

Abstract
Objective
To evaluate the long‐term safety and tolerability of ubrogepant for the acute treatment of migraine.
Background
Ubrogepant is an oral, calcitonin gene–related receptor antagonist in development for the acute treatment of migraine. The efficacy of ubrogepant was demonstrated in 2 phase 3 trials in which a significant improvement was observed in migraine headache pain, migraine‐associated symptoms, and ability to function.
Methods
This was a phase 3, multicenter, randomized, open‐label, 52‐week extension trial. Adults with migraine with or without aura entered the trial after completing one of 2 phase 3 lead‐in trials and were re‐randomized 1:1:1 to usual care, ubrogepant 50 mg, or ubrogepant 100 mg. Randomization to ubrogepant dose was blinded. Those randomized to usual care continued to treat migraine attacks with their own medication. The usual care arm was included in this trial to capture background rates of hepatic laboratory parameters and contextualize hepatic safety assessments. Safety and tolerability were the primary outcome measures. The safety population for the ubrogepant arms included all randomized participants who received at least 1 dose of treatment. All cases of alanine aminotransferase (ALT)/aspartate aminotransferase (AST) elevations of ≥3 times the upper limit of normal were adjudicated by an independent panel of liver experts who were blinded to dose.
Results
The safety population included 1230 participants (404 in the ubrogepant 50‐mg group, 409 in the ubrogepant 100‐mg group, and 417 in the usual care group). Participants were on average 42 years of age, 90% (1106/1230) female and 85% (1043/1230) white, with an average BMI of 30 kg/m2. Throughout the trial, 21,454 migraine attacks were treated with 31,968 doses of ubrogepant. Treatment‐emergent adverse events (TEAEs) were reported by 268/404 (66%) participants receiving ubrogepant 50 mg and 297/409 (73%) receiving ubrogepant 100 mg. The most commonly reported TEAE was upper respiratory tract infection (<12%); findings were similar across dose groups. Treatment‐related TEAEs were reported by 42/404 (10%) participants in the ubrogepant 50‐mg group and 43/409 (11%) in the ubrogepant 100‐mg group. Serious adverse events (SAEs) were reported by 9/404 (2%) participants in the ubrogepant 50‐mg group and 12/409 (3%) participants in the ubrogepant 100‐mg group. Twenty cases of ALT/AST levels of ≥3 times the upper limit of normal were reported and reviewed by an independent clinical adjudication committee of liver experts. There were no cases of Hy’s Law.
Conclusions
Long‐term intermittent use of ubrogepant 50 and 100 mg given as 1 or 2 doses per attack for the acute treatment of migraine was safe and well tolerated, as indicated by a low incidence of treatment‐related TEAEs and SAEs and discontinuations due to adverse events in this 1‐year trial.
Keywords: migraine, acute treatment, calcitonin gene–related peptide, safety
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- CGRP
calcitonin gene‐related peptide
- ECG
electrocardiogram
- IWRS
interactive Web response system
- NSAIDs
non‐steroidal anti‐inflammatory drugs
- SAE
serious adverse event
- TEAE
treatment‐emergent adverse event
Introduction
Migraine is a highly prevalent and burdensome chronic disease with attacks characterized by 1‐sided pulsatile pain associated with sensitivity to light and sound, and nausea in various combinations.1 Based on the recent Global Burden of Disease study, migraine ranks second among the leading causes of years lived with disability,2, 3 impacting not only a person’s daily living activities but having a negative effect on their families as well.4, 5, 6, 7, 8, 9 Effective and safe treatment options are needed to help reduce the burden of migraine.
Several acute treatment options are currently available, including analgesics, nonsteroidal anti‐inflammatory drugs (NSAIDs), ergot derivatives, and triptans.10, 11, 12 Even with the number of acute treatments available, approximately 50% of those with migraine are not satisfied with their acute treatment for migraine and most have significant unmet treatment needs.13 In the Migraine in America Symptoms and Treatment study,14 of the 15,133 adults with migraine, only 15% were using a triptan and 95.8% had at least 1 unmet treatment need.15 Among migraine‐specific acute medications, ergot derivatives and triptans, though effective, are associated with safety and tolerability issues as well as contraindications.11, 16 This supports the need for additional migraine‐specific treatment options with desirable efficacy and safety profiles.
Calcitonin gene–related peptide (CGRP) plays an important role in migraine pathophysiology and as a result, small molecule CGRP receptor antagonists, known as gepants, are being studied for their efficacy in treating migraine.17, 18 Ubrogepant is an oral gepant in development for the acute treatment of migraine. The efficacy and safety of ubrogepant have been shown in proof‐of‐concept and large, placebo‐controlled trials.19, 20, 21 The phase 3 ACHIEVE I and II single‐attack trials met their co‐primary endpoints for the 50‐ and 100‐mg doses, thereby establishing ubrogepant’s efficacy. Rates of headache pain freedom 2 hours post‐dose were significantly superior to placebo with ubrogepant 25, 50, and 100 mg (P ≤ .01).20, 21 In addition, the rates of absence of the most bothersome migraine‐associated symptom (photophobia, phonophobia, or nausea) at 2 hours were significantly greater with ubrogepant 50 and 100 mg than placebo (P ≤ .01), but not with ubrogepant 25 mg.
The primary objective of this phase 3, multicenter, randomized, 52‐week extension trial was to evaluate the long‐term safety and tolerability of intermittent treatment with ubrogepant for the acute treatment of migraine over 1 year. Treatment groups included ubrogepant 50 mg, ubrogepant 100 mg, and a usual care arm. The usual care arm was included to contextualize any hepatic safety findings over the course of this trial. Many treatments were included in the usual care arm, often treatments the patients were already using; therefore, non‐laboratory adverse events (AEs) were not captured in relation to treatment in this arm.
Methods
Trial Design
This was a phase 3, multicenter, randomized, open‐label, 52‐week extension trial conducted at 161 centers in the United States. The trial protocol and all amendments were approved by properly constituted local and central institutional review boards. All participants provided written informed consent before initiation of any trial‐specific procedures. The trial was sponsored by Allergan plc, which included provision of trial treatment for those randomized to ubrogepant treatment arms. Changes to the original protocol are in Supplementary S1.
The first participant was enrolled September 13, 2016 and the last participant completed August 2, 2018. Participants were identified by the trial investigator and entered the trial after completing one of 2 randomized, double‐blind, placebo‐controlled, single‐attack, phase 3 lead‐in trials (ACHIEVE I, NCT02828020; ACHIEVE II, NCT02867709) and were re‐randomized 1:1:1 to usual care, ubrogepant 50 mg, or ubrogepant 100 mg.
