

Abstract
Background -
Obesity and diets high in saturated fat increase the risk of arrhythmias and sudden cardiac death. However, the molecular mechanisms are not well understood. We hypothesized that an increase in dietary saturated fat could lead to abnormalities of calcium homeostasis and heart rhythm by a NADPH oxidase 2 (NOX2)-dependent mechanism.
Methods -
We investigated this hypothesis by feeding mice high fat diets. In vivo heart rhythm telemetry, optical mapping, and isolated cardiac myocyte imaging was used to quantify arrhythmias, repolarization, calcium transients, and intracellular calcium sparks.
Results -
We found that saturated fat activates NOX, whereas polyunsaturated fat does not. The high saturated fat diet increased repolarization heterogeneity and ventricular tachycardia (VT) inducibility in perfused hearts. Pharmacologic inhibition or genetic deletion of NOX2 prevented arrhythmogenic abnormalities in vivo during high statured fat diet and resulted in less inducible VT. High saturated fat diet activates Ca2+/calmodulin-dependent protein kinase (CaMK) in the heart, which contributes to abnormal calcium handling, promoting arrhythmia.
Conclusions -
We conclude that NOX2 deletion or pharmacologic inhibition prevents the arrhythmogenic effects of a high saturated fat diet, in part mediated by activation of CaMK. This work reveals a molecular mechanism linking cardiac metabolism to arrhythmia, and suggests that NOX2 inhibitors could be a novel therapy for heart rhythm abnormalities caused by cardiac lipid overload.
Keywords: NAD(P)H oxidase, repolarization, fatty acid, reactive oxygen species, arrhythmia
Journal Subject Terms: Arrhythmias, Lipids and Cholesterol, Oxidant Stress
Graphical Abstract
Introduction
Many epidemiologic studies have shown that obese patients have approximately twice the risk of sudden cardiac death (SCD) than age matched controls1–4; SCD is often caused by ventricular arrhythmias. The increased risk of SCD is greater than the increased risk of myocardial infarction, suggesting that arrhythmic events are increased more than coronary events in obese humans. Excessive lipid accumulation is found in cardiomyocytes from obese and diabetic patients, and is believed to contribute to heart failure and arrhythmia5–8. Epidemiology studies also show that higher saturated fat intake leads to an increased risk of sudden cardiac death9–12, suggesting that the effects of dietary saturated fat may be sufficient to cause abnormal heart rhythm, without obesity.
The most common electrophysiologic abnormalities found in obese patients are increased frequency of ventricular ectopy and prolongation of the QT interval13–15. We have previously shown that wild type (WT) mice with high-fat diet induced obesity (DIO) have long QT and increased ventricular ectopy16, mimicking the abnormalities found in obese humans. Oxidative stress is a plausible mechanistic link between lipid metabolism and cardiovascular pathology17–19. Mild, transient increases in cardiac reactive oxygen species (ROS) may be involved in adaptive processes, but it is postulated that long-term increases in cardiac ROS are detrimental20. There are several sources of ROS in cardiomyocytes, including NADPH oxidase (NOX), nitric oxide synthase (NOS), and byproducts of mitochondrial metabolism. We hypothesized that increasing saturated fatty acid in the diet could increase oxidative stress in cardiomyocytes by activating NOX2, resulting in abnormalities of calcium homeostasis and heart rhythm, before the onset of obesity.
Materials and Methods
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Materials
The OxyBlot Protein Oxidation Detection Kit and apocynin were purchased from Sigma. The anti-PKC antibodies were purchased from Cell Signaling (PKC alpha) and Santa Cruz (PKC beta and delta). Ion channel antibodies were purchased from Alomone. The anti-CaMK antibodies were purchased from Fisher Scientific. The anti-PKA antibodies were purchased from Cell Signaling. RyR2 blots were performed as previously described21.
Animal care and cardiomyocyte isolation
Animal protocols were approved by the Columbia University Institutional Animal Care and Use Committee and were carried out in accordance with the NIH guidelines for the care and use of laboratory animals. Wild type (WT) and B6.129S-Cybbtm1Din/J (NOX2KO) mice were purchased from Jackson labs and were used for experiments at 12–16 weeks of age. High fat diets were special orders from Research Diets Inc: D04051707, 60 kcal% from palm oil, 20% kcal protein, 20% kcal carbohydrate; and D01112603, 60 kcal% from olive oil, 20% kcal protein, 20% kcal carbohydrate. Control chow was Picolab Rodent diet 20 from LabDiet, which has 13% kcal from fat, 25% protein, and 62% carbohydrate. Animals were fed ad libitum. Isolation of cardiomyocytes was performed as previously described6. Animals were randomly assigned to treatment groups. No animals were excluded from the analysis. Echocardiograms, optical mapping, and histology quantification were performed by blinded operators.
Echocardiograms and blood glucose measurements
Echocardiograms were performed on mice using a high-resolution imaging system with a 30-MHz imaging transducer (Vevo 770; VisualSonics). Isoflurane anesthesia was used. The operator was blinded to group assignment. Whole blood glucose was measure at the time of sacrifice with one-touch ultra-glucometer (Johnson&Johnson).
Histology
Heart tissue was fixed with 4% paraformaldehyde, embedded in paraffin wax, and then sectioned. Sections were incubated with trichrome stain using standard methods by the Columbia University Pathology core facility. Fibrosis was quantified by measuring mean blue pixels in multiple slices using Adobe Photoshop 11.0.2.
NOX activity measurements
NADPH-dependent superoxide production was measured in cellular homogenates using lucigenin-enhanced chemiluminescence as previously described22. Cells were homogenized in Krebs buffer, pH 7.4 (130 mM NaCl, 5 mM KCl, 2 mM MgCl2, 1.5 mM CaCl2, 5 mM glucose, 35 mM phosphoric acid, and 20 mM HEPES). Homogenates were centrifuged at 1000 × g at 4°C for 10 min to remove the unbroken cells and debris. The pellet was resuspended in Krebs buffer containing 0.5 mM lucigenin followed by the addition of 0.1 mM NADPH as the substrate. Protein content was measured using the Bradford protein assay reagent. The samples were transferred to a 96-well plate at 50 μg of protein per well. Photon emission in terms of relative light units was measured in a Tecan 200 microplate reader after 5 min incubation in the dark. There was no measurable activity in the absence of NADPH. Superoxide anion production was expressed as relative lucigenin luminescence.
Heart rhythm telemetry
Telemetry devices (Data Sciences International, model EA-F10) were implanted in mice under sterile conditions with inhaled isoflurane for anesthesia. The two subcutaneous leads were positioned to approximate ECG limb lead II. The mice recovered for one week after surgery before initiating recordings. ECG intervals were measured manually using Ponemah 3 software from recordings with minimal artifact at approximately the same time of day. Intervals were averaged from 4 consecutive beats and QTc was calculated by the formula QTc=QT/(RR/100)23. PVC and arrhythmia counts were tallied manually by a blinded reader.
Optical mapping protocol
The operator was blinded to group assignment. Mice were injected with heparin prior to administration of isoflurane. Hearts were isolated and perfused via a Langendorff apparatus with warm oxygenated Krebs-Henseleit buffer (pH 7.4; 95% O2, 5% CO2, 36–38°C), and were placed in a glass chamber in a Tyrode bath for superfusion. One AgCl wire was attached to the metal aortic cannula, and another AgCl wire was positioned near the surface of the heart to record an ECG. Blebbistatin (5–10 μM) was perfused to reduce motion, and Di-4-ANEPPS (100 μM) was perfused to record optical membrane potentials. Hearts were uniformly shone with green excitation lasers (532 nm) to activate Di-4-ANEPPS. Emitted fluorescence was captured through a 715-nm pass filter using a complementary metal-oxide-semiconductor (CMOS) camera (MICAM Ultima, SciMedia). Movies were acquired at 1000 frames per second for a duration of 4–5 sec, with 100 × 100-pixel resolution (0.095 mm per pixel). Optical movies were acquired at different pacing cycle lengths. Susceptibility to pacing-induced VF/VT was assessed by 3 attempts of burst pacing (20 Hz, 10 s) at twice the excitation threshold of the left ventricle (Pulsar 6i, FHM, Brunswick, ME).
