

Abstract
Most often, the structures of secondary metabolites are solved using a suite of NMR techniques. However, there are times when it can be challenging to position double bonds, particularly those that are fully substituted or when there are multiple double bonds in similar chemical environments. Ozone-Induced Dissociation Mass Spectrometry (OzID-MS) serves as an orthogonal structure elucidation tool, using predictable fragmentation patterns that are generated after ozonolysis across a carbon-carbon double bond. This technique is finding growing use in the lipidomics community, suggestive of its potential value for secondary metabolites. This methodology was evaluated by confirming the double bond positions in five fungal secondary metabolites, specifically: ent-sartorypyrone E (1), sartorypyrone A (2), sorbicillin (3), trichodermic acid A (4), and AA03390 (5). This demonstrated its potential with a variety of chemotypes, ranging from polyketides to terpenoids, and including those in both conjugated and non-conjugated polyenes. In addition, the potential of using this methodology in the context of a mixture was piloted by studying Aspergillus fischeri, first examining a traditional extract and then sampling a live fungal culture in situ. While the intensity of signals varied from pure compound to extract to in situ, the utility of the technique was preserved.
Graphical Abstract
The structures of many organic compounds, particularly natural products, are typically elucidated by interpretation of a suite of NMR experiments.1–6 However, there are times when derivatization is required to finalize a structure. In the lore of drug discovery, camptothecin and taxol (paclitaxel) are two prominent examples,7–9 although there are certainly scores of others. While NMR instrumentation and pulse sequences continue to evolve,10–12 some derivatizations are used fairly often in structure elucidation, such as reductions/oxidations,13–15 reactions to assign absolute configuration, either via installing heavy atoms for X-ray crystallography or the use of Mosher’s esters,16–20 and peptide hydrolysis/Marfey’s analysis.21–23 In short, the reactions of organic chemistry remain beneficial for structural analysis.
Challenges can arise when striving to position double bonds in structurally complex natural products via NMR, particularly for fully substituted double bonds and compounds with multiple double bonds in similar chemical environments.24–26 During the initial steps of structure elucidation of secondary metabolites, many factors can lead to the assignment of an incorrect structure,27–29 and common examples include endocyclic vs. exocyclic double bonds30,31 and a change in functional groups.32,33 As new techniques are developed, this often leads to structural re-assignments.15,34–36 Again, even with the latest advancements in NMR, there is room for orthogonal techniques that serve to either assign, or at least verify, the de novo structure elucidation of natural products. In addition, with the growing popularity of metabolomics, where structures may be inferred solely from hyphenated mass spectrometry data, a tool that could position carbon-carbon double bonds could be valuable, particularly for discerning the structures of isomers.37
In a molecule containing carbon-carbon double bonds, low energy collision-induced dissociation (CID) cannot be used readily for their positioning in a polyene motif due to their high stability. To overcome this challenge, alternative approaches were introduced to derivatize such bonds, with the aim of inducing diagnostic fragment ions under CID. There are many selective reactions, and some of them were coupled with mass spectrometry to pinpoint the double bond location in a molecule, such as: dimethyl disulfate reaction,38 Paternò-Büchi reaction,39,40 ozonolysis, and meta chloroperoxybenzoic acid (m-CPBA)41 epoxidation reaction. The common requirement between these methods (with the exception of ozonolysis) is that the reaction was performed in solution phase prior to the ionization, which leads to the loss of information about the accurate mass of the intact molecule and the need to use CID to induce subsequent diagnostic product ions. Hence, when using collision energy to fragment the desired bond, additional fragments would be generated from neighboring bonds, leading to a complicated spectrum, and as a result, such data were time consuming to interpret.42
Ozonolysis, which uses ozone to cleave alkene double bonds, is a well-known reaction that is covered in nearly all introductory organic chemistry classes.43 In the past, ozone (O3) gas was employed to study the carbon-carbon double bonds of lipids in solution and at the ionization source of the mass spectrometer (a technique termed OzESI). Every unsaturated molecule would interact with ozone at the ionization source, to produce the molozonide, and this would subsequently cleave to the Criegee and carbonyl product ions (discussed further below). However, the detected ions would be the combination of the ozonolysis products and ESI products (i.e. electrospray ionization). Hence, a full scan mass spectrum of a complex sample under such conditions would be convoluted with information of precursor ions and product ions in the same scan. Moreover, one cannot assign the product ion to the original precursor ions. As a result, OzESI experiments were not recommended for complex samples, and as expected, the resulting data from an OzESI experiment were both complicated and lacked intact structural characterization information.44,45
To circumvent the aforementioned challenges, ozone-induced dissociation mass spectrometry (OzID-MS) was introduced in 2008,46 which implements the ozonolysis reaction in the gas phase inside the mass spectrometer. This approach provided the mass selection option for intact structure elucidation of double bond positional isomers of lipids before the targeted ion reacted with ozone in the trap chamber.37,47–51 The mechanism of OzID-MS is based on the fundamentals of ozonolysis, which selectively cleaves carbon-carbon double bonds, particularly those on the acyl chains of lipids. This results in unique product ions, aldehyde or ketone, depending on the double bond substitution before the cleavage, and a Criegee ion, which is a carbonyl oxide zwitterion (Figure 1).52 This mechanism has the advantages of being both reliable and predictable, thereby facilitating downstream data analysis. Recently, OzID-MS has become a prominent technique for pinpointing the location of double bonds in the fatty acyl chains of lipids exactly because of the predictable cleavage patterns and unique fragments.37,46,48,51
Figure 1.
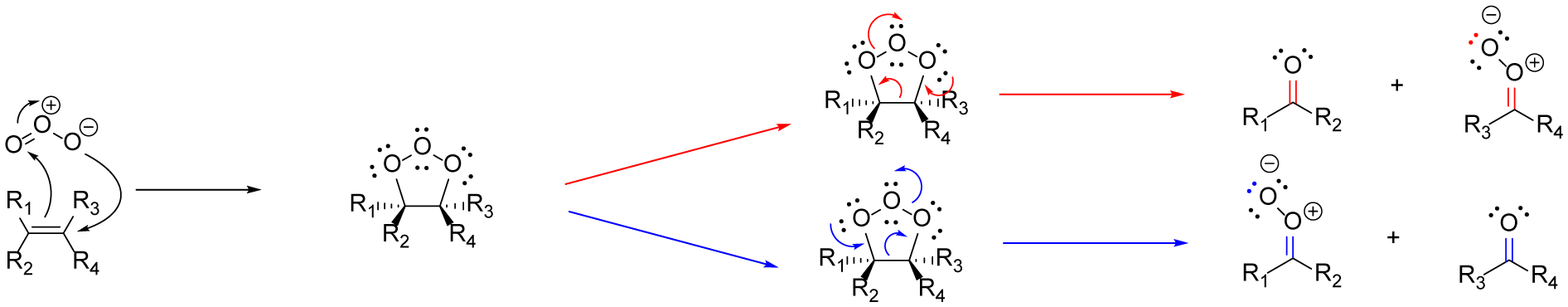
In ozonolysis carbon-carbon double bonds react with ozone via a 1,3-dipolar cycloaddition. Two products are formed via the subsequent retro 1,3-diplolar cycloaddition, resulting in a ketone/aldehyde and a Criegee intermediate. The red arrows (top) and blue arrows (bottom) represent the two routes in which the molozonide can undergo the retro 1,3-diplolar cycloaddition.
Given the promise of OzID-MS for facilitating double bond placement in lipids, it is somewhat surprising that this technique has rarely been used in natural products, with most examples being of compounds similar to fatty acids but with various head groups.53–55 For example, it aided in the elucidation of double bond locations in several bisresorcinol and hexadecenoylanthranilic acid isomers.54,55 While it is not possible to replace the tools and value of NMR characterization, OzID-MS can serve as an orthogonal structure elucidation tool. Essentially, initiation of ozonolysis across a carbon-carbon double bond in a mass spectrometer provides valuable data that complement those derived from NMR experiments. Since OzID-MS is effectively a tandem mass spectrometry technique, it has the further advantage of not requiring pure compounds, which means the technique can be applied to a complex sample matrix. With the goal of expanding the utility of this technique, we piloted its use with a range of fungal metabolites as pure compounds, in the context of an extract, and in situ.
Results and Discussion
Structure Elucidation by Traditional NMR and Mass Spectrometry Techniques.