Participants who were on a stable dose of migraine preventive medication were allowed to continue on their medication. Those with an alanine aminotransferase (ALT) or aspartate aminotransferase (AST) level ≥1.5 times the upper limit of normal (ULN) or bilirubin >1.5 mg/dL at screening were not included. Participants could not have any clinically significant abnormalities in their electrocardiogram (ECG), physical examination, or laboratory test and could not have uncontrolled hypertension. Those with clinically significant cardiovascular or cerebrovascular disease or myocardial infarction, transient ischemic attack, or stroke within 6 months prior to Visit 1 were excluded from the trial. Use of moderate to strong CYP3A4 inhibitors and inducers was prohibited during the trial.
Although this was an open‐label trial, randomization to the ubrogepant dose was blinded. Participants randomized to ubrogepant took 2 tablets to treat each migraine attack. Participants in the ubrogepant 100‐mg arm took two 50‐mg ubrogepant tablets, whereas participants in the ubrogepant 50‐mg arm took 1 ubrogepant 50‐mg tablet and 1 placebo tablet. Participants randomized to ubrogepant could treat up to 8 migraine attacks per 4‐week interval during the 1‐year trial period. If participants returned to the site and received study medication prior to the end of a designated 4‐week interval, they could treat more than 8 migraine attacks in a 4‐week interval. Participants could treat headache pain of any severity (ie, mild, moderate, or severe). A qualifying migraine attack must have met all of the following conditions: at least 48 hours had passed since the last dose of ubrogepant, the participant had at least 48 hours of pain freedom, less than 4 hours had passed since the headache started, the headache was not resolving on its own, and prohibited medications were not taken.
An optional second dose of ubrogepant or rescue medication, which could include analgesics (eg, acetaminophen, NSAIDs, opioids), anti‐emetics, or triptans, was allowed at least 2 hours after the initial dose if the participant had a nonresponding migraine or migraine recurrence. The second dose of ubrogepant was the same as the first dose and could be taken 2–48 hours after the initial ubrogepant dose. If a second dose was taken, the participant was not allowed to take rescue medication for at least 2 hours after the second dose. If the participant opted to take rescue medication instead, an optional second dose of ubrogepant was not allowed. Participants randomized to the usual care arm continued to treat their migraine attacks with medication that they had been taking historically. Their usual care medications were identified at Visit 1 and any changes were recorded in the electronic case report form.
After screening and randomization, visits occurred every 4 weeks for 1 year. The clinic visits for safety assessments were at 4, 8, 12, 16, 20, 24, 28, 32, 36, 40, 44, 48, and 52 weeks relative to the randomization visit. A safety follow‐up visit occurred 4 weeks after the 52‐week visit or after early termination. Participants who did not treat a single migraine attack in the first 6 months of the trial were discontinued.
An automated interactive Web response system (IWRS) was used to manage the randomization and treatment assignment. The sponsor’s biostatistician prepared the randomization codes. Investigational product was labeled with medication kit numbers. The IWRS provided the site with the specific medication kit number for each randomized participant in the ubrogepant arms at the time of randomization. Randomization was stratified by the participant’s historical triptan response and current preventive concomitant medication use, as determined in the lead‐in trials. All participants, site personnel, and trial sponsor personnel were blinded to ubrogepant dose in this open‐label trial.
An interim analysis was planned for when at least 300 ubrogepant‐treated participants who treated an average of ≥2 migraine attacks per month were enrolled in the trial for 6 months and 200 ubrogepant‐treated participants who treated an average of ≥2 migraine attacks per month were enrolled for 1 year. At the time of the interim database lock, the ubrogepant dosing assignments were unblinded to the sponsor; participants and investigators remained blinded. A full data analysis was conducted after trial completion and the data from the final 1‐year trial are reported here.
Outcome Measures
The primary outcomes were safety and tolerability, which included collection of AEs, clinical laboratory determinations, ECGs, vital sign measurements, physical examinations, and the Columbia‐Suicide Severity Rating Scale at each clinic visit. Investigators began each visit by querying for AEs by asking the participant a general, non‐directed question such as “How have you been feeling since the last visit?” and then followed up with directed questioning, examination, and documentation, as appropriate. All AEs were coded using the Medical Dictionary for Regulatory Activities version 20.1 by the sponsor’s medical coding group. The investigators were required to provide an assessment of the severity, the likelihood of a causal relationship to the study medication, and seriousness of the event, document all actions taken with regard to the study medication, and detail any other treatment measures taken for the AE.
An independent clinical adjudication committee of liver experts was established for blinded adjudication of posttreatment elevations of ALT and/or AST ≥3 times the ULN. Potential Hy’s Law cases were defined as an elevation of ALT or AST value ≥3× the ULN and an elevated total bilirubin value ≥2× ULN, and, at the same time, an alkaline phosphatase value <2× ULN, all based on blood draws collected within a 24‐hour period.22, 23 A data safety monitoring board monitored unblinded safety data. Exploratory efficacy assessments of headache severity, migraine‐associated symptoms (photophobia, phonophobia, nausea, and vomiting), use of rescue medications, use of optional second dose, and recurrence of headache pain were collected for ubrogepant treatment arms only. This manuscript’s focus is the safety outcomes; thus a comprehensive summary of efficacy findings will be reported separately.
Statistical Analysis
The sample size was driven by regulatory safety requirements for duration of exposure and number of participants rather than statistical considerations. The expected randomization was 1200 participants with at least 70% completing 12 months of treatment and at least 50% expected to treat on average at least 2 migraine attacks per month.
The screened population included all screened participants who signed the informed consent. The intent‐to‐treat population included all randomized participants. The safety population included all randomized participants who took at least 1 dose of ubrogepant treatment and all participants randomized to the usual care arm.
