

Abstract
Genome‐scale high‐throughput sequencing enables the detection of unprecedented numbers of sequence variants. Variant filtering and interpretation are facilitated by mutation databases, in silico tools, and population‐based reference datasets such as ExAC/gnomAD, while variants are classified using the ACMG/AMP guidelines. These methods, however, pose clinically relevant challenges. We queried the gnomAD dataset for (likely) pathogenic variants in genes causing autosomal‐dominant disorders. Furthermore, focusing on the fibrillinopathies Marfan syndrome (MFS) and congenital contractural arachnodactyly (CCA), we screened 500 genomes of our patients for co‐occurring variants in FBN1 and FBN2. In gnomAD, we detected 2653 (likely) pathogenic variants in 253 genes associated with autosomal‐dominant disorders, enabling the estimation of variant‐filtering thresholds and disease predisposition/prevalence rates. In our database, we discovered two families with hitherto unreported co‐occurrence of FBN1/FBN2 variants causing phenotypes with mixed or modified MFS/CCA clinical features. We show that (likely) pathogenic gnomAD variants may be more frequent than expected and are challenging to classify according to the ACMG/AMP guidelines as well as that fibrillinopathies are likely underdiagnosed and may co‐occur. Consequently, selection of appropriate frequency cutoffs, recognition of digenic variants, and variant classification represent considerable challenges in variant interpretation. Neglecting these challenges may lead to incomplete or missed diagnoses.
Keywords: congenital contractural arachnodactyly, digenic variants, genome sequencing, Marfan syndrome, variant interpretation
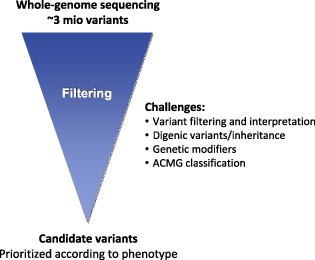
1. INTRODUCTION
Current high‐throughput sequencing (HTS) technologies such as whole‐exome (WES) and whole‐genome sequencing (WGS) enable the detection of ten thousands and millions of sequence variants, respectively. For Mendelian disorders, the identification of typically one or two disease‐causing sequence variants represents a bottleneck in the filtering and interpretation of the large number of sequence variants detected by HTS. As sequence variants causing Mendelian disorders are expected to be rare,1 filtering for sequence variants absent or infrequent in population‐based reference datasets is one of the main approaches used to discriminate (likely) pathogenic from benign variants. ExAC and its successor gnomAD are currently the largest publicly available population‐based reference datasets, providing the best variant frequency estimates.2, 3 Cutoffs of low allele frequencies are often used, including 1%, 0.1%, or 0.01%.4, 5 Recently, a gene‐specific framework has been introduced, which accounts for disease prevalence, allelic and genetic heterogeneity, inheritance mode, and penetrance, resulting in cutoff values even below 0.01%.6
Frequency cutoffs below 1%, however, may not be applicable to all clinical cases. Indeed, in apparently healthy population‐based reference datasets low‐allele‐frequency cutoffs may not appropriately account for common genetic modifiers or for the considerable number of disease‐affected individuals with atypical, late‐onset, and/or unrecognized phenotypes. Moreover, recent reports of pseudo and true digenic inheritance indicate that the identification of a single disease‐causing variant may be insufficient.7, 8, 9 Neglecting these factors may result in the exclusion of clinically relevant sequence variants, leading to incomplete or even missed diagnoses. Here, we address these challenges by assessing the presence and frequency of a priori (likely) pathogenic variants in gnomAD as well as by showing hitherto unreported cases of digenic variants causing clinically relevant complex phenotypes.
2. MATERIALS AND METHODS
2.1. Evaluation of gnomAD
We compared the gnomAD‐based relative frequencies of most likely pathogenic variants in the genes COL1A1, COL3A1, FBN1, FGFR2, JAG1, KMT2D, NSD1, SCN1A, TSC1, and TSC2 with the previously reported prevalence rates of respective autosomal‐dominant disorders. In addition, we compared the carrier frequencies of selected known pathogenic variants causing pediatric and/or adult‐onset disorders in the genes BRCA1, BRCA2, CFTR, GJB2, and HBB among ExAC, gnomAD, and currently appreciated estimates as described.10 We hypothesized that such a comparison of observed and expected frequencies enables to assess whether or not (likely) pathogenic variants are overrepresented in gnomAD and, hence, gnomAD can be considered as representative for the general population (incl. individuals with genetic predisposition to adult‐onset autosomal‐dominant disorders).
2.2. Detection of pathogenic variants in gnomAD
Variant call format files containing all sequence variants in gnomAD were downloaded from http://gnomad.broadinstitute.com/downloads (r2.0.1; 123 136 exomes and 15 496 genomes; released 2017, genome build GRCh37/hg19) and parsed using LeftAlignAndTrimVariants contained in GATK 3.5.11 Using VarSeq 2.0.1 (Golden Helix, Montana), the downloaded gnomAD sequence variants were annotated with several datasets and in silico tools (Supporting Information Table S1). To restrict the analysis to high‐confidence calls, we excluded sequence variants with a non‐PASS gnomAD filter, a 75‐mer mappability <1,12 or a position outside of the canonical transcript (Table “knownCanonical” in the track “UCSC Genes” from the UCSC Table browser; http://genome.ucsc.edu/cgi‐bin/hgTables) as well as indels with a length ≥50 bp. To assess the presence and frequency of (likely) pathogenic variants in gnomAD, we performed automated filtering using VarSeq as well as manual evaluation. Only genes exclusively associated with autosomal‐dominant disorders (status “confirmed” in OMIM version 2018.5, http://omim.org) and with a DOMINO score ≥0.9 were selected.13 To restrict our analyses to genes likely intolerant to loss‐of‐function (LOF) variants, only genes with a pLi score ≥0.9 and an “observed/expected” (o/e) metric 90% confidence interval upper bound <0.35 in gnomAD were further considered (Figure 1).2 Genes associated with disorders inherited in an autosomal‐recessive or X‐linked manner were not analyzed, because for the gnomAD dataset (r2.0.1) information on haplotype and sex is not (yet) available.
Figure 1.