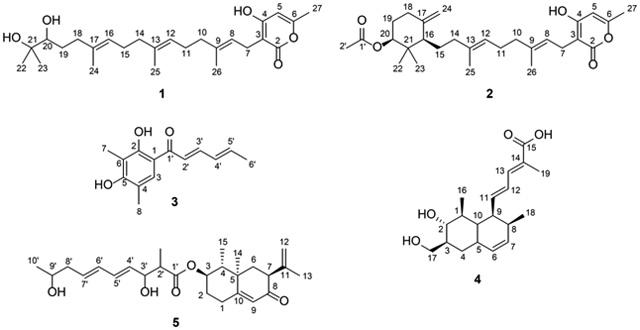
In ongoing studies of the genetic and secondary metabolite profiles of Aspergillus fischeri, which is of interest due to its close evolutionary relationship with the human pathogen A. fumigatus, we have characterized a series of fungal metabolites, including seven known compounds, one new natural product, and two new compounds.56 The structure of one of the latter, which was assigned the trivial name ent-sartorypyrone E (1), was elucidated by traditional spectroscopic and spectrometric techniques, and all of those data are discussed in the Supporting Information. Mosher’s esters analysis19 was attempted to assign the absolute configuration of position 20, but unfortunately, those data were inconclusive. This was supported by the literature, which showed that this methodology does not work well for vicinal diols.57 Instead, the Snatzke method would need to be implemented, but due to the paucity of sample, this was not conducted.58–60 During the final drafting of this manuscript, the structure of sartorypyrone E was published by Bang et al.,61 and our NMR data were in agreement. However, those authors reported performing Mosher’s ester analysis, establishing the hydroxy in the S configuration. Since the measured [α]D20 value for 1 was opposite in sign, although of greater magnitude, to the value reported for sartorypyrone E,61 we hypothesized position 20 as R, as suggested by the trivial name ent-sartorypyrone E (1).
When the structure of 1 was solved via NMR data, the double bond locations were assigned through HMBC and COSY correlations (Figure 2). The double bond between positions 8 and 9 was identified by the COSY correlation of H-26 to H-8 and the HMBC correlations of H-10 to C-8, C-9, and C-26 and H-8 to C-3, C-10, and C-26. The double bond between positions 12 and 13 was identified by the COSY correlations of H-25 to H-12 and the HMBC correlations of H-14 to C-12, C-13, and C-25 and H-12 to C-10, C-14, and C-15. The double bond between positions 16 and 17 was identified by the COSY correlation of H-24 to H-16 and the HMBC correlations of H-18 to C-16, C-17, and C-24 and H-16 to C-14, C-18, and C-24. Thus, 2D NMR data were instrumental for positioning the double bonds.
Figure 2.
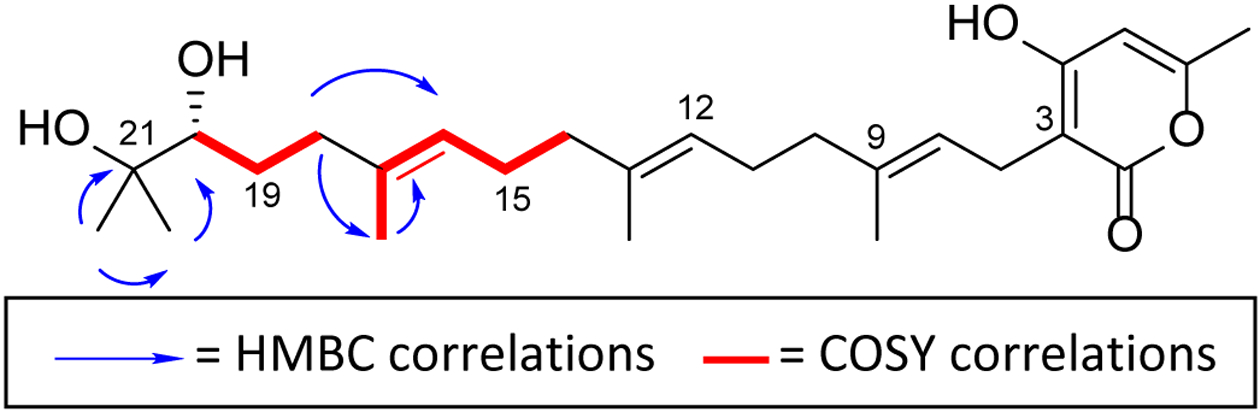
Key COSY and HMBC correlations for compound 1.
Analysis of Double Bond Positions in Pure Compounds by OzID-MS.
The OzID-MS technique was tested on the fungal metabolites ent-sartorypyrone E (1), sartorypyrone A (2), sorbicillin (3),62,63 trichodermic acid A (4),64 and AA03390 (5),65 where the observed accurate masses corresponding to the carbonyl/Criegee product ions confirmed the placement of double bonds in these molecules. Infusion of sodium acetate into the mass spectrometry system (see Experimental) was used to increase the abundance of sodiated adducts, which increase the efficiency of ozonolysis in the OzID-MS experiment.37,47,66
When performing OzID-MS on compound 1, the sodium ion can associate with the hydroxy moieties at the end of the isoprene chain (left side of molecule) or the oxygens in the lactone ring (right side of molecule), which is why all the theoretically possible carbonyl and Criegee ions were observed from both ends of the molecule. Regardless of where sodium associated with 1 (i.e. [M + Na]+), the targeted precursor ion was observed at m/z 455.2772. When sodium associated with the hydroxys at the ends of the isoprene chain, the double bonds were cleaved into ketones (previously di-substituted) and Criegee ions (Figure 3A). The ketones were observed at positions C-9 (m/z 319.2248), C-13 (m/z 251.1634), and C-17 (m/z 183.0992), and the Criegee ions were observed at positions C-9 (m/z 335.2202), C-13 (m/z 267.1579), and C-17 (m/z 199.0954) (Supporting Information Table S2). When sodium associated with the lactone ring, the double bonds were cleaved into aldehydes (previously mono-substituted) and Criegee ions (Figure 3). The aldehydes were observed at positions C-8 (m/z 191.0332), C-12 (m/z 259.0954), and C-16 (m/z 327.1578), and the Criegee ions were observed at positions C-8 (m/z 207.0287), C-12 (m/z 275.0902) and C-16 (m/z 343.1526) (Supporting Information Table S2). A key observation with this type of experiment is the formation of a presumed epoxide, due to conjugated double bonds. In this example, the conjugation is throughout the lactone ring system.37 To verify the unique product ions of OzID-MS, 1 was re-analyzed under the identical parameter settings, but with argon in replacement of ozone (Figure 3B); as expected, the OzID fragmentation patterns were not observed under these conditions.
Figure 3.