AE data were analyzed by calculating the frequency and percentage of participants reporting the event in each treatment group. No formal statistical comparisons between treatment groups were planned or executed. The usual care population was included only to contextualize background rates of ALT/AST safety findings occurring in the ubrogepant‐treated participants. Elevations of ALT/AST are often asymptomatic; thus, the usual care arm provided within‐study control of natural fluctuations in these evaluations within a migraine population. The usual care population included participants who had been on and had tolerated their medication prior to enrollment. The participants in the usual care arm selected their usual acute treatment for migraine based on their prior treatment experience; therefore, direct comparisons of AE rates between ubrogepant and the usual care arm cannot be made given this self‐selection. Many of the usual care arm participants had a history of tolerating their selected medication; therefore, AE rates in the usual care arm would not have reflected the introduction of a new treatment, as they did in the ubrogepant arms. Because previous experience with the usual care drug could have reduced the probability of experiencing/reporting safety or tolerability AEs, AEs reported in the usual care arm were not assessed for relatedness to the usual care medication. To directly evaluate the safety and tolerability profiles of acute treatments, enrolled participants should be naïve to each treatment before randomization, after which AE profiles can be compared.
AEs occurring after the screening visit were summarized for treatment‐emergent adverse events (TEAEs), treatment‐related TEAEs, serious adverse events (SAEs), and AEs leading to discontinuation. Treatment‐related TEAEs were only determined for ubrogepant treatment arms.
Clinical laboratory assessments were summarized by treatment group. The adjudication committee findings were summarized by treatment group for participants with ALT or AST elevations ≥3 times the ULN.
Statistical analyses were conducted using SAS Version 9.4 or later (Cary, NC).
Results
Participants and Extent of Exposure
In this trial, 1310 participants were screened, 1254 randomized, and 1230 included in the safety population (Fig. 1). Of the safety population, 665 participants were from the ACHIEVE I lead‐in trial and 565 from the ACHIEVE II lead‐in trial. In the ubrogepant 50‐mg group, 404 participants treated 10,323 migraine attacks with 1 or more doses of ubrogepant for a total of 15,536 doses. In the ubrogepant 100‐mg group, 409 participants treated 11,131 migraine attacks with 1 or more doses of ubrogepant for a total of 16,432 doses. Over the course of the trial, an average of 13.2 and 14.8 migraine attacks were treated with 1 dose of ubrogepant 50 and 100 mg, respectively, and an average of 12.3 and 12.4 migraine attacks were treated with 2 or more doses of ubrogepant 50 and 100 mg, respectively. The average number of doses taken per participant over the 1‐year trial period was 38.5 doses for ubrogepant 50 mg and 40.2 doses for ubrogepant 100 mg.
Figure 1.
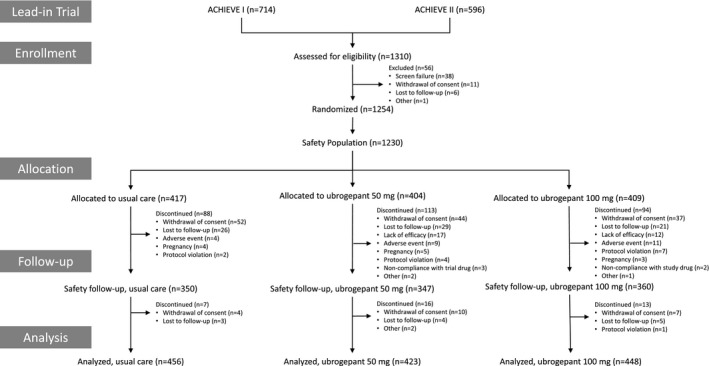
Participant disposition.
Considering the maximum number of treated attacks in any given month, a total of 233 (29%) ubrogepant‐treated participants treated 6 or more migraine attacks during at least 1 month in the trial (109 participants in the ubrogepant 50‐mg group and 124 participants in the ubrogepant 100‐mg group). Of these, a total of 72 (9%) participants treated 8 or more migraine attacks in at least 1 month (30 participants in the ubrogepant 50‐mg group and 42 in the ubrogepant 100‐mg group). Cumulatively, participants treated 6 or more migraines in a month in 740 months (334 months for those who took ubrogepant 50 mg and 406 months for those who took ubrogepant 100 mg). Participants treated 8 or more migraine attacks in a month for a total of 144 months (55 months for those who took ubrogepant 50 mg and 89 months for those who took 100 mg).
Demographics and Clinical Characteristics
In the safety population, participants were on average 41.8 years of age (SD = 11.7), predominantly female (N = 1106/1230, 90%) and white (N = 1043/1230, 85%), with an average BMI of 29.9 kg/m2 (SD = 7.7). At the time of randomization, 306 (24.9%) participants in the safety population were on a preventive treatment for migraine (eg, topiramate, onabotulinumtoxinA, propranolol, amitriptyline), and use of preventive treatment was evenly distributed across treatment groups (n = 102 [25%] in each group). In the safety population, nearly all participants (99.5%) took at least 1 concomitant medication during the treatment period. The most common were ibuprofen (54.5%), combination aspirin‐acetaminophen‐caffeine (36.4%), sumatriptan (27.6%), and acetaminophen (25.6%). Overall, participant demographics and clinical characteristics were similar in the ubrogepant and usual care arms.
Ubrogepant Adverse Events
TEAEs occurred in 268 (66%) participants in the ubrogepant 50‐mg group and in 297 (73%) participants in the ubrogepant 100‐mg group (Table 1). Most commonly reported TEAEs (≥2% of participants in any treatment group; Table 2) were upper respiratory tract infection, nasopharyngitis, sinusitis, urinary tract infection, influenza, and nausea. No increase in the incidence of TEAEs was noted with an increase in number of attacks treated per month (Supplementary S2). The incidence of TEAEs was similar across age (Supplementary S3), sex (Supplementary S4), and race subgroups (Supplementary S5).
Table 1.
Overall Summary of Adverse Events (Safety Population)
| Ubrogepant 50 mg | Ubrogepant 100 mg | |
|---|---|---|
| n = 404 | n = 409 | |
| n (%) | n (%) | |
| TEAEs† | 268 (66.3) | 297 (72.6) |
| Treatment‐related TEAEs† | 42 (10.4) | 43 (10.5) |
| On therapy SAE‡, § | 9 (2.2) | 12 (2.9) |
| Deaths‡ | 0 | 0 |
| AEs leading to discontinuation¶ | 9 (2.2) | 11 (2.7) |
Treatment‐emergent events were defined as those events that initially occurred or increased in intensity on or after the initial dose of treatment of the lead‐in trial. An event that occurred after Visit 16 for participants with Visit 16 or more than 30 days after the last visit or last treatment, whichever was later, for participants without Visit 16 was not considered to be treatment emergent.
On‐therapy events were defined as those events that occurred between the treatment start date of the lead‐in trial and Visit 16, or within 30 days after the last visit or last treatment, whichever was later, for participants without Visit 16.
