contrast to the suppression of the IMB complex in GC, a group of immunoglobulin(Ig)‐superfamily receptors involved in NK−/T‐cell mediated cytotoxicity are activated in the EBV subtype via hypo‐methylation (Fig. 4 a,b). Similar to the identification of EBV‐HEMGs, we also derived a set of EBV‐specific hypo‐methylation genes (EBV‐HOMGs) that were upregulated and hypomethylated in the EBV subtype in comparison with all the other subtypes (Supporting Material 6A, C). M102, a module associated with immune response, is highly enriched for the EBV‐HOMGs (Bonferroni corrected FET p = 1.14E‐69, 8.15 FE), and is conserved in vTCGA‐GCC and GSE84437 (Fig. 1 b; Supporting Material 2B, C). Also, cosignaling Ig receptors for T‐cell activation genes including CD96, TIGIT, CRTAM and CD226,28 were EBV‐HOMGs present in M102 (Fig. 4 a).
Figure 4.
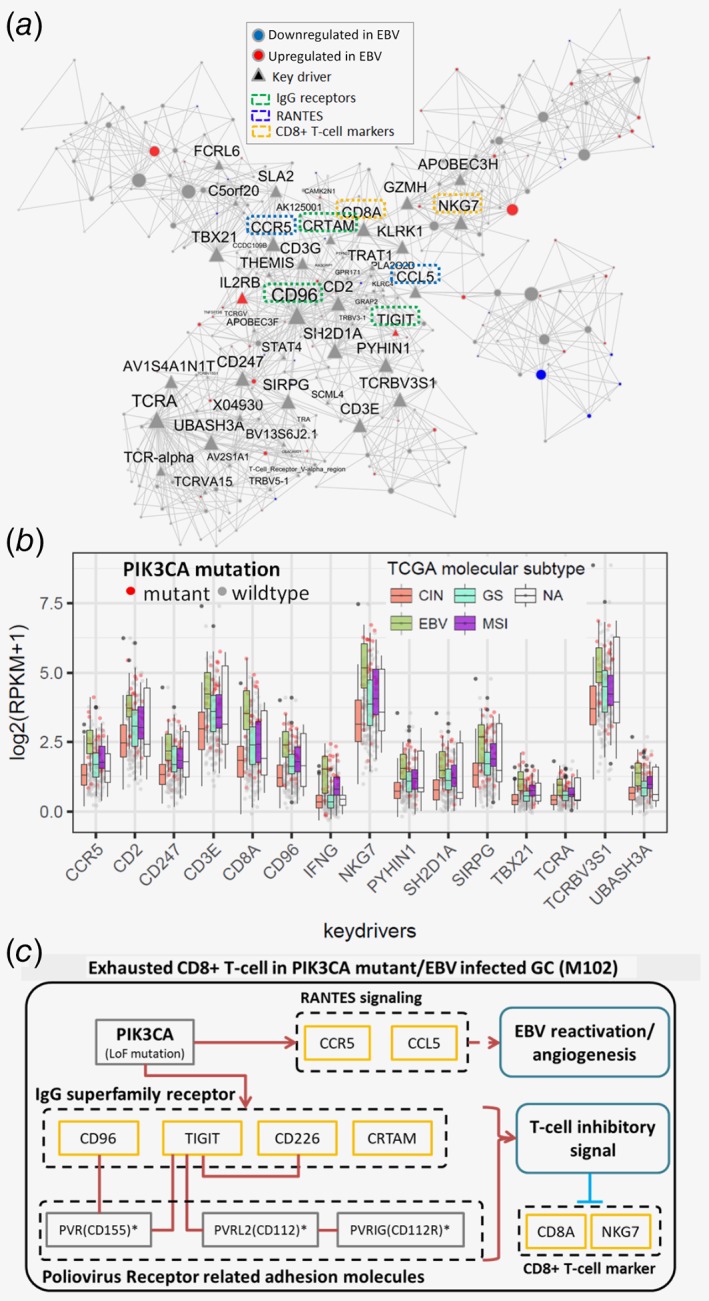
The NK‐/T‐cell cytotoxicity related module, M102, is synergistically upregulated by EBV and PIK3CA mutations. (a) Network structure of M102. The red and blue nodes are the genes up‐ and down‐regulated in the EBV subtype compared to the other subtypes. Triangle nodes are the inferred key drivers of M102. The key driver genes involved in specific signaling pathways are labeled by dotted boxes with green (Ig superfamily receptors), blue (RANTES signaling) and yellow (CD8+ T‐cell markers). (b) EBV subtype specific upregulation of M102 key drivers. (c) Exhausted CD8+ T‐cell pathway captured by M102. Cosignaling T‐cell receptors are upregulated by EBV infection/PIK3CA mutation, which may lead to CD8+ T‐cell exhaustion. [Color figure can be viewed at http://wileyonlinelibrary.com]
Using the cell type specific markers from CIBERSORT,15 we found that the key driver genes of M102 were specifically expressed in several immune cell types including CD8+ T‐cells, macrophage‐M1, activated CD4 memory T‐cells and activated NK cells (Supporting Material 5E). For instance, expression of CD96 was highly correlated with the markers in CD8+ T‐cells (Spearman ρ = 0.572, p = 2.39E‐20), activated CD4 memory T‐cells (Spearman ρ = 0.511, p = 5.04E‐12), macrophage‐M1 (Spearman ρ = 0.452, p = 1.63E‐8) and activated NK (Spearman ρ = 0.180, p = 7.56E‐3) cell populations.
Enrichment of PIK3CA mutations in the EBV subtype has been well established within TCGA‐GCC.5 Not surprisingly, the upregulated PIK3CA SMGs in the TCGA‐GCC tumors are highly enriched in M102 (Bonferroni FET p = 1.89E‐110, 14.3 FE), and M102 key drivers including TIGIT (co‐inhibitory Ig receptor) show higher expressions in EBV subtype, compared to other subtypes (Fig. 4 b). These results indicate PIK3CA mutations and EBV infection synergistically upregulate NK−/T‐cell cytotoxic pathways including cosignaling T‐cell activation receptors (Fig. 4 c).