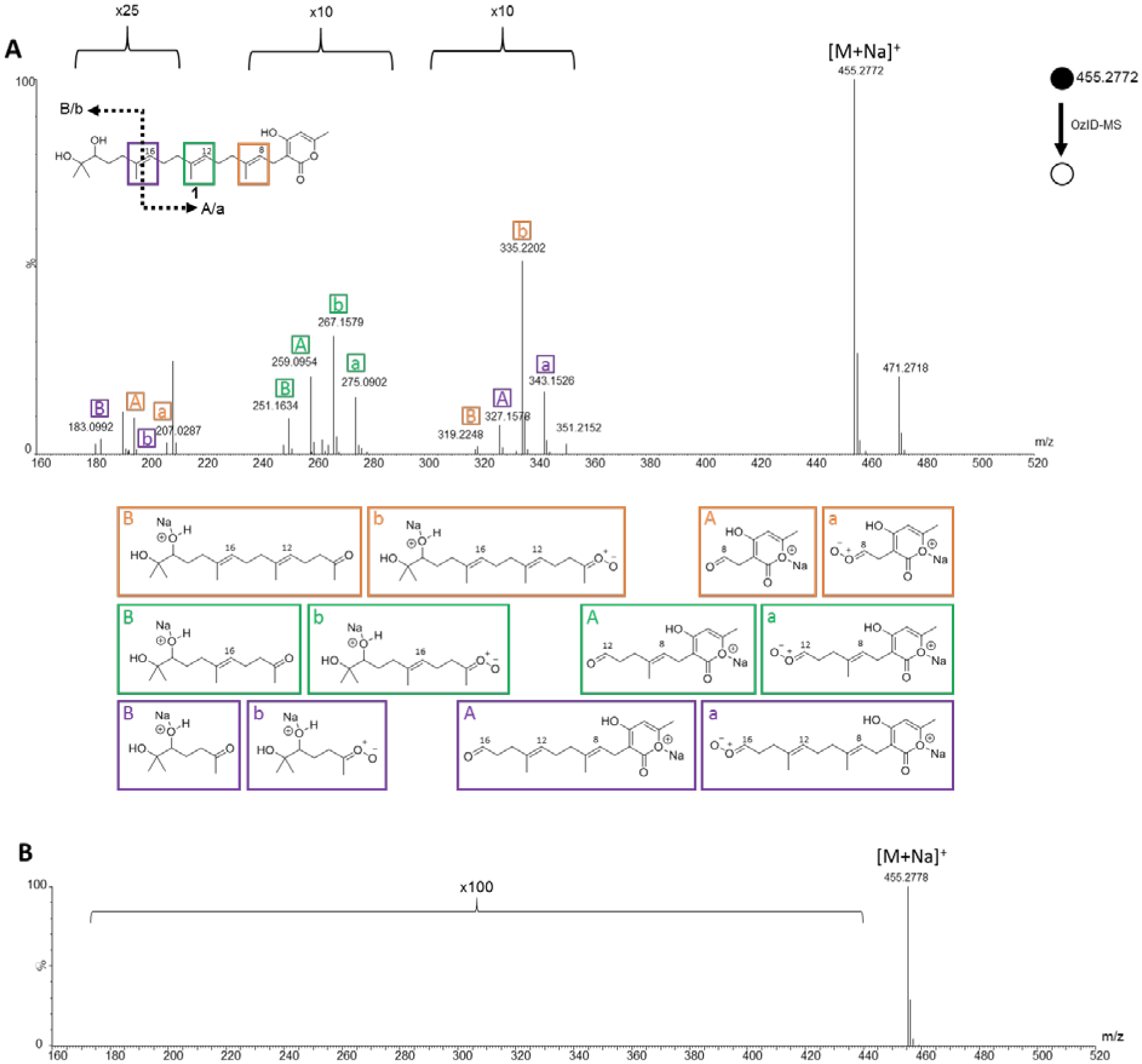
A: The OzID-MS spectrum of ent-sartorypyrone E (1) after targeted OzID fragmentation. The structure of 1 is shown on the top left. The purple, green, and orange boxes around the diterpene double bonds indicate positions where ozonolysis occurred. The informative OzID fragments are labeled with the colors corresponding to the structure. The capital letters represent the ketone fragments, and the lower-case letters represent the Criegee ion fragments. The A/a corresponds to the sodium ion associating with the lactone ring (right side of the molecule), whereas the B/b corresponds to the sodium ion associating with hydroxys at the end of the isoprene chain (left side of the molecule). The OzID-MS products (shown under spectrum) are color and letter coded to represent the cleaved double bond, and these data are tabulated in Supporting Information Table S2. B: Compound 1 being analyzed in argon, to demonstrate that the fragments in A are unique to OzID-MS
A known metabolite, sartorypyrone A (2), was also isolated from A. fischeri, and it was subjected to OzID-MS to evaluate the technique, illustrating its use as an orthogonal tool for pinpointing the location of double bonds within a chain. Given their structural similarities, sodium associated with compound 2 in a manner largely analogous to what was observed with 1. For instance, regardless of where sodium associated with the molecule (i.e. [M + Na]+), the targeted precursor ion was observed at m/z 479.2784. When the sodium associated with lactone ring, there were two pairs of OzID product ions, corresponding to double bonds at C-12 (m/z 259.0984 and 275.0936) and C-8 (m/z 191.0335 and 207.0283), recognized as aldehyde (previously mono-substituted) and Criegee ions, respectively, in each pair. However, when sodium associated with the ester, only a pair of ozonolysis product ions were observed, representing those induced at C-8 and recorded at m/z 343.1948 and 359.2246, corresponding to the ketone and Criegee ions, respectively (Figure 4 and Supporting Information Table S5). While this was slightly different than what was observed with 1, since C-12 was not cleaved, we were not surprised by these results. OzID-MS is known to be a charged-induced reaction, and as such, the distance and interaction between where the sodium adduct was formed, relative to the double bond, can hinder the efficiency of ozonolysis.66 An example of this phenomenon is sphingomyelin, where OzID-MS was able to characterize the double bonds in the fatty acyl chains, but not the double bond on the long chain base.67 This is an important distinction for OzID-MS, as the distance of the double bond(s) from where the sodium associates to form the adduct is important. Even though it was not possible to obtain product ions from ozonolysis at the C-12 position when sodium associated with the left side of molecule, the data from the OzID products from when sodium associated with the lactone ring confirmed the position of this carbon-carbon double bond.
Figure 4.
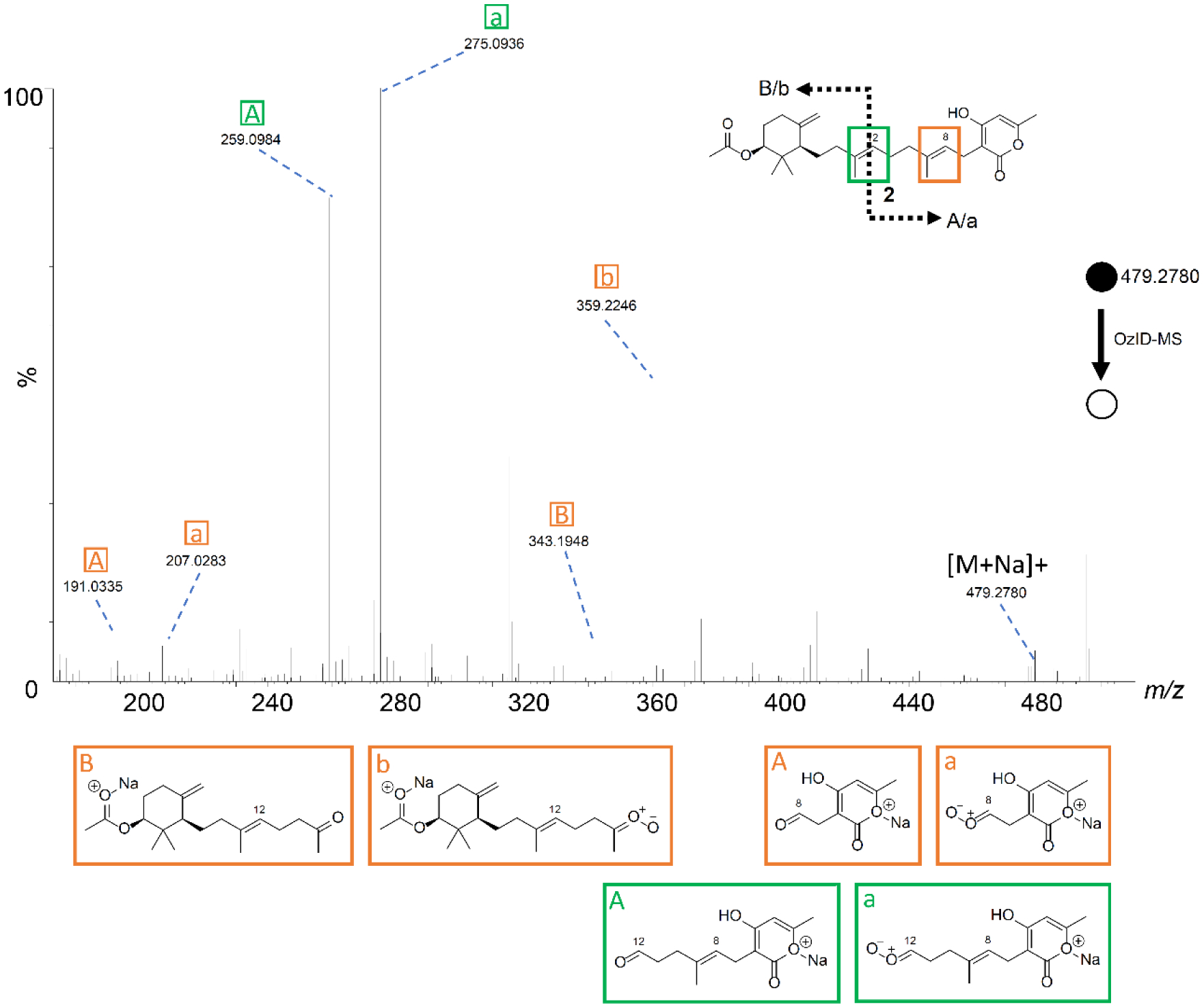
The OzID-MS spectrum of 2 after targeted OzID fragmentation. The structure of 2 is shown on the top left. The green and orange boxes around the double bonds indicate positions where the ozonolysis occurred. The informative OzID fragments are labeled with the colors corresponding to the structure, analogous to those in Figure 3. The OzID-MS products (shown under spectrum) are color and letter coded to represent the double bond that was cleaved, and these data are tabulated in Supplementary Table S5.
Next, a selection of other fungal metabolites from our compound library were examined, such as the polyketide 3, which has conjugated double bonds and was first described from Penicillium notatum.62,63 In a conjugated system, in addition to the aldehyde/ketone and Criegee ion fragments, an epoxide is often observed.37 In this example, sodium can associate with the hydroxys or ketone to form a singly charged [M + Na]+ species observed at m/z 255.1053. The double bonds at positions C-2′ (m/z 217.0475) and C-4′ (m/z 243.0625) were cleaved into aldehydes, confirming their positions (Figure 5 and Supporting Information Table S6). A highlight of this structure was the formation of the epoxide ring [M+Na+16]+ caused by the loss of dioxygen and a hydrocarbon radical from a conjugated double bond.68 This observation indicated conjugated double bonds, which was confirmed by the product ion of an epoxide ring between positions C-4′ and C-5′ (m/z 271.0966), which has been observed previously with the use of this technique to discern conjugated double bonds in fatty acids.37
Figure 5.
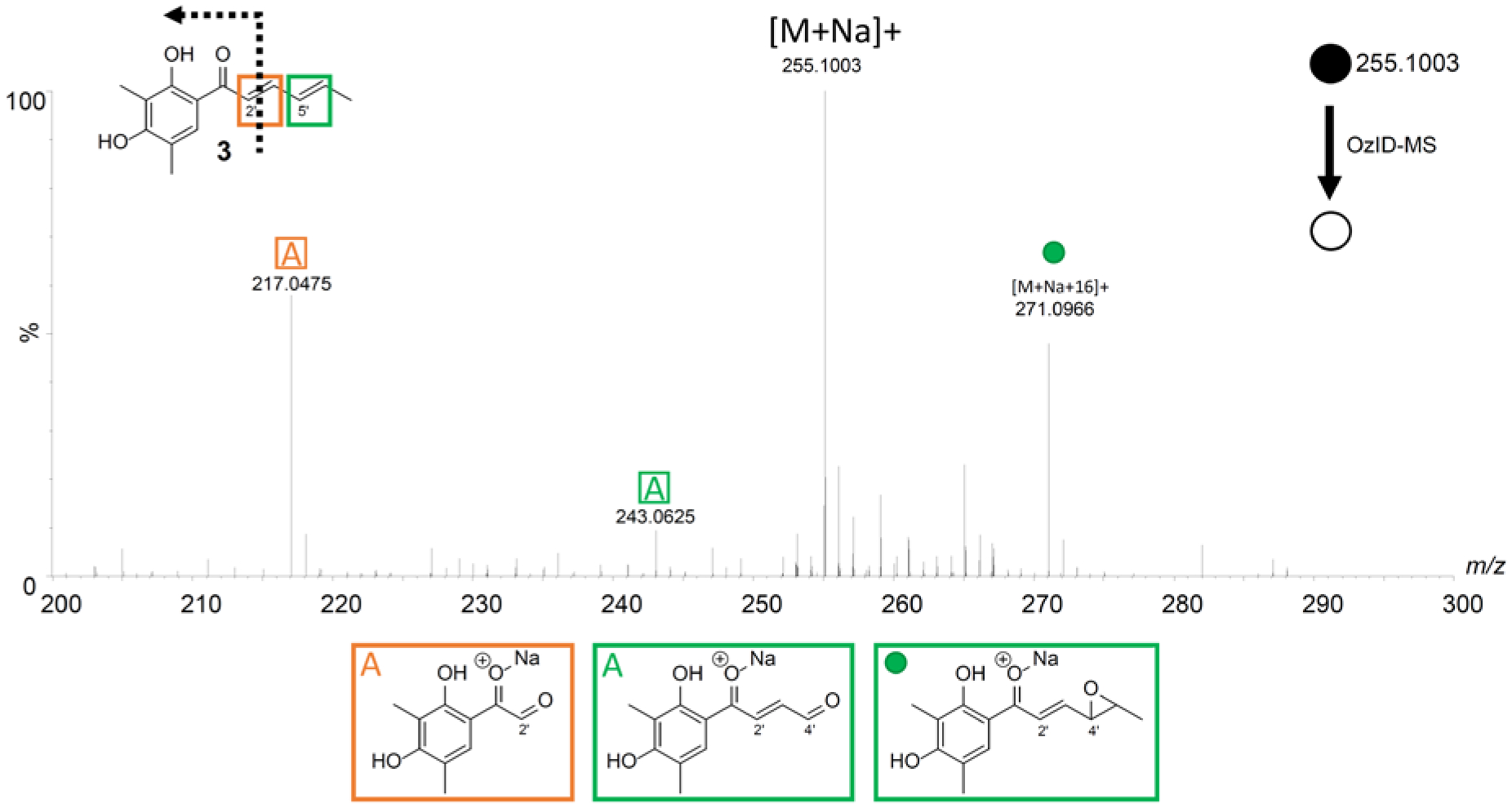
The OzID-MS spectrum of 3 after targeted OzID fragmentation. The structure of 3 is shown on the top left. The green and orange boxes around the double bonds indicate positions where ozonolysis occurred. The informative OzID fragments are labeled with the colors corresponding to the structure, analogous to those in Figure 3. The OzID-MS products (shown under spectrum) are color and letter coded to represent the double bond that was cleaved, and these data are tabulated in Supporting Information Table S6.
Compound 4 is a terpenoid with conjugated double bonds that was described originally from the endophytic fungus Trichoderma spirale.64 The sodiated adduct of this compound was found to favor the hydroxy moieties on the ring based on the recorded OzID-MS product ions. Besides the expected ozonolysis product ions observed at m/z 261.1485, 287.1632, and 303.1581, a product ion of [M+Na+16]+ was also obtained at m/z 359.1840, analogous to 3 and as expected for a conjugated system (Figure 6 and Supporting Information Table S7).
Figure 6.
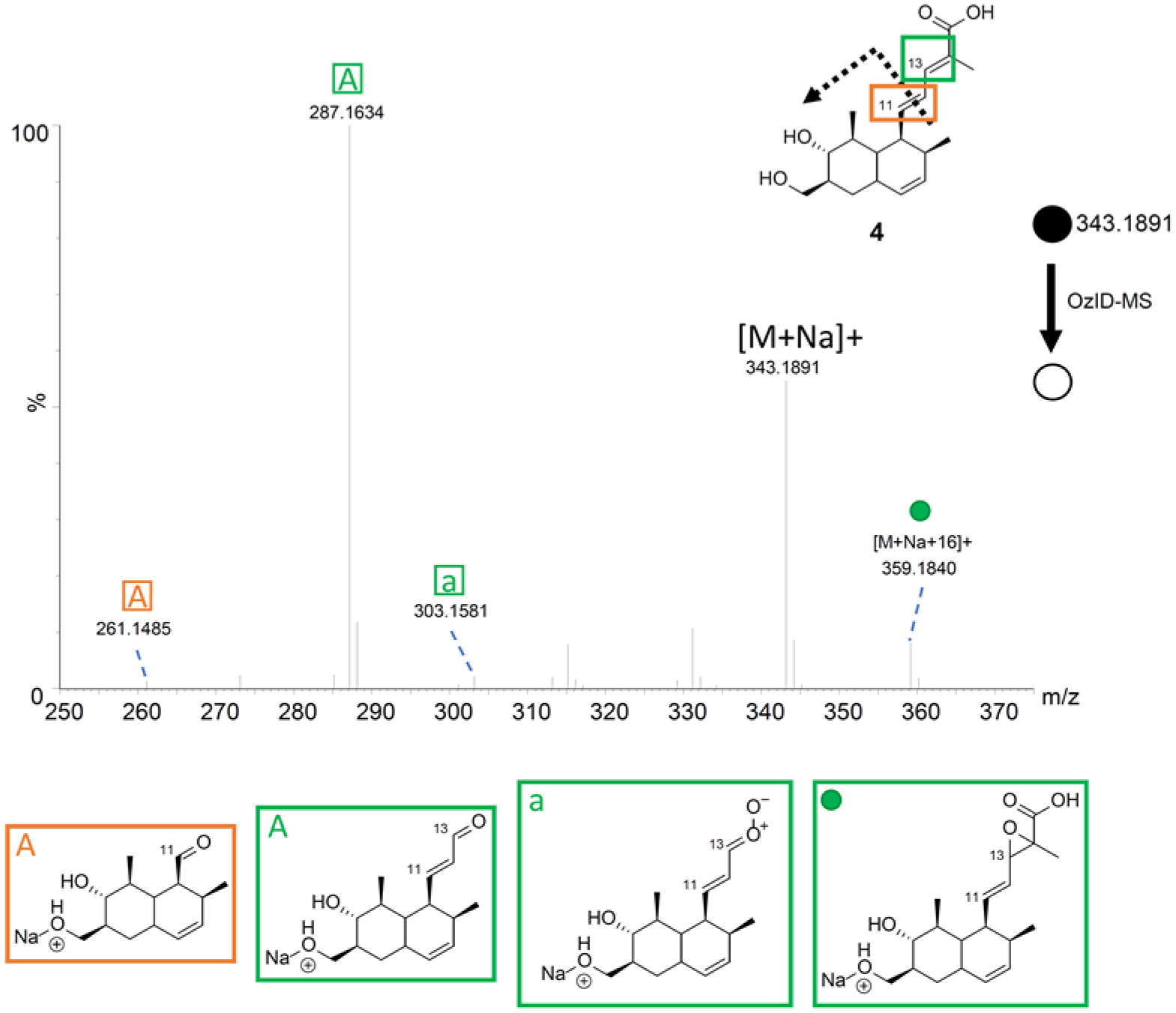
The OzID-MS spectrum of 4 after targeted OzID fragmentation. The structure of 4 is shown on the top left. The green and orange boxes around the double bonds indicate positions where ozonolysis occurred. The informative OzID fragments are labeled with the colors corresponding to the structure, analogous to those in Figure 3. The OzID-MS products (shown under spectrum) are color and letter coded to represent the double bond that was cleaved, and these data are tabulated in Supporting Information Table S7.
With compound 5 (a meroterpenoid originally described from the deep-sea fungus Phomopsis lithocarpus),65 the ion [M+Na+16]+ was observed at m/z 469.2548, suggestive of conjugation. In addition, the expected OzID-MS products, specifically the aldehyde (m/z 371.1844 and 397.1985) and Criegee ions (m/z 387.1808 and 413.2004) were observed (Figure 7 and Supporting Information Table S8), further verifying the position of the double bonds.
Figure 7.
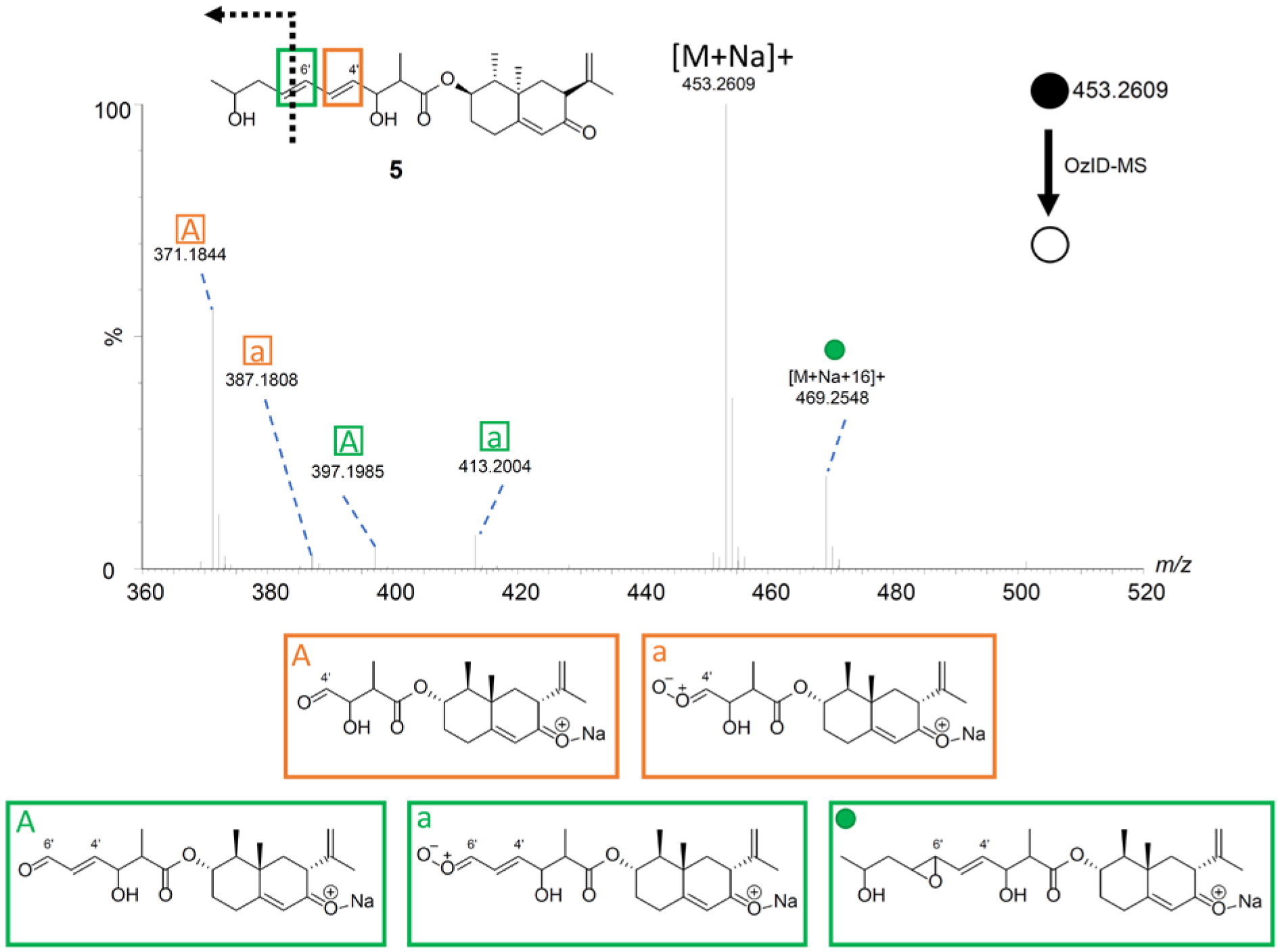
The OzID-MS spectrum of 5 after targeted OzID fragmentation. The structure of 5 is shown on the top left. The green and orange boxes around the double bonds indicate positions where the ozonolysis occurred. The informative OzID fragments are labeled with the colors corresponding to the structure, analogous to those in Figure 3. The OzID-MS products (shown under spectrum) are color and letter coded to represent the double bond that was cleaved, and these data are tabulated in Supplementary Table S8.
As could be seen from data generated by analyzing compounds 1-5, OzID-MS can be used to verify the position of carbon-carbon double bonds in secondary metabolites. Since ozonolysis is sensitive and results in predictable fragmentation patterns, the expected product ions can be calculated and matched with the acquired data. This represents an efficient and orthogonal approach to confirm the assignment of double bond positions. In addition, the signal of [M+Na+16]+ can be suggestive for conjugated double bonds. In total, OzID-MS confirmed the locations of the double bonds in a series of structurally diverse fungal secondary metabolites, serving as orthogonal confirmation of double bond positioning based on NMR experiments.
Structural Confirmation from Extract and in situ by Direct Infusion OzID-MS.
To test whether this technique could work in the context of a multi-component mixture, the organic extract of a fermentation of Aspergillus fischeri was subjected to OzID-MS. The precursor ion was targeted, and then OzID-MS was performed. The fragment ions from this experiment were identical to those observed with the pure compound (1) (Figures 3 and 8). While the intensity of the signals was not as robust, we could confidently assign the positions of the double bonds. This was of particular relevance, since 1 was only a minor component of the extract (Supporting Information Figure S6).
Figure 8.
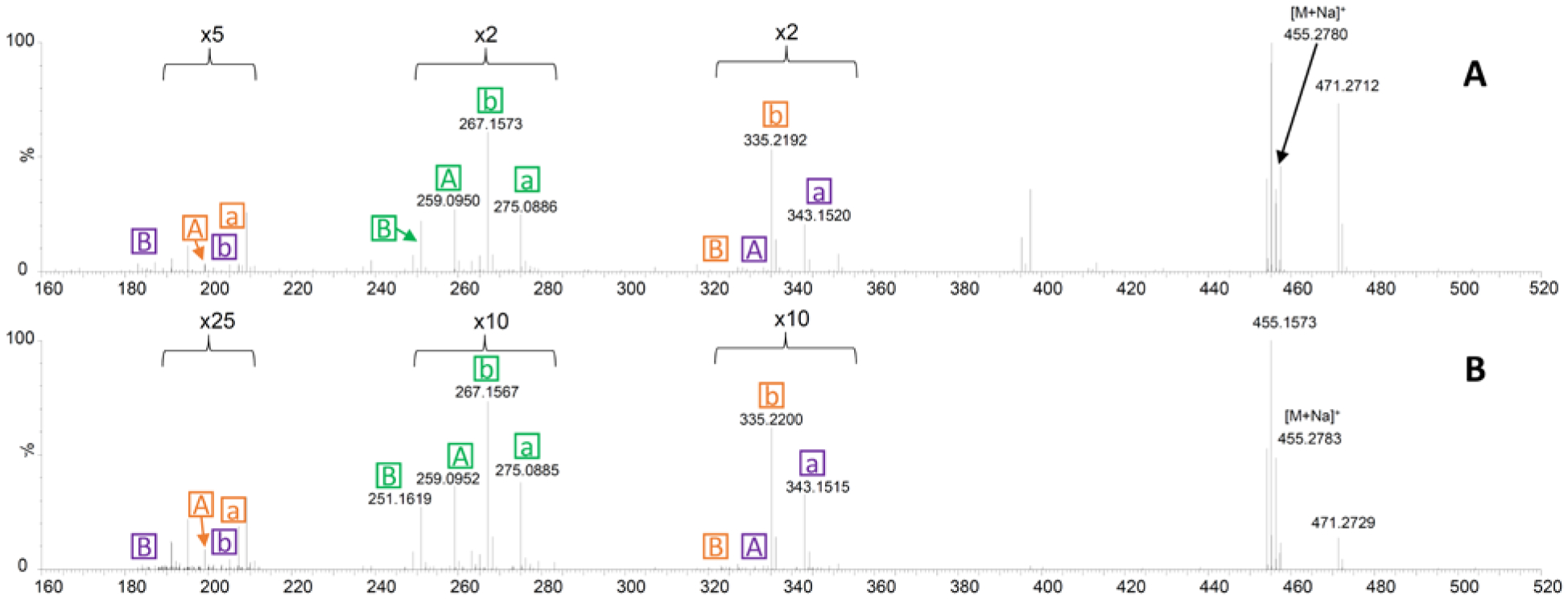
The OzID-MS studies of Aspergillus fischeri showing the analysis of compound 1 in the context of an extract (panel A) and from in situ sampling of a Petri dish of a life culture (panel B). The fragmentation patterns in both cases are analogous to those in Figure 3, and these data are tabulated in Supporting Information Tables S3 and S4.
In addition, there is growing interest in studying the chemistry of nature in situ via a variety of techniques.69,70 As such we tested if double bond placement could be elucidated by coupling OzID-MS with the droplet probe.71,72 We envisaged this could be powerful for co-culture studies, where interspecific interactions may stimulate the biosynthesis of unique metabolites.73–75 A Petri dish of A. fischeri was analyzed via this technique, and the sodiated precursor ion of compound 1 was observed in the full scan spectrum. Subsequently, OzID-MS was performed through the targeted analysis of the precursor ion. The product ions that were present in the complex and in situ extracts mirrored those of the pure compound (Figures 3 and 8). These data confirmed that OzID-MS can also aid as an orthogonal structure elucidation tool for a compound within the context of an extract and/or in situ, albeit at a lower concentration (Figure 8, Supporting Information Tables S3 and S4). In addition, these results demonstrated the sensitivity of approach, regardless of the low abundance of the targeted compound in the extract or in situ.
In summary, OzID-MS shows promise as a methodology that can be used to support the positioning of double bonds in secondary metabolites. The most straight forward approach may be to first use NMR experiments to elucidate a structure, and then employ OzID-MS fragment ions to confirm and/or pinpoint the location of carbon-carbon double bonds. While only a limited set of fungal metabolites were analyzed, we do not expect unforeseen challenges with other natural products, particularly since its application in the lipidomics community continues to grow. The predictable nature of the fragmentation patterns expected in an OzID-MS experiment may lead to emerging tools, such as artificial intelligence,76,77 and again, the omics community may take the lead in this regard, but the results should benefit natural products research too. In addition, it is very tantalizing to add this technique to the growing spectrum of tools that are being used to study natural products in situ, particularly since the sensitivity of mass spectrometry can be capitalized upon to select for ions of interest for OzID-MS analysis, even those in low abundance. Key considerations for implementing this technique have been summarized (Table 1), and it is worth noting that this system was assembled using commercially available components that has been detailed previously.37,47
Table 1.
Key considerations for performing OzID-MS analysis.
| Questions | Reasoning |
|---|---|
| Why infuse salt adducts such as Na+? | The counter ion aids in OzID fragmentation. Essentially, the increased intensity of the ozonide ions leads to an increase in the ozonolysis efficiency.66 Other salts could be used, such as those that produce Li+ ions. |
| What differentiates the observation of aldehyde vs. ketone peaks? | This is based on the double bond substitution before OzID fragmentation occurs. If the carbon was di-substituted, then it becomes a ketone. Alternatively, if it was mono-substituted, it becomes an aldehyde. |
| What is the Criegee ion? | A Criegee ion is an ion with the same mass as the carbonyl oxide zwitterion (shown below). However, structurally it could undergo rearrangement to various isomeric forms, which may differ, depending on the compound, due to the high reactivity and rapid rearrangement of the intermediate. Examples could include carboxylic acids, vinyl hydroperoxides, dioxiranes, etc.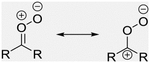
|
| Why is OzID-MS observed to occur from both sides of a molecule? | The counter ion (i.e. Na+) typically associates with oxygenation, and if two spots are available on either side of the metabolite, then both the theoretical carbonyl/Criegee ions pairs are observed. |
| Why are not all of the “potential” fragments produced for every double bond? | The distance between where the adduct associates and the double bond affects reactivity.37,67 |
| Why is [M+Na+16]+ a key observation? | This ion suggests that the metabolite has conjugated double bonds due to the formation of an epoxide.37,68 |
Experimental
General Experimental Procedures.
Optical rotation data were obtained using a Rudolph Research Autopol III polarimeter, and UV spectra were measured with a Varian Cary 100 Bio UV−vis spectrophotometer. The NMR data were collected using a JOEL ECS-400 spectrometer, which was equipped with a JOEL normal geometry broadband Royal probe, and a 24-slot autosampler, and operated at 400 MHz for 1H, a JOEL ECA-500 spectrometer operating at 500 MHz for 1H, or an Agilent 700 MHz spectrometer, equipped with a cryoprobe, operating at 700 MHz for 1H and 175 MHz for 13C. HRMS experiments utilized either a Thermo LTQ Orbitrap XL mass spectrometer or a Thermo Q Exactive Plus; both were equipped with an electrospray ionization source. A Waters Acquity UPLC was utilized for both mass spectrometers, using a BEH C18 column (1.7 μm; 50 mm x 2.1 mm) set to a column temperature of 40oC and a flow rate of 0.3 mL/min. The mobile phase consisted of a linear gradient of CH3CN-H2O (acidified with 0.1% formic acid), starting at 15% CH3CN and increasing linearly to 100% CH3CN over 8 min, with a 1.5 min hold before returning to the starting conditions. OzID-MS data were collected using a Waters Synapt G2 HDMS. The HPLC separations were performed with an Atlantis T3 C18 semi-preparative (5 μm; 10 x 250 mm) and preparative (5 μm; 19 x 250 mm) columns, at a flow rate of 4.6 mL/min and 16.9 mL/min, respectively, with a Varian Prostar HPLC system equipped with a Prostar 210 pumps and a Prostar 335 photodiode array detector (PDA), with the collection and analysis of data using Galaxie Chromatography Workstation software. Flash chromatography was performed on a Teledyne ISCO Combiflash Rf 200 and monitored by both ELSD and PDA detectors.
Fungal Material.
Aspergillus fischeri strain NRRL 181 was obtained from ARS Culture Collection (NRRL), and as described previously,56,73 the large-scale fermentation was grown on solid breakfast oatmeal (Old-fashioned breakfast Quaker oats). This was prepared by adding 10 g oatmeal to a 250 mL Erlenmeyer flask with 16 mL of DI-H2O, which was autoclaved at 121oC for 30 min. After inoculation, the culture was grown for 14 days at room temperature.
Extraction and Isolation.
To each of the solid fermentation cultures (10 flasks) of A. fischeri, 60 mL of CHCl3-MeOH (1:1, v/v) were added, and the cultures were chopped using a spatula followed by shaking overnight (~ 16 hrs.). The resulting slurries were vacuum filtered, and 90 mL of CHCl3 and 150 mL of DI H2O were added to the filtrate. This 4:1:5 CHCl3:MeOH:H2O mixture was partitioned in a separatory funnel, and the organic layer was drawn off and evaporated to dryness in vacuo. This sample was then reconstituted and partitioned with 100 mL CH3CN:MeOH (1:1, v/v) and 100 mL of hexanes. The organic layer was then evaporated to dryness to generate 440mg of the organic extract. The sample was then dissolved in CHCl3, absorbed onto celite 545 (Acros Organics, celite 545), and fractioned by normal phase flash chromatography using a gradient of hexane-CHCl3-CH3OH at an 18 mL/min flow rate and 90.0 column volumes over 24.0 min to produce five fractions. Fraction 1 was purified further via preparative HPLC using a gradient system 20:80 to 100:0 of CH3CN-H2O (with 0.1% formic acid) over 30 min at a flow rate of 16.9 mL/min to yield seven subfractions. Subfraction 4 yielded 2 (2.88 mg), which eluted at approximately 17.0 min. Fraction 3 was purified further via preparative HPLC using the same gradient system to yield ten subfractions. Subfraction 4 yielded 1 (1.17 mg), which eluted at approximately 17.2 min. ent-sartorypyrone E (1): white solid; [α]D20 = +10 (c 0.02, MeOH); UV (MeOH) λmax (log ε) 286 (2.85), 239 (2.83) nm; 1H and 13C NMR data, see Supporting Table S1; HRESIMS [M + H]+ 433.2941 (calculated for C26H41O5, 433.2948).
General Procedure for OzID-MS.
The OzID-MS experiment was performed on the modified Waters Synapt G2 HDMS as reported previously in detail.37,47 A commercially available O3 generator [O3 MEGA (MKS Inc.)] was used to convert O2 to O3 (6%), and it was plumbed to the trap and transfer cell in the TRIWAVE region through a t-valve, essentially replacing the commonly used collision gas (argon) (Supporting Information, Figure S10). Previously, we developed a method, coined “ion dam,” to enhance the ozonolysis efficiency of unsaturated molecules with ozone inside the high-resolution Traveling wave MS.47 This works by having ions indirectly prolonged in the trap cell to extend their reaction time with ozone. O3 was introduced to the system at 4 mL/min, creating a pressure of 1.32x10−2 mbar in the trap cell, which is the reaction chamber for the ozonolysis gas phase reaction. The excess or unused O3 is delivered to the converting chamber, creating O2, which is sent to the exhaust system. A commonly voiced concern is that O3 can be highly corrosive due to its oxidative properties; however, in over five years of operation, such damage has not been observed on this system, likely because only a small amount of O3 is needed to induce ozonolysis. Similar experiments have been reported in the literature using several different mass spectrometry platforms and/or vendors.46,68,78,79
Pure Compound and Extract Analysis via Direct Infusion of OzID-MS.
The Synapt G2 HDMS was directly infused with 10 mmol sodium acetate at 8 μL/min using an automated syringe pump at the beginning of every injection series to increase the relative abundance of sodium ions. Pure compounds were prepared at a concentration of 0.05 mg/mL and the organic extract at 2.0 mg/mL, both in MeOH, and kept at -20°C until analyzed. Pure compounds and an organic extract were directly infused at 8 μL/min using an automated syringe pump in the positive mode; due to the system being flushed with sodium acetate the predominant ions were sodiated adducts.
The ESI source conditions were finalized as: spray voltage, 3.0 kV; sampling cone, 45 V; extraction cone, 6 V; source temperature, 100 °C; desolvation temperature, 200 °C; cone gas flow, 50 L/h; desolvation gas flow, 500 L/h. Isolation of precursor ions was carried out in the quadrupole at ~1Th isolation width (LM=16, HM =15). All spectra were acquired for 30s at 0.5s/scan. For pure compound OzID-MS, the traveling wave in the trap was optimized and operated accordingly to the “ion dam” setting,47 i.e. trap entrance, 5.0V; bias, 2.0V; trap dc, 0.1V; exit, 0V; trap wave velocity, 8m/s; wave height, 0.2V; for extract, same parameters were applied except trap entrance was set to 3V and dc to -0.5V to increase signal intensity. Default instrument settings for all the remaining parameters were applied. The instrument was calibrated daily in Resolution mode using sodium iodide following manufacturer’s instructions obtaining less than 5 ppm RMS residual mass. Both the full scan and MS/MS levels were mass-corrected traceable to the reference lock mass using MassLynx v4.1 instrument control software (Waters Corp.).
In situ OzID-MS.
Sampling of the surface of the fungus in situ was performed by a CTC/LEAP HTC PAL auto-sampler (LEAP Technologies Inc.) that was converted to an automated droplet probe system, as detailed previously.71,72 Briefly, the microextractions (5 μL) were performed by using MeOH:H2O (1:1, v/v), with water being for droplet retention and MeOH for the microextraction, on the surface of the fungus. The droplet was dispensed at a rate of 2 μL/s from the needle, held on the surface for 2 s, then withdrawn at the same rate with each droplet extraction performed thrice to concentrate the sample. Traditionally, the droplet probe is connected to a UPLC-MS system, which is how the droplet is then analyzed.16,80–82 However, currently software is not available to monitor the CTC/LEAP HTC PAL autosampler with the Synapt G2 HDMS. Thus, after conducting the microextraction via droplet probe, the solution was loaded into an offline 20 μL sample loop. A solvent system MeOH:H2O (1:1, v/v) controlled by the Synapt G2 HDMS was connected to the sample loop to carry the sample to the ESI source at 5 μL/min. The OzID-MS analysis instrument settings were the same as described above for analysis of extract.
Supplementary Material
Acknowledgements
The authors thank Dr. Vilmos Kertesz from Oak Ridge National Laboratory for guidance and assistance with the droplet probe. This research was supported in part via the National Institutes of Health through the National Center for Complementary and Integrative Health under award numbers T32 AT008938 and F31 AT010558, through the National Institute of General Medical Sciences under award number R21 GM104678, and through the National Cancer Institute under award number P01 CA125066. Partial support was also through a Vanderbilt University Discovery Grant (to A.R.).
References
- (1).Martin GE; Hilton BD; Blinov KA, J. Nat. Prod 2011, 74, 2400–2407. [DOI] [PubMed] [Google Scholar]
- (2).Elyashberg M, Trends Analyt. Chem 2015, 69, 88–97. [Google Scholar]
- (3).Sauri J; Liu Y; Parella T; Williamson RT; Martin GE, J. Nat. Prod 2016, 79, 1400–1406. [DOI] [PubMed] [Google Scholar]
- (4).Buevich AV; Williamson RT; Martin GE, J. Nat. Prod 2014, 77, 1942–1947. [DOI] [PubMed] [Google Scholar]
- (5).Sauri J; Frédérich M; Tchinda AT; Parella T; Williamson RT; Martin GE, J. Nat. Prod 2015, 78, 2236–2241. [DOI] [PubMed] [Google Scholar]
- (6).McAlpine JB; Chen S-N; Kutateladze A; MacMillan JB; Appendino G; Barison A; Beniddir MA; Biavatti MW; Bluml S; Boufridi A; Butler MS; Capon RJ; Choi YH; Coppage D; Crews P; Crimmins MT; Csete M; Dewapriya P; Egan JM; Garson MJ; Genta-Jouve G; Gerwick WH; Gross H; Harper MK; Hermanto P; Hook JM; Hunter L; Jeannerat D; Ji N-Y; Johnson TA; Kingston DGI; Koshino H; Lee H-W; Lewin G; Li J; Linington RG; Liu M; McPhail KL; Molinski TF; Moore BS; Nam J-W; Neupane RP; Niemitz M; Nuzillard J-M; Oberlies NH; Ocampos FMM; Pan G; Quinn RJ; Reddy DS; Renault J-H; Rivera-Chávez J; Robien W; Saunders CM; Schmidt TJ; Seger C; Shen B; Steinbeck C; Stuppner H; Sturm S; Taglialatela-Scafati O; Tantillo DJ; Verpoorte R; Wang B-G; Williams CM; Williams PG; Wist J; Yue J-M; Zhang C; Xu Z; Simmler C; Lankin DC; Bissona J; Pauli GF, Nat. Prod. Rep 2018, 36, 35–107, 10.1039/c7np00064b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Wani MC; Taylor HL; Wall ME; Coggon P; McPhail AT, J. Am. Chem. Soc 1971, 93, 2325–2327. [DOI] [PubMed] [Google Scholar]
- (8).Oberlies NH; Kroll DJ, J. Nat. Prod 2004, 67, 129–135. [DOI] [PubMed] [Google Scholar]
- (9).Wall ME; Wani MC; Cook C; Palmer KH; McPhail A. a.; Sim G, J. Am. Chem. Soc 1966, 88, 3888–3890. [Google Scholar]
- (10).Liu Y; Cohen RD; Martin GE; Williamson RT, J. Magn. Reson 2018, 291, 63–72. [DOI] [PubMed] [Google Scholar]
- (11).Saurí J; Bermel W; Parella T; Thomas Williamson R; Martin GE, Magn. Reson. Chem 2018, 56, 1029–1036. [DOI] [PubMed] [Google Scholar]
- (12).Milanowski DJ; Oku N; Cartner LK; Bokesch HR; Williamson RT; Saurí J; Liu Y; Blinov KA; Ding Y; Li X-C, Chem. Sci 2018, 9, 307–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ospina CA; Rodríguez AD, J. Nat. Prod 2006, 69, 1721–1727. [DOI] [PubMed] [Google Scholar]
- (14).Wegerski CJ; Sonnenschein RN; Cabriales F; Valeriote FA; Matainaho T; Crews P, Tetrahedron 2006, 62, 10393–10399. [Google Scholar]
- (15).Shen YC; Shih PS; Lin YS; Lin YC; Kuo YH; Kuo YC; Khalil AT, Helv. Chim. Acta 2009, 92, 2101–2110. [Google Scholar]
- (16).Rivera-Chávez J; Raja HA; Graf TN; Gallagher JM; Metri P; Xue D; Pearce CJ; Oberlies NH, RSC Adv. 2017, 7, 45733–45741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).El-Elimat T; Raja HA; Day CS; Chen W-L; Swanson SM; Oberlies NH, J. Nat. Prod 2014, 77, 2088–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Sy-Cordero AA; Day CS; Oberlies NH, J. Nat. Prod 2012, 75, 1879–1881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Hoye TR; Jeffrey CS; Shao F, Nat. Protoc 2007, 2, 2451. [DOI] [PubMed] [Google Scholar]
- (20).Deyrup ST; Swenson DC; Gloer JB; Wicklow DT, J. Nat. Prod 2006, 69, 608–611. [DOI] [PubMed] [Google Scholar]
- (21).Kumari G; Serra A; Shin J; Nguyen PQ; Sze SK; Yoon HS; Tam JP, J. Nat. Prod 2015, 78, 2791–2799. [DOI] [PubMed] [Google Scholar]
- (22).Von Bargen KW; Niehaus E-M; Bergander K; Brun R; Tudzynski B; Humpf H-U, J. Nat. Prod 2013, 76, 2136–2140. [DOI] [PubMed] [Google Scholar]
- (23).Bhushan R; Brückner H, Amino acids 2004, 27, 231–247. [DOI] [PubMed] [Google Scholar]
- (24).Kubo I; Tanis S; Lee Y; Miura I; Nakanishi K; Chapya A, Heterocycles 1976, 5, 485–498. [Google Scholar]
- (25).Rajab MS; Rugutt JK; Fronczek FR; Fischer NH, J. Nat. Prod 1997, 60, 822–825. [Google Scholar]
- (26).Senior MM; Williamson RT; Martin GE, J. Nat. Prod 2013, 76, 2088–2093. [DOI] [PubMed] [Google Scholar]
- (27).Suyama TL; Gerwick WH; McPhail KL, Bioorg. Med. Chem 2011, 19, 6675–6701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Robien W, A Critical Evaluation of the Quality of Published 13 C NMR Data in Natural Product Chemistry In Progress in the Chemistry of Organic Natural Products 105, Springer: 2017; pp 137–215. [DOI] [PubMed] [Google Scholar]
- (29).Brown PD; Lawrence AL, Nat. Prod. Rep 2017, 34, 1193–1202. [DOI] [PubMed] [Google Scholar]
- (30).Choudhary MI; Hayat S; Khan AM; Ahmed A, Chem. Pharm. Bull 2001, 49, 105–107. [DOI] [PubMed] [Google Scholar]
- (31).Lin W; Brauers G; Ebel R; Wray V; Berg A; Sudarsono a.; Proksch P, J. Nat. Prod 2003, 66, 57–61. [DOI] [PubMed] [Google Scholar]
- (32).Shen Y-C; Lo K-L; Lin Y-C; Khalil AT; Kuo Y-H; Shih P-S, Tetrahedron Lett. 2006, 47, 4007–4010. [Google Scholar]
- (33).Jain S; Laphookhieo S; Shi Z; Fu L.-w.; Akiyama S.-i.; Chen Z-S; Youssef DT; van Soest RW; El Sayed KA, J. Nat. Prod 2007, 70, 928–931. [DOI] [PubMed] [Google Scholar]