All SAEs reported, by preferred term, ubrogepant 50 mg: cholecystitis acute, cholelithiasis, pneumonia, device allergy (allergic reaction due to Essure® implant), gait disturbance, hypertensive crisis, intestinal obstruction, pelvic inflammatory disease, postprocedural infection, sinus tachycardia, substance‐induced mood disorder; ubrogepant 100 mg: abortion, abortion spontaneous, acute respiratory failure, cholecystitis acute, cholelithiasis, colitis, diabetic ketoacidosis, ectopic pregnancy, gastroenteritis norovirus, hemiparesis, hiatus hernia, non‐cardiac chest pain, pancreatitis acute, pneumonia, sepsis, subdural hematoma, suicidal ideation.
Includes events that occurred from screening to Visit 16, or within 30 days after the last visit or last treatment, whichever was later, for participants without Visit 16.
Only events that occurred on or after the randomization date are included except for AEs leading to discontinuation. Participants are counted only once within each category.
AE = adverse event; SAE = serious adverse event; TEAE = treatment‐emergent adverse event.
Table 2.
Common (≥2%)† Treatment‐Emergent Adverse Events by Preferred Term
| Ubrogepant 50 mg | Ubrogepant 100 mg | |
|---|---|---|
| n = 404 | n = 409 | |
| Preferred Term‡ | n (%) | n (%) |
| Nasopharyngitis | 33 (8.2) | 47 (11.5) |
| Upper respiratory tract infection | 47 (11.6) | 44 (10.8) |
| Sinusitis | 28 (6.9) | 26 (6.4) |
| Urinary tract infection | 22 (5.4) | 26 (6.4) |
| Influenza | 17 (4.2) | 25 (6.1) |
| Nausea | 19 (4.7) | 19 (4.6) |
| Bronchitis | 13 (3.2) | 18 (4.4) |
| Blood creatine phosphokinase increased | 10 (2.5) | 16 (3.9) |
| Alanine aminotransferase increased | 9 (2.2) | 15 (3.7) |
| Aspartate aminotransferase increased | 7 (1.7) | 14 (3.4) |
| Back pain | 14 (3.5) | 14 (3.4) |
| Nephrolithiasis | 4 (1.0) | 13 (3.2) |
| Dizziness | 5 (1.2) | 12 (2.9) |
| Anxiety | 6 (1.5) | 11 (2.7) |
| Cough | 8 (2.0) | 11 (2.7) |
| Diarrhea | 10 (2.5) | 11 (2.7) |
| Arthralgia | 11 (2.7) | 10 (2.4) |
| Gastroenteritis viral | 7 (1.7) | 10 (2.4) |
| Muscle strain | 2 (0.5) | 10 (2.4) |
| Depression | 1 (3.2) | 5 (1.2) |
| Migraine | 6 (1.5) | 4 (1.0) |
| Abdominal pain | 4 (1.0) | 2 (0.5) |
| Epididymitis§ | 1 (3.2) | 0 (0) |
| Testicular pain§ | 1 (3.2) | 0 (0) |
Only adverse events reported in ≥2% of participants (after rounding) in any treatment group are shown. Participants are counted only once within each preferred term.
Preferred terms were coded according to the Medical Dictionary of Regulatory Activity (MedDRA) Version 20.1.
For sex‐specific adverse events, percentages are relative to the number of participants of the appropriate sex.
Of the participants who had TEAEs, approximately 10% were considered related to ubrogepant treatment by the investigator. The most commonly reported treatment‐related TEAEs (≥1%) in any ubrogepant dose group were nausea (1.5 and 1.7% with ubrogepant 50 and 100 mg, respectively), dizziness (0.5 and 1.5%), and somnolence (1.5 and 1.2%). ALT increases (0.7 and 1.0%) and AST increases (0.5 and 1.0%) also occurred with ubrogepant 50 and 100 mg.
Most TEAEs were mild or moderate in severity. Severe TEAEs were reported for 6.2 and 8.1% of participants in ubrogepant 50‐mg and ubrogepant 100‐mg groups, respectively. Severe TEAEs that were reported for 3 or more participants in the ubrogepant groups were AST increase, cholelithiasis, nephrolithiasis, pneumonia, and sinusitis (each reported for 3 ubrogepant‐treated participants); influenza, migraine, and nausea were each reported for 4 ubrogepant‐treated participants.
SAEs were reported for 9/404 (2%) participants in the ubrogepant 50‐mg group and 12/409 (3%) participants in the ubrogepant 100‐mg group (Table 1). Only 1 SAE was considered related to treatment by the investigator (sinus tachycardia), and this event occurred in a participant with a history of supraventricular tachycardia with ablation; the participant continued to take ubrogepant after this event without further complications. No deaths occurred during the trial. Discontinuations due to AEs were reported for 2–3% of the ubrogepant‐treated participants. TEAEs leading to discontinuation assessed as related to treatment by the investigator were reported for 4 participants in the ubrogepant 50‐mg group (nausea, thrombocytopenia, urticaria, and vomiting) and 4 in the ubrogepant 100‐mg group (abnormal ECG and extrasystolic beats; nausea and palpitations; pruritus; ALT and AST increase). The event of thrombocytopenia was reported as unknown in terms of relatedness and imputed as related.
Cardiovascular Safety
Frequency of cardiovascular TEAEs in predefined categories of cardiac arrhythmia, central nervous system (CNS) vascular disorders, embolic and thrombotic events, hypertension, and ischemic heart disease were generally balanced between ubrogepant treatment groups (Table 3). No cardiovascular TEAEs occurred at a rate >2% in either ubrogepant treatment arm; few cardiovascular SAEs were reported. Two SAEs were reported in the category of CNS vascular disorders, both considered not related to study medication. One event was an SAE of hemiparesis; vascular etiology (ie, stroke) was ruled out and the investigator considered the event to be due to poorly controlled hypothyroidism (Supplementary S6). The other SAE was a traumatic subdural hematoma due to a head injury. One cardiovascular SAE was considered related to treatment by the investigator (sinus tachycardia, details presented in Ubrogepant Adverse Events section). No cardiovascular events related to myocardial infarction or stroke were reported in either ubrogepant treatment arm.
Table 3.