Discussion
Our integrative network approach not only revealed robust network structures and key regulators of GC, but also presented detailed signaling circuits underlying survival‐associated GC microenvironments involving cross‐talks among epithelial, stromal and immune cells (Table 1; Fig. 5 a,b). We validated GKN2, an emerging driver of gastric mucosa, through in silico and in vitro perturbation experiments as well as pathological examination of a large number of human gastric cancer specimens.
Figure 5.
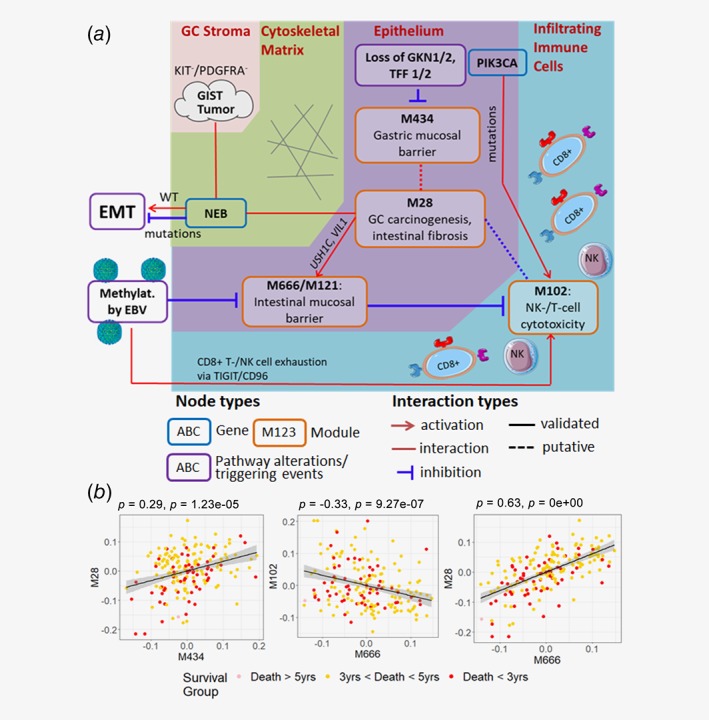
Molecular interactions among stromal, epithelial and immune cells in GC microenvironment. (a) Key prognostic pathways in GC: The most prognostic network modules (M666/M121, M434 and M28) and their interacting partner M102 capture complex signaling circuits among stromal, epithelial and immune cells in GC microenvironment, involving genetic (somatic mutations in NEB and PIK3CA), epigenetic (altered methylation by EBV infection) and transcriptomic alterations (loss of GKN1, GKN2, TFF1 and TFF2) as triggering events. Respective cellular domains/cell types are bordered by different background colors. Activation, interaction and inhibition relationships are shown. Interactions between genes with different mutation status and pathways are further labeled with WT (wildtype) and mutations (mutated). Putative interactions identified by the TCGA‐GCC network, but without literature support or independent validations are marked in short‐dashed lines. (b) Cross‐talks across key prognostic pathways. Spearman's correlations between eigen‐genes of key prognostic modules were computed to evaluate interactions/cross‐talks between modules. Coefficients and significance p‐values are shown on the top. Colors represent overall survival of respective patients. [Color figure can be viewed at http://wileyonlinelibrary.com]
Gastrointestinal mucosal barrier: different modes of protections for different contexts of GC progression
Several modules involved gastric‐specific mucosa (M434) and intestinal‐specific mucosa (M121 and M666) emerged as key tumor suppressive pathways in GC. Our analysis reveals different modes of operations for these mucosal protective functions. The gastric mucosal barrier (GMB) module is specifically associated with GC proliferation and invasion while the intestinal mucosal barrier (IMB) niche is specifically associated with the EBV infection.
GKN2 is an independent regulator of GC proliferation and invasion in GMB niche