- (34).Venkateswarlu S; Panchagnula GK; Gottumukkala AL; Subbaraju GV, Tetrahedron 2007, 63, 6909–6914. [Google Scholar]
- (35).Kuramochi K; Saito F; Nakazaki A; Takeuchi T; Tsubaki K; Sugawara F; Kobayashi S, Biosci. Biotechnol. Biochem 2010, 74, 1635–1640. [DOI] [PubMed] [Google Scholar]
- (36).Jain S; Abraham I; Carvalho P; Kuang Y-H; Shaala LA; Youssef DT; Avery MA; Chen Z-S; El Sayed KA, J. Nat. Prod 2009, 72, 1291–1298. [DOI] [PubMed] [Google Scholar]
- (37).Barrientos RC; Vu N; Zhang Q, J. Am. Soc. Mass Spectrom 2017, 28, 2330–2343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Carlson DA; Roan CS; Yost RA; Hector J, Anal. Chem 1989, 61, 1564–1571. [Google Scholar]
- (39).Ma X; Xia Y, Angew. Chem. Int. Ed. Engl 2014, 53, 2592–2596. [DOI] [PubMed] [Google Scholar]
- (40).Xie X; Xia Y, Anal. Chem 2019. [DOI] [PubMed] [Google Scholar]
- (41).Feng Y; Chen B; Yu Q; Li L, Anal. Chem 2019, 91, 1791–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Hancock SE; Poad BL; Batarseh A; Abbott SK; Mitchell TW, Anal. Biochem 2017, 524, 45–55. [DOI] [PubMed] [Google Scholar]
- (43).Bruice PY, Organic Chemistry eighth edition Pearson: 2017. [Google Scholar]
- (44).Kishimoto Y; Radin NS, J Lipid Res 1963, 4, 437–43. [PubMed] [Google Scholar]
- (45).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Murphy RC; Blanksby SJ, Anal. Chem 2007, 79, 5013–22, 10.1021/ac0702185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Thomas MC; Mitchell TW; Harman DG; Deeley JM; Nealon JR; Blanksby SJ, Anal. Chem 2008, 80, 303–311. [DOI] [PubMed] [Google Scholar]
- (47).Vu N; Brown J; Giles K; Zhang Q, Rapid Commun. Mass Spectrom 2017, 31, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Mitchell TW; Pham H; Thomas MC; Blanksby SJ, J. Chromatogr. B 2009, 877, 2722–2735. [DOI] [PubMed] [Google Scholar]
- (49).Poad BL; Green MR; Kirk JM; Tomczyk N; Mitchell TW; Blanksby SJ, Anal. Chem 2017, 89, 4223–4229. [DOI] [PubMed] [Google Scholar]
- (50).Poad BL; Zheng X; Mitchell TW; Smith RD; Baker ES; Blanksby SJ, Anal. Chem 2017, 90, 1292–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cui Z; Thomas MJ, J. Chromatogr. B 2009, 877, 2709–2715. [DOI] [PubMed] [Google Scholar]
- (52).Criegee R, Angew. Chem. Int. Ed. Engl 1975, 14, 745–752. [Google Scholar]
- (53).Auranwiwat C; Maccarone AT; Carroll AW; Rattanajak R; Kamchonwongpaisan S; Blanksby SJ; Pyne SG; Limtharakul T, Tetrahedron 2019, 75, 2336–2342. [Google Scholar]
- (54).Wang H; Leach DN; Thomas MC; Blanksby SJ; Forster PI; Waterman PG, Nat. Prod. Commun 2009, 4, 951–958. [PubMed] [Google Scholar]
- (55).Shou Q; Banbury LK; Maccarone AT; Renshaw DE; Mon H; Griesser S; Griesser HJ; Blanksby SJ; Smith JE; Wohlmuth H, Fitoterapia 2014, 93, 62–66. [DOI] [PubMed] [Google Scholar]
- (56).Mead ME; Knowles SL; Raja HA; Beattie SR; Kowalski CH; Steenwyk JL; Silva LP; Chiaratto J; Ries LN; Goldman GH; Oberlies NH; Rokas A, mSphere 2019, 4, e00018–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (57).Shi G; Gu Z.-m.; He K; Wood KV; Zeng L; Ye Q; MacDougal JM; McLaughlin JL, Bioorg. Med. Chem 1996, 4, 1281–1286. [DOI] [PubMed] [Google Scholar]
- (58).Liu YX; Ma SG; Wang XJ; Zhao N; Qu J; Yu SS; Dai JG; Wang YH; Si YK, Helv. Chim. Acta 2012, 95, 1401–1408. [Google Scholar]
- (59).Wang Y; Zheng Z; Liu S; Zhang H; Li E; Guo L; Che Y, J. Nat. Prod 2010, 73, 920–924. [DOI] [PubMed] [Google Scholar]
- (60).Zhang Y; Wang J-S; Wei D-D; Gu Y-C; Wang X-B; Kong L-Y, J. Nat. Prod 2013, 76, 1191–1195. [DOI] [PubMed] [Google Scholar]
- (61).Bang S; Song JH; Lee D; Lee C; Kim S; Kang KS; Lee JH; Shim SH, J. Agric. Food Chem 2019, 67, 1831–1838. [DOI] [PubMed] [Google Scholar]
- (62).Bigi F; Casiraghi G; Casnati G; Marchesi S; Sartori G; Vignali C, Tetrahedron 1984, 40, 4081–4084. [Google Scholar]
- (63).Cram DJ, J. Am. Chem. Soc 1948, 70, 4240–4243. [DOI] [PubMed] [Google Scholar]
- (64).Dong‐Li L; Yu‐Chan C; Mei‐Hua T; Hao‐Hua L; Wei‐Min Z, Helv. Chim. Acta 2012, 95, 805–809. [Google Scholar]
- (65).Jian-Lin X; Hong-Xin L; Yu-Chan C; Hai-Bo T; Heng G; Li-Qiong X; Sai-Ni L; Zi-Lei H; Hao-Hua L; Xiao-Xia G; Wei-Min Z, Mar. Drugs 2018, 16, 329. [Google Scholar]
- (66).Barrientos RC; Zhang Q, J. Am. Soc. Mass Spectrom 2019, 10.1007/s13361-019-02267-7. [DOI] [Google Scholar]
- (67).Marshall DL; Criscuolo A; Young RS; Poad BL; Zeller M; Reid GE; Mitchell TW; Blanksby SJ, J. Am. Soc. Mass Spectrom 2019, 1–10. [DOI] [PubMed] [Google Scholar]
- (68).Pham HT; Maccarone AT; Campbell JL; Mitchell TW; Blanksby SJ, J. Am. Soc. Mass Spectrom 2013, 24, 286–296. [DOI] [PubMed] [Google Scholar]
- (69).Spraker JE; Luu GT; Sanchez LM, Nat. Prod. Rep 2019, 10.1039/C9NP00038K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Wu C; Dill AL; Eberlin LS; Cooks RG; Ifa DR, Mass Spectrom Rev 2013, 32, 218–43, 10.1002/mas.21360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Sica VP; Raja HA; El-Elimat T; Kertesz V; Van Berkel GJ; Pearce CJ; Oberlies NH, J. Nat. Prod 2015, 78, 1926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Oberlies NH; Knowles SL; Amrine CSM; Kao D; Kertesz V; Raja HA, Nat. Prod. Rep 2019, 36, 944–959, 10.1039/C9NP00019D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).Knowles SL; Raja HA; Wright AJ; Lee AML; Caesar LK; Cech NB; Mead ME; Steenwyk JL; Ries L; Goldman GH; Rokas A; Oberlies NH, Front. Microbiol 2019, 10, 285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Adnani N; Chevrette MG; Adibhatla SN; Zhang F; Yu Q; Braun DR; Nelson J; Simpkins SW; McDonald BR; Myers CL, ACS Chem. Biol 2017, 12, 3093–3102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Xu X-Y; Shen X-T; Yuan X-J; Zhou Y-M; Fan H; Zhu L-P; Du F-Y; Sadilek M; Yang J; Qiao B, Front. Microbiol 2018, 8, 2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Bahado-Singh RO; Yilmaz A; Bisgin H; Turkoglu O; Kumar P; Sherman E; Mrazik A; Odibo A; Graham SF, PLoS ONE 2019, 14, e0214121, 10.1371/journal.pone.0214121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Vijayan V; Rouillard AD; Rajpal DK; Agarwal P, Expert Opin. Drug Discov 2019, 14, 191–194, 10.1080/17460441.2019.1573811. [DOI] [PubMed] [Google Scholar]
- (78).Poad BL; Pham HT; Thomas MC; Nealon JR; Campbell JL; Mitchell TW; Blanksby SJ, J. Am. Soc. Mass Spectrom 2010, 21, 1989–1999. [DOI] [PubMed] [Google Scholar]
- (79).Paine MR; Poad BL; Eijkel GB; Marshall DL; Blanksby SJ; Heeren RM; Ellis SR, Angew. Chem. Int. Ed. Engl 2018, 57, 10530–10534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Amrine CSM; Raja HA; Darveaux BA; Pearce CJ; Oberlies NH, J. Ind. Microbiol. Biotechnol 2018, 45, 1053–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Sica V; El-Elimat T; Oberlies N, Anal. Methods 2016, 8, 6143–6149. [Google Scholar]
- (82).Crnkovic CM; Krunic A; May DS; Wilson TA; Kao D; Burdette JE; Fuchs JR; Oberlies NH; Orjala J, J. Nat. Prod 2018, 81, 2083–2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.