Treatment‐Emergent Cardiovascular Adverse Events of Special Interest
| TEAE Category† | Ubrogepant 50 mg | Ubrogepant 100 mg |
|---|---|---|
| n = 404 | n = 409 | |
| Preferred Term | n (%) | n (%) |
| Cardiac arrhythmias | 4 (1.0) | 5 (1.2) |
| Arrhythmia | 0 | 1 (0.2) |
| ECG QRS complex prolonged | 0 | 1 (0.2) |
| Extrasystoles | 0 | 1 (0.2) |
| Sinus tachycardia‡ | 1 (0.2) | 1 (0.2) |
| Ventricular extrasystoles | 0 | 1 (0.5) |
| Atrioventricular block first degree | 1 (0.2) | 0 |
| Cardiac flutter | 1 (0.2) | 0 |
| Sinus bradycardia | 1 (0.2) | 0 |
| CNS vascular disorder/embolic and thrombotic events | 0 | 2 (0.5) |
| Hemiparesis†, ‡, § | 0 | 1 (0.2) |
| Traumatic subdural hematoma‡ | 0 | 1 (0.2) |
| Hypertension | 13 (3.2) | 11 (2.7) |
| Hypertension | 8 (2.0) | 7 (1.7) |
| Blood pressure diastolic increased | 1 (0.2) | 2 (0.5) |
| Blood pressure increased | 3 (0.7) | 2 (0.5) |
| Hypertensive crisis‡ | 1 (0.2) | 0 |
| Prehypertension | 1 (0.2) | 0 |
| Ischemic heart disease | 2 (0.5) | 2 (0.5) |
| Angina pectoris | 2 (0.5) | 2 (0.5) |
Each category is based on predefined standard groups of Preferred Terms (narrow Standard MedDRA Queries).
Reported as a serious adverse event.
The participant with hemiparesis had negative imaging for any vascular events and stroke was ruled out as a possible cause. The investigator attributed the event to severe uncontrolled hypothyroidism.
CNS = central nervous system; TEAE = treatment‐emergent adverse event.
Hepatic Safety
The mean changes in ALT and AST from baseline to postbaseline visits were similar across the ubrogepant treatment groups and the usual care arm (Supplementary S7). Postbaseline hepatic laboratory values of clinical interest are presented in Table 4. There were no cases of confirmed Hy’s Law. One participant, while hospitalized with recurrent acute cholecystitis due to gallstones seen in the common bile duct, had rapid rises in ALT/AST and bilirubin levels. Following gallbladder removal, enzymes returned to normal (Supplementary S8). As there was an underlying cause for these ALT/AST and bilirubin elevations, this case did not meet criteria for Hy’s Law. Twenty cases of ALT or AST elevations of ≥3× ULN were reported, as follows: 4/398 (1.0%) in the usual care arm, 5/399 (1.3%) in the ubrogepant 50 mg arm, and 11/406 (2.7%) in the ubrogepant 100 mg arm. All ≥3× ULN ALT or AST cases were adjudicated by an independent panel of liver experts blinded to ubrogepant dose (Supplementary S8). Out of the 20 cases, 17 (4 usual care, 3 ubrogepant 50 mg, and 10 ubrogepant 100 mg) were judged Unlikely Related to study medication. Two cases (both ubrogepant 50 mg) were judged Possibly Related with confounding factors reported (increased alcohol and acetaminophen use; dilated bile duct). Only 1 case of ALT or AST elevations of ≥3× ULN (ubrogepant 100 mg) was judged Probably Related; however, confounding factors were noted (prednisone use for exacerbation of psoriasis prior to rise in ALT/AST). All cases of ALT or AST elevation ≥3× ULN resolved in those who continued ubrogepant dosing.
Table 4.
Hepatic Laboratory Parameters: Postbaseline Values of Clinical Interest (Safety Population)
| Usual Care | Ubrogepant 50 mg | Ubrogepant 100 mg | |
|---|---|---|---|
| n = 417 | n = 404 | n = 409 | |
| Preferred Term | n (%) | n (%) | n (%) |
| ALT (U/L) | |||
| ≥1 × ULN | 114/397 (28.7) | 117/399 (29.3) | 122/406 (30.0) |
| ≥1.5 × ULN | 38/397 (9.6) | 38/399 (9.5) | 55/406 (13.5) |
| ≥2 × ULN | 16/397 (4.0) | 14/399 (3.5) | 33/406 (8.1) |
| ≥3 × ULN | 4/397 (1.0) | 4/399 (1.0) | 8/406 (2.0) |
| ≥5 × ULN | 1/397 (0.3) | 2/399 (0.5) | 4/406 (1.0) |
| ≥10 × ULN | 0/397 | 0/399 | 1/406 (0.2) |
| ≥20 × ULN | 0/397 | 0/399 | 0/406 |
| AST (U/L) | |||
| ≥1 × ULN | 60/397 (15.1) | 55/399 (13.8) | 70/406 (17.2) |
| ≥1.5 × ULN | 13/397 (3.3) | 16/399 (4.0) | 28/406 (6.9) |
| ≥2 × ULN | 5/397 (1.3) | 9/399 (2.3) | 10/406 (2.5) |
| ≥3 × ULN | 2/397 (0.5) | 2/399 (0.5) | 9/406 (2.2) |
| ≥5 × ULN | 1/397 (0.3) | 1/399 (0.3) | 4/406 (1.0) |
| ≥10 × ULN | 0/397 | 0/399 | 2/406 (0.5) |
| ≥20 × ULN | 0/397 | 0/399 | 1/406 (0.2) |
| ALT or AST (U/L) | |||
| ≥1 × ULN | 123/397 (31.0) | 124/399 (31.1) | 134/406 (33.0) |
| ≥1.5 × ULN | 42/397 (10.6) | 41/399 (10.3) | 59/406 (14.5) |
| ≥2 × ULN | 19/397 (4.8) | 20/399 (5.0) | 35/406 (8.6) |
| ≥3 × ULN | 4/397 (1.0) | 5/399 (1.3) | 11/406 (2.7) |
| ≥5 × ULN | 1/397 (0.3) | 2/399 (0.5) | 6/406 (1.5) |
| ≥10 × ULN | 0/397 | 0/399 | 2/406 (0.5) |
| ≥20 × ULN | 0/397 | 0/399 | 1/406 (0.2) |
| Bilirubin total (μmol/L) | |||
| ≥1 × ULN | 14/397 (3.5) | 16/399 (4.0) | 18/406 (4.4) |
| ≥1.5 × ULN | 3/397 (0.8) | 5/399 (1.3) | 2/406 (0.5) |
| ≥2 × ULN | 1/397 (0.3) | 3/399 (0.8) | 2/406 (0.5) |
| ≥3 × ULN | 0/397 | 1/399 (0.3) | 1/406 (0.2) |
| ≥5 × ULN | 0/397 | 0/399 | 0/406 |
| ≥10 × ULN | 0/397 | 0/399 | 0/406 |
| ≥20 × ULN | 0/397 | 0/399 | 0/406 |
| Alkaline phosphatase (U/L) | |||
| ≥1 × ULN | 35/397 (8.8) | 48/399 (12.0) | 36/406 (8.9) |
| ≥1.5 × ULN | 3/397 (0.8) | 1/399 (0.3) | 1/406 (0.2) |
| ≥2 × ULN | 1/397 (0.3) | 1/399 (0.3) | 0/406 |
| ≥3 × ULN | 0/397 | 0/399 | 0/406 |
| ≥5 × ULN | 0/397 | 0/399 | 0/406 |
| ≥10 × ULN | 0/397 | 0/399 | 0/406 |
| ≥20 × ULN | 0/397 | 0/399 | 0/406 |
| Concurrent elevations† | |||
| ALT or AST ≥3× ULN and bilirubin | 0/397 | 0/399 | 1/406 (0.2) |
| total ≥1.5× ULN | |||
| ALT or AST ≥3× ULN and bilirubin | 0/397 | 0/399 | 1/406 (0.2) |
| total ≥2× ULN | |||
| Potential Hy's law‡ | |||
| ALT or AST ≥3× ULN and bilirubin | 0/397 | 0/399 | 1/406 (0.2) |
| total ≥2× ULN and ALP <2× ULN |
Concurrent elevations are from the same day.