The GMB module (M434), ranked as the second module most predictive of survival time, reveals the antitumor effect of the TFF1‐TFF2‐GKN1‐GKN2 axis. These genes are robustly downregulated in GC compared to normal gastric tissue29 and their downregulation in tumor is associated with increased tumor size and poor overall survival in GC. These findings are confirmed in the independent Fujian cohort.
The TFF1‐TFF2‐GKN1‐GKN2 axis exerts pressures on GC tumorigenesis via gastric mucosal defense30 silencing oncogenic PTEN/PI3K/AKT/mTOR 31 JAK/STAT pathway32 ERK1/2 axes33 and eventually inhibits GC proliferation and invasion.19, 32 GKN1 is known to play an important role in the progression of gastric cancers via inhibition of EMT and cancer cell migration34 and TFF2 forms heteromer with MUC6, which assembles and stabilizes the laminated structure of gastric mucus and has antibiotic activity against Helicobacter pylori.30 Heterodimeric interaction between GKN2 and TFF1 has synergistic antiproliferative and proapoptotic effects in GC.19
In our study, in vitro overexpression of GKN2 in GC cells silences PTEN/PI3K/AKT/mTOR and JAK/STAT pathway, and inhibits GC proliferation and invasion as previously reported. Interestingly, overexpression of GKN2 alone recovered expression of its heterodimeric interaction partner, TFF1 to confer antitumor effects.
On the contrary, GKN2 overexpression upregulated cholesterol synthesis module, M293, includes HMGCR, which promotes GC growth and migration.35 Although we observed a clear antitumor effect of GKN2 overexpression in our clinical and in vitro studies, the unexpected activation of a GC oncogenic pathway requires further in‐depth investigation to clarify its roles conditioned on recovered GKN2 functions in vivo.
EBV infection silences the IMB niche
Two top ranked modules, M121 and M666, include many of intestinal epithelia specific genes in the IMB niche and are specifically downregulated by hypermethylation in the EBV subtype.
The IMB niche in GC acquires several intestinal features such as brush border formation and intestinal cell specific goblet cells. For instance, IMAC is a critical adhesion complex for brush border formation, and consists of USH1C, MYO7B, ANKS4B and intestinal specific protocadherins CDHR2/5 as essential components for the complex formation.25 USH1C belongs to GC epithelial specific module M28 while MYO7B, ANKS4B and CDHR2/5 fall into M121. These data suggest that IMAC formation requires contributions from both actively expressed intestinal components and GC specific epithelium to become fully functional.
On the other hand, the IMB niche involving M121 and M666 is not limited to the intestinal subtype in TCGA‐GCC. For instance, an intestinal brush border specific gene, ESPN, which falls into both M121 and M666, is not differentially expressed between intestinal and diffuse subtypes in TCGA‐GCC (t‐test p = 0.93). Many other genes in the modules show similar results. Although intestinal phenotypes such as gland formation and fibrosis are observed in early stages of GC and atropic gastritis36 and constitute a major subtype of GC as intestinal subtype by Lauren classification,12 the IMB niche defined by M121 and M666 is generally applicable to a broader spectrum of GC beyond the intestinal subtype.
In summary, the IMB niche represents an emergent protective mechanism in advanced GC. With the known protective roles of IMB in host defense in intestine against gut microbiome37 the observed suppression of gastric IMB may indicate a prerequisite for EBV infection to function in GC development and progression.
Nebulin mutations: blocking stromal–epithelial cell interactions in GC
Stromal cells constitute large portions of GC tumors, and cancer‐stromal cell interactions promote tumor growth and metastasis.38 Coexistence of often benign gastro‐stromal intestinal tumors (GISTs) with gastric adenocarcinoma has been frequently observed.23 In our TCGA‐GCC analysis, a KRAS dependent GC tumorigenic module (M28) associates to varying degree of “stromal‐ness.” This stromal‐ness corresponded to GIST specific oncogenic mutations, KIT and PDGFRA,23 pertaining stromal–epithelial interactions in GC.
In our study, somatic nonsynonymous mutations in an actin‐binding protein, Nebulin (NEB) is associated to this stromal–epithelial interactions. Nebulin is a giant 600‐ to 900‐kDa filamentous protein constituting cytoskeletal matrix that coexists with the thick and thin filaments within the sarcomeres of skeletal muscle39 and is an overexpressed protein in gastric cancer tissues compared to normal.40 However, its roles in GC progression have remained largely unknown.
We present NEB‐centered molecular mechanisms of GC invasion by leveraging differentially expressed genes in NEB somatic nonsynonymous mutation. Specifically, upregulated genes in NEB mutants coincided with GC epithelial specific M28, and the respective downregulated genes were associated to EMT module (M109), which bears ZEB1 as a top key driver, a key transcription factor in EMT and metastasis41 (Supporting Information Fig. S4a,b ). Another interesting key driver of M109 is MYLK (myosin light chain kinase), which phosphorylates myosin regulatory light chains to facilitate myosin interaction with actin filaments to produce contractile activity,42 and involved in cell motility and morphology.
Together with NEB functions in maintaining cytoskeletal matrix, the downregulation of M109 in NEB mutants strongly suggests disruption of dynamic changes in actin cytoskeleton via loss‐of‐function mutation in NEB, a necessary process of EMT for cancerous cells to acquire cell motility and morphological changes.22 This also explains the underrepresentation of N3 patients in NEB mutant samples in TCGA‐GCC (Supporting Information Fig. S4c ).
Overall, NEB‐centered GC mechanisms illustrate the complex cross‐talks between GC stroma and epithelium via gastric actin cytoskeleton, and propose NEB mutation as a protective mechanism by disrupting EMT.
PIK3CA and EBV driven methylation: recipes for GC immune evasion
M102 is NK−/T‐cell cytotoxic pathway module, including key receptors for T‐cell and NK‐cell activation for immune surveillance of GC tumor. Interestingly, M102 is subject to combination of genetic alteration (PIK3CA mutation) and epigenetic alteration (hypomethylation in EBV subtype), which synergistically upregulated M102 compared to other GC subtypes in TCGA‐GCC.
Among the key driver genes of M102, TIGIT is a poliovirus receptor (PVR)–like protein, an immunoreceptor expressed in T‐cells that acts as inhibitory checkpoint on both of T‐cells and NK‐cells.43 Recently, TIGIT emerged as a promising immune checkpoint blockade target in GC. TIGIT+ CD8+ T‐cell population increases in GC, and these cells show functional exhaustions impairing antitumoral activities.44 Blocking TIGIT showed synergistic effect in recovering antitumoral CD8+ T‐cell functions with anti PD‐L1 treatment.44 As M102 also includes CD8+ T‐cell markers such as CD8A and NKG7 as key drivers, these strongly suggest M102 depicts exhausted CD8+ T‐cells by exploiting coinhibitory TIGIT signaling.
The top key driver of M102, CD96 (also known as T Cell‐Activated Increased Late Expression Protein), is expressed in T‐ and NK‐cells with adhesive functions to modulate their interactions and enhance cytotoxicity.45 In HIV‐1 infected adults, CD96 is associated with different cell effector functions of CD8+ T cells.45 It has been shown that targeting NK cell activity via CD96 has promising anticancer potential, complementary to the existing PD‐1 and PD‐L1 targeted therapeutics46 and suppresses metastasis in melanoma lung metastasis mouse model (B16F10).47 To our knowledge, the effectiveness of CD96 blockade, thereby recovering NK‐cell antitumoral activity in GC has not been explored.
These results suggest targeted immunotherapy for PIK3CA‐mutant and/or EBV‐infected GC patients, via targeting adaptive and innate immune response systems. Especially, CD96 and TIGIT are attractive blockade targets in such GC patients though future investigation is needed to confirm the finding.
In summary, the most prognostic coexpression network modules reveal a series of pathways representing complex cross‐talks among epithelial, stromal and immune cells in GC (Fig. 5 a), which are potentially driven by environmental factor (i.e., EBV infection), genetic alterations (i.e., NEB and PIK3CA mutations) and epigenetic alterations (i.e. hypo‐/hyper‐methylations). We further systematically investigate key regulators and proposed interactions of the top modules. In particular, we validate antitumor effects of one key network driver GKN2 overexpression and identified downstream pathways of GKN2 in GC. These findings enable generation of novel hypotheses regarding complex interplay among multifaceted axes (e.g., different pathways and different cell types) in GC progression. For instance, we propose the IMB niche in M121 and M666 as a suppressor of EBV driven immune evasion in M102, where host defense function of the IMB niche may be capable of inhibiting EBV activation.
Our integrative network analysis of TCGA‐GCC reveals some fundamental patterns of molecular interactions and specific mechanisms in GC progression. The network models and the key regulators identified here pave a way for defining novel therapeutic strategies. Future work will include validation of prioritized key drivers and subnetworks in GC and develop network‐based biomarkers for stratifying GC patients.
Supporting information
Supporting Material 1. Supporting Materials and Methods Descriptions, Supporting Results and Supporting Figures 1–10.
Supporting Material 2. A. Ranked table of gene modules identified by MEGENA. B. Module preservation statistics of TCGA‐GCC modules in GSE84437 and vTCGA‐GCC cohorts. C. Patient barcodes of primary tumor samples used in TCGA‐GCC and vTCGA‐GCC.
Supporting Material 3. A. Somatic mutation oncomatrix generated using maftools R package. B. Significant enrichment of somatic mutation gene signatures in MEGENA modules with BH FDR corrected FET p‐value <0.05.
Supporting Material 4. Differentially expressed genes in GKN2 over‐expressed AGS cells compared to the mock control.
Supporting Material 5. Computationally inferred cell types in TCGA‐GCC primary tumor samples. A. ESTIMATE inferred stromal/immune cell scores. B. CIBERSORT inferred immune cell type compositions. C. ESTIMATE inferred stromal cell correlated gene signature in TCGA‐GCC. D. ESTIMATE inferred stromal cell correlated gene signature in vTCGA‐GCC. E. Correlation between hub genes of M102 and CIBERSORT inferred cell type compositions.
Supporting Material 6. Differentially expressed genes (DEGs) and differentially methylated CpG sites (DMR) in EBV subtype, compared to other primary GC in TCGA‐GCC. A. Differential expression between EBV subtype and the rest of primary GC in TCGA‐GCC. B. List of genes in EBV‐specific hyper‐methylation gene signature (EBV‐HEMG) by intersecting significantly down‐regulated genes and hyper‐methylated genes in cis in EBV subtype. C. List of genes in EBV‐specific hypo‐methylation gene signature (EBV‐HOMG) by intersecting significantly up‐regulated genes and hypo‐methylated genes in cis in EBV subtype.
Supporting Material 7. Supporting results for correlation analysis between methylation and gene expression profiles. A. cis−/trans‐eMSG signatures identified by Bonferroni corrected p‐value <0.05 by Spearman correlation analysis. B. Table of all pairwise tests between eMSGs and MSigDB signatures. C. Table of all pairwise tests between eMSGs and gene modules of TCGA‐GCC.
Author contributions: Perceived the concept and designed the study: Zhang B; Curated data and performed statistical and network analyses: Song W; Designed and oversaw the validation experiments: Lin X, Zheng X; Performed validation experiments: Liao X, Hu D, Lin J; Drafted the manuscript: Song W, Lin X, Zhang B; Editing of manuscript: All authors.
Conflict of Interest: The authors declare that they have no competing financial interests.
References
- 1. Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA: A Cancer Journal for Clinicians 2012;62:10–29. [DOI] [PubMed] [Google Scholar]
- 2. Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell 2011;144:646–74. [DOI] [PubMed] [Google Scholar]
- 3. Lin X, Zhao Y, Song WM, et al. Molecular classification and prediction in gastric cancer. Comput Struct Biotechnol J 2015;13:448–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Brettingham‐Moore KH, Duong CP, Heriot AG, et al. Using gene expression profiling to predict response and prognosis in gastrointestinal cancers‐the promise and the perils. Ann Surg Oncol 2011;18:1484–91. [DOI] [PubMed] [Google Scholar]
- 5. Cancer Genome Atlas Research N . Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Barabasi AL, Oltvai ZN. Network biology: understanding the cell's functional organization. Nat Rev Genet 2004;5:101–13. [DOI] [PubMed] [Google Scholar]
- 7. Song WM, Zhang B. Multiscale embedded gene co‐expression network analysis. PLoS Comput Biol 2015;11:e1004574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005;102:15545–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ritchie ME, Phipson B, Wu D, et al. Limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Langfelder P, Horvath S. Eigengene networks for studying the relationships between co‐expression modules. BMC Syst Biol 2007;1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cox DR. Regression models and life‐tables. J R Stat Soc B 1972;34:187–220. [Google Scholar]
- 12. Ma J, Shen H, Kapesa L, et al. Lauren classification and individualized chemotherapy in gastric cancer. Oncol Lett 2016;11:2959–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Langfelder P, Luo R, Oldham MC, et al. Is my network module preserved and reproducible? PLoS Comput Biol 2011;7:e1001057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Yoshihara K, Shahmoradgoli M, Martinez E, et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat Commun 2013;4:2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Newman AM, Liu CL, Green MR, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Szklarczyk D, Franceschini A, Wyder S, et al. STRING v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Szasz AM, Lanczky A, Nagy A, et al. Cross‐validation of survival associated biomarkers in gastric cancer using transcriptomic data of 1,065 patients. Oncotarget 2016;7:49322–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. McCarthy DJ, Chen Y, Smyth GK. Differential expression analysis of multifactor RNA‐Seq experiments with respect to biological variation. Nucleic Acids Res 2012;40:4288–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kim O, Yoon JH, Choi WS, et al. Heterodimeric interaction between GKN2 and TFF1 entails synergistic antiproliferative and pro‐apoptotic effects on gastric cancer cells. Gastric Cancer 2017;20:772–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh A, Greninger P, Rhodes D, et al. A gene expression signature associated with “K‐Ras addiction” reveals regulators of EMT and tumor cell survival. Cancer Cell 2009;15:489–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Warzecha CC, Shen S, Xing Y, et al. The epithelial splicing factors ESRP1 and ESRP2 positively and negatively regulate diverse types of alternative splicing events. RNA Biol 2009;6:546–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sun BO, Fang Y, Li Z, et al. Role of cellular cytoskeleton in epithelial‐mesenchymal transition process during cancer progression. Biomed Rep 2015;3:603–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yan Y, Li Z, Liu Y, et al. Coexistence of gastrointestinal stromal tumors and gastric adenocarcinomas. Tumour Biol 2013;34:919–27. [DOI] [PubMed] [Google Scholar]
- 24. Wang Z, Monteiro CD, Jagodnik KM, et al. Extraction and analysis of signatures from the gene expression omnibus by the crowd. Nat Commun 2016;7:12846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Crawley SW, Weck ML, Grega‐Larson NE, et al. ANKS4B is essential for Intermicrovillar adhesion complex formation. Dev Cell 2016;36:190–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Bartles JR. Parallel actin bundles and their multiple actin‐bundling proteins. Curr Opin Cell Biol 2000;12:72–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kim YS, Ho SB. Intestinal goblet cells and mucins in health and disease: recent insights and progress. Curr Gastroenterol Rep 2010;12:319–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen L, Flies DB. Molecular mechanisms of T cell co‐stimulation and co‐inhibition. Nat Rev Immunol 2013;13:227–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Moss SF, Lee JW, Sabo E, et al. Decreased expression of gastrokine 1 and the trefoil factor interacting protein TFIZ1/GKN2 in gastric cancer: influence of tumor histology and relationship to prognosis. Clin Cancer Res 2008;14:4161–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Hanisch FG, Bonar D, Schloerer N, et al. Human trefoil factor 2 is a lectin that binds alpha‐GlcNAc‐capped mucin glycans with antibiotic activity against helicobacter pylori. J Biol Chem 2014;289:27363–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Dai J, Qian C, Su M, et al. Gastrokine‐2 suppresses epithelial mesenchymal transition through PI3K/AKT/GSK3beta signaling in gastric cancer. Tumour Biol 2016;37:12403–10. [DOI] [PubMed] [Google Scholar]
- 32. Ouyang J, Pan X, Lin H, et al. GKN2 increases apoptosis, reduces the proliferation and invasion ability of gastric cancer cells through down‐regulating the JAK/STAT signaling pathway. Am J Transl Res 2017;9:803–11. [PMC free article] [PubMed] [Google Scholar]
- 33. Dubeykovskaya Z, Dubeykovskiy A, Solal‐Cohen J, et al. Secreted trefoil factor 2 activates the CXCR4 receptor in epithelial and lymphocytic cancer cell lines. J Biolog Chem 2009;284:3650–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Yoon JH, Kang YH, Choi YJ, et al. Gastrokine 1 functions as a tumor suppressor by inhibition of epithelial‐mesenchymal transition in gastric cancers. J Cancer Res Clin Oncol 2011;137:1697–704. [DOI] [PubMed] [Google Scholar]
- 35. Chushi L, Wei W, Kangkang X, et al. HMGCR is up‐regulated in gastric cancer and promotes the growth and migration of the cancer cells. Gene 2016;587:42–7. [DOI] [PubMed] [Google Scholar]
- 36. Giannakis M, Chen SL, Karam SM, et al. Helicobacter pylori evolution during progression from chronic atrophic gastritis to gastric cancer and its impact on gastric stem cells. Proc Natl Acad Sci U S A 2008;105:4358–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Okumura R, Takeda K. Maintenance of intestinal homeostasis by mucosal barriers. Inflamm Regen 2018;38:5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kitadai Y. Cancer‐stromal cell interaction and tumor angiogenesis in gastric cancer. Cancer Microenviron 2010;3:109–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Labeit S, Ottenheijm CA, Granzier H. Nebulin, a major player in muscle health and disease. FASEB J 2011;25:822–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li W, Li JF, Qu Y, et al. Comparative proteomics analysis of human gastric cancer. World journal of gastroenterology: WJG 2008;14:5657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Zhang P, Sun Y, Ma L. ZEB1: at the crossroads of epithelial‐mesenchymal transition, metastasis and therapy resistance. Cell Cycle 2015;14:481–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stroka KM, Aranda‐Espinoza H. Endothelial cell substrate stiffness influences neutrophil transmigration via myosin light chain kinase‐dependent cell contraction. Blood 2011;118:1632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Blake SJ, Dougall WC, Miles JJ, et al. Molecular pathways: targeting CD96 and TIGIT for cancer immunotherapy. Clin Cancer Res 2016;22:5183–8. [DOI] [PubMed] [Google Scholar]