Optical mapping data processing and analysis
Recorded optical movies were processed using custom software based on PV-WAVE (Precision Visuals - Workstation Analysis and Visualization Environment, Visual Numerics, Inc)24. The background fluorescence was subtracted from each frame, and spatial (5 × 5 pixels) and temporal (9 frames) conical convolution filters were used to increase signal-to-noise ratio. Optical mapping movies were spatio-temporarily filtered to reduce noise. Phase movies were obtained after Hilbert transformation of the fluorescent signal and action potential duration (APD) maps were generated as previously reported25. APD dispersion (APDmax –APDmin) was calculated after drawing a custom 10×10 pixel area.
Cardiomyocyte contractility and calcium spark recording and analysis
Cardiomyocyte isolations were performed as previously described26. Isolated cardiomyocyte contractility was measured using an Ionoptix system as previously described26. Spontaneous contractions were measured for 40 seconds after a brief pacing interval. Calcium transients were measured using Fura2 (Invitrogen). SR calcium load was evoked with rapid application of caffeine (10 mM). The operator was blinded to group assignment. Sparks were recorded with a Leica SP2 confocal microscope (Wetzlar, Germany) equipped with a 63×x1.4 NA objective. Isolated cardiac myocytes experiments were performed at room temperature. Cardiomyocytes were loaded with fluo-4 (5 μM for 10 minutes) in modified Tyrode solution containing 1 mM calcium. Line scan images were recorded for 10 seconds with Leica TCS software and quantified with the Sparkmaster plugin of ImageJ.
Statistical analysis
Results are given as mean ± SEM. The unpaired t test was used for comparisons of 2 means. a 2-tailed value of P <.05 was considered statistically significant. For groups of 2 or more, analysis of variance (ANOVA) was used with post hoc testing (Prism v5, GraphPad Software Inc., La Jolla, CA).
Results
High saturated fat diet rapidly causes ventricular ectopy and long QT without obesity
To determine the effects of high-fat diet on cardiac function, we fed adult WT C57Bl6 mice regular chow or one of two high fat diets: 60 kcal% from palm oil (42% saturated fat, high saturated fat diet = HSFD); and 60 kcal% from olive oil (14% saturated fat, high unsaturated fat diet = HUFD). Palm oil is commonly used as a cooking oil in many parts of the world and is composed of approximately 44% palmitate (C16, a saturated fat) and 37% oleate (C18:1, a monounsaturated fat). The olive oil diet was used as a control because olive oil has generally been found to have neutral or benign effects on cardiovascular health in humans. The 4-week duration of high-fat diet did not result in obesity (mean increase in weight 3.2–3.8 g for all groups, supplemental table 1), nor did we observe significant differences in serum glucose (supplemental Fig1A).
Serum free-fatty acids (FFA) were significantly increased after 4 weeks of the HSFD (Fig1A). Serum lipidomic analysis showed that mice on the HSFD had 2.6 times as much palmitate in the bloodstream as regular chow controls, which was statistically significant (Fig 1A). The HSFD also caused several significant changes in the ventricular tissue lipid content compared to regular chow. Lipidomic analysis of ventricular tissue samples showed that both palmitate and oleate levels were significantly increased, consistent with the composition of palm oil (supplemental Fig1B).
Figure 1. High saturated fat diet causes activation of NOX and ventricular ectopy.
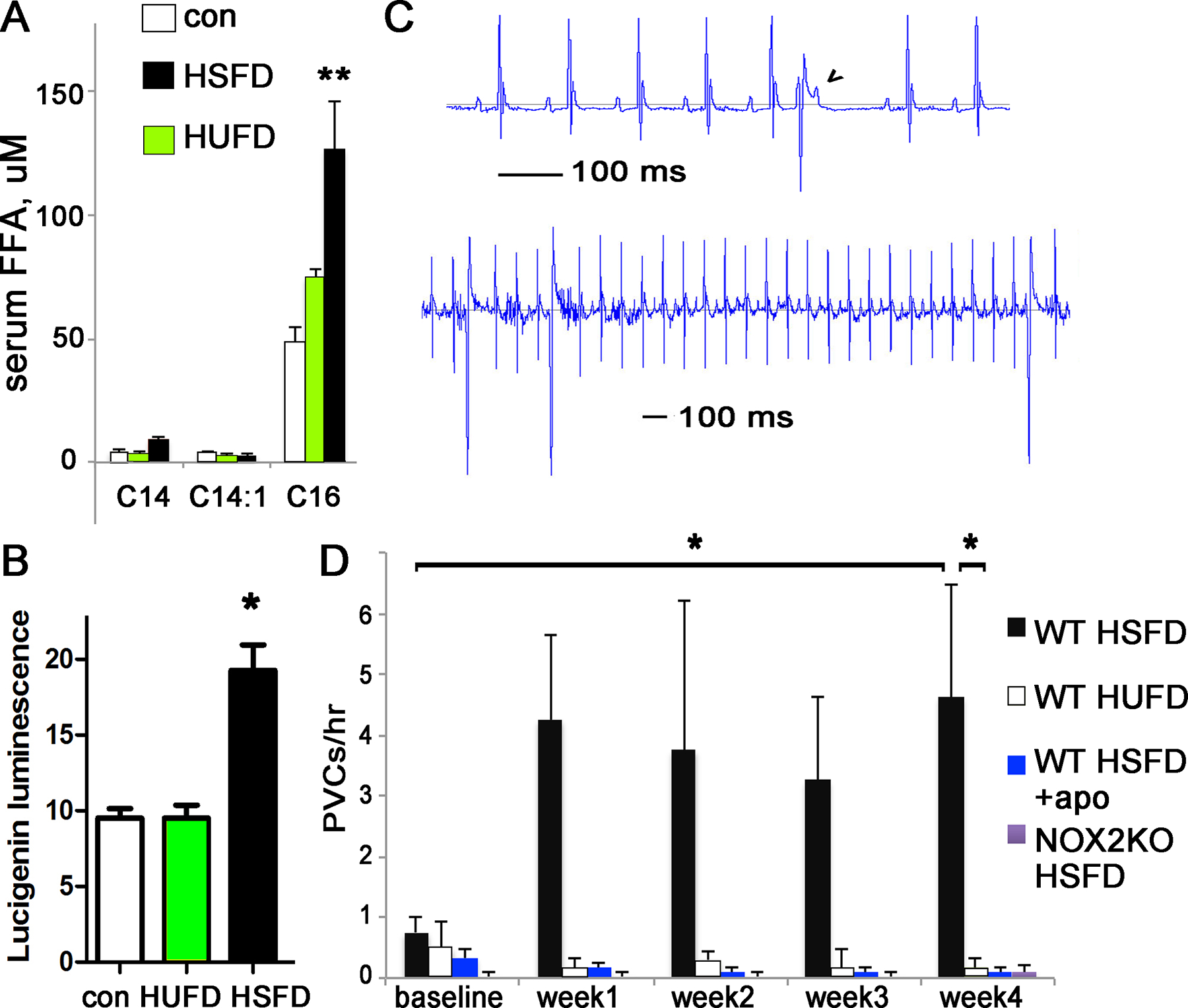
A. Graph of serum palmitate after 4 weeks of diet. B. NOX2 activity is increased in ventricular lysates from mice on high fat diets. C. Example of PVCs in a WT mouse fed PA diet; note the p-waves continue at the same rate despite the PVC (wide early QRS). Lower section shows frequent monomorphic PVCs in a mouse fed PA diet; note the time scale is compressed compared to upper section. D. Graph of PVCs/hour in unrestrained mice, before and during palm oil or olive oil diet.