One participant met biochemical Hy’s Law criteria due to an episode of acute cholecystitis; however, there were no confirmed Hy’s Law cases.
ALT = alanine aminotransferase; AST = aspartate aminotransferase; ULN = upper limit of normal value.
Usual Care Adverse Events
As noted, the usual care population was included to examine variability in hepatic laboratory parameters to help contextualize the hepatic safety data. The trial was not designed to specifically compare AEs between these groups due to the differences in the usual care and ubrogepant‐treated populations, as outlined above. A total of 271/417 participants (65.0%) reported a TEAE; relatedness was not assessed for the usual care arm. The most common (≥2% participants in any group; Supplementary S9) were upper respiratory tract infection (n = 48/417, 11.5%), nasopharyngitis (n = 33, 7.9%), sinusitis (n = 25, 6.0%), urinary tract infection (n = 23, 5.5%), and influenza (n = 21, 5.0%). Severe TEAEs were reported for 6.2% of participants. Serious AEs were reported by 17 participants (4.1%) (Supplementary S10). No deaths were reported and 4 participants (1.0%) reported TEAEs that led to discontinuation.
Discussion
This phase 3, long‐term safety evaluation followed 813 participants treated intermittently with ubrogepant 50 or 100 mg over the course of 1 year. A total of 21,454 migraine attacks were treated with 31,968 doses of ubrogepant. Participants treated ≥8 migraine attacks with ubrogepant 50 or 100 mg in a total of 144 months. Overall, there was a low incidence of TEAEs, which were mostly mild to moderate in severity with the majority considered unrelated to treatment by the investigator. There was no apparent relationship between TEAE incidence and the number of attacks treated per month. In addition, no hepatic or cardiovascular safety issues were noted. Additionally the rates of SAEs and discontinuations due to AEs were low, with less than 3% of participants treated with ubrogepant experiencing either. Based on these results, long‐term, intermittent use of ubrogepant for the acute treatment of migraine appears to be safe and well tolerated.
Previously, the gepant class had been evaluated for their efficacy and safety in the acute treatment of migraine and showed promising efficacy, but safety concerns halted their development.24, 25, 26, 27 The results of 1 phase 2B19 and 2 phase 320, 21 clinical trials demonstrated that ubrogepant is well tolerated and effective for the acute treatment of migraine attacks. The most common (≥2% of participants in any treatment group) TEAEs in the ACHIEVE I phase 3 trial were nausea, somnolence, and dry mouth and in the phase 3 ACHIEVE II trial were nausea and dizziness.20, 21 No serious AEs were reported within 48 hours postdose in either trial. No participants discontinued because of AEs and no hepatic safety signals were identified in either trial.
Overall, data from this trial further support the safety profile of ubrogepant. With regard to hepatic safety, 2% (16/805) of participants randomized to ubrogepant had increases in their ALT/AST ≥3× the ULN versus 1% (4/397) of participants in the usual care arm. There were no concerning hepatic safety findings and 13 of the 16 cases in the ubrogepant groups were adjudicated as Unlikely Related to trial treatment. Two cases were judged Possibly Related and 1 case was judged Probably Related; however, confounding factors were noted in each case (increased alcohol and acetaminophen use; dilated bile duct; and prednisone use). These findings, along with the observation that all aminotransferase elevations ≥3× ULN resolved in participants who continued ubrogepant dosing, indicate that there were no clinically meaningful hepatic safety signals for ubrogepant in this trial. These data also help to confirm findings from the dedicated hepatic safety trial in healthy volunteers, where ubrogepant was found to be safe and well tolerated, with no hepatic safety signals noted following high‐frequency dosing for 12 weeks.28
The efficacy of ubrogepant has been demonstrated in 2 phase 3, multicenter, randomized, double‐blind, placebo‐controlled single‐attack trials (ACHIEVE I and II).20, 21 The percentage of participants reporting pain freedom 2 hours post‐initial dose was significantly greater in the ubrogepant arms than the placebo arms of both trials. In addition, the proportion of participants reporting absence of the most bothersome migraine‐associated symptom (photophobia, phonophobia, or nausea) at 2 hours was greater in the ubrogepant arms than the placebo arms of both trials. While the present trial was designed as a long‐term safety trial, efficacy measures were collected for the ubrogepant treatment arms.29 Participants reported pain freedom at 2 hours for an average of 23% of attacks treated with ubrogepant 50 mg and 25% of attacks treated with ubrogepant 100 mg. Pain relief at 2 hours was achieved for 65% of attacks treated with ubrogepant 50 mg and 68% treated with ubrogepant 100 mg. Efficacy was maintained over the 1‐year trial period.
