

Abstract
Purpose of the review
The complex multistep life-cycle of HIV allows it to proliferate within the host and integrate its genome in to the host chromosomal DNA. This provirus can remain dormant for an indefinite period. The process of integration, governed by integrase (IN), is highly conserved across the Retroviridae family. Hence, targeting integration is not only expected to block HIV replication but may also reveal new therapeutic strategies to treat HIV as well as other retrovirus infections.
Recent findings
HIV integrase (IN) has gained attention as the most promising therapeutic target as there are no equivalent homologues of IN that has been discovered in humans. Although current nano-formulated long-acting IN inhibitors have demonstrated the phenomenal ability to block HIV integration and replication with extraordinary half-life, they also have certain limitations.
Summary
In this review, we have summarized the current literature on clinically established IN inhibitors, their mechanism of action, the advantages and disadvantages associated with their therapeutic application, and finally current HIV cure strategies using these inhibitors.
Keywords: HIV Integration, Integrase inhibitors, Latent provirus, Retroviridae, Therapeutic application
Introduction
Human Immunodeficiency Virus (HIV) is an etiological agent of the Acquired Immunodeficiency Syndrome (AIDS)(1–3), which has caused major devastation across the globe from past 4 decades. After the introduction of combination antiretroviral therapy (cART), HIV-1 induced AIDS-related morbidity and mortality has dropped significantly. However, HIV infected long-lived CD4+ T-cells and macrophages in different anatomical sanctuaries represent a major obstacle for a cure since cART fails to completely eliminate the virus from immunologically privileged tissues and organs(4). There are more than 20 antiretroviral drug formulations approved by the USFDA that are in clinical use for the treatment of HIV exploiting different modes of action as summarized in Fig. 1. HIV RT (reverse transcriptase) enzyme governs the conversion of viral ssRNA genome to dsDNA and lacks 3’−5’ exonuclease proofreading activity. Additionally, selective drug pressure, suboptimal penetrance of the cART drug candidates in anatomical sanctuaries, limited ability to cross blood brain barrier and accumulation of accrued mutations confer resistance to different classes of anti-retroviral drugs. In certain scenarios, the resistant mutations are observed even in cART naïve individuals due to horizontal transmission of drug resistance viral strains(5, 6). Moreover, a lifelong cART regimen comes with various undesired effects such as drug fatigue, chronic systemic toxicity, persistent immune inflammation, and premature ageing. These side effects can accelerate non-AIDS related complications like chronic obstructive pulmonary disease (COPD), and cardiovascular diseases. Therefore, more effective strategies are of the urgent need to combat these drawbacks of the current anti-HIV therapeutic arsenal and to eventually eradicate HIV.
Fig. 1.

Schematic representation of FDA approved anti-HIV drugs inhibiting different stages of the HIV-1 life cycle.
With limited resources of its own, HIV is inefficient to complete its life-cycle without exploiting the host. Furthermore, to escape from the host anti-viral immune response, HIV-1 hijacks the host’s cellular machinery, thus proliferating throughout the host while simultaneously seizing the means of propagation. (7, 8•). One key step in the HIV-1 replication cycle involves the conversion of ssRNA genome into cDNA which further associates with multiple hosts as well as viral factors and the formation of the pre-integration complex (PIC). PIC is transported into the nucleus, and cDNA gets integrated into the host genome(9••, 10•). Once integrated, the virus establishes latent reservoirs in the host cells and persists there forever through clonal expansion. With present therapeutic interventions, it is virtually impossible to eliminate the entire pool of latent reservoirs. Hence, it is important to develop therapeutic strategies to block HIV-1 before integration.
RT, IN, and Protease (PR) enzymes of HIV-1 participate in three rate-limiting steps of the HIV-1 replication cycle, including reverse transcription, integration, and maturation, respectively. For this reason, these enzymes are major targets for the development of anti-retroviral drugs. Nucleoside Reverse Transcriptase Inhibitors (NRTIs) and Non-Nucleoside Reverse Transcriptase Inhibitors (NNRTIs) block HIV-1 replication during the reverse transcription. Unfortunately, HIV-1 tends to develop resistance to both NRTIs and NNRTIs faster than any other class of inhibitors due to the extremely fast error prone kinetics(11, 12). As nucleotide analogues, NRTIs also pose a disadvantage by causing mitochondrial toxicity; these inhibitors get incorporated into the host mitochondrial DNA, thus hampering cellular metabolism(12) and causing toxicity. In comparison to RT inhibitors, PR inhibitors are less desirable because PR inhibitors hamper viral replication at the very late stage of the viral life-cycle. This means that PR inhibitors cannot prevent the death of infected cells as reviewed by Eron 2017(13). Also, due to poor specificity, PR inhibitors also act against host cell proteases resulting in cellular toxicity of the host(14, 15). Targeting IN for anti-HIV drug development is both advantageous and important because IN plays a pivotal role in the integration of the viral genome in the host cell chromosome(16, 17). Due to the absence of any HIV-1 IN ortholog in host cells, the off-target effects of IN inhibitors are relatively limited when compared to RT and PR inhibitors.
Current combination therapy uses formulations that contain two or more classes of anti-retroviral drugs that include NRTIs, NNRTIs, PIs, INIs, and entry inhibitors. A list of US FDA approved fixed-dose combinational anti-retroviral drug formulations is described in Table 1.
Table 1:
Timeline of FDA approved formulation [Information adapted from www.aidsinfo.nih.gov accession date: July 13, 2019]
| FDA Approved combinations | Generic Name | Dosage | Approval Date |
|---|---|---|---|
| lamivudine and zidovudine [3TC / ZDV] | Combivir | Twice a day | September 27, 1997 |
| lopinavir and ritonavir [ritonavir-boosted lopinavir, LPV/r, LPV / RTV] | Kaletra | Twice a day | September 15, 2000 |
| abacavir, lamivudine, and zidovudine [abacavir sulfate / lamivudine / zidovudine, ABC / 3TC / ZDV] | Trizivir | Twice a day | November 14, 2000 |
| abacavir and lamivudine [abacavir sulfate / lamivudine, ABC / 3TC] | Epzicom | Once daily | August 2, 2004 |
| emtricitabine and tenofovir disoproxil fumarate [emtricitabine / tenofovir DF, FTC / TDF] | Truvada | Once daily | August 2, 2004 |
| efavirenz, emtricitabine, and tenofovir disoproxil fumarate [efavirenz / emtricitabine / tenofovir DF, EFV / FTC / TDF] | Atripla | Once daily | July 12, 2006 |
| emtricitabine, rilpivirine, and tenofovir disoproxil fumarate [emtricitabine / rilpivirine hydrochloride / tenofovir disoproxil fumarate, emtricitabine / rilpivirine / tenofovir, FTC / RPV / TDF] | Complera | Once daily | August 10, 2011 |
| elvitegravir, cobicistat, emtricitabine, and tenofovir disoproxil fumarate [QUAD, EVG / COBI / FTC / TDF] | Stribild | Once daily | August 27, 2012 |
| abacavir, dolutegravir, and lamivudine [abacavir sulfate / dolutegravir sodium / lamivudine, ABC / DTG / 3TC] | Triumeq | Once daily | August 22, 2014 |
| atazanavir and cobicistat (atazanavir sulfate / cobicistat, ATV / COBI) | Evotaz | Once daily | January 29, 2015 |
| darunavir and cobicistat [darunavir ethanolate / cobicistat, DRV / COBI] | Prezcobix | Once daily | January 29, 2015 |
| elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide fumarate [elvitegravir / cobicistat / emtricitabine / tenofovir alafenamide, EVG / COBI / FTC / TAF] | Genvoya | Once daily | November 5, 2015 |
| emtricitabine, rilpivirine, and tenofovir alafenamide [emtricitabine / rilpivirine / tenofovir AF, emtricitabine / rilpivirine / tenofovir alafenamide fumarate, emtricitabine / rilpivirine hydrochloride / tenofovir AF, emtricitabine / rilpivirine hydrochloride / tenofovir alafenamide, emtricitabine / rilpivirine hydrochloride / tenofovir alafenamide fumarate, FTC / RPV / TAF] | Odefsey | Once daily | March 1, 2016 |
| emtricitabine and tenofovir alafenamide [emtricitabine / tenofovir AF, emtricitabine / tenofovir alafenamide fumarate, FTC / TAF] | Descovy | Once daily | April 4, 2016 |
| dolutegravir and rilpivirine [dolutegravir sodium / rilpivirine hydrochloride, DTG / RPV] | Juluca | Once daily | November 21, 2017 |
| efavirenz, lamivudine, and tenofovir disoproxil fumarate [EFV / 3TC / TDF] | Symfi Lo | Once daily | February 5, 2018 |
| bictegravir, emtricitabine, and tenofovir alafenamide [bictegravir sodium / emtricitabine / tenofovir alafenamide fumarate, BIC / FTC / TAF] | Biktarvy | Once daily | February 7, 2018 |
| lamivudine and tenofovir disoproxil fumarate [3TC / TDF] | Cimduo | Once daily | February 28, 2018 |
| efavirenz, lamivudine, and tenofovir disoproxil fumarate [EFV / 3TC / TDF] | Symfi | Once daily | March 22, 2018 |
| darunavir, cobicistat, emtricitabine, and tenofovir alafenamide [darunavir ethanolate / cobicistat / emtricitabine / tenofovir AF, darunavir ethanolate / cobicistat / emtricitabine / tenofovir alafenamide, darunavir / cobicistat / emtricitabine / tenofovir AF, darunavir / cobicistat / emtricitabine / tenofovir alafenamide fumarate] | Symtuza | Once daily | July 17, 2018 |
| doravirine, lamivudine, and tenofovir disoproxil fumarate (doravirine / lamivudine / TDF, doravirine / lamivudine / tenofovir] | Delstrigo | Once daily | August 30, 2018 |
In the next section, we will discuss USFDA approved IN inhibitors (INIs) that are used to treat HIV infection as well as the INIs drug candidates at various stages of development. We will also highlight the mechanistic details of these inhibitors, as well as their advantages and disadvantages as anti-HIV therapeutic agents.