- 44. He W, Zhang H, Han F, et al. CD155T/TIGIT signaling regulates CD8(+) T‐cell metabolism and promotes tumor progression in human gastric cancer. Cancer Res 2017;77:6375–88. [DOI] [PubMed] [Google Scholar]
- 45. Eriksson EM, Keh CE, Deeks SG, et al. Differential expression of CD96 surface molecule represents CD8(+) T cells with dissimilar effector function during HIV‐1 infection. PLoS One 2012;7:e51696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Guillerey C, Huntington ND, Smyth MJ. Targeting natural killer cells in cancer immunotherapy. Nature Immunol 2016;17:1025–36. [DOI] [PubMed] [Google Scholar]
- 47. Blake SJ, Stannard K, Liu J, et al. Suppression of metastases using a new lymphocyte checkpoint target for cancer immunotherapy. Cancer Discov 2016;6:446–59. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Material 1. Supporting Materials and Methods Descriptions, Supporting Results and Supporting Figures 1–10.
Supporting Material 2. A. Ranked table of gene modules identified by MEGENA. B. Module preservation statistics of TCGA‐GCC modules in GSE84437 and vTCGA‐GCC cohorts. C. Patient barcodes of primary tumor samples used in TCGA‐GCC and vTCGA‐GCC.
Supporting Material 3. A. Somatic mutation oncomatrix generated using maftools R package. B. Significant enrichment of somatic mutation gene signatures in MEGENA modules with BH FDR corrected FET p‐value <0.05.
Supporting Material 4. Differentially expressed genes in GKN2 over‐expressed AGS cells compared to the mock control.
Supporting Material 5. Computationally inferred cell types in TCGA‐GCC primary tumor samples. A. ESTIMATE inferred stromal/immune cell scores. B. CIBERSORT inferred immune cell type compositions. C. ESTIMATE inferred stromal cell correlated gene signature in TCGA‐GCC. D. ESTIMATE inferred stromal cell correlated gene signature in vTCGA‐GCC. E. Correlation between hub genes of M102 and CIBERSORT inferred cell type compositions.
Supporting Material 6. Differentially expressed genes (DEGs) and differentially methylated CpG sites (DMR) in EBV subtype, compared to other primary GC in TCGA‐GCC. A. Differential expression between EBV subtype and the rest of primary GC in TCGA‐GCC. B. List of genes in EBV‐specific hyper‐methylation gene signature (EBV‐HEMG) by intersecting significantly down‐regulated genes and hyper‐methylated genes in cis in EBV subtype. C. List of genes in EBV‐specific hypo‐methylation gene signature (EBV‐HOMG) by intersecting significantly up‐regulated genes and hypo‐methylated genes in cis in EBV subtype.
Supporting Material 7. Supporting results for correlation analysis between methylation and gene expression profiles. A. cis−/trans‐eMSG signatures identified by Bonferroni corrected p‐value <0.05 by Spearman correlation analysis. B. Table of all pairwise tests between eMSGs and MSigDB signatures. C. Table of all pairwise tests between eMSGs and gene modules of TCGA‐GCC.
Data Availability Statement
The original gene expression data, analyzed as “TCGA‐GCC” and “vTCGA‐GCC,” that support findings of our study are available from TCGA data portal (https://portal.gdc.cancer.gov/). Respective patient barcodes for TCGA‐GCC and vTCGA‐GCC are provided in Supporting Material 2C within this manuscript. Another independent validation data, GSE84437, that support findings our study, are available from Gene Expression Omnibus (GEO) with the accession code “GSE84437.”
The primary software MEGENA (version 1.3.7) for network analysis is publicly available as R package in The Comprehensive R Archive Network (CRAN) (https://cran.r-project.org/web/packages/MEGENA/index.html). The development version of MEGENA is available from GitHub repository (https://github.com/songw01/MEGENA).