For all panels n=4 animals for each group, error bars are SEM, * indicates p<0.05, ** indicates p<0.01
We have previously demonstrated increased ventricular ectopy and long QT in obese mice16. To determine if the HSFD was sufficient to cause similar heart rhythm abnormalities without obesity, we recorded heart rhythm in WT mice before and after the HSFD or HUFD. Mice were implanted with heart rhythm telemetry monitors and allowed to recover for one week. Following baseline heart rhythm recordings on regular chow, mice were fed the HSFD or HUFD diets. Telemetry recordings showed that mice developed a statistically significant increase in QT on the HSFD diet (Table 1). The mice also had a significant increase in ventricular ectopy in the form of premature ventricular complexes (PVCs) (Fig1D). No atrial arrhythmias were detected during this time. In contrast, mice fed the HUFD diet did not show increased PVCs or QT duration (Fig1D). These findings indicate that a HSFD can cause the same arrhythmogenic abnormalities as diet-induced obesity without substantial weight gain, and that a high fat diet does not cause cardiac abnormalities if it is composed of olive oil.
Table 1. QT prolongs with HSFD diet in WT mice;
intervals are in milliseconds,
| RR | PR | QRS | QT | ||
|---|---|---|---|---|---|
| WT HUFD | basal | 104.3 ± 0.9 | 35.1 ± 0.6 | 11.3 ± 0.3 | 42.8 ± 0.4 |
| 4 wks | 105.7 ± 0.3 | 35.7 ± 1.5 | 12.2 ± 0.2 | 43.0 ± 0.6 | |
| WT HSFD | basal | 104.5 ± 1.0 | 33.8 ± 0.3 | 11.8 ± 0.3 | 43.8 ± 1.4 |
| 4 wks | 105.3 ± 1.3 | 36.3 ± 0.5* | 12.5 ± 0.3 | 49.5 ± 1.0* | |
| WT HSFD +apo | basal | 104.0 ± 1.5 | 33.0 ± 0.4 | 11.5 ± 0.3 | 45.0 ± 0.4 |
| 4 wks | 103.5 ± 0.6 | 34.0 ± 0.4 | 11.6 ± 0.4 | 43.9 ± 0.4 | |
| NOX2KO HSFD | basal | 107.0 ± 0.9 | 32.8 ± 0.2 | 12.0 ± 0.2 | 42.9 ± 0.7 |
| 4 wks | 106.4 ± 1.2 | 33.2 ± 0.8 | 11.2 ± 0.1 | 40.9 ± 1.1 |
= p<0.05 compared to baseline.
Pharmacologic NOX2 inhibition during high saturated fat diet prevents heart rhythm abnormalities and NOX2KO mice are protected from arrhythmogenic changes of saturated fat
To determine if the high-fat diets increased oxidative stress, we measured NOX activity in ventricular tissue and found that it was increased by the HSFD, but not by the HUFD (Fig1B). NOX2 protein levels in the heart were not changed significantly by diet (supplemental Fig1C). Total cellular oxidative stress, as measured by protein carbonyl groups in ventricular lysates, showed no significant difference between HSFD and regular chow (supplemental Fig1D). This suggests that the oxidative stress caused by NOX activation does not cause an increase in total protein oxidation. Since the HUFD did not cause abnormalities, we focused the remainder of our study on the effects of the HSFD.
To test the in vivo relevance of NOX activation, we quantified NOX activity in ventricular lysates from mice given regular chow or HSFD, with or without the NOX inhibitor apocynin in the drinking water. As hypothesized, NOX activity is increased in ventricular lysates from mice on high fat saturated fat diet and apocynin prevents the increase in NOX activity (supplemental Fig2). After telemetry surgery, we treated another group of WT mice with apocynin (5mM in drinking water) to block NOX2 activity. Drug treatment was started one day before switching mice to the HSFD and we recorded heart rhythm for four weeks. Apocynin prevented the increase in ventricular ectopy caused by HSFD; PVC frequency was unchanged from baseline recordings on regular chow for these mice (less than 0.3 PVC/hour for all mice at all time points) (Fig1D). The QT interval is also preserved by apocynin (Table 1). This supports NOX2 activation as a critical factor in arrhythmia caused by dietary saturated fat.
We also tested the role of NOX2 with NOX2 KO mice. ECG parameters at baseline, on regular chow, were the same at WT mice (Table 1). With HSFD, however, the NOX2KO mice did not exhibit PVCs or long QT on the high saturated fat diet (Table 1 and Fig1D), demonstrating that deletion of NOX2 is protective in vivo.
High saturated fat diet promotes ventricular arrhythmia and repolarization dispersion in a NOX2-dependent manner