As with most trials, this trial does have some limitations that may impact the interpretation and generalizability of the data. First, exclusion of those with certain diseases, disease states, and clinically significant conditions as outlined in the methods section may limit these data from being reflective of real‐world experience. However, inclusion of participants with ALT/AST elevations ≤1.5× the ULN and bilirubin <1.5 mg/dL increases the generalizability of the data. Second, differences in participant populations and trial designs limit the ability to compare safety profiles of different acute treatment options for migraine. In particular, headache pain of any severity was allowed to be treated in this trial, which may not be consistent with other reported trials. Third, commonly reported AEs of nausea and dizziness can be symptoms associated with a migraine attack, making it is difficult to discern the causality of these AEs. Finally, the usual care arm was included to identify possible fluctuations in hepatic laboratory parameters to help contextualize the hepatic safety data. Interpretation of the AE data in the usual care arm, and comparison to the ubrogepant arms, is limited as the usual care arm consisted of participants who had been on and had tolerated their medication; therefore, AE rates do not reflect the introduction of a new treatment. The usual care arm of the present study does, however, reflect similar AE rates observed in other long‐term safety studies conducted on alternative acute treatments for migraines, in which overall AE rates ranged from 45 to 68% of patients for up to an 18‐month period.30, 31, 32
Conclusions
Long‐term intermittent use of ubrogepant 50 and 100 mg given as 1 or 2 doses per attack for the acute treatment of migraine was safe and well tolerated, as indicated by a low incidence of TEAEs and low rate of SAEs and discontinuations due to AEs, in this 1‐year trial.
Statement of Authorship
Category 1
(a) Conception and Design
Sung Yun Yu, Michelle Finnegan, Lawrence Severt, Joel M. Trugman
(b) Acquisition of Data
Matthew Butler, Joel M. Trugman
(c) Analysis and Interpretation of Data
Jessica Ailani, Richard B. Lipton, Susan Hutchinson, Kerry Knievel, Kaifeng Lu, Matthew Butler, Sung Yun Yu, Michelle Finnegan, Lawrence Severt, Joel M. Trugman
Category 2
(a) Drafting the Manuscript
Jessica Ailani, Richard B. Lipton, Susan Hutchinson, Kerry Knievel, Kaifeng Lu, Matthew Butler, Sung Yun Yu, Michelle Finnegan, Lawrence Severt, Joel M. Trugman
(b) Revising It for Intellectual Content
Jessica Ailani, Richard B. Lipton, Susan Hutchinson, Kerry Knievel, Matthew Butler, Sung Yun Yu, Michelle Finnegan, Lawrence Severt, Joel M. Trugman
Category 3
(a) Final Approval of the Completed Manuscript
Jessica Ailani, Richard B. Lipton, Susan Hutchinson, Kerry Knievel, Kaifeng Lu, Matthew Butler, Sung Yun Yu, Michelle Finnegan, Lawrence Severt, Joel M. Trugman
Supporting information
Acknowledgments
The authors and trial sponsor would like to thank all the participants and investigators for their participation in and dedication to this trial. We also thank Armin Szegedi, MD, PhD, for substantial contributions to the study design and Amy Kuang, PhD, of Allergan plc, for medical writing support.
Conflict of Interest: Jessica Ailani, MD, has received research grants from Theranica, Allergan, American Migraine Foundation, and Biohaven, and has received honoraria for the following: Alder (speaking, consulting), Allergan (speaking, consulting), Amgen (speaking, consulting), Biohaven (consulting), Eli Lilly and company (speaking, consulting), Electrocore (speaking, consulting), Promius (speaking, consulting), Teva (speaking, consulting), Impel (consulting), Satsuma (consulting), Supernus (consulting), and Current Pain and Headache Reports (section editor). Richard B. Lipton, MD, serves on the editorial board of Neurology and Cephalalgia and as senior advisor to Headache but does not get paid for his roles on Neurology or Headache. He has received research support from the NIH. He also receives support from the Migraine Research Foundation and the National Headache Foundation. He receives research grants from Allergan, Amgen, Dr Reddy’s, and Novartis. He has reviewed for the NIA and NINDS, and serves as consultant, advisory board member, or has received honoraria from Alder, Allergan, Amgen, Autonomic Technologies, Avanir, Boston Scientific, Colucid, Dr. Reddy’s Laboratories, Electrocore, Eli Lilly, eNeura Therapeutics, GlaxoSmithKline, Merck, Novartis, Teva, and Vedanta. He receives royalties from Wolff’s Headache (8th Edition, Oxford University Press, 2009) and Informa. He holds stock options in eNeura Therapeutics and Biohaven. Susan Hutchinson, MD, has served on advisory boards for Alder, Allergan, Amgen, Avanir, Biohaven, Electrocore, Eli Lily, Supernus, and Teva. She is on the speaker’s bureau for Allergan, Amgen, Avanir, Electrocore, Eli Lilly, Promius, Supernus, and Teva. Kerry Knievel, DO, has served as a consultant for Allergan, Amgen, Eli Lilly, and Biohaven; conducted research with Allergan, Amgen, and Eli Lilly; and is on speaker programs with Allergan and Amgen. Kaifeng Lu, PhD, is a full‐time employee of Allergan plc. Matthew Butler, MD, is a full‐time employee of Allergan plc. Sung Yun Yu, BA, is a full‐time employee of Allergan plc. Michelle Finnegan, MPH, is a full‐time employee of Allergan plc. Lawrence Severt, MD, is a full‐time employee of Allergan plc. Joel M. Trugman, MD, is a full‐time employee and stockholder of Allergan plc.
Trial Sponsor/Funding Source: Allergan plc.
ClinicalTrials.gov NCT02873221.