HIV-1 Integrase
Integration, like reverse transcription, is conserved and unique to retroviruses. Both integration and transcription, collectively govern mutagenic activities, oncogenic activities, and the establishment of latent reservoirs of retroviruses(18). Once the provirus is established in the host genome, it would pass to any progeny cell as a Mendelian locus. As a result, removing the hidden provirus from host cell is virtually impossible using current anti-HIV drugs(19, 20).
Structurally, HIV-1 IN is a 288-amino acid long, 32 kDa protein composed of three domains: the N-terminal HH-CC zinc-binding domain, the RNaseH fold containing catalytic core domain, and the DNA binding C-terminal domain(21). IN forms a tetramer of two dimers(22), which binds to both 3’ ends of viral DNA and uses the same active site for catalytic activity. This active site contains a triad of carboxylates comprising the canonical D, DX35E active site motif. These carboxylates coordinate Mg+2 or Mn+2, a pair of divalent cations, which are critical for catalytic activity(23–25). Notably, although IN can function with either of these cations, it is largely dependent on Mg+2 over Mn+2 for in vivo functioning. These findings were supported by the study in which pharmacological inhibitors for Mn+2-based carboxylates failed to inhibit HIV-1 integration in vivo (26•). Once assembled with viral DNA, IN immediately cleaves the conserved sequences of two nucleotides. Removal of two nucleotides from both 3’ ends generates pre-integration substrates with 3’ sticky ends. This is followed by strand transfer reaction that occurs inside the nucleus where IN governs the cleavage of host DNA and 3’ sticky ends are joined to the 5’ overhangs of host DNA. Finally, the cellular repair machinery fills the gaps leading to the completion of the integration process(27, 28).
Integrase inhibitors (INIs)
Since INIs likely have reduced off-target effects compared to other classes of anti-retrovirals, HIV-1 IN emerged as a high-value therapeutic target. INIs belong to two major categories: i) IN strand transfer inhibitors (INSTIs) that bind to the catalytic core domain of the enzyme to block the binding of the enzyme to dsDNA, and ii) IN binding inhibitors (INBIs) that bind to the allosteric pocket of the IN and hamper the conformational changes required for strand transfer reaction. Currently, all USFDA-approved INIs belong to the group of INSTIs; whereas, therapeutic use of INBIs is still under the developmental phase (Table 2).
Table 2:
Summary of INIs (approved and in development)
| Integrase Inhibitor | Chemical Structure | In Vitro Efficacy | In Vivo Dosage | Estimated Half-life | Resistant Mutations |
|---|---|---|---|---|---|
| BI224436 |  |
<1 nM | NA | NA | A128T, A128N, L102F |
| Bictegravir (BIC) GS-9883 | 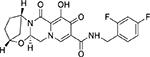 |
1.5 – 2.4 nM | NA | ∼35 h | Q148-E138, Q148-G140, R263L, R263K/M50I |
| BMS-707035 |  |
2–20 nM | NA | NA | NR |
| Cabotegravir (GSK744) | 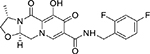 |
3 nM | 400 mg (or 200 mg split injection) once a month | ∼40 d | NR |
| Dolutegravir (DTG) (GSK-1349572) | 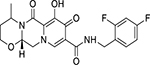 |
0.51 – 2 nM | 50 mg per day | ∼ 14 d | R263K, S230R, Q148-E138, Q148-G140 |
| Elvitegravir (EVG) (GS-9137) |  |
0.7 – 1.5 nM | 85 mg and 150 ng twice per day | 3–9 h | E92Q, T66I/A/K, T124A, P145S, Q148K/R |
| MK-2048 | 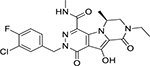 |
1.5–2.6 nM | 30 mg once daily | 1–1.5 h | G118R, E138K |
| Raltegravir (RAL) (MK-0518) | 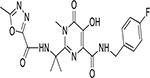 |
2–7 nM | 400 mg thrice per day | 9 h | N155H, Q148K/R/H |
NA = Not Available NR = Not Reported
FDA approved INIs
Raltegravir (RAL)
RAL (MK-0518; Table 2) was the first IN inhibitor approved by USFDA in 2007 following the evidence of rapid, potent, and sustained anti-HIV activity in clinical trials(29). While it was initially approved for salvage therapy of patients who had developed resistance to first- and second-line cART, it has since been promoted to be a first-line drug(30, 31). RAL demonstrates significant anti-HIV activity in vitro, with an IC50 value of 2 to 7 nM; whereas, the IC90 value is 19 to 31 nM. Notably, RAL is reported to be active against multidrug-resistant isolates of HIV(32–34). In a randomised, double-blind, 156-week STARTMRK trial, RAL revealed significant durability in combination with Tenofovir disoproxil fumarate (TDF) in treatment naïve patients. RAL is also reported to alter the kinetics of HIV-1 decay and reduce the second phase viral load remarkably(35, 36).
Docking studies provided a better insight into INSTI’s mechanism of action by revealing a crucial binding site in the active IN site. Specifically, RAL directly interacts with the catalytic residues D64, D116, E152, T66, E92, Y143, Q148, and N155 of HIV-1 IN. Additionally, sequencing data from clinical studies confirmed that mutations in E92 and Y143 led to reduced RAL susceptibility(37). The plasma half-life of RAL is approximately 9 hours in adults, with a rapid elimination stage lasting approximately 1 hour, thus making it possible to administer this drug twice a day(38, 39). RAL also demonstrated a relatively minimal level of interaction with NNRTIs and PR inhibitors (PIs), meaning that these drugs could potentially be used in combination(40). The phase-III ONCEMRK trial reported a higher bioavailability of the 600 mg tablet over the 400 mg RAL formulation twice daily post 48 and 96 hours of treatment(29). Currently, RAL is administered as a once daily 1200 mg dose with emtricitabine and TDF.
A major drawback associated with RAL based therapy is the development of drug associated resistance mutations. RAL associated minor INI-resistance mutations are frequently observed in drug naïve patients due to horizontal transfer of HIV-1 strains harbouring the mutations(41). Over 40 mutations are currently associated with INSTI inhibitors, with 14 associated with RAL resistance(42, 43). In particular, N155H and Q148K/R/H mutations are associated with almost complete resistance to RAL in infected individuals(31, 44). Phenotypic analysis of viral isolates from patients revealed that Q148K/R/Hs confer greater resistance than N155H mutation. However, none of the transmitted viral isolates carried both these mutations simultaneously. This indicates that resistance to INSTIs may be developed through two or more independent pathways(45). Importantly, some mutations, such as E92, E157, and Y143, have a debated role in resistance against RAL due to the fact that they reduce INSTI susceptibility to the IN enzyme that occur significantly less frequently than the other mutations(46). RAL has a relatively shorter half-life compared to other INSTIs. Development of rapid INI resistance mutations against RAL makes it a less effective therapeutic option in salvage therapy for patients failing second-line cART. For this reason, nowadays RAL is best used as a component of the first-line cART for treatment naïve individuals.
Elvitegravir (EVG)
EVG (GS-9137) was the first anti-HIV drug developed by Gilead and Japan Tobacco Company in early 2008 and received FDA approval in late 2012(47). EVG demonstrated significant anti-HIV activity in in vitro assays on various laboratory strains of both HIV-1 and HIV-2, with an IC50 value ranging from 0.7 to 1.5 nM. EVG also inhibited the IN strand transfer reaction in vitro with an EC50 value of 54 nM. Compared to RAL, EVG demonstrated superior therapeutic efficacy due to its blocking ability against multidrug-resistant strains of HIV, Murine Leukaemia Virus (MLV), and Simian Immunodeficiency Virus (SIV)(48). Moreover, EVG absorption increases if taken with food and interestingly, ritonavir booster can increase its elimination half-life from 3 hours to a maximum of 9 hours(49, 50). Mechanistically, EVG recognises the IN-DNA complex and binds to the interface between the 3’ end of the viral DNA and the IN enzyme. EVG interacts strongly with D64 and E152 amino acids in the catalytic core domains that contain ß-hydroxy carboxylates, chelates metal ions, and hydroxylic oxygen(51) leading to the inhibition of strand transfer reaction as well as the integration of viral dsDNA into the host genome.
A series of clinical trials and cohort studies have identified multiple mutations that govern EVG treatment failure. Five different substitution mutations were reported, and among these, E92Q alone was found to be capable of reducing EVG susceptibility by 33-fold(52). The other substitution mutations T66I/A/K, T124A, P145S, Q148K/R can reduce EVG susceptibility by a range of 3 to 30 folds(53). EVG has a low barrier to resistance compared to other INIs as it overlaps with some of RAL’s resistance mutations including Q148K(54, 55) which limits its therapeutic efficiencies.
Dolutegravir (DTG)
DTG (GSK-1349572) belongs to the World Health Organisation’s (WHO) List of Essential Medicines and is currently thought to be the single most promising long-term anti-HIV medication available. In 2013, DTG received USFDA approval for third-line cART in patients who developed resistance to first- and second-line cART. However, DTG is now preferred in combination with abacavir and lamivudine, collectively known as Triumeq(56). Mechanistically, DTG binds to the active site of HIV-1 IN which brings the β4-α2 loop of the enzyme into close contact with the extended linker region. This interaction leads to conformational changes that disengage IN from the deoxyadenosine present at the 3’ end of the virus. As a result, DTG chelates the divalent cations that are required for the enzyme function leading to inhibition of strand transfer reaction. The presence of a halobenzyl group in the structure allows DTG to reach even farther in the active site, resulting in more stable interaction with the enzyme then RAL and EVG. This enables DTG to re-adjust to the conformational changes observed in RAL and EVG resistant mutants that make DTG capable of inhibiting IN mutant strains more effectively than other INIs(57).
In in vitro studies, DTG demonstrated superiority compared to RAL and EVG, with an IC50 value of 2.7 nM, EC50 value of 0.51 nM, and a Therapeutic Index of > 9400 in human peripheral blood mononuclear cells (PBMCs); all of which are remarkably higher than in either RAL or EVG(58). Randomised double-blind phase-II and phase-III trials were conducted on 208 patients as a part of the SPRING study. In this study, patients received DTG in combination with Abacavir (ABC) and Lamivudine (3TC) for 96 weeks. DTG maintained significant bioavailability without any dose-related clinical adverse effect. The subjects were also found to maintain CD4+ cell counts with an average of 301 cells/μL after 96 weeks of therapy(59). Additionally, treatment of DTG in combination of TDF and emtricitabine (FTC) did not significantly alter the time of virologic failure as compared to the patients treated with TDF/3TC. These results collectively indicate that the difference in the events of viral failure upon different regimen is not due to the intrinsic difference in the anti-HIV activity of these drugs(60, 61). FLAMINGO, SINGLE and VIKING trials suggest that DTG is the best available INSTI, and the WHO’s current guideline recommends the addition of DTG in combination with 3TC/FTC and ABC in first-line drug therapy for cART naïve patients(62–64). DTG has a plasma half-life of ~ 14 days, and recent reports suggest that DTG may persist for up to 72 hours after the last dose with 90% protein binding concentration(65). DTG is administered without any pharmacokinetic boosters such as cobicistat (66); its consistent efficacy, excellent tolerability, and lack of reported drug-drug interactions make it an excellent option and the preferred choice to couple with two NNRTIs/NRTIs for cART.
Although DTG has demonstrated promising results over other INI regimens, DTG-resistance mutations have been observed in the viruses isolated from DTG treated individuals. Surprisingly, these mutations are different from the resistance mutations that are frequently associated with RAL and EVG, thus implying that DTG may be inhibited through a unique pathway of its own. R263K substitution, for example, was the first documented DTG-resistance mutation (67). Pham et al. recently reported that mutation at S230R of IN impaired HIV-1 IN function as well as reduced DTG susceptibility by approximately 4 fold (68). In the DOMONO trial, due to its high genetic barrier, DTG was used as an HIV maintenance monotherapy in patients who attained complete virologic control with cART (69). In this study, several individuals reported virologic failure who carried HIV-1 strains without any mutations in the integrase gene. A comprehensive analysis of the viral genome revealed that mutations in the 3′-polypurine tract (3′-PPT) confer high-level resistance to DTG(69, 70). This finding further validates one previous report where Malet et.al., showed in vitro that high concentrations of prolonged DTG exposure confers mutations in 3′-PPT of wildtype HIV-1 strains(71••). This mutant strain of HIV-1 selected by high DTG exposure were also highly resistant to RAL and EVG. These reports reveal cautionary signals for a shift to DTG based monotherapy as well as DTG based combination therapy. An in vitro study reported that de novo selection of Env mutations confer resistance to DTG(72). These findings suggest that in order to evaluate virologic failure and drug resistance mutation, the complete IN gene needs to be screened to detect genetic alterations that may be responsible for therapeutic failure of INIs. Furthermore, the TSEPAMO study from Botswana reported a greater risk for development of infant neural tube defects in women receiving DTG at the time of conception compared to other classes of anti-retrovirals(73). Based on these findings, WHO revised its guideline of use of DTG in women of childbearing age (https://www.who.int/hiv/mediacentre/news/dtg-qa/en/).
Bictegravir (BIC)
Bictegravir (BIC), also known as GS-9883, was developed by Gilead in 2016(74). BIC is one of the latest INIs to receive US FDA-approval and is being used in clinic as a single tablet fixed-dose combination with bictegravir 50mg, emtricitabine 200mg and tenofovir alafenamide 25 mg (Biktarvy). Clinical data and pharmacokinetic studies revealed that BIC is a more efficient INSTI compared to RAL or EVG(74). A rapid absorption rate and an elimination half-life of 18 hours mean that BIC is supportive of a once-daily administration plan without a pharmacokinetic booster(76). Notably, BIC also possesses the longest integrase/DNA dissociation half-life of approximately ~ 35 hours which is best among this class(77).
In vitro, BIC demonstrated the strong, specific, anti-HIV activity with an IC50 value ranging between 1.5 – 2.4 nM in both T-cell lines and primary cells. BIC inhibits IN strand transfer activity with an EC90 of ~ 8.3 nM in the cell-free assay system. Additionally, BIC demonstrated improved anti-HIV potential against various INSTIs resistant clinical isolates of HIV-1 compared to DTG. BIC also demonstrated a slower rate of resistance mutations’ accumulation compared to DTG and EVG, even after 150 days in culture(75–77).
Despite these advantages, a Q148 substitution mutation was reported to reduce the efficiency of BIC by ~ 63 folds, while an R263L single mutation or R263K/M50I dual mutation have also been reported to reduced BIC efficiency by 2.5 – 3.0 folds in vitro(78). Notably, no other mutations have yet been reported in patients, and further studies on viral resistance to BIC are urgently needed.
Drugs in various stages of development
Cabotegravir (CAB)
CAB (GSK744), with its carbamoyl pyridone backbone, is structurally and mechanistically similar to DTG(79) also demonstrating very promising results in Phase-IIa trials(80). In vitro, CAB displayed significant anti-HIV activity with an IC50 value of 3 nM and an IC90 value of ~ 400 nM(81). Due to its extremely high efficacy, rapid absorption rate upon intramuscular injection, and elimination half-life of ~ 40 days, CAB has attracted significant attention as an ideal candidate for monthly or even quarterly administration(82). CAB’s unique properties allow it to be administered as either an oral tablet or a long-acting nano-formulated injectable. Currently, CAB combined with rilpivirine is being investigated through phase-IIb trials in antiviral naïve patients by the ÉCLAIR, LATTE, and LATTE-2 studies(83–85). Recently, preliminary outcomes of ATLAS (Antiretroviral Therapy as Long-Acting Suppression: NCT02951052) and FLAIR (First Long-Acting Injectable Regimen: NCT02938520) trials were reported. The ATLAS and FLAIR were Phase III randomized, open-label trials involving 616 and 566 active participants, respectively. In both trials, during viral suppression, study participants were given oral CAB 30 mg + RPV 25 mg once daily for 4 weeks followed by intramuscular (IM) injections of a loading dose of CAB LA (600 mg) and RPV LA (900 mg). From week eight, the participants received IM injection of CAB LA 400 mg and RPV LA 600 mg every four weeks up to 48 weeks. The other arm received daily oral dose of 2 NRTIs plus an INI, NNRTI, or PI for 48 weeks (ATLAS) and daily oral dose of ABC / DTG / 3TC (600 mg/50mg/300mg) once daily for 48 weeks (FLAIR). In the ATLAS trial, at the end of 48 weeks, the CAB LA and RPV LA was non-inferior in maintaining viral suppression in treated participants (5 [1.6%] out of 313 had plasma viral loads above 50 copies/ml) when compared with the arm receiving conventional oral daily ART regimen (3 [1.0%] participants out of 313 had plasma viral loads above 50 copies/ml) (adjusted difference: 0.6%, 95% confidence interval [CI]: −1.2, 2.5). The CAB LA and RPV LA arm of the FLAIR trial was also non-inferior in maintaining viral suppression (6 [2.1%] out of 283 participants had plasma viral loads above 50 copies/ml) when compared with the arm receiving conventional oral daily ART regimen (7 [2.5%] out of 283 participants had plasma viral loads above 50 copies/ml) (adjusted difference: −0.4%, 95% CI: −2.8, 2.1). Overall the long acting formulation is well-tolerated, with low rates of serious adverse events (SAEs) of (13/308 [4.2%]) and (18/283 [6.4%]) in ATLAS and FLAIR trials, respectively. These results demonstrated a proof-of-principle that a once monthly dose of a long acting CAB+RPV formulation can achieve long-term virologic control. Furthermore, a recent study using a SHIV infected macaque model of HIV infection showed a risk of drug resistance to CAB-LA when used as a post-exposure prophylaxis. Radzio-Basu et al., found selection of multiple mutations in integrase gene conferring resistance to CAB, several of which also have cross-resistance to all other INIs(86); and these mutations potentially reduce the option of using INSTIs in follow-up therapy. This study warrants detailed sequencing and use of frequent serological tests while using CAB LA for post-exposure prophylaxis therapy. In in vitro studies, CAB shows a high genetic barrier. To better predict the clinical efficacy of CAB in in vivo studies, long term follow up of the patients in the above-mentioned trials is required to monitor development of virologic failure due to the emergence of drug-associated mutations.
Due to longer biological half-life and sustained release that is achieved by nano-formulations, CAB has the potential to reduce the daily anti-retroviral pill burden to just once a month administration. Also, both CAB, as well as nano-formulated CAB, does not show any major drug-drug interactions in the short term as well as long term usage. CAB is also administered without pharmacokinetic boosters. These characteristics collectively make CAB a potential candidate for both pre- and post-exposure prophylaxis (PrEP/PEP).
BMS-707035
BMS-707035, a pyrimidinone carboxamide, is structurally similar to RAL and differs only by the substitution of an oxadiazole group for the sulphonamide group in the scaffold(87). BMS-707035 also interacts with many of the same amino acid residues in the catalytic core domain as RAL, including D64, D116, E152, T66, E92, Y143, Q148, and N155(88), displaying significant anti-HIV potential in vitro, with an IC50 value of ~ 2–20 nM(18, 89). Although BMS-707035 displays similar efficacy as RAL, BMS-707035’s efficacy can be significantly reduced by mutations in HIV-1 LTR(90). Additionally, the toxicity studies and results of a 12-month dog safety assessment study showed multiple acute as well as chronic systemic toxicities that ultimately halted the development of BMS-707035 for higher studies and further therapeutics. Recent studies by Naidu et al. revealed several structural analogues with improved specificity and anti-HIV activity(91). The EC50 values of these analogues range from 2 nM to 19 nM in a cell-free assay system, while the in vitro IC50 values range from 1 nM to 206 nM. Some of these molecules also displayed an improved toxicity profile in dogs, and additional animal studies are currently being investigated as pre-clinical trials(91).
MK-2048
MK-2048 was first developed by Merck & Co. in early 2009. The first phase of the phase-II trial of MK-2048 was completed in 2014. One advantage of MK-2048 is that it inhibits viral isolates with R263K resistant mutation more efficiently than wild type virions (IC50 value of 1.5 nM and 2.6 nM, respectively) in the cell-free integrase strand transfer assay(43). A daily MK-2048 dosage of 30 mg is currently being evaluated for safety and efficacy in combination with vicriviroc in phase-I trials(92). These initial studies suggest that MK-2048 does not show cross-resistance to RAL and EVG resistance mutations, making it one of the most potent second generation INSTIs(93).
In vitro, MK-2048 demonstrates significant anti-HIV activity in both cell lines and human PBMCs with an IC50 value of 0.9 nM and an IC90 value of 57 nM(94). However, it has an extremely low half-life of only 1–1.5 hours, and its efficacy can be reduced more than 8 fold by the G118R and E138K mutations, which do not affect either RAL or EVG(95, 96). As a result, additional research is necessary to develop more specific, efficient, and longer lasting analogues of MK-2048.
BI224436
BI224436, a non-catalytic site integrase inhibitor, binds to the allosteric pocket of IN enzyme and blocks the conformational changes required by the strand transfer and recombination steps of integration(97). BI224436 is a 3-quinolineacetic acid derivative with a significantly higher therapeutic index (TI > 10,000) than other INIs against various laboratory strains of HIV-1, as shown by in vitro assay systems(97). The binding of BI224436 with IN enzyme interferes with the interaction of IN with LEDGF (Lens Epithelial Derived Growth Factor) on the allosteric pocket of the enzyme, thus preventing the formation of PIC and its nuclear import. Initially, BI224436 was thought to be a potent inhibitor of IN as it targets the conserved non-catalytic domain of IN and is unlikely to develop resistant mutations. However, three mutations A128T, A128N and L102F in the conserved region of IN, which reduced the binding affinity of BI224436 with the IN by 2.9, 64, and 61 folds, respectively, were reported in HIV-1 infected patients(98), limiting the advancement of BI224436 to phase-I trials.
Nano-formulations
Nano-formulations have recently gained popularity for AIDS-related neurological complications due to cART’s suboptimal penetrance to cross the blood-brain barrier(99–101•). The primary purpose of nano-formulations is to implement a sustained release of the drug in the host along with a target-specific delivery of the drug(s). Both of these outcomes can potentially result in a specific and long-term therapeutic effect(102). Many cART candidate drugs, as described earlier, are designed in gold and diamond-based nano-formulations in order to increase the specificity and efficacy of the drug(103). Both gold and diamond nano-formulations have a high surface area to volume ratio providing a large surface for drug binding and interaction. This property results in a more sustained release of the drug and may eventually be applied to reduce the burden of daily administration. This also assists in overcoming the issues of uneven tissue distribution and toxicity to the host(104).
The nano-suspension of GSK1265744 with polysorbate 20 and polyethylene glycol 3350 is currently being evaluated in phase II trials, both for sustained delivery as well as reduced toxicity (NCT02076178). In these trials, GSK1265744 was detected in the plasma of seronegative donors even after 48 weeks post-exposure, thus demonstrating the drug to be a promising long-acting IN inhibitor. Additionally, recently Sillman et al. developed a long-acting parenteral nano-formulation of DTG prodrug by encapsulating it in a poloxamer (NMDTG). Pharmacokinetic profiling of NMDTG revealed an IC90 concentration of nearly 64 ng/mL in the plasma for 56 days, with a similar concentration obtained from lymphoid tissues after 28 days(105). However, in a recent study the combination of sequential long-acting slow-effective release antiviral therapy and CRISPR-Cas 9 treatment eliminated complete HIV in few of humanized mice (106••). Recent exploitation of nano-formulated DTG and CAB, given once in two-month administration formulation, are currently under critical investigations(105, 107, 108••). These studies have opened new horizons of long-acting antiretroviral therapy and may potentially change the future of anti-HIV therapeutics.
To conclude, nano-formulations of drugs have comparatively longer half-lives, and can easily cross cellular barriers, such as the BBB and the portal systems of the body, thus serving as potential therapy for the central nervous system viral reservoir and other immune-privileged areas. Nano-formulated medicines are also metabolized slightly differently than conventional cytochrome mediated metabolism. However, additional studies are necessary to gain a better understanding of nano-formulated medicines as well as nanoparticles. Therefore, long term toxicity of nano-formulations should be understood in more detail. It is also important to examine the metabolism that is mediated by the cytochrome machinery of nano-formulations in vivo because the conjugation of gold nanoparticles can potentially affect the metabolism of the prodrug in the long term.
Future considerations
Development of drug resistant mutations is a major challenge in achieving a functional cure of HIV-1. Current therapeutics has greatly reduced the lethality of the disease; but nevertheless, we are still far from the true cure. Additionally, the issue of resistance mutations increases the complexity of treatment. For this reason, current treatment strategies must be continuously updated. Integration is a point of no return and is of unique importance because a provirus that has been integrated into the host genome is virtually impossible to eliminate from host cells. IN inhibitors present a promising option to this end. However, since all current INSTIs bind directly to the active site of the enzyme, which is more prone to the point mutations, the development of drug resistance can occur. For this reason, other sites on the IN enzyme should be explored as well, including the allosteric pocket of enzymes that are less sensitive to mutations. Additional studies on the structural perspective of PIC formation will provide new insights regarding the HIV dependency factors (HDFs) that are required for IN function. Understanding the role of these HDFs in host cells regulating the viral life cycle can potentially assist in identifying new HDFs as well as better treatment options.
Another promising area of anti-retroviral drug discovery is the development of dual inhibitors that will simultaneously act against two different targets like IN and RT enzymes using a single drug molecule. Several small molecules that act against IN and RNase H domain of RT are at different stages of pre-clinical development(109, 110). A new class of dual inhibitors that target IN and LEDGF/p75 interaction is also in development(111).
Nano-formulations and sustained release long-acting (LA) formulations of both RT as well as IN inhibitors are at various stages of their clinical trials which are expected to improve the efficiency of anti-HIV therapeutics overcoming the drawbacks of present arsenal (112••). Nano-formulations of the current IN inhibitors and other investigational drugs require further evaluation in both treatment-naïve and ART-experienced patient populations. If a sustained release can be achieved without any significant adverse effect, these nano-formulations could potentially be considered for PrEP. Design of new INIs with pharmacological properties that consider the drug-resistant IN mutations should be a priority for anti-HIV therapeutic development. Also, designing of the INIs that can also hamper the function of RT as well as PR might revolutionize the antiretroviral therapeutic arsenal.
Acknowledgements
We thank Robin Taylor for editorial help. This work is partially supported by National Institute of Allergy and Infectious Diseases Grant R01 AI129745.
Footnotes
Conflicts of Interest
No potential conflicts of interest relevant to this article were reported.
Human and Animal rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
References
- 1.Barre-Sinoussi F, Chermann JC, Rey F, Nugeyre MT, Chamaret S, Gruest J, et al. Isolation of a T-lymphotropic retrovirus from a patient at risk for acquired immune deficiency syndrome (AIDS). 1983. Rev Invest Clin. 2004;56(2):126–9. doi: 10.1126/science.6189183. [DOI] [PubMed] [Google Scholar]
- 2.Gallo RC, Sarin PS, Gelmann EP, Robert-Guroff M, Richardson E, Kalyanaraman VS, et al. Isolation of human T-cell leukemia virus in acquired immune deficiency syndrome (AIDS). Science. 1983;220(4599):865–7. doi: 10.1126/science.6601823. [DOI] [PubMed] [Google Scholar]
- 3.Levy JA, Hoffman AD, Kramer SM, Landis JA, Shimabukuro JM, Oshiro LS.Isolation of lymphocytopathic retroviruses from San Francisco patients with AIDS. Science. 1984;225(4664):840–2. doi: 10.1126/science.6206563 [DOI] [PubMed] [Google Scholar]
- 4.Prieto P, Podzamczer D. Switching strategies in the recent era of antiretroviral therapy. Expert Rev Clin Pharmacol. 2019;12(3):235–47. doi: 10.1080/17512433.2019.1575728. [DOI] [PubMed] [Google Scholar]
- 5.Nanfack AJ, Redd AD, Bimela JS, Ncham G, Achem E, Banin AN, et al. Multimethod Longitudinal HIV Drug Resistance Analysis in Antiretroviral-Therapy-Naive Patients. J Clin Microbiol. 2017;55(9):2785–800. doi: 10.1128/JCM.00634-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schauer G, Sluis-Cremer N. HIV-1 Resistance to Reverse Transcriptase Inhibitors In: Berghuis A, Matlashewski G, Wainberg MA, Sheppard D, editors. Handbook of Antimicrobial Resistance. New York, NY: Springer New York; 2017. p. 523–42. [Google Scholar]
- 7.Holmes M, Zhang F, Bieniasz PD. Single-Cell and Single-Cycle Analysis of HIV-1 Replication. PLoS Pathog. 2015;11(6):e1004961. doi: 10.1371/journal.ppat.1004961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schott K, Konig R. Picking the Survivor! CRISPR Reveals HIV Dependency Factors. Trends Microbiol. 2017;25(4):243–5. doi: 10.1016/j.tim.2017.02.004.•. This interesting study highlights three HIV dependency factors by utilisation of loss-of-function CRISPR/Cas9 technology. These HIV dependency factors are vital for virus replication cycle; their knock out attenuates virus replication even in primary cells without being lethal to cells.
- 9.Engelman AN, Singh PK. Cellular and molecular mechanisms of HIV-1 integration targeting. Cell Mol Life Sci. 2018;75(14):2491–507. doi: 10.1007/s00018-018-2772-5.••. Outstanding elaboration about the events that take place during integration of viral genome in to the “gene-rich” area of the chromosome. The review also briefs about the development of small molecules that block these events and ultimately hamper the virus replication cycle.
- 10.Hu H, Xiao A, Zhang S, Li Y, Shi X, Jiang T, et al. DeepHINT: understanding HIV-1 integration via deep learning with attention. Bioinformatics. 2019;35(10):1660–7. doi: 10.1093/bioinformatics/bty842.•. Interesting new approach of attention based deep learning network, DeepHINT, that predicts the integration site of provirus into the host chromosome.
- 11.Koczor CA, Lewis W. Nucleoside reverse transcriptase inhibitor toxicity and mitochondrial DNA. Expert Opin Drug Metab Toxicol. 2010;6(12):1493–504. doi: 10.1517/17425255.2010.526602. [DOI] [PubMed] [Google Scholar]
- 12.Margolis AM, Heverling H, Pham PA, Stolbach A. A review of the toxicity of HIV medications. J Med Toxicol. 2014;10(1):26–39. doi: 10.1007/s13181-013-0325-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Eron JJ Jr. HIV-1 protease inhibitors. Clin Infect Dis. 2000;30 Suppl 2:S160–70. doi: 10.1086/313853 [DOI] [PubMed] [Google Scholar]
- 14.Echecopar-Sabogal J, D’Angelo-Piaggio L, Chaname-Baca DM, Ugarte-Gil C. Association between the use of protease inhibitors in highly active antiretroviral therapy and incidence of diabetes mellitus and/or metabolic syndrome in HIV-infected patients: A systematic review and meta-analysis. Int J STD AIDS. 2018;29(5):443–52. doi: 10.1177/0956462417732226. [DOI] [PubMed] [Google Scholar]
- 15.Soriano V, Fernandez-Montero JV, Benitez-Gutierrez L, Mendoza C, Arias A, Barreiro P, et al. Dual antiretroviral therapy for HIV infection. Expert Opin Drug Saf. 2017;16(8):923–32. doi: 10.1080/14740338.2017.1343300. [DOI] [PubMed] [Google Scholar]
- 16.Engelman A, Cherepanov P. Retroviral Integrase Structure and DNA Recombination Mechanism. Microbiol Spectr. 2014;2(6):1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pommier Y, Johnson AA, Marchand C. Integrase inhibitors to treat HIV/AIDS. Nat Rev Drug Discov. 2005;4(3):236–48. doi: 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- 18.Al-Mawsawi LQ, Al-Safi RI, Neamati N. Anti-infectives: clinical progress of HIV-1 integrase inhibitors. Expert Opin Emerg Drugs. 2008;13(2):213–25. doi: 10.1517/14728214.13.2.213. [DOI] [PubMed] [Google Scholar]
- 19.Archin NM, Liberty AL, Kashuba AD, Choudhary SK, Kuruc JD, Crooks AM, et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature. 2012;487(7408):482–5. doi: 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coiras M, Lopez-Huertas MR, Perez-Olmeda M, Alcami J. Understanding HIV-1 latency provides clues for the eradication of long-term reservoirs. Nat Rev Microbiol. 2009;7(11):798–812. doi: 10.1038/nrmicro2223. [DOI] [PubMed] [Google Scholar]
- 21.Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, et al. Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding. Proc Natl Acad Sci U S A. 2000;97(15):8233–8. doi: 10.1073/pnas.150220297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yuan P, Gupta K, Van Duyne GD. Tetrameric structure of a serine integrase catalytic domain. Structure. 2008;16(8):1275–86. doi: 10.1016/j.str.2008.04.018. [DOI] [PubMed] [Google Scholar]
- 23.Engelman A, Cherepanov P. The structural biology of HIV-1: mechanistic and therapeutic insights. Nat Rev Microbiol. 2012;10(4):279–90. doi: 10.1038/nrmicro2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P. Retroviral intasome assembly and inhibition of DNA strand transfer. Nature. 2010;464(7286):232–6. doi: 10.1038/nature08784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM. Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases. Mol Cell Biol. 1992;12(5):2331–8. doi: 10.1128/mcb.12.5.2331 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Choi E, Mallareddy JR, Lu D, Kolluru S. Recent advances in the discovery of small-molecule inhibitors of HIV-1 integrase. Future Sci OA. 2018;4(9):Fso338.•. Interesting review about the development of novel small molecules that inhibit HIV-1 IN by targeting different sites of the enzyme which are less sensitive to point mutations.
- 27.Li X, Krishnan L, Cherepanov P, Engelman A. Structural biology of retroviral DNA integration. Virology. 2011;411(2):194–205. doi: 10.1016/j.virol.2010.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Schroder AR, Shinn P, Chen H, Berry C, Ecker JR, Bushman F. HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 2002;110(4):521–9. doi: 10.1016/S0092-8674(02)00864-4 [DOI] [PubMed] [Google Scholar]
- 29.Deeks ED. Raltegravir Once-Daily Tablet: A Review in HIV-1 Infection. Drugs. 2017;77(16):1789–95. [DOI] [PubMed] [Google Scholar]
- 30.Croxtall JD, Lyseng-Williamson KA, Perry CM. Raltegravir. Drugs. 2008;68(1):131–8. doi: 10.2165/00003495-200868010-00009 [DOI] [PubMed] [Google Scholar]
- 31.Steigbigel RT, Cooper DA, Kumar PN, Eron JE, Schechter M, Markowitz M, et al. Raltegravir with optimized background therapy for resistant HIV-1 infection. N Engl J Med. 2008;359(4):339–54. doi: 10.1056/NEJMoa0708975. [DOI] [PubMed] [Google Scholar]
- 32.Pace P, Di Francesco ME, Gardelli C, Harper S, Muraglia E, Nizi E, et al. Dihydroxypyrimidine-4-carboxamides as novel potent and selective HIV integrase inhibitors. J Med Chem. 2007;50(9):2225–39. doi: 10.1021/jm070027u [DOI] [PubMed] [Google Scholar]
- 33.Summa V, Petrocchi A, Bonelli F, Crescenzi B, Donghi M, Ferrara M, et al. Discovery of raltegravir, a potent, selective orally bioavailable HIV-integrase inhibitor for the treatment of HIV-AIDS infection. J Med Chem. 2008;51(18):5843–55. doi: 10.1021/jm800245z. [DOI] [PubMed] [Google Scholar]
- 34.Roquebert B, Damond F, Collin G, Matheron S, Peytavin G, Benard A, et al. HIV-2 integrase gene polymorphism and phenotypic susceptibility of HIV-2 clinical isolates to the integrase inhibitors raltegravir and elvitegravir in vitro. J Antimicrob Chemother. 2008;62(5):914–20. doi: 10.1093/jac/dkn335. [DOI] [PubMed] [Google Scholar]
- 35.Markowitz M, Nguyen BY, Gotuzzo E, Mendo F, Ratanasuwan W, Kovacs C, et al. Rapid and durable antiretroviral effect of the HIV-1 Integrase inhibitor raltegravir as part of combination therapy in treatment-naive patients with HIV-1 infection: results of a 48-week controlled study. J Acquir Immune Defic Syndr. 2007;46(2):125–33. doi: 10.1097/QAI.0b013e318157131c. [DOI] [PubMed] [Google Scholar]
- 36.Rockstroh JK, Lennox JL, Dejesus E, Saag MS, Lazzarin A, Wan H, et al. Long-term treatment with raltegravir or efavirenz combined with tenofovir/emtricitabine for treatment-naive human immunodeficiency virus-1-infected patients: 156-week results from STARTMRK. Clin Infect Dis. 2011;53(8):807–16. doi: 10.1093/cid/cir510. [DOI] [PubMed] [Google Scholar]
- 37.Serrao E, Odde S, Ramkumar K, Neamati N. Raltegravir, elvitegravir, and metoogravir: the birth of “me-too” HIV-1 integrase inhibitors. Retrovirology. 2009;6:25. doi: 10.1186/1742-4690-6-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cocohoba J, Dong BJ. Raltegravir: the first HIV integrase inhibitor. Clin Ther. 2008;30(10):1747–65. doi: 10.1097/QAI.0000000000002065. [DOI] [PubMed] [Google Scholar]
- 39.Iwamoto M, Kassahun K, Troyer MD, Hanley WD, Lu P, Rhoton A, et al. Lack of a pharmacokinetic effect of raltegravir on midazolam: in vitro/in vivo correlation. J Clin Pharmacol. 2008;48(2):209–14. doi: 10.1177/0091270007310382 [DOI] [PubMed] [Google Scholar]
- 40.Wenning LA, Friedman EJ, Kost JT, Breidinger SA, Stek JE, Lasseter KC, et al. Lack of a significant drug interaction between raltegravir and tenofovir. Antimicrob Agents Chemother. 2008;52(9):3253–8. doi: 10.1128/AAC.00005-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu J, Miller MD, Danovich RM, Vandergrift N, Cai F, Hicks CB, et al. Analysis of low-frequency mutations associated with drug resistance to raltegravir before antiretroviral treatment. Antimicrob Agents Chemother. 2011;55(3):1114–9. doi: 10.1128/AAC.01492-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ceccherini-Silberstein F, Malet I, D’Arrigo R, Antinori A, Marcelin AG, Perno CF. Characterization and structural analysis of HIV-1 integrase conservation. AIDS Rev. 2009;11(1):17–29. doi: 10.1128/AAC.01492-10. [DOI] [PubMed] [Google Scholar]
- 43.Quashie PK, Mesplede T, Han YS, Oliveira M, Singhroy DN, Fujiwara T, et al. Characterization of the R263K mutation in HIV-1 integrase that confers low-level resistance to the second-generation integrase strand transfer inhibitor dolutegravir. J Virol. 2012;86(5):2696–705. doi: 10.1128/JVI.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cooper DA, Steigbigel RT, Gatell JM, Rockstroh JK, Katlama C, Yeni P, et al. Subgroup and resistance analyses of raltegravir for resistant HIV-1 infection. N Engl J Med. 2008;359(4):355–65. doi: 10.1056/NEJMoa0708978. [DOI] [PubMed] [Google Scholar]
- 45.Armenia D, Vandenbroucke I, Fabeni L, Van Marck H, Cento V, D’Arrigo R, et al. Study of genotypic and phenotypic HIV-1 dynamics of integrase mutations during raltegravir treatment: a refined analysis by ultra-deep 454 pyrosequencing. J Infect Dis. 2012;205(4):557–67. doi: 10.1093/infdis/jir821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croxtall JD, Keam SJ. Raltegravir: a review of its use in the management of HIV infection in treatment-experienced patients. Drugs. 2009;69(8):1059–75. doi: 10.2165/00003495-200969080-00007. [DOI] [PubMed] [Google Scholar]
- 47.Stellbrink HJ. Antiviral drugs in the treatment of AIDS: what is in the pipeline? Eur J Med Res. 2007;12(9):483–95. [PubMed] [Google Scholar]
- 48.Shimura K, Kodama E, Sakagami Y, Matsuzaki Y, Watanabe W, Yamataka K, et al. Broad antiretroviral activity and resistance profile of the novel human immunodeficiency virus integrase inhibitor elvitegravir (JTK-303/GS-9137). J Virol. 2008;82(2):764–74. doi: 10.1128/JVI.01534-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.DeJesus E, Berger D, Markowitz M, Cohen C, Hawkins T, Ruane P, et al. Antiviral activity, pharmacokinetics, and dose response of the HIV-1 integrase inhibitor GS-9137 (JTK-303) in treatment-naive and treatment-experienced patients. J Acquir Immune Defic Syndr. 2006;43(1):1–5. doi: 10.1097/01.qai.0000233308.82860.2f [DOI] [PubMed] [Google Scholar]
- 50.Natukunda E, Gaur AH, Kosalaraksa P, Batra J, Rakhmanina N, Porter D, et al. Safety, efficacy, and pharmacokinetics of single-tablet elvitegravir, cobicistat, emtricitabine, and tenofovir alafenamide in virologically suppressed, HIV-infected children: a single-arm, open-label trial. Lancet Child Adolesc Health. 2017;1(1):27–34. doi: 10.1016/S2352-4642(17)30009-3. [DOI] [PubMed] [Google Scholar]
- 51.Savarino A In-Silico docking of HIV-1 integrase inhibitors reveals a novel drug type acting on an enzyme/DNA reaction intermediate. Retrovirology. 2007;4:21. doi: 10.1186/1742-4690-4-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Margot NA, Hluhanich RM, Jones GS, Andreatta KN, Tsiang M, McColl DJ, et al. In vitro resistance selections using elvitegravir, raltegravir, and two metabolites of elvitegravir M1 and M4. Antiviral Res. 2012;93(2):288–96. doi: 10.1016/j.antiviral.2011.12.008. [DOI] [PubMed] [Google Scholar]
- 53.Kobayashi M, Nakahara K, Seki T, Miki S, Kawauchi S, Suyama A, et al. Selection of diverse and clinically relevant integrase inhibitor-resistant human immunodeficiency virus type 1 mutants. Antiviral Res. 2008;80(2):213–22. doi: 10.1016/j.antiviral.2008.06.012. [DOI] [PubMed] [Google Scholar]
- 54.Garrido C, Villacian J, Zahonero N, Pattery T, Garcia F, Gutierrez F, et al. Broad phenotypic cross-resistance to elvitegravir in HIV-infected patients failing on raltegravir-containing regimens. Antimicrob Agents Chemother. 2012;56(6):2873–8. doi: 10.1128/AAC.06170-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Malet I, Delelis O, Valantin MA, Montes B, Soulie C, Wirden M, et al. Mutations associated with failure of raltegravir treatment affect integrase sensitivity to the inhibitor in vitro. Antimicrob Agents Chemother. 2008;52(4):1351–8. doi: 10.1128/AAC.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Comi L, Maggiolo F. Abacavir + dolutegravir + lamivudine for the treatment of HIV. Expert Opin Pharmacother. 2016;17(15):2097–106. doi: 10.1080/14656566.2016. [DOI] [PubMed] [Google Scholar]
- 57.Hare S, Smith SJ, Metifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, et al. Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572). Mol Pharmacol. 2011;80(4):565–72. doi: 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kobayashi M, Yoshinaga T, Seki T, Wakasa-Morimoto C, Brown KW, Ferris R, et al. In Vitro antiretroviral properties of S/GSK1349572, a next-generation HIV integrase inhibitor. Antimicrob Agents Chemother. 2011;55(2):813–21. doi: 10.1128/AAC.01209-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Stellbrink HJ, Reynes J, Lazzarin A, Voronin E, Pulido F, Felizarta F, et al. Dolutegravir in antiretroviral-naive adults with HIV-1: 96-week results from a randomized dose-ranging study. Aids. 2013;27(11):1771–8. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Grant PM, Tierney C, Budhathoki C, Daar ES, Sax PE, Collier AC, et al. Early virologic response to abacavir/lamivudine and tenofovir/emtricitabine during ACTG A5202. HIV Clin Trials. 2013;14(6):284–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sax PE, Tierney C, Collier AC, Fischl MA, Mollan K, Peeples L, et al. Abacavir-lamivudine versus tenofovir-emtricitabine for initial HIV-1 therapy. N Engl J Med. 2009;361(23):2230–40. doi: 10.1056/NEJMoa0906768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Castagna A, Maggiolo F, Penco G, Wright D, Mills A, Grossberg R, et al. Dolutegravir in antiretroviral-experienced patients with raltegravir- and/or elvitegravir-resistant HIV-1: 24-week results of the phase III VIKING-3 study. J Infect Dis. 2014;210(3):354–62. doi: 10.1093/infdis/jiu051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Molina JM, Clotet B, van Lunzen J, Lazzarin A, Cavassini M, Henry K, et al. Once-daily dolutegravir versus darunavir plus ritonavir for treatment-naive adults with HIV-1 infection (FLAMINGO): 96 week results from a randomised, open-label, phase 3b study. Lancet HIV. 2015;2(4):e127–36. doi: 10.1016/S2352-3018(15)00027-2. [DOI] [PubMed] [Google Scholar]
- 64.Raffi F, Jaeger H, Quiros-Roldan E, Albrecht H, Belonosova E, Gatell JM, et al. Once-daily dolutegravir versus twice-daily raltegravir in antiretroviral-naive adults with HIV-1 infection (SPRING-2 study): 96 week results from a randomised, double-blind, non-inferiority trial. Lancet Infect Dis. 2013;13(11):927–35. doi: 10.1016/S1473-3099(13)70257-3. [DOI] [PubMed] [Google Scholar]
- 65.Elliot E, Amara A, Jackson A, Moyle G, Else L, Khoo S, et al. Dolutegravir and elvitegravir plasma concentrations following cessation of drug intake. J Antimicrob Chemother. 2016;71(4):1031–6. doi: 10.1093/jac/dkv425. [DOI] [PubMed] [Google Scholar]
- 66.Shah BM, Schafer JJ, Desimone JA Jr. Dolutegravir: a new integrase strand transfer inhibitor for the treatment of HIV. Pharmacotherapy. 2014;34(5):506–20. doi: 10.1002/phar.1386. [DOI] [PubMed] [Google Scholar]
- 67.Cahn P, Pozniak AL, Mingrone H, Shuldyakov A, Brites C, Andrade-Villanueva JF, et al. Dolutegravir versus raltegravir in antiretroviral-experienced, integrase-inhibitor-naive adults with HIV: week 48 results from the randomised, double-blind, non-inferiority SAILING study. Lancet. 2013;382(9893):700–8. doi: 10.1016/S0140-6736(13)61221-0. [DOI] [PubMed] [Google Scholar]
- 68.Pham HT, Labrie L, Wijting IEA, Hassounah S, Lok KY, Portna I, et al. The S230R Integrase Substitution Associated With Virus Load Rebound During Dolutegravir Monotherapy Confers Low-Level Resistance to Integrase Strand-Transfer Inhibitors. J Infect Dis. 2018;218(5):698–706. doi: 10.1093/infdis/jiy175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wijting I, Rokx C, Boucher C, van Kampen J, Pas S, de Vries-Sluijs T, et al. Dolutegravir as maintenance monotherapy for HIV (DOMONO): a phase 2, randomised non-inferiority trial. Lancet HIV. 2017;4(12):e547–e54. doi: 10.1016/S2352-3018(17)30152-2. [DOI] [PubMed] [Google Scholar]
- 70.Wijting IEA, Lungu C, Rijnders BJA, van der Ende ME, Pham HT, Mesplede T, et al. HIV-1 Resistance Dynamics in Patients With Virologic Failure to Dolutegravir Maintenance Monotherapy. J Infect Dis. 2018;218(5):688–97. doi: 10.1093/infdis/jiy176. [DOI] [PubMed] [Google Scholar]
- 71.Malet I, Subra F, Charpentier C, Collin G, Descamps D, Calvez V, et al. Mutations Located outside the Integrase Gene Can Confer Resistance to HIV-1 Integrase Strand Transfer Inhibitors. MBio. 2017;8(5). doi: 10.1128/mBio.00922-17.••. This report is an alarming signal showing that a mutation that occurred outside the integrase gene can also contribute to the resistance to INSTIs.
- 72.Van Duyne R, Kuo LS, Pham P, Fujii K, Freed EO. Mutations in the HIV-1 envelope glycoprotein can broadly rescue blocks at multiple steps in the virus replication cycle. Proc Natl Acad Sci U S A. 2019;116(18):9040–9. doi: 10.1073/pnas.1820333116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Zash R, Makhema J, Shapiro RL. Neural-Tube Defects with Dolutegravir Treatment from the Time of Conception. N Engl J Med. 2018;379(10):979–81. doi: 10.1056/NEJMc1807653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Sax PE, DeJesus E, Crofoot G, Ward D, Benson P, Dretler R, et al. Bictegravir versus dolutegravir, each with emtricitabine and tenofovir alafenamide, for initial treatment of HIV-1 infection: a randomised, double-blind, phase 2 trial. Lancet HIV. 2017;4(4):e154–e60. doi: 10.1016/S2352-3018(17)30016-4. [DOI] [PubMed] [Google Scholar]
- 75.Hassounah SA, Alikhani A, Oliveira M, Bharaj S, Ibanescu RI, Osman N, et al. Antiviral Activity of Bictegravir and Cabotegravir against Integrase Inhibitor-Resistant SIVmac239 and HIV-1. Antimicrob Agents Chemother. 2017;61(12). doi: 10.1128/AAC.01695-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Smith SJ, Zhao XZ, Burke TR Jr., Hughes SH. Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants. Retrovirology. 2018;15(1):37. doi: 10.1186/s12977-018-0420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tsiang M, Jones GS, Goldsmith J, Mulato A, Hansen D, Kan E, et al. Antiviral Activity of Bictegravir (GS-9883), a Novel Potent HIV-1 Integrase Strand Transfer Inhibitor with an Improved Resistance Profile. Antimicrob Agents Chemother. 2016;60(12):7086–97. doi: 10.1128/AAC.01474-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Anstett K, Brenner B, Mesplede T, Wainberg MA. HIV drug resistance against strand transfer integrase inhibitors. Retrovirology. 2017;14(1):36. doi: 10.1186/s12977-017-0360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zhou T, Su H, Dash P, Lin Z, Dyavar Shetty BL, Kocher T, et al. Creation of a nanoformulated cabotegravir prodrug with improved antiretroviral profiles. Biomaterials. 2018;151:53–65. doi: 10.1016/j.biomaterials.2017.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Markowitz M, Frank I, Grant RM, Mayer KH, Elion R, Goldstein D, et al. Safety and tolerability of long-acting cabotegravir injections in HIV-uninfected men (ECLAIR): a multicentre, double-blind, randomised, placebo-controlled, phase 2a trial. Lancet HIV. 2017;4(8):e331–e40. doi: 10.1016/S2352-3018(17)30068-1. [DOI] [PubMed] [Google Scholar]
- 81.Yoshinaga T, Kobayashi M, Seki T, Miki S, Wakasa-Morimoto C, Suyama-Kagitani A, et al. Antiviral characteristics of GSK1265744, an HIV integrase inhibitor dosed orally or by long-acting injection. Antimicrob Agents Chemother. 2015;59(1):397–406. doi: 10.1128/AAC.03909-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rusconi S, Marcotullio S, Cingolani A. Long-acting agents for HIV infection: biological aspects, role in treatment and prevention, and patient’s perspective. New Microbiol. 2017;40(2):75–9. [PubMed] [Google Scholar]
- 83.Margolis DA, Brinson CC, Smith GHR, de Vente J, Hagins DP, Eron JJ, et al. Cabotegravir plus rilpivirine, once a day, after induction with cabotegravir plus nucleoside reverse transcriptase inhibitors in antiretroviral-naive adults with HIV-1 infection (LATTE): a randomised, phase 2b, dose-ranging trial. Lancet Infect Dis. 2015;15(10):1145–55. doi: 10.1016/S1473-3099(15)00152-8. [DOI] [PubMed] [Google Scholar]
- 84.Margolis DA, Gonzalez-Garcia J, Stellbrink HJ, Eron JJ, Yazdanpanah Y, Podzamczer D, et al. Long-acting intramuscular cabotegravir and rilpivirine in adults with HIV-1 infection (LATTE-2): 96-week results of a randomised, open-label, phase 2b, non-inferiority trial. Lancet. 2017;390(10101):1499–510. doi: 10.1016/S0140-6736(17)31917-7. [DOI] [PubMed] [Google Scholar]
- 85.Whitfield T, Torkington A, van Halsema C. Profile of cabotegravir and its potential in the treatment and prevention of HIV-1 infection: evidence to date. HIV AIDS (Auckl). 2016;8:157–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Radzio-Basu J, Council O, Cong ME, Ruone S, Newton A, Wei X, et al. Drug resistance emergence in macaques administered cabotegravir long-acting for pre-exposure prophylaxis during acute SHIV infection. Nat Commun. 2019;10(1):2005. doi: 10.1038/s41467-019-10047-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Korolev SP, Agapkina YY, Gottikh MB. Clinical Use of Inhibitors of HIV-1 Integration: Problems and Prospects. Acta Naturae. 2011;3(3):12–28. [PMC free article] [PubMed] [Google Scholar]
- 88.Langley DR, Samanta HK, Lin Z, Walker MA, Krystal MR, Dicker IB. The terminal (catalytic) adenosine of the HIV LTR controls the kinetics of binding and dissociation of HIV integrase strand transfer inhibitors. Biochemistry. 2008;47(51):13481–8. doi: 10.1021/bi801372d. [DOI] [PubMed] [Google Scholar]
- 89.Smith SJ, Zhao XZ, Burke TR Jr., Hughes SH. HIV-1 Integrase Inhibitors That Are Broadly Effective against Drug-Resistant Mutants. Antimicrob Agents Chemother. 2018;62(9). doi: 10.1128/AAC.01035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Dicker IB, Samanta HK, Li Z, Hong Y, Tian Y, Banville J, et al. Changes to the HIV long terminal repeat and to HIV integrase differentially impact HIV integrase assembly, activity, and the binding of strand transfer inhibitors. J Biol Chem. 2007;282(43):31186–96. doi: 10.1074/jbc.M704935200 [DOI] [PubMed] [Google Scholar]
- 91.Naidu BN, Walker MA, Sorenson ME, Ueda Y, Matiskella JD, Connolly TP, et al. The discovery and preclinical evaluation of BMS-707035, a potent HIV-1 integrase strand transfer inhibitor. Bioorg Med Chem Lett. 2018;28(12):2124–30. doi: 10.1016/j.bmcl.2018.05.027. [DOI] [PubMed] [Google Scholar]
- 92.Hoesley CJ, Chen BA, Anderson PL, Dezzutti CS, Strizki J, Sprinkle C, et al. Phase 1 Safety and Pharmacokinetics Study of MK-2048/Vicriviroc (MK-4176)/MK-2048A Intravaginal Rings. Clin Infect Dis. 2019;68(7):1136–43. doi: 10.1093/cid/ciy653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P. Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance. Proc Natl Acad Sci U S A. 2010;107(46):20057–62. doi: 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Scopelliti F, Pollicita M, Ceccherini-Silberstein F, Di Santo F, Surdo M, Aquaro S, et al. Comparative antiviral activity of integrase inhibitors in human monocyte-derived macrophages and lymphocytes. Antiviral Res. 2011;92(2):255–61. doi: 10.1016/j.antiviral.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 95.Bar-Magen T, Sloan RD, Donahue DA, Kuhl BD, Zabeida A, Xu H, et al. Identification of novel mutations responsible for resistance to MK-2048, a second-generation HIV-1 integrase inhibitor. J Virol. 2010;84(18):9210–6. doi: 10.1128/JVI.01164-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Van Wesenbeeck L, Rondelez E, Feyaerts M, Verheyen A, Van der Borght K, Smits V, et al. Cross-resistance profile determination of two second-generation HIV-1 integrase inhibitors using a panel of recombinant viruses derived from raltegravir-treated clinical isolates. Antimicrob Agents Chemother. 2011;55(1):321–5. doi: 10.1128/AAC.01733-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, et al. Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1. ACS Med Chem Lett. 2014;5(4):422–7. doi: 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Fenwick C, Amad M, Bailey MD, Bethell R, Bos M, Bonneau P, et al. Preclinical profile of BI 224436, a novel HIV-1 non-catalytic-site integrase inhibitor. Antimicrob Agents Chemother. 2014;58(6):3233–44. doi: 10.1128/AAC.02719-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Kaushik A, Jayant RD, Nair M. Advancements in nano-enabled therapeutics for neuroHIV management. Int J Nanomedicine. 2016;11:4317–25. doi: 10.2147/IJN.S109943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Li W, Gorantla S, Gendelman HE, Poluektova LY. Systemic HIV-1 infection produces a unique glial footprint in humanized mouse brains. Dis Model Mech. 2017;10(12):1489–502. doi: 10.1242/dmm.031773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Montenegro-Burke JR, Woldstad CJ, Fang M, Bade AN, McMillan J, Edagwa B, et al. Nanoformulated Antiretroviral Therapy Attenuates Brain Metabolic Oxidative Stress. Mol Neurobiol. 2019;56(4):2896–907. doi: 10.1007/s12035-018-1273-8.•. Interesting discovery that highlights the critical importance of formulation design by convincingly demonstrating the metabolic stress in rodent brain upon administration of nano-DTG as compared to the native drug treatment.
- 102.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13(9):655–72. doi: 10.1038/nrd4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Kevadiya BD, Woldstad C, Ottemann BM, Dash P, Sajja BR, Lamberty B, et al. Multimodal Theranostic Nanoformulations Permit Magnetic Resonance Bioimaging of Antiretroviral Drug Particle Tissue-Cell Biodistribution. Theranostics. 2018;8(1):256–76. doi: 10.7150/thno.22764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Roy U, Drozd V, Durygin A, Rodriguez J, Barber P, Atluri V, et al. Characterization of Nanodiamond-based anti-HIV drug Delivery to the Brain. Sci Rep. 2018;8(1):1603. doi: 10.1038/s41598-017-16703-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.McMillan J, Szlachetka A, Slack L, Sillman B, Lamberty B, Morsey B, et al. Pharmacokinetics of a Long-Acting Nanoformulated Dolutegravir Prodrug in Rhesus Macaques. Antimicrob Agents Chemother. 2018;62(1). doi: 10.1128/AAC.01316-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dash PK, Kaminski R, Bella R, Su H, Mathews S, Ahooyi TM, et al. Sequential LASER ART and CRISPR Treatments Eliminate HIV-1 in a Subset of Infected Humanized Mice. Nat Commun. 2019;10(1):2753. doi: 10.1038/s41467-019-10366-y.••. Excellent first ever report that demonstrates elimination of latent HIV-1 proviral reservoirs from humanized mice.
- 107.McMillan J, Szlachetka A, Zhou T, Morsey B, Lamberty B, Callen S, et al. Pharmacokinetic testing of a first-generation cabotegravir prodrug in rhesus macaques. Aids. 2019;33(3):585–8. doi: 10.1097/QAD.0000000000002032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sillman B, Bade AN, Dash PK, Bhargavan B, Kocher T, Mathews S, et al. Creation of a long-acting nanoformulated dolutegravir. Nat Commun. 2018;9(1):443. doi: 10.1038/s41467-018-02885-x.••. Excellent first report of nano-formulated DTG with hydrophobic and lipophilic modifications in DTG prodrug.
- 109.Tang J, Vernekar SKV, Chen YL, Miller L, Huber AD, Myshakina N, et al. Synthesis, biological evaluation and molecular modeling of 2-Hydroxyisoquinoline-1,3-dione analogues as inhibitors of HIV reverse transcriptase associated ribonuclease H and polymerase. Eur J Med Chem. 2017;133:85–96. doi: 10.1016/j.ejmech.2017.03.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang Z, Bennett EM, Wilson DJ, Salomon C, Vince R. Rationally designed dual inhibitors of HIV reverse transcriptase and integrase. J Med Chem. 2007;50(15):3416–9. doi. 10.1021/jm070512p [DOI] [PubMed] [Google Scholar]
- 111.Rogolino D, Carcelli M, Compari C, De Luca L, Ferro S, Fisicaro E, et al. Diketoacid chelating ligands as dual inhibitors of HIV-1 integration process. Eur J Med Chem. 2014;78:425–30. doi: 10.1016/j.ejmech.2014.03.070 [DOI] [PubMed] [Google Scholar]
- 112.Singh K, Sarafianos SG, Sonnerborg A. Long-Acting Anti-HIV Drugs Targeting HIV-1 Reverse Transcriptase and Integrase. Pharmaceuticals (Basel). 2019;12(2). doi: 10.3390/ph12020062.••. Excellent detailed information provided on long acting forms of RT and IN inhibitors that are at various stages of clinical trials.