To determine if dietary saturated fat increases the substrate for arrhythmia we used a pacing protocol with perfused hearts to quantify ventricular arrhythmia susceptibility. These experiments showed an increase in ventricular tachycardia and ventricular fibrillation (VT/VF) inducibility in WT hearts after 4 weeks of HSFD (Fig2A,B,C and supplemental movie). None of the control chow hearts had inducible VT/VF, indicating that this was not an aggressive pacing protocol, whereas 5/6 HSFD hearts had inducible VT/VF. Both monomorphic VT and VF with rotor activity were induced in HSFD hearts. To further evaluate the substrate, we quantified repolarization heterogeneity with action potential dispersion in the LV epicardium. HSFD significantly increased repolarization heterogeneity. Hearts from NOX2 KO mice fed the same HSFD exhibited a small number of VT/VF episodes, and did not exhibit increased repolarization heterogeneity compared to control hearts (Fig2D,E), indicating that NOX2 is an important component of the arrhythmia mechanism.
Figure 2. VT/VF in high saturated fat diet (HSFD diet) hearts.
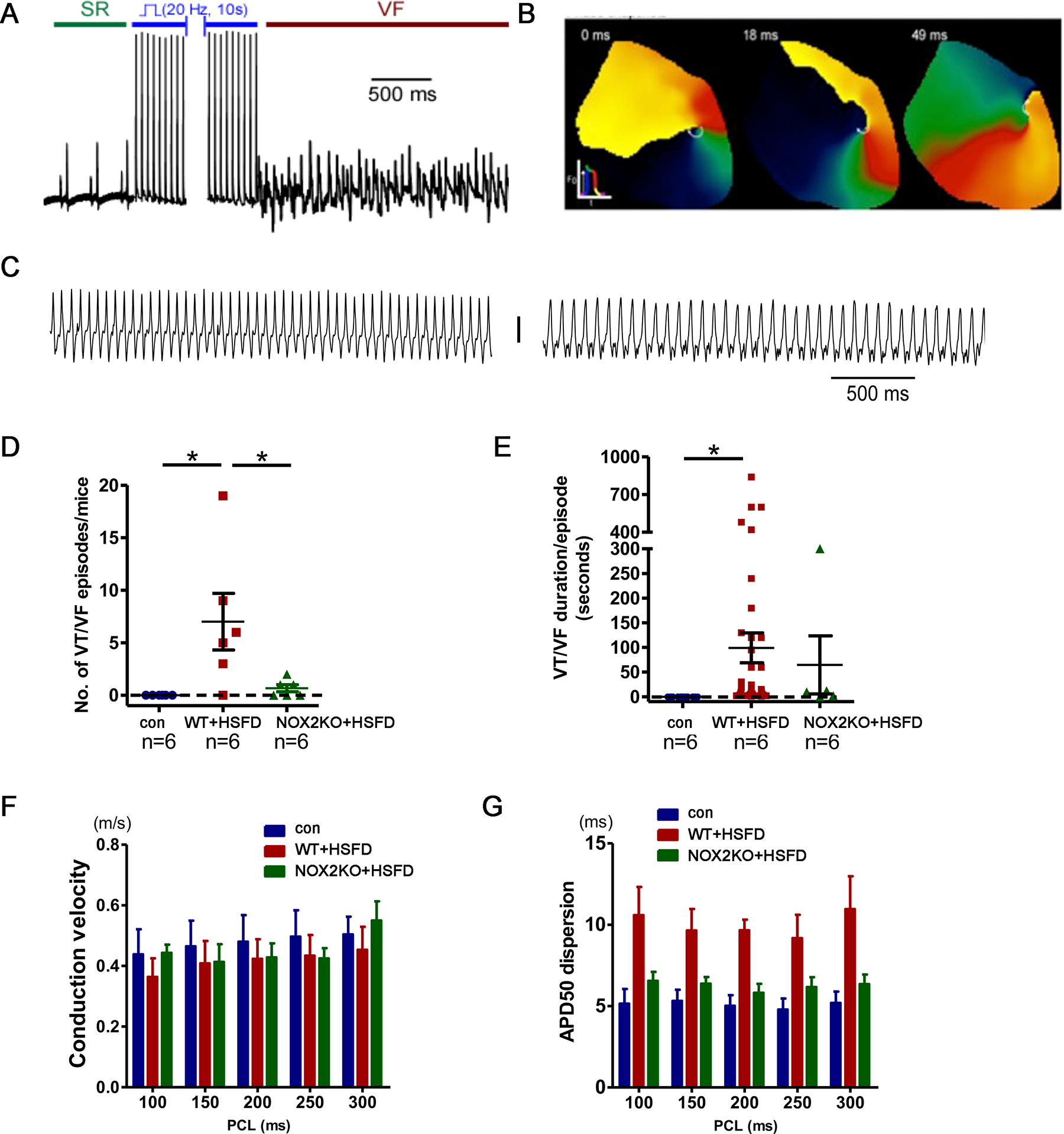
A. Example of VF in an HSFD heart after pacing. B. Optical mapping phase snapshots showing a rotor during VF. C. Examples of monomorphic VT after HSFD. D. Quantification of induced VT/VF, the means are different by ANOVA, * indicates significant difference by post-hoc testing. E. Quantification of duration of induced VT/VF, the means are different by ANOVA, * indicates significant difference by post-hoc testing. F. Conduction velocity is not significantly different between groups. G. APD dispersion is increased in WT hearts after HSFD but not NOX2KO hearts. Control and HSFD groups are significantly different by 2-way ANOVA with post-hoc testing. PCL = pacing cycle length
Four weeks of high saturated fat diet does not change cardiac histology or cause cardiac hypertrophy
The histologic appearance of left ventricular (LV) cardiac tissue was normal after 4 weeks of HSFD. Trichrome staining showed only minimal perivascular fibrosis in both control and HSFD hearts and quantification of trichrome staining indicated no significant difference in cardiac fibrosis. (Fig3A,B). Total ventricular weight and heart weight to body weight ratios were not significantly different, demonstrating that this duration of HSFD does not cause cardiac hypertrophy (Fig3C). Echocardiography showed that there was no statistically significant change in ejection fraction or end-diastolic volume, indicating that this duration of HSFD does not cause systolic heart failure (Fig3D,E and supplemental table 2). In summary, the heart is structurally and histologically normal after HSFD.
Figure 3. Four weeks of high saturated fat diet does not cause fibrosis, hypertrophy, or systolic dysfunction.
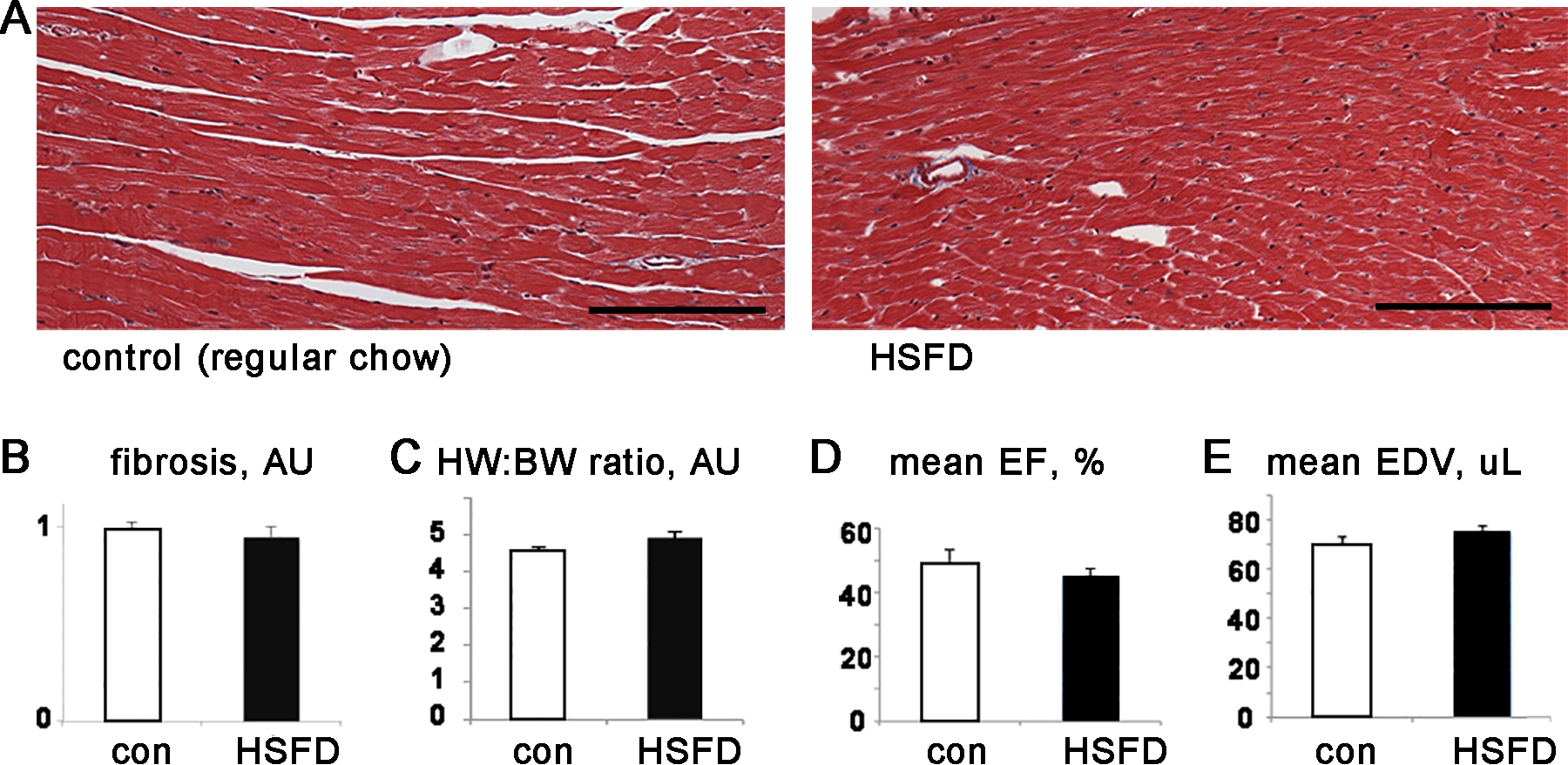
A. Representative histology, trichrome stain of LV tissue, scale bar = 100 μm. B. LV fibrosis quantification. C. Graph of heart weight to body weight ratio. D. Graph of mean ejection fraction (EF). E. Graph of LV end-diastolic volume (EDV). For B, n=4 each group, C,D,E n=7–8 each group, error bars are SEM, none of the comparisons are statistically significant
High saturated fat diet causes spontaneous contractions from abnormal sarcoplasmic reticulum calcium leak in cardiomyocytes.
To determine if high saturated fat diet could increase ectopic beats in a cell-autonomous manner, we isolated cardiac myocytes from adult mice fed the high saturated fat diet (or regular chow for controls). We recorded contractions in isolated adult mouse ventricular myocytes, selecting cardiomyocytes with normal morphology that were not spontaneously contracting at baseline. After a short period of pacing at 1Hz (20–30 seconds to achieve steady-state contraction), pacing was stopped and spontaneous contractions were counted. Cardiomyocytes from high saturated fat diet mice had significantly more contractions than cardiomyocytes from control mice (Fig4A). Spontaneous contractions can be caused by abnormal intracellular calcium handling. To examine calcium handling, we measured calcium sparks, which were significantly increased in cardiomyocytes from WT mouse hearts after HSFD. Sparks in NOX2KO cardiomyocytes were not increased after high saturated fat diet (Fig4).
Figure 4. Spontaneous contractions and increased calcium sparks in cardiomyocytes from high saturated fat diet hearts.
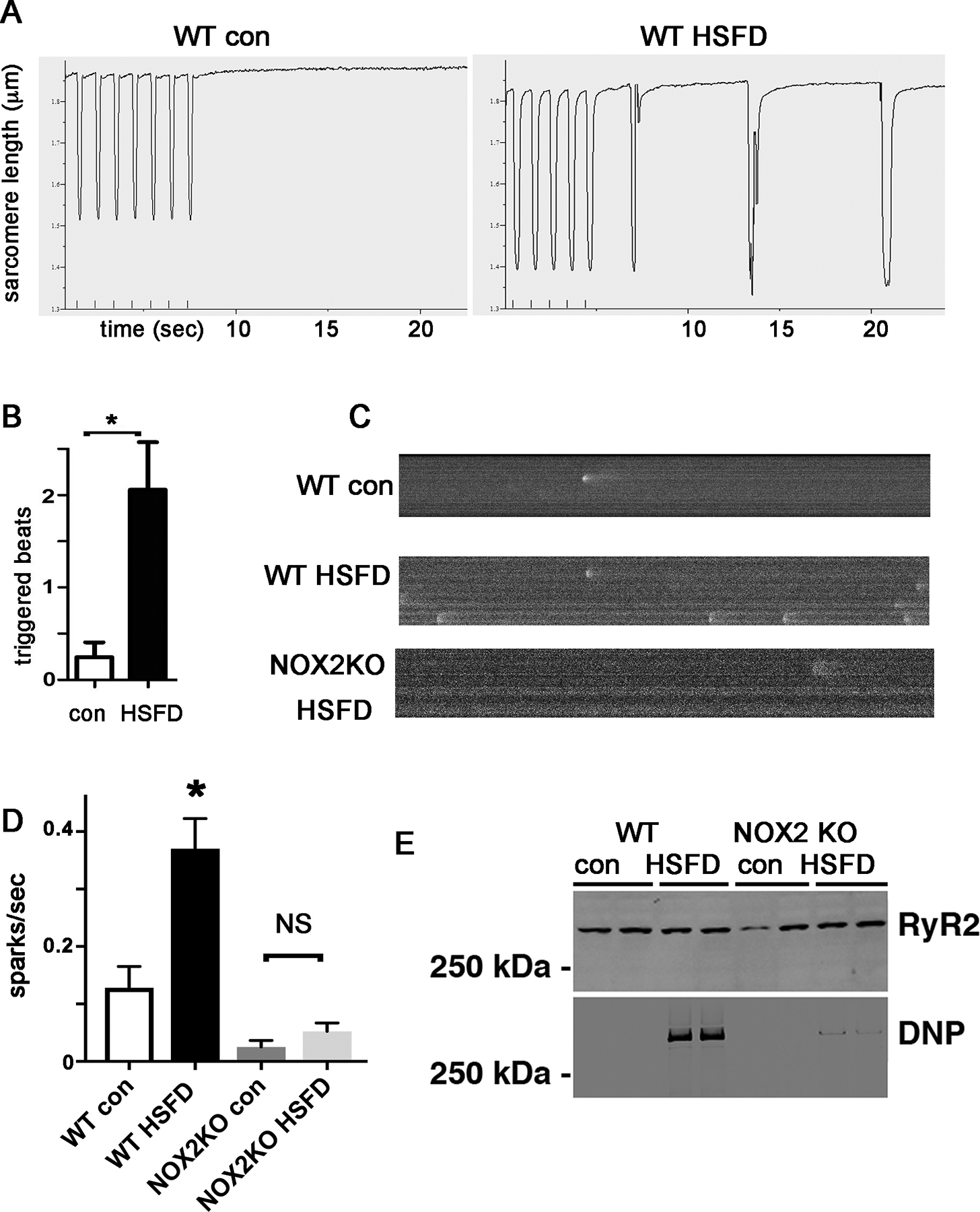
A. Raw data from isolated cardiac myocytes, pacing indicated by small vertical marks at the bottom of the rectangle, followed by cessation of pacing. Note that the high-fat diet cardiomyocyte demonstrates spontaneous contractions but the control does not. B. Graph of triggered beats from HSFD cardiomyocyte. N= 29–36 cells, from 3 hearts each group, error bars are SEM. C. Representative images of sparks from control and HSFD cardiomyocytes. D. Graph of calcium sparks/second from control and HSFD cardiomyocytes. N= 15–18 cells, from 3 hearts each group. * indicates p<0.05, error bars are SEM. E. Blots of RyR2 and oxidized RyR2 from mouse hearts. DNP = dinitrophenylhydrazine, indicating carbonyl from protein oxidation.
Since calcium leak generally involves post-translation modification of RYR2, we examined RYR2 proteins. RyR2 was oxidized in the WT hearts after HSFD (Fig4E). In contrast, NOX2KO hearts had minimal oxidation of RyR2 after HSFD, corresponding to the lack of significant increase in sparks.
To characterize cellular calcium handling, we also evaluated single-cell contractility, calcium transients, and sarcoplasmic reticulum (SR) calcium load. There were no significant differences in these measurements with the exception of a mild decrease in the return velocity of the calcium transients in the cardiomyocytes from HSFD hearts compared to chow control, which may suggest early stage diastolic dysfunction (table 2). The NOX2 cardiomyocytes did not show any significant differences compared to WT. Calcium transient amplitude and SR calcium content were similar comparing WT HSFD to WT chow and comparing WT to NOX2 KO (table 2, sup fig. 3).
Table 2.
Contractility and calcium transient measurements from of WT chow, WT HSFD, and NOX2 KO mice on chow.
| CONTRACTILITY | CALCIUM TRANSIENT | SR CALCIUM | |||||
|---|---|---|---|---|---|---|---|
| AMP | RETv | AMP | DEPv | RETv | AMP | Ratio (transient vs total) |
|
| WT CHOW | 14.5 ± 0.5 | 3.6 ± 0.2 | 45.2 ± 3.8 | 19.8 ± 2.3 | −1.8 ± 0.2 | 64.4 ± 4.2 | 0.7 ± 0.04 |
| WT HSFD | 14.2 ± 0.6 | 4.5 ± 0.3 | 39.2 ± 2.5 | 19.0 ± 2.0 | −1.4 ± 0.1* | 60.5 ± 4.8 | 0.7 ± 0.04 |
| NOX2KO CHOW | 14.6 ± 0.4 | 3.7 ± 0.2 | 39.3 ± 2.5 | 24.7 ± 1.9 | −1.7 ± 0.1 | 63.1 ± 4.4 | 0.7 ± 0.03 |
AMP= amplitude, DEPv = depolarization velocity, RETv = return velocity
N=3 hearts, n=29 cells for WT chow; N=4, n=44 for WT HSFD; N=3, n=30 for NOX2 KO CHOW (*p<0.05 vs control).
High saturated fat diet activates cardiac PKC isoforms and CaMKIIdelta
Our group and others have shown that saturated fat activates NOX2 via PKC isoforms in cardiomyocytes27, 28. We performed western blots from membrane preparations of ventricular tissue, since activated PKC translocates to the membrane, which showed that PKC beta and delta isoforms are activated in the heart after 4 weeks of HSFD (supplemental Fig3). PKC alpha was not activated. Because previous reports have shown that the kinase Src is activated in the heart by disease states associated with oxidative stress, causing arrhythmogenic remodeling17, 29, we also examined Src, but did not find significant activation (supplemental Fig4). In summary, dietary saturated fat activates PKC isoforms in the heart, which are known to activate NOX2.
CaMKIIdelta activation, caused by calcium or oxidative stress, is a well-established pathway in several forms of heart disease30, 31. Phosphorylation of CaMKII at threonine 286, which indicates activation, was significantly increased in mouse hearts after 4 weeks of high saturated fat diet (Fig5). The olive oil diet did not activate CaMKII. Oxidation of CaMKII as detected by immunoblot was not significantly increased by either high fat diet. To investigate the protective effect of NOX2 deletion in the heart, we examined CaMKII activation, which demonstrates that NOX2 deletion reduces CaMKII activation by HSFD (Fig5). Since oxidation of CaMKII was not increased, the activation of CaMK by NOX is more likely an indirect effect mediated by increased calcium leak from SR caused by oxidation of RyR2.
Figure 5. CaMKII is hyperphosphorylated in the WT heart after high saturated fat diet, but not after high unsaturated fat diet.
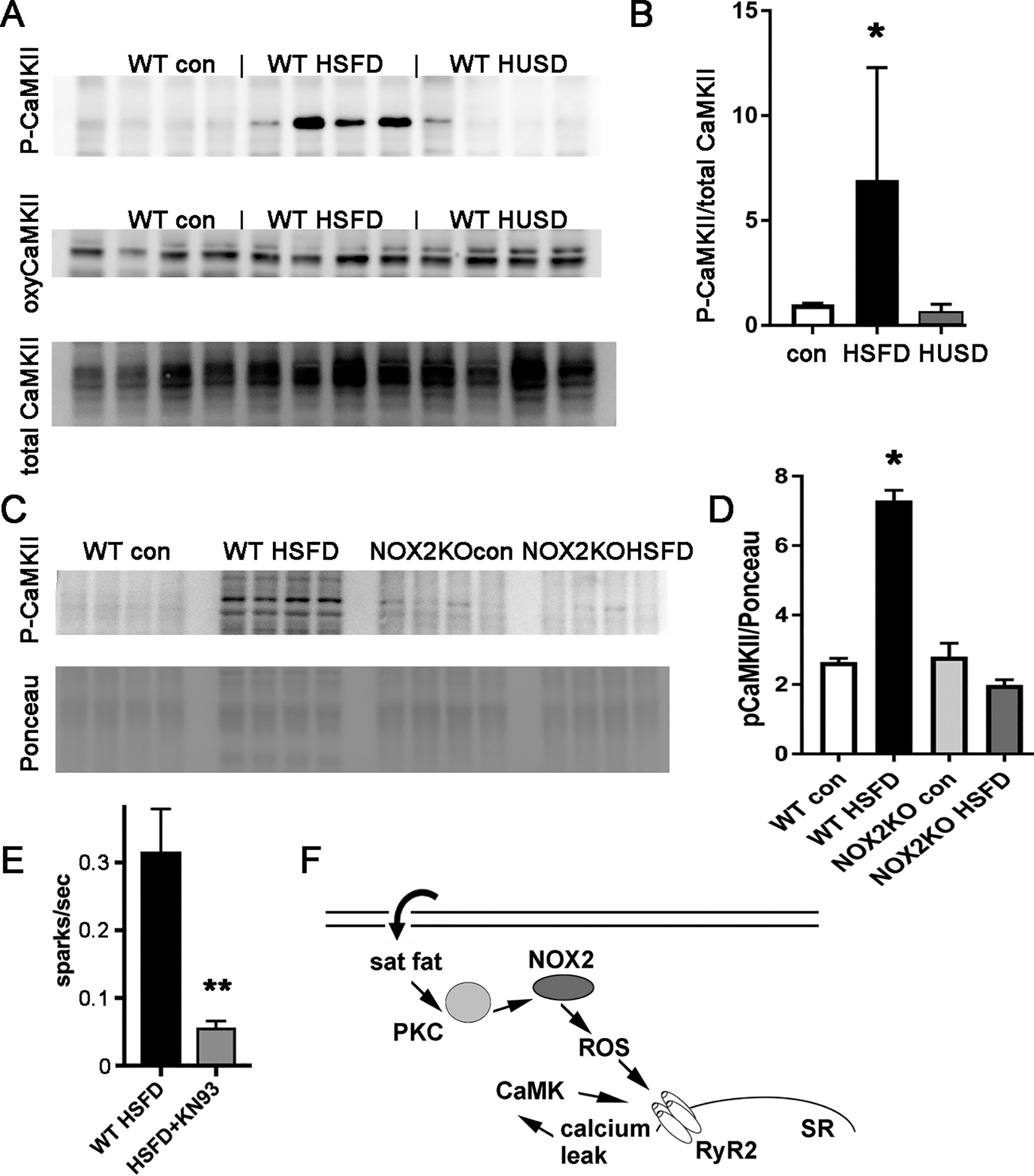
A. Representative images of immunoblots. B. Graph of immunoblots of P-CaMKII, normalized to total CaMK, from heart lysates; n=4 each group, error bars are SEM, the means are significantly different by ANOVA, * indicates significantly different from chow control by post-hoc test. C. Representative immunoblots showing that NOX2 KO hearts do not have a significant increase in P-CaMK after saturated fat diet. D. Graph of immunoblots of P-CaMKII, normalized to Ponceau, * indicates significantly different from WT control. E. Sparks quantification from WT cardiomyocytes after 4 weeks HSFD under control conditions or with the CaMK inhibitor KN93. ** indicates P <0.01 by t-test, N=31–36 cells from 3 different cardiac isolations. F. Schematic of proposed pathways: dietary saturated fat activates NOX2 via PKC. NOX2 activation increases reactive oxygen species (ROS) and causes SR calcium leak, activating CaMK, which worsens SR calcium leak. CaMK = calcium/calmodulin kinase, PKC = protein kinase C, RyR2 = ryanodine receptor 2, SR = sarcoplasmic reticulum.
To determine if CaMK activation plays a role in the abnormal calcium handling, we isolated cardiomyocytes from mice fed HSFD and treated them with the CaMK inhibitor KN93 for one hour. CaMK inhibition reduced calcium sparks in cardiomyocytes, indicating that CaMK activation does have a significant role in the calcium handling abnormalities caused by high fat diet (fig 5E).
Potential roles for PKA and NOX4
Since the PKA pathway has been shown to have interactions with CaMK and NOX232, we examined PKA activation by quantifying phosphorylation. We found a nonsignificant trend toward activation (sup fig 5A,B). Although we cannot exclude a role for PKA in this model system, it seems less likely that PKA is a major component.
NOX2 and NOX4 are both present in the rodent heart. We found that there is a small, nonsignificant increase in NOX4 in the hearts of WT mice after HSFD (sup fig 5C,D). We also found that NOX2 KO mice have a significant increase in NOX4 at baseline, which does not increase after HSFD (sup fig 5E,F). Since the NOX2 KO mice have more NOX4 protein, and these mice are protected, it seems unlikely that the increase in NOX4 is pathologic in this context but there could be some contribution to oxidative stress.
Discussion
Overall, these results support an arrhythmogenic effect of a high saturated fat diet, prior to the onset of obesity, due to NOX2 activation. Our findings demonstrate that pharmacologic NOX2 inhibition is anti-arrhythmic. Mechanistically, we show that dietary saturated fat changes the lipidomic profile of cardiac muscle, activating PKC isoforms in the heart. It is likely that PKC activation is upstream of NOX activation, based on prior work27.
Our study shows the saturated fat is sufficient to promote arrhythmia, without fibrosis, hypertrophy or a decrease in contractility. The absence of contractile dysfunction and hypertrophy after a short duration of high-fat diet is consistent with prior literature33, 34. In contrast to saturated fat, unsaturated fat (olive oil diet) does not promote arrhythmia at the four-week time-point. Thus the type of fat is critically important, at least at this shorter time-point.
NOX2 activation in turn causes both abnormal calcium handling and abnormal repolarization (figure 5F). These findings may explain the mechanism of increased risk of sudden cardiac death in humans with obesity and/or high saturated fat intake, which is a consistent finding in several epidemiologic studies1–4. One issue regarding the nutritional aspect of the project is that the diets are not matched for carbohydrates. It is impossible to adjust percent fat without also changing percent carbohydrate and/or percent protein. Early the project we used two high-fat diets and the diet made from olive oil did not cause heart rhythm abnormalities in vivo, as shown in figure 1. This makes it very unlikely that reducing carbohydrates has an effect on heart rhythm, at least at this time-point.
Abnormal calcium homeostasis and heart rhythm
We show that a high saturated fat diet is sufficient to induce QT prolongation and ventricular ectopy. Prolongation of the QT interval and ventricular ectopy are both associated with increased mortality in the general population35, 36. PVCs themselves are probably not a cause of mortality, but appear to be a predictor of sudden cardiac death from sustained ventricular arrhythmias, which we induced in hearts from WT mice after feeding them the saturated fat diet. The increase in PVC frequency and QT interval caused by high saturated fat diet in our animal model mimics human heart disease caused by obesity. Frequent PVCs may be an early-warning sign of abnormal cardiac calcium homeostasis. We show that a high saturated fat diet increases sarcoplasmic reticulum calcium leak by causing post-translational modification of RyR2, which is known to promote arrhythmia.
Saturated fat, NOX2, and CaMK
Our data indicate that CaMK is activated in the heart by dietary saturated fat and CaMK activation is increased by NOX2 activation in vivo. Although our findings are novel, these data are consistent with prior work which has shown that NOX2 can be upstream of CaMK activation caused by angiotensin in isolated cardiomyocytes32. The strong protective effect in the NOX2KO mouse indicates that this is the major isoform that is activated in the rodent heart during high fat diet.
CaMK activation is known to increase SR calcium leak which could promote ventricular ectopy and our sparks experiments are consistent with this mechanism. The fact that CaMK is phosphorylated, but not oxidized, in our animal model indicates that NOX2 activation does not activate CaMK directly through increased ROS. Rather, CaMK activation is probably mediated by increased SR calcium leak from oxidized RyR2, but then CaMK further increases SR calcium leak in a positive feedback loop.
Abnormal repolarization
Abnormal calcium handling is probably the trigger for VT/VF in this model, but abnormal repolarization may be required as a substrate for sustained VT/VF. Our work shows that a high saturated fat diet is sufficient to cause abnormal repolarization in WT mice, without structural abnormalities. The pathways connecting saturated fat to abnormal repolarization may involve CaMK activation, which is known to modulate the function of the cardiac sodium channel (including late sodium current) and several potassium channels37. More research will be needed to examine the complex interactions of CaMK and repolarization in the context of high fat diet.
Translational potential
Although clinical trials of antioxidants for cardiovascular disease have been disappointing, for the most part, there is some clinical evidence supporting beneficial effects on heart rhythm38, 39. It may be that antioxidants are only beneficial in certain forms of cardiovascular disease, where increased ROS has a central role in the pathophysiology. Pharmacotherapy that specifically targets the source of cardiac ROS could be more effective. Our results demonstrate that pharmacologic inhibition of NOX2 is anti-arrhythmic in vivo.
Conclusion
We conclude that dietary saturated fat activates NOX2 which results in arrhythmia by causing increased sarcoplasmic reticulum calcium leak and abnormal ventricular repolarization. Genetic deletion of NOX2 or pharmacologic NOX2 inhibition prevents the pro-arrhythmic effect of high saturated fat diet in vivo. The mechanisms revealed by this work may have therapeutic implication for heart disease caused by diabetes and obesity and support NOX2 inhibition as a potential anti-arrhythmic therapy.
Supplementary Material
What is known:
Clinical research demonstrates that obese people have an increased risk of sudden cardiac death
Other studies indicate that high-fat diets increase the risk of sudden cardiac death. The mechanisms are not understood.
What the study adds:
In mice a, high saturated fat diet causes increased ventricular ectopy (PVCs), prolongation of the QT interval, and increased susceptibility to induced ventricular tachycardia.
These abnormalities are dependent on oxidative stress from activation of NOX2, by promoting abnormal calcium handling and repolarization heterogeneity.
CaMK activation appears to be a downstream component of the pathophysiology and could contribute to both abnormal calcium handling and abnormal repolarization.
Sources of Funding:
This work was supported by T32 HL007343 to LCJ. JPM is supported by NIH R01 HL136758. Images were collected in the Confocal and Specialized Microscopy Shared Resource of the Herbert Irving Comprehensive Cancer Center at Columbia University, supported by NIH grant #P30 CA013696 (National Cancer Institute). The lipidomics experiments in this publication were supported by the National Center for Advancing Translational Sciences, National Institutes of Health, through Grant Number 1UL1 TR001873. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH.
Non-standard Abbreviations and Acronyms:
- APD
action potential duration
- CaMK
calcium/calmodulin-dependent protein kinase
- DIO
diet induced obesity
- HSFD
high saturated fat diet
- HUFD
high unsaturated fat diet
- NOX2
NADPH oxidase 2
- PVC
premature ventricular complexes
- ROS
reactive oxygen species
- SCD
sudden cardiac death
Footnotes
Disclosures: Conflict of interest statement: ARM is a consultant and member of the board of ARMGO, which is targeting RyR channels for therapeutic purposes.
References:
- 1.Albert CM, Chae CU, Grodstein F, Rose LM, Rexrode KM, Ruskin JN, Stampfer MJ, Manson JE. Prospective study of sudden cardiac death among women in the United States. Circulation. 2003;107:2096–101. [DOI] [PubMed] [Google Scholar]
- 2.Jouven X, Desnos M, Guerot C, Ducimetiere P. Predicting sudden death in the population: the Paris Prospective Study I. Circulation. 1999;99:1978–83. [DOI] [PubMed] [Google Scholar]
- 3.Filippi A, Sessa E Jr., Mazzaglia G, Pecchioli S Jr., Capocchi R Jr., Caprari F, Scivales A, Cricelli C. Out of hospital sudden cardiac death in Italy: a population-based case-control study. J Cardiovasc Med (Hagerstown). 2008;9:595–600. [DOI] [PubMed] [Google Scholar]
- 4.Hookana E, Junttila MJ, Puurunen VP, Tikkanen JT, Kaikkonen KS, Kortelainen ML, Myerburg RJ, Huikuri HV. Causes of nonischemic sudden cardiac death in the current era. Heart Rhythm. 2011;8:1570–5. [DOI] [PubMed] [Google Scholar]
- 5.Szczepaniak LS, Victor RG, Orci L, Unger RH. Forgotten but not gone: the rediscovery of fatty heart, the most common unrecognized disease in America. Circ Res. 2007;101:759–67. [DOI] [PubMed] [Google Scholar]
- 6.Morrow JP, Katchman A, Son NH, Trent CM, Khan R, Shiomi T, Huang H, Amin V, Lader JM, Vasquez C, et al. Mice With Cardiac Overexpression of Peroxisome Proliferator-Activated Receptor gamma Have Impaired Repolarization and Spontaneous Fatal Ventricular Arrhythmias. Circulation. 2011;124:2812–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Son NH, Park TS, Yamashita H, Yokoyama M, Huggins LA, Okajima K, Homma S, Szabolcs MJ, Huang LS, Goldberg IJ. Cardiomyocyte expression of PPARgamma leads to cardiac dysfunction in mice. J Clin Invest. 2007;117:2791–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sharma S, Adrogue JV, Golfman L, Uray I, Lemm J, Youker K, Noon GP, Frazier OH, Taegtmeyer H. Intramyocardial lipid accumulation in the failing human heart resembles the lipotoxic rat heart. FASEB J. 2004;18:1692–700. [DOI] [PubMed] [Google Scholar]
- 9.Jouven X, Charles MA, Desnos M, Ducimetiere P. Circulating nonesterified fatty acid level as a predictive risk factor for sudden death in the population. Circulation. 2001;104:756–61. [DOI] [PubMed] [Google Scholar]
- 10.Oliver MF, Kurien VA, Greenwood TW. Relation between serum-free-fatty acids and arrhythmias and death after acute myocardial infarction. Lancet. 1968;1:710–4. [DOI] [PubMed] [Google Scholar]
- 11.Chiuve SE, Rimm EB, Sandhu RK, Bernstein AM, Rexrode KM, Manson JE, Willett WC, Albert CM. Dietary fat quality and risk of sudden cardiac death in women. Am J Clin Nutr. 2012;96:498–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lemaitre RN, King IB, Sotoodehnia N, Knopp RH, Mozaffarian D, McKnight B, Rea TD, Rice K, Friedlander Y, Lumley TS, et al. Endogenous red blood cell membrane fatty acids and sudden cardiac arrest. Metabolism. 2010;59:1029–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ramirez AH, Schildcrout JS, Blakemore DL, Masys DR, Pulley JM, Basford MA, Roden DM, Denny JC. Modulators of normal electrocardiographic intervals identified in a large electronic medical record. Heart Rhythm. 2011;8:271–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Messerli FH, Nunez BD, Ventura HO, Snyder DW. Overweight and sudden death. Increased ventricular ectopy in cardiopathy of obesity. Arch Intern Med. 1987;147:1725–8. [DOI] [PubMed] [Google Scholar]
- 15.Zemva A, Zemva Z. Ventricular ectopic activity, left ventricular mass, hyperinsulinemia, and intracellular magnesium in normotensive patients with obesity. Angiology. 2000;51:101–6. [DOI] [PubMed] [Google Scholar]
- 16.Huang H, Amin V, Gurin M, Wan E, Thorp E, Homma S, Morrow JP. Diet-induced obesity causes long QT and reduces transcription of voltage-gated potassium channels. J Mol Cell Cardiol. 2013;59C:151–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Luo M, Guan X, Luczak ED, Lang D, Kutschke W, Gao Z, Yang J, Glynn P, Sossalla S, Swaminathan PD, et al. Diabetes increases mortality after myocardial infarction by oxidizing CaMKII. J Clin Invest. 2013;123:1262–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang KC, Dudley SC Jr., Oxidative stress and atrial fibrillation: finding a missing piece to the puzzle. Circulation. 2013;128:1724–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steinberg SF. Oxidative stress and sarcomeric proteins. Circ Res. 2013;112:393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Santos CX, Anilkumar N, Zhang M, Brewer AC, Shah AM. Redox signaling in cardiac myocytes. Free Radic Biol Med. 2011;50:777–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xie W, Santulli G, Reiken SR, Yuan Q, Osborne BW, Chen BX, Marks AR. Mitochondrial oxidative stress promotes atrial fibrillation. Sci Rep. 2015;5:11427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bendall JK, Cave AC, Heymes C, Gall N, Shah AM. Pivotal role of a gp91(phox)-containing NADPH oxidase in angiotensin II-induced cardiac hypertrophy in mice. Circulation. 2002;105:293–6. [DOI] [PubMed] [Google Scholar]
- 23.Mitchell GF, Jeron A, Koren G. Measurement of heart rate and Q-T interval in the conscious mouse. Am J Physiol. 1998;274:H747–51. [DOI] [PubMed] [Google Scholar]
- 24.Yamazaki M, Avula UM, Bandaru K, Atreya A, Boppana VS, Honjo H, Kodama I, Kamiya K, Kalifa J. Acute regional left atrial ischemia causes acceleration of atrial drivers during atrial fibrillation. Heart Rhythm. 2013;10:901–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Martins RP, Kaur K, Hwang E, Ramirez RJ, Willis BC, Filgueiras-Rama D, Ennis SR, Takemoto Y, Ponce-Balbuena D, Zarzoso M, et al. Dominant frequency increase rate predicts transition from paroxysmal to long-term persistent atrial fibrillation. Circulation. 2014;129:1472–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Joseph LC, Kokkinaki D, Valenti MC, Kim GJ, Barca E, Tomar D, Hoffman NE, Subramanyam P, Colecraft HM, Hirano M, et al. Inhibition of NADPH oxidase 2 (NOX2) prevents sepsis-induced cardiomyopathy by improving calcium handling and mitochondrial function. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Joseph LC, Barca E, Subramanyam P, Komrowski M, Pajvani U, Colecraft HM, Hirano M, Morrow JP. Inhibition of NADPH Oxidase 2 (NOX2) Prevents Oxidative Stress and Mitochondrial Abnormalities Caused by Saturated Fat in Cardiomyocytes. PLoS One. 2016;11:e0145750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jaishy B, Zhang Q, Chung HS, Riehle C, Soto J, Jenkins S, Abel P, Cowart LA, Van Eyk JE, Abel ED. Lipid-induced NOX2 activation inhibits autophagic flux by impairing lysosomal enzyme activity. J Lipid Res. 2015;56:546–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sovari AA, Iravanian S, Dolmatova E, Jiao Z, Liu H, Zandieh S, Kumar V, Wang K, Bernstein KE, Bonini MG, et al. Inhibition of c-Src tyrosine kinase prevents angiotensin II-mediated connexin-43 remodeling and sudden cardiac death. J Am Coll Cardiol. 2011;58:2332–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Swaminathan PD, Purohit A, Soni S, Voigt N, Singh MV, Glukhov AV, Gao Z, He BJ, Luczak ED, Joiner ML, et al. Oxidized CaMKII causes cardiac sinus node dysfunction in mice. J Clin Invest. 2011;121:3277–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Zhang R, Khoo MS, Wu Y, Yang Y, Grueter CE, Ni G, Price EE Jr., Thiel W, Guatimosim S, Song LS, et al. Calmodulin kinase II inhibition protects against structural heart disease. Nat Med. 2005;11:409–17. [DOI] [PubMed] [Google Scholar]
- 32.Wagner S, Dantz C, Flebbe H, Azizian A, Sag CM, Engels S, Mollencamp J, Dybkova N, Islam T, Shah AM, et al. NADPH oxidase 2 mediates angiotensin II-dependent cellular arrhythmias via PKA and CaMKII. J Mol Cell Cardiol. 2014;75:206–15. [DOI] [PubMed] [Google Scholar]
- 33.Okere IC, Chess DJ, McElfresh TA, Johnson J, Rennison J, Ernsberger P, Hoit BD, Chandler MP, Stanley WC. High-fat diet prevents cardiac hypertrophy and improves contractile function in the hypertensive dahl salt-sensitive rat. Clin Exp Pharmacol Physiol. 2005;32:825–31. [DOI] [PubMed] [Google Scholar]
- 34.Cheng Y, Li W, McElfresh TA, Chen X, Berthiaume JM, Castel L, Yu X, Van Wagoner DR, Chandler MP. Changes in myofilament proteins, but not Ca(2)(+) regulation, are associated with a high-fat diet-induced improvement in contractile function in heart failure. Am J Physiol Heart Circ Physiol. 2011;301:H1438–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Schouten EG, Dekker JM, Meppelink P, Kok FJ, Vandenbroucke JP, Pool J. QT interval prolongation predicts cardiovascular mortality in an apparently healthy population. Circulation. 1991;84:1516–23. [DOI] [PubMed] [Google Scholar]
- 36.Bikkina M, Larson MG, Levy D. Prognostic implications of asymptomatic ventricular arrhythmias: the Framingham Heart Study. Ann Intern Med. 1992;117:990–6. [DOI] [PubMed] [Google Scholar]
- 37.Maier LS, Bers DM. Role of Ca2+/calmodulin-dependent protein kinase (CaMK) in excitation-contraction coupling in the heart. Cardiovasc Res. 2007;73:631–40. [DOI] [PubMed] [Google Scholar]
- 38.Harling L, Rasoli S, Vecht JA, Ashrafian H, Kourliouros A, Athanasiou T. Do antioxidant vitamins have an anti-arrhythmic effect following cardiac surgery? A meta-analysis of randomised controlled trials. Heart. 2011;97:1636–42. [DOI] [PubMed] [Google Scholar]
- 39.Rodrigo R, Korantzopoulos P, Cereceda M, Asenjo R, Zamorano J, Villalabeitia E, Baeza C, Aguayo R, Castillo R, Carrasco R, et al. A randomized controlled trial to prevent post-operative atrial fibrillation by antioxidant reinforcement. J Am Coll Cardiol. 2013;62:1457–65. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.