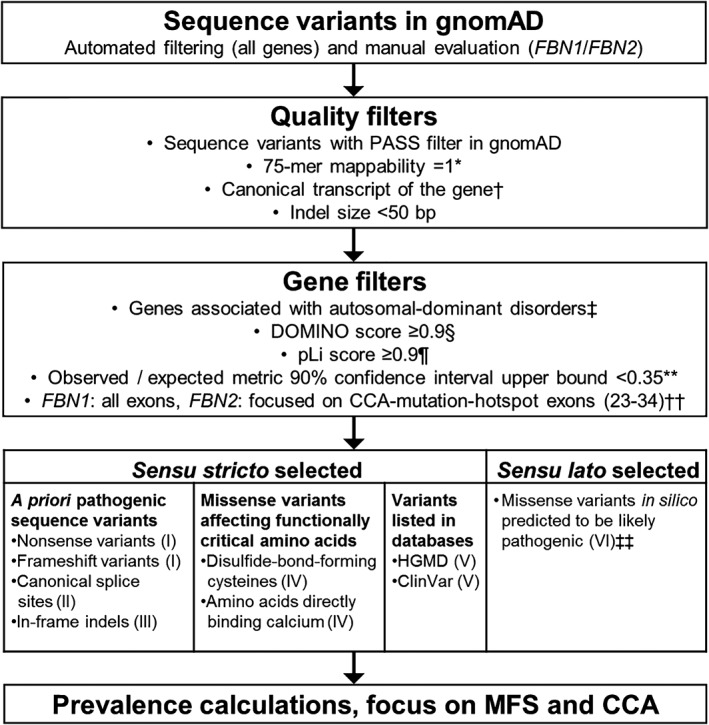
Overview of used workflow. Categories I and II were applied to all genes associated with autosomal‐dominant disorders in gnomAD, whereas categories III‐VI were only applied to FBN1 and FBN2. *,† Data obtained from the Tables “wgEncodeCrgMapabilityAlign75mer” (*) and “knownCanonical” (†) from the UCSC Table browser; http://genome.ucsc.edu/cgi-bin/hgTables. ‡ Data obtained from the Online Mendelian Inheritance in Man (OMIM) dataset, 05.2018; http://omim.org. § According to Quinodoz et al. (2017).13 ¶ According to Lek et al. (2016).2 ** According to Karczewski et al. (2019).3 †† In FBN2 the prevalence calculation was limited to the CCA‐mutation‐hotspot region (exons 23‐34) for categories II‐VI, while for category I (nonsense and frameshift) all exons were considered. ‡‡ Sequence variants predicted “damaging” or “deleterious” by all six used in silico prediction tools (FATHMM, FATHMM‐MKL, MutationAssessor, MutationTaster, Polyphen2, SIFT; see also Supporting Information Table S1). CCA, congenital contractural arachnodactyly; gnomAD, Genome Aggregation Consortium; HGMD, Human Gene Mutation Database; indel, insertion/deletion; MFS, Marfan syndrome
As prime examples, we focused on the genes FBN1 and FBN2, which cause the autosomal‐dominant fibrillinopathies Marfan syndrome (MFS, OMIM #154700) and congenital contractural arachnodactyly (CCA, OMIM #121050), respectively.14, 15 In addition to nonsense, frameshift, and splicing variants, FBN1 and FBN2 sequence variants disrupting the consensus calcium‐binding sequence as well as disrupting or creating disulfide‐bond‐forming cysteines are known to be damaging and MFS/CCA‐causing due to increased proteolytic degradation.16, 17, 18, 19 Thus, such FBN1 and FBN2 sequence variants may be considered as a priori (likely) pathogenic, allowing the more elaborate gnomAD‐based predisposition/prevalence assessment of fibrillinopathies.
We considered following sequence variants as most likely pathogenic in six (I‐VI) categories (Figure 1): (I) Nonsense and frameshift variants with or without expected nonsense‐mediated mRNA decay, because both may cause disease but via separate mechanisms;20, 21 (II) Single nucleotide variants located at the canonical splice sites (exonic ±1 bp, intronic ±1‐2 bp) and in silico predicted to alter splicing (Supporting Information Table S1); (III) In‐frame indels;22, 23 (IV) Missense variants disrupting functionally critical amino acids in the calcium‐binding epidermal growth factor domains of FBN1 and FBN2 such as disulfide‐bond‐forming cysteines as well as amino acids Asn, Asp, and Glu directly binding calcium (http://uniprot.org/uniprot/P35555; http://uniprot.org/uniprot/P35556);16, 17, 18, 19 (V) Sequence variants not included in categories I‐IV but described as disease‐causing (DM) in the Human Gene Mutation Database (HGMD) professional (v2019.1; http://portal.biobase-international.com) and/or as pathogenic in ClinVar (v2019.5; http://ncbi.nlm.nih.gov/clinvar) in association with MFS or CCA and with clear evidence for pathogenicity (segregation analysis, functional assays); (VI) Missense variants in FBN1 and FBN2 not included in categories I‐V but classified as “damaging” or “deleterious” by all six corresponding in silico prediction tools (FATHMM, FATHMM‐MKL, MutationAssessor, MutationTaster, Polyphen2, SIFT) without additional evidence (Supporting Information Table S1).
Subsequently, categories I‐II were applied to the entire gnomAD dataset, whereas categories III‐VI were only applied to FBN1 (NM_000138.4, all exons) and FBN2 (NM_001999.3, CCA‐mutation‐hotspot exons 23‐34).24 Categories I‐V were defined as sensu stricto selected, that is, sequence variants with clear evidence for pathogenicity, while category VI contains sensu lato selected sequence variants, that is, additional, potentially pathogenic variants requiring manual expert review and interpretation. To largely exclude false‐positive results (ie, indeed non‐pathogenic variants), we manually evaluated all gnomAD variants in categories I and II occurring above a low‐frequency cutoff value of 0.01% or affecting genes with a total frequency of >1:2000.5 Accordingly, variants were excluded if one of the following criteria was met: (a) ≥1 “benign” classifications in HGMD and/or ClinVar; (b) >10% allele frequency in any gnomAD subpopulation; (c) variant exclusively affects weakly expressed exon (<10% of the highest expressed exon in disease‐relevant tissue according to http://gtexportal.org).
2.3. Detection of FBN1/FBN2 dual variants in WGS data
WGS (PCR‐free, 60× 150PE) of 500 individuals with rare, mainly cardiovascular or connective tissue disorders was performed as described.25 FASTQ files were aligned using GENALICE MAP (Genalice, Nijkerk, The Netherlands).26 Variant calling was performed using the Population Calling module of GENALICE MAP to simultaneously extract all FBN1 and FBN2 sequence variants in our database.26 Using VarSeq, called sequence variants were annotated and filtered for individuals harboring (likely) pathogenic variants in both FBN1 and FBN2 (ie, dual variants). As a second interpretation platform, the artificial‐intelligence‐driven interpretation software Moon (Diploid, Leuven, Belgium) was used to independently detect FBN1/FBN2 dual variants. For the confirmation of detected FBN1 and FBN2 variants and segregation analyses, Sanger sequencing of the corresponding region was performed as described.27 Data on clinical phenotypes were collected from medical records and/or during physical examination by one of the authors (A.N., M.R., or B.S.). Written informed consent was obtained from patients and family members.
2.4. ACMG/AMP classifications
Automated classifications using the American College of Medical Genetics and Genomics (ACMG) and the Association for Molecular Pathology (AMP) published guidelines were obtained from InterVar v.201904/hg19.28 For FBN1 variants disrupting disulfide‐bond‐forming cysteines, the ACMG/AMP criterion PM1 (moderate evidence for pathogenicity) was manually adjusted to PS3 (strong evidence for pathogenicity) as previously suggested.29 To avoid a bias caused by the ACMG/AMP criterion PM2 (moderate evidence for pathogenicity) fulfilled for variants absent from control populations, the automated classification was manually adapted by applying the criterion PM2 for extremely rare gnomAD variants as well (allele count ≤2). Likewise, for the two families with FBN1/FBN2 dual variants, segregation data (PP1) were manually adjusted in the classification.
2.5. Statistical analysis
The upper and lower limits of the 95% confidence interval (CI) of a proportion were calculated using the online tool VassarStats with a correction for continuity (http://vassarstats.net/prop1.html) or the software GraphPad Prism 8 (GraphPad Software, California). Proportion of genetic predisposition and disease prevalence was calculated under the assumption that carrier individuals in gnomAD harbor no more than one (likely) pathogenic sequence variant in the considered gene(s).
3. RESULTS
3.1. Detection of pathogenic variants in gnomAD
Our comparison revealed that the evaluated carrier frequencies in the genes BRCA1, BRCA2, CFTR, GJB2, and HBB are not overrepresented in gnomAD (Supporting Information Table S2), confirming ExAC‐based previous results.10 In categories I and II (Figure 1), considering all gnomAD sequence variants in genes associated with autosomal‐dominant disorders (pLi ≥0.9, upper bound of the o/e metric 90% CI <0.35) we identified by software filtering and manual evaluation a total of 2653 a priori (likely) pathogenic variants in 253 genes. Ten of these genes are the major cause of disorders with previously reported prevalence but we found no clear evidence that gnomAD‐based predisposition/prevalence rates are significantly higher than reported estimates (Supporting Information Figure S1). Per gene, 1 up to 130 gnomAD individuals harbor an a priori (likely) pathogenic variant with relative allele frequencies ranging from 1/246 272 (0.0004%) to 82/183 872 (0.0446%; ATXN7 c.2673delA; Supporting Information Figure S2), resulting in disease predisposition/prevalence estimates ranging from approximately 1:100 000 to approximately 110:100 000 not associating with pLi, o/e, or DOMINO values (Supporting Information Table S3, Supporting Information Figure S2). During manual evaluation, we detected 16 apparently a priori (likely) pathogenic variants, which we subsequently reclassified as (likely) non‐pathogenic and thus excluded from the analysis. This reclassification was due to the less frequent allele being the reference allele in GRCH37/hg19 or to miscalled deletion/insertion variants or because the sequence variants are exclusively present in a weakly expressed exon (Supporting Information Table S4 and Table S5).
For the more elaborate predisposition/prevalence assessment of fibrillinopathies, we considered sequence variants of categories I‐V (sensu stricto selected, Figure 1) in the genes FBN1 (all exons) and FBN2 (CCA‐mutation‐hotspot exons 23‐34), thereby identifying most likely MFS‐ and CCA‐causing variants in 67 and 39 gnomAD individuals, respectively (Table 1, Figure 2, Supporting Information Table S6). Furthermore, from category VI (sensu lato selected, Figure 1) we added additional 880 FBN1 and 321 FBN2 missense variants that were in silico predicted to be (likely) deleterious, leading to a total of 947 and 360 potentially MFS‐ and CCA‐causing variants, respectively. Accordingly, the gnomAD‐based predisposition to MFS and CCA ranges from 4.8:10 000 (95% CI: 4‐6:10 000) up to 68:10 000 (95% CI: 64‐72:10 000) and 2.8:10 000 (95% CI: 2‐4:10 000) up to 26:10 000 (95% CI: 23‐29:10 000), respectively, depending whether sensu stricto and/or lato selected variants were considered (Table 1). In gnomAD, the majority of the sensu stricto selected FBN1 and FBN2 sequence variants were detected in individuals aged ≥50 years and were rare, with a non‐reference allele count of 1. However, the three most frequent sensu lato selected FBN1 variants, c.3890A>G (289 of 299 non‐reference alleles in Latino), c.3896C>T (70 of 73 non‐reference alleles in Latino), and c.3089A>G (64 of 69 non‐reference alleles in South Asian) were predominant in one gnomAD subpopulation, indicating a founder effect or selection bias (Supporting Information Table S6).
Table 1.
Overview of likely disease‐causing FBN1 and FBN2 variants in gnomAD
| FBN1 exons 23‐34 | FBN1 all exons | FBN2 exons 23‐34 | FBN2 all exons | |
|---|---|---|---|---|
| Nonsense and frameshift (category I) | 2 | 8 | 5 | 29a (27) |
| Splicing (category II) | 0 | 26 (10) | 0 | 36 (25) |
| In‐frame indels (category III) | 1 | 9 (6) | 0 | 8 (5) |
| Disulfide bonds (category IV) | 1 | 8 | 4 | 36 (31) |
| Calcium binding (category IV) | 4 | 12 (12) | 6 (5) | 49 (23) |
| HGMD 2019.1/ClinVar 2019.5 (category V)b | 1 | 4 (3) | 0 | 0 |
|
Sensu stricto selected sequence variants (categories I‐V) / (Prevalence) |
9:138 632 / (0.65:10 000) |
67:138 632 / (4.83:10 000) |
39:138 632 / (2.81:10 000) |
158:138 632 / (11.40:10 000) |
| Sensu lato selected sequence variants (category VI)c | 566 (40) | 880 (162) | 321 (71) | 1.999 (301) |
| All variants (categories I‐VI) / (Prevalence) |
575:138 632 / (41.48:10 000) |
947:138 632 / (68.31:10 000) |
360:138 632 / (25.97:10 000) |
2,157:138 632 / (155.59:10 000) |
| All individuals in gnomAD (exomes and genomes) | 138 632 | |||
Note: Numbers indicate the total number of variants, while numbers in parenthesis indicate the number of unique variants.
Abbreviations: gnomAD, Genome Aggregation Consortium; HGMD, Human Gene Mutation Database; indel, small insertion/deletion.
Nonsense/frameshift variants were counted regardless of their position in FBN2.
Only sequence variants not already included in categories I‐IV and passing manual evaluation were counted.
Sequence variants predicted as "damaging" or "deleterious" by all of the six used in silico prediction tools (FATHMM, FATHMM‐MKL, MutationAsessor, MutationTaster, PolyPhen2, SIFT) not contained in I‐V were counted; see also Supporting Information Table S1.
Figure 2.
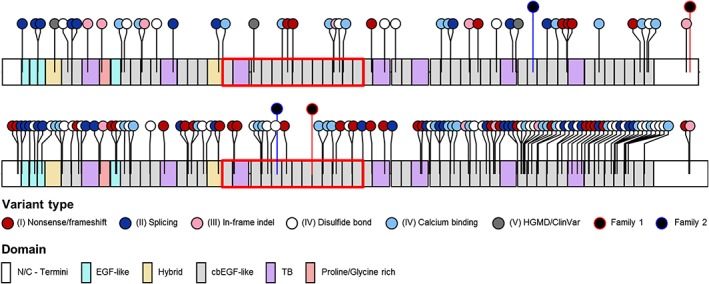
FBN1 and FBN2 a priori (likely) pathogenic variants in gnomAD and FBN1/FBN2 dual variants in Family 1 and Family 2. Lollipops show the type and position of variants in relation to protein domain structure. Red boxes indicate the severe/neonatal region in FBN1 (exons 23‐34) and the comparable congenital contractural arachnodactyly (CCA)‐mutation‐hotspot region in FBN2 (exons 23‐34). cbEGF, calcium‐binding epidermal growth factor; EGF, epidermal growth factor; HGMD, Human Gene Mutation Database; indel, small insertion/deletion; TB, transforming growth factor β binding. Information on protein domains was obtained from umd.be/FBN1 and umd.be/FBN2. Lollipop diagrams were generated using the R package “trackViewer,” available from http://bioconductor.org/packages/release/bioc/html/trackViewer.html [Colour figure can be viewed at http://wileyonlinelibrary.com]
3.2. Evaluation of in silico prediction tools and ACMG/AMP classifications
Splicing (category II) and functionally critical missense (category IV) variants in FBN1 and FBN2 served as a set of positive controls for the used in silico prediction tools (Supporting Information Table S1). All but two missense variants disrupting disulfide‐bond‐forming cysteines were correctly identified as “damaging” or “deleterious” by all six used in silico missense prediction tools, whereas five sequence variants disrupting calcium binding were missed by at least one tool. In contrast, 41, 38, and 21 of the 47 sensu stricto selected FBN1 variants were classified as variant of unknown significance (VUS) according to the ACMG/AMP guidelines by using automated classification, manually adjusting variants disrupting disulfide‐bond‐forming cysteines from PM1 to PS3,29 and applying the criterion PM2 for extremely rare (allele count ≤2 in gnomAD) variants, respectively (Supporting Information Table S6). All splicing (category II) and functionally critical missense (category IV) variants were identified as phylogenetically conserved by at least one of the three used conservation scores (PhastCons, PhyloP, and SiPhy; Supporting Information Table S1), of which the SiPhy score generated the fewest apparently false‐negative results (Supporting Information Table S6). The CADD algorithm, capable of predicting the effect of all types of sequence variants, assigned high values to the majority of the sensu stricto selected sequence variants, but it also scored six FBN1/FBN2 sequence variants in categories I‐V below a conservative PHRED‐scaled CADD cutoff score of 20 (Supporting Information Table S6).
3.3. Evaluation of HGMD and ClinVar entries
Sixty‐six and two entries not contained in categories I‐IV were classified as DM (ie, disease‐causing mutations; not to be confused with the ACMG/AMP classification pathogenic and likely pathogenic) in HGMD (v2019.1), while six and none were classified as pathogenic in ClinVar (v2019.5) for MFS and CCA, respectively. By manual evaluation of corresponding publications/entries, segregation or functional analyses provided proof of pathogenicity for three of the FBN1 HGMD DM entries, which, but no FBN2 variant, could be included in category V (Table 1).
3.4. Families with pathogenic variants in both FBN1 and FBN2
In our cohort of 500 genomes, screening for individuals harboring (likely) pathogenic variants in both FBN1 and FBN2 resulted in two unrelated individuals (index patients with dual variants). Segregation analysis by Sanger sequencing of family members revealed a further dual variant carrier as well as individuals harboring only one or none of the sequence variants (Table 2). Affected family members were physically examined, revealing overlapping as well as distinguishing MFS and CCA clinical features (Table 2, Supporting Information Table S7).
Table 2.
Clinical features observed in Family 1 and Family 2 harboring sequence variants in FBN1 and FBN2
| Family 1 | Family 2 | |||||||
|---|---|---|---|---|---|---|---|---|
| Clinical features | Ab6 | Ab1 | Ab2 | Ab4 | Ab3 | Ac3 | Ac1 | Ac2 |
| Age at examination (years) / Sex | 70 / M | 15 / F | 12 / F | 45 / M | 46 / F | 58 / F | 21 / M | 53 / M |
| Affected gene | FBN1 | FBN1 | FBN1/FBN2 | FBN1/FBN2 | FBN2 | FBN1 | FBN1/FBN2 | FBN2 |
| MFS‐associated features 55 | ||||||||
| Pes valgus | + | − | − | + | − | − | − | (+) |
| Pes planus | (+) | + | + | + | + | + | + | − |
| Pneumothorax | − | − | − | − | − | − | − | +a |
| Reduced elbow extension (r / l) | + / + | >180° / >180° | + / + | + / + | – / – | >180° / >180° | >180° / >180° | – / – |
| 3 of 5 facial features (dolichocephaly, enophthalmus, malar hypoplasia, retrognathia, downslanting fissures) | − | − | − | − | − | − | + | − |
| Skin striae | + | − | − | − | (+) | − | − | + |
| Myopia (>3 diopters) | − | + | − | − | − | − | + | + |
| Aortic dilatation (cm) | n/a | − | − |
+ (4.9) (Z.score: >2.0) |
− | − |
+ (4.1) (Z‐score: >2.0) |
− |
| Ghent nosology systemic score | <7 | <5 | 7 | >7 | <7 | <5 | >7 | <7 |
| CCA‐associated features 24 | ||||||||
| Contractures | ||||||||
| Fingers (r / l) | – / – | – / – | + / + | + / + | + / + | – / – | – / – | + / + |
| Elbow (r / l) | + / +b | – / – | + / + | + / + | – / – | – / ‐ | ‐ / ‐ | – / ‐ |
| Hip | n/a | − | − | n/a | n/a | − | − | − |
| Knee (r / l) | – / – | – / ‐ | ‐ / ‐ | – / ‐ | ‐ / ‐ | – / ‐ | ‐ / ‐ | – / ‐ |
| Crumpled ears (remark) | − | − | – (small ears) | + | − | − | − | − |
| MFS‐ and CCA‐associated features | ||||||||
| Palatal arch | n/a | narrow | high | n/a | n/a | normal | high | high |
| Mitral valve prolapse | − | − | − | − | (+) | − | − | − |
| US/LS ratio < 0.85 (height, cm) | – (172) | – (163.5) | – (159) | + (179.5) | + (171) | – (168) | – (188) | – (195) |
| Armspan/height ratio > 1.05 (armspan, cm) | – (180.5) | – (163.5) | – (159) | – (188.5) | – (179.5) | – (169) | + (205) | – (198.5) |
| Scoliosis or thoracolumbar kyphosis | (+) | − | + | − | (+) | (+) | + | − |
| Wrist sign | − | − | + | + | − | − | + | + |
| Thumb sign | − | − | + | + | − | − | + | + |
| Pectus abnormity | − | + (carinatum) | + (excavatum) | + (carinatum & excavatum) | (+) (carinatum) | − | + (carinatum) | − |
Note: Sequence variants detected in Family 1: FBN1 (NM_000138.4) c.8489A>G p.(Gln2830Arg) and FBN2 (NM_001999.3) c.3974‐26T>G p.(Asn1327_Val1368del), sequence variants detected in Family 2: FBN1 c.6595G>A p.(Gly2199Ser) and FBN2 c.3481G>A p.(Glu1161Lys).
Abbreviations: +, present; −, absent; (+), mildly present; CCA, congenital contractural arachnodactyly; F, female; l, left; LS, lower segment; M, male; MFS, Marfan syndrome; n/a, information not available; r, right; US, upper segment.
Pneumothorax due to tuberculosis infection during adolescence.
Likely explained by his old age of 70 years at examination.
The female index patient of Family 1 (Ab2) (Figure 3A), who was referred with suspected CCA, harbors the heterozygous FBN2 branch‐point variant c.3974‐26T>G causing in‐frame exon skipping (p.Asn1327_Val1368del) first described as segregating with CCA in the large family of Ab2.30 In addition, she harbors the heterozygous FBN1 variant c.8489A>G, p.(Gln2830Arg), not detected in gnomAD and in silico predicted as (likely) deleterious. According to the ACMG/AMP guidelines, these FBN2 and FBN1 variants can be classified as likely pathogenic (LP) and VUS, respectively (Supporting Information Table S8). Ab2 presented with CCA‐associated clinical features such as elbow and finger contractures, but no MFS‐associated cardiovascular involvement (likely due to her young age of 12 years at examination). In addition, she presented with MFS/CCA‐overlapping features such as wrist and thumb sign (arachnodactyly), pectus excavatum, and scoliosis (Table 2, Supporting Information Figure S3). The father of Ab2 (Ab4; first described as IV‐9)30 harbors the same FBN1/FBN2 variants and presented with similar clinical features, but also with CCA‐associated crumpled ears and MFS‐associated aortic dilatation (4.9 cm diameter, Z‐score > 2.0). Ab1, the sister of Ab2, only harbors the FBN1 variant and, as expected, presented with MFS‐associated features such as elbow hyperextension, myopia, and pectus carinatum. Ab6, the grandfather of Ab2, only harbors the FBN1 variant as well but presented with mild MFS/CCA‐overlapping scoliosis and CCA‐associated elbow contractures at age of 70 years at examination. Ab3, the aunt of Ab2, only harbors the FBN2 variant and presented with CCA‐associated finger contractures and mild MFS/CCA‐overlapping mitral valve prolapse and pectus carinatum. Ab7, the grandmother of Ab2, also only harboring the FBN2 variant was not available for clinical examination. Ab5, the mother of Ab2, likely harbors neither sequence variant but was not available for genetic testing and examination.
Figure 3.
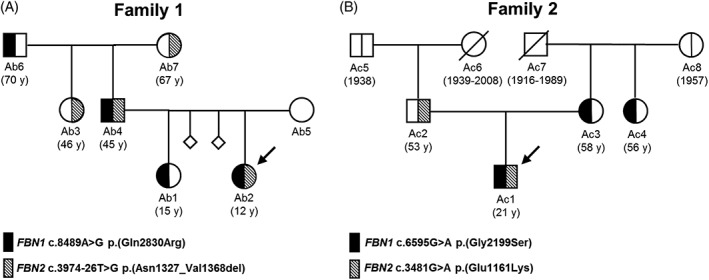
Pedigrees of Family 1 (A) and Family 2 (B). Arrows denote index patients harboring FBN1/FBN2 dual variants. The vertical line in the symbols (circle, female; square, male) denotes molecular genetic testing for the respective variants. Black halves (left) represent an FBN1 variant, striped halves (right) represent an FBN2 variant, and white halves represent absence of variant. The diagonal line through a symbol indicates deceased family members. The age at examination (y, years) or the year of birth and, where applicable, death is given in parentheses
The male index patient in Family 2 (Ac1) (Figure 3B), who was referred to us with suspected MFS, harbors the described (DM? in HGMD)31 heterozygous FBN1 variant c.6595G>A, p.(Gly2199Ser), which is in silico predicted as deleterious but detected three times in gnomAD and thus classified as VUS according to the ACMG/AMP guidelines. In addition, he harbors the reported (DM in HGMD)32 heterozygous FBN2 variant c.3481G>A, p.(Glu1161Lys), which is absent from gnomAD, affects a calcium binding residue, and is predicted as deleterious and classified as LP by in silico algorithms and the ACMG/AMP guidelines, respectively (Table 2, Supporting Information Table S8). Ac1 presented with MFS‐associated features such as elbow hyperextension, high myopia (−23/−23 dpt), and mild aortic dilatation (4.1 cm; Z‐score > 2.0) as well as MFS/CCA‐overlapping features such as severe lumbar scoliosis (surgically corrected), wrist and thumb sign (arachnodactyly), high‐arched palate, and marfanoid habitus. However, he showed no CCA‐associated contractures (Table 2). Ac2, the father of Ac1, only harbors the FBN2 variant and presented with CCA‐associated features such as finger contractures, MFS/CCA‐overlapping features like arachnodactyly and high‐arched palate, as well as high myopia (−12/−9 dpt), which is frequent in MFS and may also occur in CCA.33, 34 Both Ac3, the mother of Ac1, and Ac4, the aunt of Ac1, only harbor the FBN1 variant and presented with mild MFS phenotypes and non‐marfanoid habitus. The mother (Ac3) presented with MFS‐associated elbow hyperextension, mild scoliosis, and no cardiovascular involvement (Table 2), while the aunt (Ac4) reportedly is of short stature with no cardiovascular involvement, but was not available for clinical examination.
4. DISCUSSION
Our data show that, with appropriate consideration, gnomAD, the largest publicly available dataset for population‐based allele frequencies, can be used not only for variant filtering, interpretation, and carrier screening but also for the estimation of disease predisposition/prevalence. Accordingly, we calculated gnomAD‐based predisposition/prevalence estimates for autosomal‐dominant disorders at genome scale, thereby providing a novel estimate for MFS and the first estimate for CCA. Using the example of these fibrillinopathies, we also showed that the genetic predisposition to highly‐penetrant autosomal‐dominant disorders may occur more frequently than previously assumed. Moreover, by presenting two families with hitherto unreported co‐occurrence of FBN1/FBN2 variants causing increased or hidden clinical features of MFS and CCA, we exemplified that segregating (ie, not‐linked) pathogenic variants in more than one gene can underlie the clinical phenotype of patients and the clinical variability of affected family members. Thus, several challenges and conclusions emerge.
First, although WGS is the most comprehensive HTS method offering advantages over targeted sequencing and WES,25, 35 data interpretation remains a challenge.12 Currently, there is no generally accepted gold standard software solution for variant interpretation comparable to BWA/GATK for alignment and variant calling.11, 12 According to the ACMG/AMP guidelines, allele frequencies higher than expected for the disorder (criterion BS1) or reported >5% in population‐based reference datasets (criterion BA1) count as strong or stand‐alone evidence of benign impact of a variant.1 Accordingly, low‐frequency cutoffs are often used, down to <0.01%.6 Although such cutoffs help to reduce or minimize the number of candidate variants, they may remove clinically relevant variants from consideration. Examples include the sensu stricto selected FBN1 variant c.863A>G, p.(Asp288Gly) (allele frequency: 12/277 018; 0.0043%) and the sensu lato selected FBN1 variant c.3026C>G, p.(Pro1009Arg) (allele frequency: 16/246 218; 0.0065%) (Supporting Information Table S6), which are more frequent in gnomAD than the recently introduced maximum tolerated allele count of 2 in approximately 120 000 alleles (approximately 4/240 000; approximately 0.0017%) for MFS.6 In addition, low‐frequency cutoffs exclude weaker variants such as the homozygous Ehlers‐Danlos syndrome causing but heterozygous phenotype‐modifying COL5A1 variant c.1588C>T, p.(Gly530Ser)36 and clinically relevant but common genetic modifiers like chr1:203138970A>G (rs2250509)37 modifying the expressivity of MYH7‐related cardiomyopathy with gnomAD allele frequency of 3.4% and 18.8%, respectively. Moreover, due to a founder effect or selection bias, certain potentially pathogenic variants requiring manual expert review, including the three most frequently occurring FBN1 variants in category VI (c.3089A>G, c.3890A>G, and c.3896C>T in Supporting Information Table S6), may occur rather frequently in one ethnicity (up to 0.12% allele frequency) and thus may be filtered out by using too low cutoff frequencies (ie, below 0.12%). Thus, filtering for sequence variants extremely rare or absent in population‐based reference datasets is helpful to identify variants that should not be missed in a first step, while variants with an increased frequency should not be a priori disregarded without an appropriate consideration and literature review, especially in complex cases.38 Additional rare exonic variants may pose challenges for interpretation, such as variants solely affecting weakly expressed exons or variants miscalled as two independent variants instead of a small insertion/deletion (Supporting Information Table S5). To avoid missed or incomplete diagnoses, there is an evident demand for more advanced filtering methods that may consider more elaborated algorithms, adapted cutoffs, paralogue annotations,39 facial phenotyping algorithms,40 or data sharing platforms.41
Second, although current widely‐used in silico tools and disease‐associated databases are powerful, they are not without fault. In this study, not all in silico tools correctly classified all of the sensu stricto selected FBN1 and FBN2 variants. By individual use, these tools are therefore not capable of depicting all of the underlying disease‐causing mechanisms and, hence, we recommend combining multiple tools for the highest accuracy and the robust application of the ACMG/AMP criteria PP3 and BP4.38 Moreover, several sequence variants classified as DM in HGMD or pathogenic in ClinVar were considered for category V but were excluded, because manual evaluation of the corresponding publication/entry revealed no convincing evidence of pathogenicity (ie, functional characterization or segregation analyses). Thus, even in curated datasets such as HGMD and ClinVar unreliable variant‐disease associations might be present, which is why they need to be used with care and evaluated by experts.42, 43
Third, the ACMG/AMP guidelines provide a framework for clinical variant classification.1 To support implementation and increase inter‐laboratory concordance,44 (semi‐)automated open source (eg, InterVar, Genetic Variant Interpretation Tool)28, 45 and commercial (eg, http://varsome.org; http://goldenhelix.com) classification tools have been developed. Furthermore, a refinement of the ACMG/AMP guidelines into 108 criteria46 as well as several gene‐ and disease‐specific adaptations have been introduced.29, 47, 48 However, because 10 of the 28 ACMG/AMP criteria need manual adjustment (eg, by using functional or segregation data), the result obtained by (semi‐)automated classification is often incomplete, regardless of the applied tool.28 Unfortunately, information for manual adjustment is unavailable in gnomAD and thus the ACMG classifications in this study are based on automated classification, explaining why the majority of our positive control variants in FBN1 were classified as VUS. This, however, would likely be resolved using additional information. For instance, the FBN1‐specific refinement of the ACMG/AMP criteria led to a more appropriate classification of disulfide‐bond‐disrupting FBN1 variants as LP instead of VUS (Supporting Information Table S6).29 In addition, not only because gnomAD contains pathogenic variants but also because larger population datasets will emerge in the future, the criterion PM2 (moderate evidence for pathogenicity; fulfilled for variants absent from control populations) may require adjustment. As the application of population‐based data is a major source of inter‐laboratory discordances,38 increased number of population‐based criteria (eg, 5 criteria instead of the existing 3, PM2, BA1, BS1),46 or gene‐specific frequency thresholds6, 48 could help to appropriately implement the PM2 criterion. Indeed, in the absence of additional information, application of the PM2 criterion to extremely rare variants (allele count ≤2 in gnomAD) may lead to a reclassification, for example, from VUS (PVS1, PP3) to pathogenic (PVS1, PM2, PP3), representing a major caveat. Moreover, the ACMG/AMP guidelines were developed for highly‐penetrant monogenic disorders, complicating their implementation in cases with (pseudo) digenic inheritance or with genetic modifiers (eg, because of the application of segregation data). As the ACMG/AMP guidelines are widely used, it is crucial to use classification tools prudently and to understand their limitations. Depending on the used tool, the thresholds for several criteria may vary, leading to inconsistent or VUS classifications, which may be pivotal as the ACMG/AMP guidelines are frequently invoked for clinical decision support47 and because VUS are not recommended to be reported to patients.49
Fourth, as we detected 2653 a priori (likely) pathogenic gnomAD variants in genes associated with autosomal‐dominant disorders, this dataset should be regarded as apparently, rather than completely, non‐affected. Nevertheless, our data suggest that (likely) pathogenic variants are not overrepresented and we found no clear evidence for considerable bias due to included disease‐specific studies. Thus, gnomAD can be considered as an appropriate representation of the general population, thereby expanding the knowledge of pathogenic variants in ExAC to the much larger gnomAD database,10 enabling (re‐)estimation of the population prevalence of genomic disorders. In general, gnomAD genes of categories I‐II (Figure 1) with an increased number of (likely) pathogenic variants are not infrequently associated with highly‐penetrant diseases with late onset and/or unapparent phenotype, which may occur in fibrillinopathies as well. Indeed, without appropriate imaging of the aorta, even old (≥50 years) carriers of pathogenic FBN1 and/or FBN2 variant(s) may be considered as apparently non‐affected in gnomAD. Our gnomAD‐based MFS predisposition rate of approximately 5:10 000 (FBN1 categories I‐V) is higher than the previously reported prevalence of approximately 1‐3:10 000,33, 50 suggesting that highly‐penetrant fibrillinopathies may be underdiagnosed, especially in mild cases. However, FBN1 mutations have been reported in other non‐MFS disorders as well (cf. HGMD professional; http://portal.biobase-international.com). As for FBN2 we only considered the CCA‐mutation‐hotspot exons 23‐34, the real predisposition to CCA might be even higher than our estimate of approximately 3:10 000. In contrast, some of the detected a priori (likely) pathogenic variants in gnomAD, although de facto (likely) pathogenic, might not lead to disease in certain resilient individuals.51 Resilience may be due to protective genetic modifiers or absence of expression of the affected allele due to random monoallelic expression.52, 53 Because we only considered PASS gnomAD variants (ie, genotype quality ≥20, allelic balance >0.2, and read depth ≥10)2 with a mappability =1,12 the variants included in this study are most likely true variants and do not result from mosaicism or alignment difficulties.
Fifth, the presented two families with hitherto unreported FBN1/FBN2 dual variants showing mixed phenotypes of MFS and CCA exemplify the importance of considering clinically relevant pathogenic variants in more than one gene. The presented cases neither fulfill the definition of true digenic inheritance nor of classic genetic modifiers, because the sequence variants alone also cause at least a mild phenotype and the dual variant carriers do not show novel but rather a combination of MFS‐ and CCA‐associated features.9, 37 Thus, the presented cases are most accurately described as two diseases segregating independently according to classic Mendelian inheritance that underlie the phenotype of dual variant carriers.9 According to our data, the probability of the co‐occurrence of FBN1 and FBN2 variants in one individual might be at least approximately 1.36 × 10−7, implying that additional, unrecognized cases may exist.
As expected, only individuals harboring an FBN1 variant (with or without the FBN2 variant) show MFS‐associated aortic dilatation, whereas only individuals harboring an FBN2 variant (with or without the FBN1 variant) show CCA‐associated contractures (Table 2). Individuals carrying only the familial FBN1 variant (Ab1, Ab6, and Ac3) show several features of MFS including joint laxity, indicating the clinically relevant effect of both FBN1 variants classified as VUS according to the ACMG/AMP guidelines (Table 2). The effect of dual variants appears synergistic for some clinical features, which has also been observed in an Fbn1/Fbn2 double knock‐out mouse model.54 For instance, severe kyphoscoliosis is exclusively present in the dual‐variant carrier (Ac1) in Family 2 (FBN1 c.6595G>A, FBN2 c.3481G>A) but in neither of the parents (Ac2, Ac3). In addition, myopia is more pronounced in the dual‐variant carrier Ac1 (−23/−23 dpt) than in the FBN2 variant carrier Ac2 (−12/−8 dpt), suggesting a synergistic effect of both variants regarding the ocular phenotype as well. In contrast, for other distinguishing clinical features, the effect of one variant appears to be more pronounced, because the dual‐variant carriers (Ab2, Ab4) in Family 1 show CCA‐specific contractures, while the dual‐variant carrier (Ac1) in Family 2 shows MFS‐associated elbow hyperextension. If pathogenic variants in more than one gene underlie the disease phenotype such as in the presented cases, molecular genetic diagnosis is crucial for etiology‐oriented treatment and particularly for genetic counseling regarding family planning. Thus, information on possible digenic inheritance is clinically highly relevant and known cases are listed in the Digenic Diseases Database (http://dida.ibsquare.be).
The main limitation of our study is that for gnomAD phenotypic data of the participants is (yet) unavailable and thus the clinically relevant effect of detected variants can not be directly assessed. Furthermore, information whether multiple sequence variants are harbored by one individual is also unavailable in gnomAD, which would be necessary to assess digenic inheritance and genetic modifiers. Moreover, as we restricted our genome‐scale analysis to categories I and II containing a priori (likely) pathogenic variants (nonsense, frameshift, and canonical splice sites), we potentially missed other (likely) pathogenic variants. We considered that for some genes (eg, GFAP, TGFBR1, TGFBR2) LOF is not (yet) established as a disease mechanism. Thus, to exclude such genes, we restricted our analyses to genes likely intolerant to LOF as predicted by both the pLi score and the o/e metric.2, 3 Indeed, for all but one (ATXN7) of seven genes with (likely) pathogenic gnomAD‐based allele frequencies >1:2000 (Supporting Information Table S4), our manual evaluation revealed LOF as an established disease mechanism. On the other hand, although our data suggest that variants causing severe (pediatric) diseases are likely not overrepresented in gnomAD, the dataset includes disease‐associated projects, potentially biasing our gnomAD‐based disease prevalence calculations. Thus, considering incomplete penetrance, variable expressivity and late disease onset as well, the genome‐scale prevalence values presented here should be regarded as predisposition estimates, aiming to highlight challenges in allele‐frequency‐based variant filtering and HTS data interpretation.
Taken together, in the current genomics era using appropriate short‐ and/or long‐read HTS platforms, the bottleneck on the path to accurate diagnosis is the interpretation of variants rather than the generation of data.12 Current interpretation approaches are challenged by VUS, inappropriate variant filtering, and unrecognized digenic inheritance/variants or genetic modifiers, warranting improvement. Hence, instead of hard filtering, variant interpretation should include large‐scale deeply geno‐ and phenotyped databases, powerful in silico tools, and data sharing as well as, if available, appropriate segregation and functional analyses to aid expert evaluation and enable accurate diagnosis.
CONFLICT OF INTEREST
The authors declare that they have no conflict of interest.
AUTHOR CONTRIBUTIONS
All authors revised the work critically for important intellectual content and gave approval for the final version of the manuscript. G.M. conceptualized this study. A.N., B.S., and M.R. performed physical examination of the patients. A.N., S.C., and J.M. analyzed clinical data. S.C. performed the analysis of database‐derived data. S.C., A.N., J.M., and G.M. wrote the manuscript and B.S. and M.R. critically reviewed the manuscript.
ETHICS APPROVAL
The employed procedures were reviewed and approved by the local institutional review committee, following the principles outlined in the Helsinki Declaration. Informed consent was obtained from all individual participants included in the study or their guardians.
Supporting information
Appendix S1. Supporting Information.
Appendix S2. Supporting Information.
Appendix S3. Supporting Information.
ACKNOWLEDGEMENTS
The authors would like to thank the patients for their participation in this study. This work was supported by funding from the Ernst Göhner Stiftung, Gebauer Stiftung, Hirzel‐Callegari Stiftung, Stiftung Suyana, Palatin‐Stiftung, Gemeinnützige Stiftung der ehemaligen Sparkasse Limmattal, and the Stiftung “Perspektiven” of Swiss Life.
Najafi A, Caspar SM, Meienberg J, Rohrbach M, Steinmann B, Matyas G. Variant filtering, digenic variants, and other challenges in clinical sequencing: a lesson from fibrillinopathies. Clin Genet. 2020;97:235–245. 10.1111/cge.13640
Arash Najafi and Sylvan M. Caspar contributed equally to this work.
Peer Review The peer review history for this article is available at https://publons.com/publon/10.1111/cge.13640.
Funding information Ernst Göhner Stiftung; Gebauer Stiftung; Gemeinnützige Stiftung der ehemaligen Sparkasse Limmattal; Hirzel‐Callegari Stiftung; Palatin‐Stiftung; Stiftung "Perspektiven" of Swiss Life; Stiftung Suyana
DATA ACCESSIBILITY
The data that support the findings of this study are available on request from the corresponding author.
REFERENCES
- 1. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lek M, Karczewski KJ, Minikel EV, et al. Analysis of protein‐coding genetic variation in 60,706 humans. Nature. 2016;536:285‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Karczewski KJ, Francioli LC, Tiao G, et al. Variation across 141,456 human exomes and genomes reveals the spectrum of loss‐of‐function intolerance across human protein‐coding genes. bioRxiv. 2019. 10.1101/531210. [DOI] [Google Scholar]
- 4. Bamshad MJ, Ng SB, Bigham AW, et al. Exome sequencing as a tool for Mendelian disease gene discovery. Nat Rev Genet. 2011;12:745‐755. [DOI] [PubMed] [Google Scholar]
- 5. Kobayashi Y, Yang S, Nykamp K, Garcia J, Lincoln SE, Topper SE. Pathogenic variant burden in the ExAC dataset: an empirical approach to evaluating population data for clinical variant interpretation. Genome Med. 2017;9:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Whiffin N, Minikel E, Walsh R, et al. Using high‐resolution variant frequencies to empower clinical genome interpretation. Genet Med. 2017;19:1151‐1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Lupski JR. Digenic inheritance and Mendelian disease. Nat Genet. 2012;44:1291‐1292. [DOI] [PubMed] [Google Scholar]
- 8. Brehm A, Liu Y, Sheikh A, et al. Additive loss‐of‐function proteasome subunit mutations in CANDLE/PRAAS patients promote type I IFN production. J Clin Invest. 2015;125:4196‐4211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Deltas C. Digenic inheritance and genetic modifiers. Clin Genet. 2018;93:429‐438. [DOI] [PubMed] [Google Scholar]
- 10. Song W, Gardner SA, Hovhannisyan H, et al. Exploring the landscape of pathogenic genetic variation in the ExAC population dataset: insights of relevance to variant classification. Genet Med. 2016;18:850‐854. [DOI] [PubMed] [Google Scholar]
- 11. Van der Auwera GA, Carneiro MO, Hartl C, et al. From FastQ data to high confidence variant calls: the Genome Analysis Toolkit best practices pipeline. Curr Protoc Bioinformatics. 2013;43:11.10.1‐11.10.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Caspar SM, Dubacher N, Kopps AM, Meienberg J, Henggeler C, Matyas G. Clinical sequencing: from raw data to diagnosis with lifetime value. Clin Genet. 2018;93:508‐519. [DOI] [PubMed] [Google Scholar]
- 13. Quinodoz M, Royer‐Bertrand B, Cisarova K, Di Gioia SA, Superti‐Furga A, Rivolta C. DOMINO: using machine learning to predict genes associated with dominant disorders. Am J Hum Genet. 2017;101:623‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dietz HC, Cutting GR, Pyeritz RE, et al. Marfan syndrome caused by a recurrent de novo missense mutation in the fibrillin gene. Nature. 1991;352:337‐339. [DOI] [PubMed] [Google Scholar]
- 15. Putnam EA, Zhang H, Ramirez F, Milewicz DM. Fibrillin‐2 (FBN2) mutations result in the Marfan‐like disorder, congenital contractural arachnodactyly. Nat Genet. 1995;11:473‐458. [DOI] [PubMed] [Google Scholar]
- 16. Schrijver I, Liu W, Brenn T, Furthmayr H, Francke U. Cysteine substitutions in epidermal growth factor–like domains of fibrillin‐1: distinct effects on biochemical and clinical phenotypes. Am J Hum Genet. 1999;65:1007‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reinhardt DP, Ono RN, Notbohm H, Müller PK, Bächinger HP, Sakai LY. Mutations in calcium‐binding epidermal growth factor modules render fibrillin‐1 susceptible to proteolysis. A potential disease‐causing mechanism in Marfan syndrome. J Biol Chem. 2000;275:12339‐13345. [DOI] [PubMed] [Google Scholar]
- 18. Vollbrandt T, Tiedemann K, El‐Hallous E, et al. Consequences of cysteine mutations in calcium‐binding epidermal growth factor modules of fibrillin‐1. J Biol Chem. 2004;279:32924‐32931. [DOI] [PubMed] [Google Scholar]
- 19. Frédéric MY, Monino C, Marschall C, et al. The FBN2 gene: new mutations, locus‐specific dataset (Universal Mutation Dataset FBN2), and genotype‐phenotype correlations. Hum Mutat. 2009;30:181‐190. [DOI] [PubMed] [Google Scholar]
- 20. Miller JN, Pearce DA. Nonsense‐mediated decay in genetic disease: friend or foe? Mutat Res Rev Mutat Res. 2014;762:52‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kurosaki T, Maquat LE. Nonsense‐mediated mRNA decay in humans at a glance. J Cell Sci. 2016;129:461‐467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Collod‐Béroud G, Le Bourdelles S, Ades L, et al. Update of the UMD‐FBN1 mutation dataset and creation of an FBN1 polymorphism dataset. Hum Mutat. 2003;22:199‐208. [DOI] [PubMed] [Google Scholar]
- 23. Faivre L, Collod‐Beroud G, Loeys BL, et al. Effect of mutation type and location on clinical outcome in 1,013 probands with Marfan syndrome or related phenotypes and FBN1 mutations: an international study. Am J Hum Genet. 2007;81:454‐466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Godfrey M. Congenital contractural arachnodactyly In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle, WA: University of Washington, 1993–2018; 2001. [Updated 2012 Feb 23]. http://ncbi.nlm.nih.gov/books/NBK1386. Accessed June 15, 2018. [PubMed] [Google Scholar]
- 25. Meienberg J, Bruggmann R, Oexle K, Matyas G. Clinical sequencing: is WGS the better WES? Hum Genet. 2016;135:359‐362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Plüss M, Kopps AM, Keller I, et al. Need for speed in accurate whole‐genome data analysis: GENALICE MAP challenges BWA/GATK more than PEMapper/PECaller and Isaac. Proc Natl Acad Sci U S A. 2017;114:E8320‐E8322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Magyar I, Colman D, Arnold E, et al. Quantitative sequence analysis of FBN1 premature termination codons provides evidence for incomplete NMD in leukocytes. Hum Mutat. 2009;30:1355‐1364. [DOI] [PubMed] [Google Scholar]
- 28. Li Q, Wang K. InterVar: clinical interpretation of genetic variants by the 2015 ACMG‐AMP guidelines. Am J Hum Genet. 2017;100:267‐280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Muiño‐Mosquera L, Steijns F, Audenaert T, et al. Tailoring the American College of Medical Genetics and Genomics and the Association for Molecular Pathology Guidelines for the interpretation of sequenced variants in the FBN1 gene for Marfan syndrome: proposal for a disease‐ and gene‐specific guideline. Circ Genom Precis Med. 2018;11:e002039. [DOI] [PubMed] [Google Scholar]
- 30. Maslen C, Babcock D, Raghunath M, Steinmann B. A rare branch‐point mutation is associated with missplicing of fibrillin‐2 in a large family with congenital contractural arachnodactyly. Am J Hum Genet. 1997;60:1389‐1398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tan L, Li Z, Zhou C, et al. FBN1 mutations largely contribute to sporadic non‐syndromic aortic dissection. Hum Mol Genet. 2017;26:4814‐4822. [DOI] [PubMed] [Google Scholar]
- 32. Callewaert BL, Loeys BL, Ficcadenti A, et al. Comprehensive clinical and molecular assessment of 32 probands with congenital contractural arachnodactyly: report of 14 novel mutations and review of the literature. Hum Mutat. 2009;30:334‐341. [DOI] [PubMed] [Google Scholar]
- 33. Dietz H. Marfan Syndrome In: Adam MP, Ardinger HH, Pagon RA, et al., eds. GeneReviews. Seattle, WA: University of Washington; 2001. [Updated 2017 Oct 12]:1993‐2018. http://ncbi.nlm.nih.gov/books/NBK1335. Accessed June 15, 2018. [Google Scholar]
- 34. Viljoen D. Congenital contractural arachnodactyly (Beals syndrome). J Med Genet. 1994;31:640‐643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meienberg J, Zerjavic K, Keller I, et al. New insights into the performance of human whole‐exome capture platforms. Nucleic Acids Res. 2015;43:e76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giunta C, Nuytinck L, Raghunath M, Hausser I, De Paepe A, Steinmann B. Homozygous Gly530Ser substitution in COL5A1 causes mild classical Ehlers‐Danlos syndrome. Am J Med Genet. 2002;109:284‐290. [DOI] [PubMed] [Google Scholar]
- 37. Riordan JD, Nadeau JH. From peas to disease: modifier genes, network resilience, and the genetics of health. Am J Hum Genet. 2017;101:177‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kim YE, Ki CS, Jang MA. Challenges and considerations in sequence variant interpretation for Mendelian disorders. Ann Lab Med. 2019;39:421‐429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Walsh R, Peters NS, Cook SA, Ware JS. Paralogue annotation identifies novel pathogenic variants in patients with Brugada syndrome and catecholaminergic polymorphic ventricular tachycardia. J Med Genet. 2014;51:35‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gurovich Y, Hanani Y, Bar O, et al. Identifying facial phenotypes of genetic disorders using deep learning. Nat Med. 2019;25:60‐64. [DOI] [PubMed] [Google Scholar]
- 41. Sobreira NLM, Arachchi H, Buske OJ, et al. Matchmaker exchange. Curr Protoc Hum Genet. 2017;95:9.31.1‐9.31.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Groth KA, Von Kodolitsch Y, Kutsche K, et al. Evaluating the quality of Marfan genotype‐phenotype correlations in existing FBN1 datasets. Genet Med. 2017;19:772‐777. [DOI] [PubMed] [Google Scholar]
- 43. Shah N, Hou YC, Yu HC, et al. Identification of misclassified ClinVar variants via disease population prevalence. Am J Hum Genet. 2018;102:609‐619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Amendola LM, Jarvik G, Leo MC, et al. Performance of ACMG‐AMP variant‐interpretation guidelines among nine laboratories in the Clinical Sequencing Exploratory Research Consortium. Am J Hum Genet. 2016;98:1067‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kleinberger J, Maloney KA, Pollin TI, Jeng LJ. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet Med. 2016;18:1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Nykamp K, Anderson M, Powers M, et al. Sherloc: a comprehensive refinement of the ACMG‐AMP variant classification criteria. Genet Med. 2017;19:1105‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Nicora G, Limongelli I, Gambelli P, et al. CardioVAI: an automatic implementation of ACMG‐AMP variant interpretation guidelines in the diagnosis of cardiovascular diseases. Hum Mutat. 2018;39:1835‐1846. [DOI] [PubMed] [Google Scholar]
- 48. Whiffin N, Walsh R, Govind R, et al. CardioClassifier: disease‐ and gene‐specific computational decision support for clinical genome interpretation. Genet Med. 2018;20:1246‐1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Vears DF, Sénécal K, Borry P. Reporting practices for variants of uncertain significance from next generation sequencing technologies. Eur J Med Genet. 2017;60:553‐558. [DOI] [PubMed] [Google Scholar]
- 50. Judge DP, Dietz HC. Marfan's syndrome. Lancet. 2005;366:1965‐1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen R, Shi L, Hakenberg J, et al. Analysis of 589,306 genomes identifies individuals resilient to severe Mendelian childhood diseases. Nat Biotechnol. 2016;34:531‐538. [DOI] [PubMed] [Google Scholar]
- 52. Savova V, Chun S, Sohail M, et al. Genes with monoallelic expression contribute disproportionately to genetic diversity in humans. Nat Genet. 2016;48:231‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Caspar SM, Dubacher N, Matyas G. More genes for thoracic aortic aneurysms and dissections. J Am Coll Cardiol. 2019;73:528‐529. [DOI] [PubMed] [Google Scholar]
- 54. Carta L, Pereira L, Arteaga‐Solis E, et al. Fibrillins 1 and 2 perform partially overlapping functions during aortic development. J Biol Chem. 2006;281:8016‐8023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Loeys BL, Dietz HC, Braverman AC, et al. The revised Ghent nosology for the Marfan syndrome. J Med Genet. 2010;47:476‐485. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting Information.
Appendix S2. Supporting Information.
Appendix S3. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available on request from the corresponding author.