References
- 1. Headache Classification Committee of the International Headache Society (IHS) . The International Classification of Headache Disorders, 3rd edition. Cephalalgia. 2018;38:1‐211. [DOI] [PubMed] [Google Scholar]
- 2. Group GNDC . Global, regional, and national burden of neurological disorders during 1990‐2015: A systematic analysis for the Global Burden of Disease Study 2015. Lancet Neurol. 2017;16:877‐897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. GBD 2016 Disease and Injury Incidence and Prevalence Collaborators . Global, regional, and national incidence, prevalence, and years lived with disability for 328 diseases and injuries for 195 countries, 1990‐2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet. 2017;390:1211‐1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Agosti R. Migraine burden of disease: From the patient's experience to a socio‐economic view. Headache. 2018;58(Suppl. 1):17‐32. [DOI] [PubMed] [Google Scholar]
- 5. Buse DC, Powers SW, Gelfand AA, et al. Adolescent perspectives on the burden of a parent's migraine: Results from the CaMEO study. Headache. 2018;58:512‐524. [DOI] [PubMed] [Google Scholar]
- 6. Buse DC, Scher AI, Dodick DW, et al. Impact of migraine on the family: Perspectives of people with migraine and their spouse/domestic partner in the CaMEO study. Mayo Clin Proc. 2016;91:596‐611. [DOI] [PubMed] [Google Scholar]
- 7. MacGregor EA, Brandes J, Eikermann A, Giammarco R. Impact of migraine on patients and their families: The Migraine and Zolmitriptan Evaluation (MAZE) survey – Phase III. Curr Med Res Opin. 2004;20:1143‐1150. [DOI] [PubMed] [Google Scholar]
- 8. Seng EK, Mauser ED, Marzouk M, Patel ZS, Rosen N, Buse DC. When mom has migraine: An observational study of the impact of parental migraine on adolescent children. Headache. 2019;59:224‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Steiner TJ, Gururaj G, Andree C, et al. Diagnosis, prevalence estimation and burden measurement in population surveys of headache: Presenting the HARDSHIP questionnaire. J Headache Pain. 2014;15:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Becker WJ. Acute migraine treatment in adults. Headache. 2015;55:778‐793. [DOI] [PubMed] [Google Scholar]
- 11. Goadsby PJ, Sprenger T. Current practice and future directions in the prevention and acute management of migraine. Lancet Neurol. 2010;9:285‐298. [DOI] [PubMed] [Google Scholar]
- 12. Tajti J, Majlath Z, Szok D, Csati A, Vecsei L. Drug safety in acute migraine treatment. Expert Opin Drug Saf. 2015;14:891‐909. [DOI] [PubMed] [Google Scholar]
- 13. Bigal M, Rapoport A, Aurora S, Sheftell F, Tepper S, Dahlof C. Satisfaction with current migraine therapy: Experience from 3 centers in US and Sweden. Headache. 2007;47:475‐479. [DOI] [PubMed] [Google Scholar]
- 14. Lipton RB, Munjal S, Alam A, et al. Migraine in America Symptoms and Treatment (MAST) study: Baseline study methods, treatment patterns, and gender differences. Headache. 2018;58:1408‐1426. [DOI] [PubMed] [Google Scholar]
- 15. Lipton RB, Munjal S, Alam A, et al. Assessing unmet treatment needs and associated disability in persons with migraine: Results from migraine in America symptoms and treatment (MAST) study [abstract OR02]. Headache. 2018;58(Suppl. 2):63‐64. [Google Scholar]
- 16. Silberstein SD, McCrory DC. Ergotamine and dihydroergotamine: History, pharmacology, and efficacy. Headache. 2003;43:144‐166. [DOI] [PubMed] [Google Scholar]
- 17. Edvinsson L, Haanes KA, Warfvinge K, Krause DN. CGRP as the target of new migraine therapies—Successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338‐350. [DOI] [PubMed] [Google Scholar]
- 18. Ho TW, Edvinsson L, Goadsby PJ. CGRP and its receptors provide new insights into migraine pathophysiology. Nat Rev Neurol. 2010;6:573‐582. [DOI] [PubMed] [Google Scholar]
- 19. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double‐blind, placebo‐controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887‐898. [DOI] [PubMed] [Google Scholar]
- 20. Dodick DW, Lipton RB, Ailani J, et al. Ubrogepant for the acute treatment of migraine: Efficacy, safety, tolerability, and functional impact outcomes from a single attack phase III study, ACHIEVE 1 [abstract IOR‐01LB]. Headache. 2018;58:1287‐1288. [Google Scholar]
- 21. Lipton RB, Dodick DW, Ailani J, et al. Efficacy, safety, and tolerability of ubrogepant for the acute treatment of migraine: Results from a single attack phase III study, ACHIEVE II [abstract PS111LB]. Headache. 2018;58:1315‐1316. [Google Scholar]
- 22. Temple R. Hy's law: Predicting serious hepatotoxicity. Pharmacoepidemiol Drug Saf. 2006;15:241‐243. [DOI] [PubMed] [Google Scholar]
- 23. Guidance for Industry Drug‐Induced Liver Injury: Premarketing Clinical Evaluation. Silver Spring, MD: U.S. Department of Health and Human Services, Food and Drug Administration; 2009. [Google Scholar]
- 24. Holland PR, Goadsby PJ. Targeted CGRP small molecule antagonists for acute migraine therapy. Neurotherapeutics. 2018;15:304‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ho TW, Ho AP, Ge YJ, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for prevention of headache in women with perimenstrual migraine. Cephalalgia. 2016;36:148‐161. [DOI] [PubMed] [Google Scholar]
- 26. Ho TW, Connor KM, Zhang Y, et al. Randomized controlled trial of the CGRP receptor antagonist telcagepant for migraine prevention. Neurology. 2014;83:958‐966. [DOI] [PubMed] [Google Scholar]
- 27. Hewitt DJ, Aurora SK, Dodick DW, et al. Randomized controlled trial of the CGRP receptor antagonist MK‐3207 in the acute treatment of migraine. Cephalalgia. 2011;31:712‐722. [DOI] [PubMed] [Google Scholar]
- 28. Goadsby PJ, Tepper SJ, Watkins P, et al. Safety and tolerability of ubrogepant following intermittent, high‐frequency dosing [abstract P88]. Headache. 2019;59:80‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Lipton RB, Ailani J, Hutchinson S, et al. Efficacy is maintained with long‐term intermittent use of ubrogepant for the acute treatment of migraine [abstract P135]. Headache. 2019;59(Suppl. 1):110.30575970 [Google Scholar]
- 30. Gobel H, Heinze A, Stolze H, Heinze‐Kuhn K, Lindner V. Open‐labeled long‐term study of the efficacy, safety, and tolerability of subcutaneous sumatriptan in acute migraine treatment. Cephalalgia. 1999;19:676‐683; discussion 626. [DOI] [PubMed] [Google Scholar]
- 31. Cabarrocas X, Esbri R, Peris F, Ferrer P. Long‐term efficacy and safety of oral almotriptan: Interim analysis of a 1‐year open study. Headache. 2001;41:57‐62. [DOI] [PubMed] [Google Scholar]
- 32. Grosberg B, Bigal ME, Ricci J, et al. Epidemiology of chronic daily headache in adolescents [abstract]. Neurology. 2007;68:A180. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials