

Abstract
Rationale:
Genome-wide association studies (GWAS) have identified hundreds of genetic loci associated with atrial fibrillation (AF). However, these loci explain only a small proportion of AF heritability.
Objective:
To develop an approach to identify additional AF-related genes by integrating multiple omics data.
Methods and Results:
Three types of omics data were integrated: 1) summary statistics from the AFGen 2017 GWAS; 2) a whole blood epigenome-wide association study (EWAS) of AF; and 3) a whole blood transcriptome-wide association study (TWAS) of AF. The variant-level GWAS results were collapsed into gene-level associations using fast set-based association analysis (fastBAT). The CpG-level EWAS results were also collapsed into gene-level associations by an adapted SNP-set Kernel Association Test (SKAT) approach. Both GWAS and EWAS gene-based associations were then meta-analyzed with TWAS using a fixed-effects model weighted by the sample size of each data set. A tissue-specific network was subsequently constructed using the Network-wide association study (NetWAS). The identified genes were then compared with the AFGen 2018 GWAS that contained twice the number of AF cases compared to AFGen 2017 GWAS.
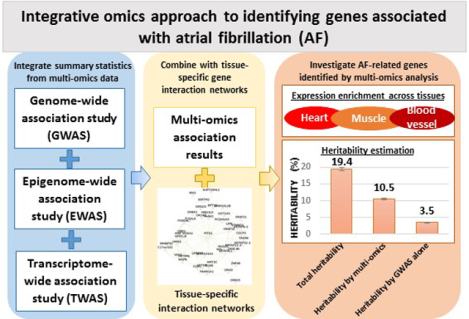
We observed that the multi-omics approach identified many more relevant AF-related genes than using AFGen 2018 GWAS alone (1931 vs. 206 genes). Many of these genes are involved in the development and regulation of heart and muscle related biological processes. Moreover, the gene set identified by multi-omics approach explained much more AF variance than those identified by GWAS alone (10.4% vs 3.5%).
Conclusions:
We developed a strategy to integrate multiple omics data to identify AF-related genes. Our integrative approach may be useful to improve the power of traditional GWAS, which might be particularly useful for rare traits and diseases with limited sample size.
Keywords: Multi-omics, atrial fibrillation, data integration, genes, genomics
Subject Terms: Atrial Fibrillation; Genetics, Association Studies; Gene Expression and Regulation; Genetics; Epigenetics
Graphical Abstract
INTRODUCTION
Atrial fibrillation (AF) is the most common heart arrhythmia. In the US, the prevalence of AF was 2.7 to 6.1 million in 2010, and this number is expected to increase to 9.3 to 12.1 million in 2030.1 AF is heritable with an estimated heritability of 22%.2 Large-scale genome-wide association studies (GWAS) have identified more than one hundred loci associated with AF.3–5 However, these loci explain only 6.4% of heritability,2 suggesting that more AF-related genetic loci remain to be identified.
The most straightforward approach to identify additional AF-related loci is to increase the sample size and thus the statistical power. In the past few years, we and others have published a series of papers with increasing sample size and number of identified loci: 6 novel AF loci from 6,707 AF cases and 52,426 referents in 2012,6 12 additional AF loci from 17,931 cases and 115,142 referents in 2017,7 and 67 new loci from more than 65,000 AF cases and over 522,000 referents in 2018.3 When additional samples are not available, alternative methods, such as simultaneous analysis of multiple related traits (for example, with MTAG8) can also increase the statistical power. Another solution is to integrate information from multi-omics data.9 The underlying assumption is that multi-omics data could provide complementary information, which is helpful to reveal the underlying biology of the associations.
Beyond GWAS, we have previously performed a whole blood transcriptome-wide association study (TWAS)10 and a whole blood epigenome-wide association study (EWAS)11 with AF. Integrating the information from these single-omics studies provides an opportunity to improve our understanding of AF. The objective of our current study is to develop a strategy to identify AF-related genes by integrating these multi-omics data. We validate our strategy using the latest AF GWAS, and investigate potential biological mechanisms underlying the associations.
METHODS
The data, analytic methods, and study materials will be made available to other researchers for purposes of reproducing the results. The summary results are available at the Broad Cardiovascular Disease Knowledge Portal (www.broadcvdi.org) and the Online Materials.
Multi-omics studies of AF.
Three types of omics data were integrated in the current study, including GWAS, EWAS and TWAS.
The GWAS data consisted of the summary statistics from the Atrial Fibrillation Genetics (AFGen) Consortium GWAS published in 2017 (referred as AFGen 2017 GWAS),7 which included 17,931 AF cases and 115,142 referents from 31 studies. The majority of participants were of European descent (89.1% for AF cases and 89.3% for referents). The remaining participants were African Americans, Japanese, Hispanic, or Brazilians.
The EWAS result consisted of the whole blood epigenome-wide association study with AF, which included 183 AF cases and 2,236 referents from the Framingham Heart Study (FHS).11 FHS is a community-based cohort consisting of three generations of participants.12–14 The EWAS samples were collected from the Offspring participants who attended the eighth examination. The DNA methylation profiling was performed using Illumina Infinium Human Methylation 450K BeadChip as described previously.11 The association between AF and the methylation level of each CpG site was tested by linear mixed effects regression models to account for the familial relatedness inferred from the FHS pedigree structure.
The TWAS result consisted of the whole blood gene transcriptome-wide association study with AF, which included 177 AF cases and 2,126 referents also from the FHS Offspring cohort who attended the eighth examination. Whole blood gene expression was measured by the Affymetrix Human Exon 1.0st Array. The association of 17,873 transcripts (representing 17,562 unique genes) with AF was also tested by linear mixed effects regression models similar to EWAS.10
Gene-based association analysis for GWAS, EWAS, and TWAS.
Prior to integrating three types of omics data, we converted all summary statistics to gene-level association statistics. For GWAS, we used the fast set-based association analysis (FastBAT) implemented in the GCTA 1.26.0 software package15 to combine the SNP-based association summary statistics into a single gene-based statistic. Gene regions were defined as 50 kb away from the gene boundary. Variants located within the gene region were jointly tested by an approximated estimation of sum of chi-squares statistics. Under the null hypothesis of no association, the z-scores follow a multivariate normal distribution with zero mean. The covariance matrix between variants within the gene region was estimated from 1,724 unrelated FHS individuals. For EWAS, we used an adapted SNP-set Kernel Association Test (SKAT) approach16 to collapse CpG-level associations into gene-level associations, which was implemented as a modified version of R seqMeta package.19 We previously demonstrated that the method could provide increased power to jointly test the association of multiple CpG sites with phenotypes of interest.17,18 Individual CpG sites were first mapped to genes based on their chromosomal locations. The SKAT analysis was then performed for all CpG sites within each gene region, and the models were adjusted for age, sex, assay sites, and accounted for familial relatedness. For TWAS, transcripts were mapped to genes based on the transcript annotation. For genes with multiple transcripts, an adjusted minimum P value was used, which was defined as min Padjusted = 1 – (1 – min P)n, in which n is the number of transcripts within a gene.20 The models for TWAS were adjusted for age, sex, batch effects, and accounted for family relatedness.
Meta-analysis of multi-omics data.
After creating gene-based summary statistics for each data type, we performed a P value based meta-analysis with a fixed-effect model irrespective of the directions of the associations. We also assigned weights to each type of omics data to reflect their different sample sizes. Details of implementation are provided in Online Methods. The pairwise correlations of the gene-based tests between omics data sets were all less than 0.1 (Online Table I) and therefore ignored.21
Tissue-specific gene prioritization.
We further examined the effect of tissue-specific expression by the Network-wide association study (NetWAS).22 NetWAS is a machine learning-based method that combines gene-level associations with the tissue-specific interaction network. The network was built using over 14,000 publications and low-throughput tissue-specific expression data,22 which could describe gene-gene functional interactions within a specific tissue (Online Methods). Genes showing association with AF with a pre-specified cutoff (default P <0.01) were treated as “pre-positive” genes, in a network built from general heart tissues which might include all heart tissue types (e.g., atrium, appendage, or ventricle).22 A heart-specific score was then assigned to each gene. The score was calculated by support vector machine to represent how far the gene was away from the hyperplane that maximally distinguished pre-positive and pre-negative genes.22 If a “pre-positive” gene was predicted positive (referred as “post-positive”), the gene was considered as an AF-related genes.
For sensitivity analyses, we also explored different significance thresholds (P <0.1, P<0.0001, or P<0.05/N, where N is the number of shared genes), and we examined another network built from the whole blood to assess the robustness of tissue-specific gene prioritization.
Method validation.
To validate our findings, we used the latest AFGen GWAS published in 2018 (referred as 2018 GWAS) as the reference gene set.3 Most samples from AFGen 2017 GWAS were also included in 2018 GWAS. The summary statistics of 2018 GWAS were also collapsed into gene-based associations by FastBAT. We then compared the overlapping genes with those identified by single-omics or multi-omics integration methods measured by the C-statistics as implemented in the pROC R package.23
The number of AF cases of GWAS (n=17,931) overwhelmed that of EWAS (n=183) and TWAS (n=177). In order to understand the effects of sample size imbalance, we examined if a decrease in GWAS sample size would significantly affect our results. We used the AFGen 2012 GWAS (6,707 cases and 52,426 referents)6 and the AF GWAS from FHS participants only (1,104 cases and 7,268 referents)3 for comparison.
Strategy application to the integration of AFGen 2018 GWAS, EWAS, and TWAS.
We applied our strategy with the optimal weights to integrate 2018 GWAS, EWAS, and TWAS, which would generate a list of candidate genes that might be related to AF. We further examined their functions using the enrichment analysis, tissue-specific expression analysis, and heritability estimations as follows.
Gene ontology and pathway enrichment analysis.
The enrichment of AF-related genes in biological processes and pathways was assessed by WebGestalt.24 Fisher’s exact test was used to calculate enrichment P values. False discovery rate (FDR) was calculated by Benjamini and Hochberg method to account for multiple testing.25 Significant pathways were defined as those with FDR<0.05.
Gene expression across multiple heart tissues.
We also examined the expression of AF-related genes across two heart tissues, left atria and right atrial appendage (Online Methods). Gene expression across tissues were obtained from the Genotype-Tissue Expression (GTEx) project26 and the Myocardial Applied Genomics Network repository.3 Two-sample Kolmogorov-Smirnov test was used to compare the percentiles between independent AF-related genes and non-AF-related genes (pairwise Spearman’s r2 < 0.25).27
Tissue-specific expression analysis.
We calculated a pSI score for each gene to represent its enrichment in a specific tissue as implemented in the pSI R package.29 Details of pSI calculation are described in Online Methods. For each tissue, genes with pSI <0.05 were considered as significantly enriched in the tissue. The overlap between AF-related genes and the genes enriched in each tissue was estimated by Fisher’s exact test. We used the Bonferroni correction to adjust for multiple testing, and the significance cutoff was set as P<0.05/25=0.002, because 25 tissue groups were tested (Online Table II). We assessed significance using 1,000 random gene sets, each with the same number of genes as the AF-related gene set.28
Estimation of AF heritability.
We also estimated AF heritability explained by regions containing AF-related genes using HESS.30 The genome was first divided into linkage disequilibrium (LD)-independent regions,31 and the summary statistics of individual variants within each region were used to estimate the local heritability taking into account LD between variants. The 1000 Genomes Phase 3 European samples were used as the LD reference panel. Variants with minor allele frequency less than 0.01 were excluded. We also estimated the heritability from participants of European ancestry in the UK Biobank, which included more than 300,000 independent participants (3,818 cases and 333,381 referents).32 Details about the HESS method were provided in Online Methods.
RESULTS
Gene-based association results based on GWAS, EWAS, and TWAS.
Figure 1 shows the overall flowchart of our approach. The summary statistics from GWAS, EWAS, and TWAS were first converted into gene-based associations. The analysis was restricted to 14,364 genes shared by three omics platforms. Bonferroni correction was used to adjust for multiple testing, and the significance cutoff was set as 0.05/14,364=3.5×10−6. As shown in Figure 2, we found 44 genes by GWAS, 41 genes by EWAS, and 1 gene by TWAS that were significantly associated with AF after adjusting for multiple testing. However, none of AF-related genes were shared between any two types of omics data.
Figure 1.

Analysis flowchart of multi-omics data integration. “n” is the number of AF cases/referents. “AFGen” is the Atrial Fibrillation Genetics Consortium.
Figure 2.

Gene-based association results of GWAS (A), EWAS (B), and TWAS (C) of AF. “n” is the number of AF cases/referents. Y-axis is the -log10 (P) of gene-based association analysis. The shared gene set was included in the analysis and grey lines represent the Bonferroni significance cutoff after adjusting for multiple testing.
We then performed meta-analysis to integrate omics data with different weighting schemes. A variety of weights between two most extreme weights were taken into consideration, one with GWAS only, and the other one with equal weights for all omics data. As shown in Online Table III, the number of significant genes first increased and then decreased with reducing weights of GWAS. The resulting gene sets were further mapped to tissue-specific network and classified by machine learning models as implemented by NetWAS.22 Genes remained significant after machine learning classification were defined as AF-related genes.
Validations for AF-related genes by AFGen 2018 GWAS.
In order to validate the utility of our multi-omics approach, we compared the predicted AF-related genes with the latest AFGen GWAS results (referred as 2018 GWAS).3 The summary statistics of 2018 GWAS were also collapsed into gene-level associations (referred as reference gene set). With the relative weights of 0.599, 0.201, and 0.200 for GWAS, EWAS, and TWAS, we achieved the best C-statistics of 0.813 (95% CI: 0.781~0.845) with the sensitivity 0.784 and specificity 0.941. In comparison, the C-statistics for GWAS alone was only 0.740 (Online Figure I and Online Table III).
We then performed a simulation with 1,000 replicates for each omics data set (see Online Method) to estimate type 1 error. As shown in Online Figure II, less than 10% inflation of type I error was observed with α=0.05 across all weights (relative type 1 error ranging from 1.00 to 1.10). With more conservative Bonferroni significance cutoff α=0.05/14,364=3.5×10−6, the relative type 1 error was higher (relative type 1 error ranging from 1.18 to 1.74), which might be partly due to the loss of directionality information during meta-analysis.
We also examined the effect of choosing different significance cutoffs from NetWAS to define “pre-positive” genes, ranging from 3.5×10−6 to 0.1. As shown in Figure 3, the C-statistics first increased and then decreased with increasing weights for EWAS and TWAS across all significance cutoffs. The optimal C-statistics were reached when P=0.01 and w_g=0.599. Online Table IV shows the number of significant genes for each cutoff. The findings suggested that not only lenient gene thresholds, but also the additional information from EWAS and TWAS helped to identify AF-related genes. We also performed the multi-omics integration analysis on the network built from whole blood, and observed a similar pattern but with relatively lower C-statistics.
Figure 3.

C-statistics for multi-omics integration approach across different weighting schemes. The stacked bar chart at the bottom represents different weight compositions for 2017 GWAS, EWAS, and TWAS. w_g is the relative weight of GWAS in the multi-omics integration, where w_g=1.00 represents that only GWAS was included in the multi-omics integration, whereas w_g=0.333 represents all three omics have the same weight. The lines above indicate the C-statistics with 95% confidence intervals, corresponding to different NetWAS significance thresholds and tissues.
Effect of sample sizes for GWAS, EWAS, and TWAS.
Given that the sample size of GWAS was much larger than that of EWAS or TWAS, the integrative results could be dominated by the GWAS results. To understand potential effect of different GWAS sample sizes, we explored an earlier published AF GWAS that only included 6,707 AF cases (referred as 2012 GWAS).6 After integrating with EWAS and TWAS, the results were largely similar although with a lower C-statistics (0.738 from 2012 GWAS vs. 0.813 from 2017 GWAS, Online Figure I and Online Figure III). However, if we further reduced GWAS sample size to include only samples from FHS (1,104 AF cases), the performance of our integrative omics analysis deteriorated significantly (Online Figure IV).
Identification of additional AF-related genes by integrating AFGen 2018 GWAS, EWAS, and TWAS.
In order to identify additional AF-related genes, we integrated AFGen 2018 GWAS together with EWAS and TWAS using the optimal weight derived from the validation analysis. Our analysis found that 1,931 genes were predicted “positive” and passed the lenient significance cutoff, and therefore referred as AF-related genes, of which 288 were previously reported in three external disease-related databases: GWAS catalog (https://www.ebi.ac.uk/gwas/), Open Targets Platform (https://www.targetvalidation.org/), and OMIM (https://www.omim.org/) (Online Table V).
We then assessed potential biological functions of AF-related genes by pathway enrichment analysis. The top 10 Gene ontology (GO) biological processes33,34 and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways35 are shown in Tables 1 and 2, respectively. Many AF-related genes were involved in cardiac muscle tissue development or adrenergic signaling in cardiomyocytes.
Table 1.
Top 10 Gene Ontology biological processes enriched with AF-related genes identified by the multi-omics integration analysis.
| Gene set | Description | Total genes | Observed genes | P value | FDR |
|---|---|---|---|---|---|
| GO:0060537 | Muscle tissue development | 371 | 94 | 3.0×10−17 | 5.7×10−14 |
| GO:0014706 | Striated muscle tissue development | 357 | 92 | 3.7×10−17 | 7.1×10−14 |
| GO:0048738 | Cardiac muscle tissue development | 200 | 63 | 9.0×10−17 | 1.7×10−13 |
| GO:0010628 | Positive regulation of gene expression | 1911 | 302 | 2.2×10−16 | 5.0×10−13 |
| GO:0071495 | Cellular response to endogenous stimulus | 1347 | 226 | 4.9×10−15 | 8.9×10−12 |
| GO:0009719 | Response to endogenous stimulus | 1595 | 257 | 7.1×10−15 | 1.1×10−11 |
| GO:0045893 | Positive regulation of transcription, DNA-templated | 1499 | 244 | 1.2×10−14 | 1.6×10−11 |
| GO:0051240 | Positive regulation of multicellular organismal process | 1661 | 261 | 8.6×10−14 | 9.8×10−11 |
| GO:0061061 | Muscle structure development | 610 | 122 | 1.2×10−13 | 1.3×10−10 |
| GO:1903508 | Positive regulation of nucleic acid-templated transcription | 1583 | 250 | 1.8×10−13 | 1.5×10−10 |
Table 2.
Top 10 KEGG biological pathways enriched with AF-related genes identified by the multi-omics integration analysis.
| Gene set | Description | Total genes | Observed genes | P value | FDR |
|---|---|---|---|---|---|
| hsa04261 | Adrenergic signaling in cardiomyocytes | 144 | 38 | 4.3×10−7 | 1.4×10−4 |
| hsa04022 | cGMP-PKG signaling pathway | 163 | 37 | 2.7×10−5 | 3.0×10−3 |
| hsa04933 | AGE-RAGE signaling pathway in diabetic complications | 99 | 26 | 3.1×10−5 | 3.0×10−3 |
| hsa04010 | MAPK signaling pathway | 295 | 57 | 3.7×10−5 | 3.0×10−3 |
| hsa04934 | Cushing syndrome | 154 | 34 | 1.0×10−4 | 6.6×10−3 |
| hsa04152 | AMPK signaling pathway | 120 | 28 | 1.5×10−4 | 8.3×10−3 |
| hsa01522 | Endocrine resistance | 98 | 24 | 2.0×10−4 | 9.1×10−3 |
| hsa04917 | Prolactin signaling pathway | 70 | 19 | 2.2×10−4 | 9.1×10−3 |
| hsa04728 | Dopaminergic synapse | 131 | 29 | 3.1×10−4 | 1.0×10−2 |
| hsa05206 | MicroRNAs in cancer | 150 | 32 | 3.1×10−4 | 1.0×10−2 |
Abbreviation: KEGG: Kyoto Encyclopedia of Genes and Genomes
We then examined the expression of AF-related genes in human left atrial and right atrial appendage tissues, which are the most relevant tissues for AF. As shown in Figure 4, these genes tended to have higher expression than other genes (both with P<2.2×10−16), suggesting their potential functions in left atrial and right atrial appendage tissues.
Figure 4.

Expression of AF-related genes and non-AF-related genes in left atria (left) and right atrial appendage (right). Each grey dot represents the expression level for each gene. Kolmogorov-Smirnov test on independent genes in each set (Spearman’s r2< 0.25) showed that AF-related genes tended to have higher expression than non-AF-related genes in both tissues (P<2.2×10−16).
We also examined the expression of these genes across 53 tissue types available from GTEx. As shown in Online Figure V, these genes showed the highest expression in heart (left ventricle and atrial appendage) and skeletal muscle. A relatively high co-expression was also observed between heart tissues and skeletal muscle for AF-related genes (Online Figure VI). We also compared AF-related genes with 1,000 randomly selected gene sets with the equal number of genes. As shown in Figure 5, the AF-related genes were significantly overexpressed in muscle (P=4.0×10−9), heart (P=1.5×10−5), and blood vessel (P=9.9×10−4).
Figure 5.

Tissue specific expression enrichment of AF-related genes. The 53 tissues in GTEx were grouped into 25 broad tissue types. Red dots represent the enrichment significance of 1,931 multi-omics AF-related genes in each tissue, whereas grey dots represent the enrichment significance of 1,000 randomly selected gene sets with the equal number of genes as AF-related genes. The blue line indicates the top 5% enrichment significant gene sets among 1,000 random gene sets. The yellow dashed line indicates the nominal significance level of 0.05. The red dashed line indicates the significance level after Bonferroni correction, P=0.05/25=0.002.
A few potential drug targets were identified, including ADORA1, ATP1A3, ATP1B2, CACNA1D, KCNQ4, NR3C2, and THRA. For example, NR3C2 encodes a receptor for both mineralocorticoids (MC) and glucocorticoids (GC).36 An observational study reported that patients with insufficient mineralocorticoid receptor blockade had a higher risk of AF incidence (OR 1.14, 95% CI: 1.07~1.22),37 which suggested the inhibition of NR3C2 was a potential therapy for AF. We further validated the candidate genes in 79 genes that were previously reported as AF pathogenesis genes.38,39 Many of these genes were either ion channels or involved in cardiac developments (Online Table VI). Among them, 39 genes were also our candidate genes (enrichment significance P= 4.9×10−15).
We next estimated the heritability explained by AF-related genes using HESS.30 The genome was divided into 1,703 LD-independent regions. Using the AFGen 2018 GWAS summary statistics,3 the AF heritability explained by all genetic variants was 19.4% (95% CI: 19.0%~19.8%). The 1,931 AF-related genes could be mapped to 675 regions, which explained 10.4% (95% CI: 10.2%~10.7%) of AF variance, more than half of the total AF heritability explained by all genetic variants. In comparison, the top 1,931 genes from 2018 GWAS could explain only 8.2% (95% CI: 8.0%~8.5%) of AF variance. We further validated the heritability estimation by another dataset from the United Kingdom Biobank (UKBB).32 Similarly the total AF heritability was estimated 18.9% (95% CI 18.2%~19.5%) after considering all genetic variants (Online Table VII). The 1,931 AF-related genes explained 8.0% (95% CI 7.6%~8.4%) of AF variance, whereas the top 1,931 genes from 2018 GWAS could explain only 5.5% (95% CI 5.2%~5.8%).
DISCUSSION
We developed a strategy to integrate association results from multi-omics data to study complex diseases. As more omics data become available, the integration of multi-omics data is expected to identify additional disease-related genes beyond GWAS. We applied the strategy to study AF and identified 1,931 potential AF-related genes, which explained more than half of AF heritability, much higher than genes identified by GWAS alone. Many of the identified genes were involved in the development of cardiac muscle structure, which might be one mechanism in AF pathogenesis. One example is PLN, which encodes a major substrate for the cAMP-dependent protein kinase in cardiac muscle. The gene was previously suggested to be involved in the AF pathogenesis40 and was identified as an AF-related gene by the multi-omics integration analysis, but it was not among the top genes identified by GWAS, EWAS, or TWAS individually.
We observed that the combination of tissue-specific gene interaction networks with multi-omics data significantly improved the power to identify AF-related genes. The most relevant tissue for AF is the left atrium. However, it is impractical to collect heart tissue in community-based observational cohorts. We thus used gene expression and DNA methylation measured from whole blood for both the EWAS and TWAS. The multi-omics integration results were then combined with gene interaction networks built from different tissues to understand their tissue-specific interactions. On the other hand, it is crucial to choose the relevant tissue type to build gene interaction networks. Otherwise limited or even no improvement of statistical power could be achieved.
For multi-omics data with a reference gene set, we recommend searching the optimum weights based on the maximum C-statistics with the reference gene set, as we did in the current study. However, if such a reference gene set is unavailable, we recommend using the square root of the sample size for each omics data set as the weights. Whereas it might not be the optimum weights for the data integration, it would still reach a reasonable accuracy from our experience (C-statistics 0.800 vs. the best C-statistics 0.813).
Our current EWAS results were derived only from samples collected in FHS. With a reference covariance matrix between CpG sites, our method potentially could be extended to use summary statistics from multiple cohorts by jointly testing CpG sites in each gene region. As data sharing is becoming a standard practice required by peer-reviewed journals and funding agencies,41,42 we anticipate more summary statistics of methylation data will be available in the future. One caveat is that whereas genetic variations largely remain unchanged over the lifetime, methylation levels could change over time with different environmental factors. Therefore, multiple representative covariance matrices might be needed to incorporate methylation data from diverse ethnicities or health conditions.
In collapsing CpG-level associations into gene-level associations, we included all CpG sites within the gene regions. CpG sites in promoter regions have traditionally been considered to be more important, but recent studies also showed that CpG sites within gene bodies other than promoter regions could be also functional.11, 43 Moderate correlation between two approaches was observed (r = 0.65, Online Figure VII). The inclusion of irrelevant CpG sites for the association analysis could dilute the true signals, which, however, might be rescued after integrating other omics data.
We have previously performed GWAS meta-analysis of AF in multiple stages, each stage adding additional cohorts. Therefore, almost all the samples in the 2017 GWAS were included in 2018 GWAS.
This way, we were able to demonstrate how the increase in sample size would enhance the identification of new AF-related genes. We found that 141 genes identified by the multi-omics approach with 2017 GWAS were also identified by the 2018 GWAS (Online Table III). On the other hand, our current study also indicated that the integration of multi-omics data would help identify additional AF-related genes (Online Table V). The underlying assumption is that multiple biological pathways are involved in AF’s pathogenesis, which could be interrogated by different omics technologies. However, further statistical simulations would be needed to thoroughly investigate the increased statistical power with multi-omics data under different scenarios. In addition, we used the summary statistics of association results instead of individual-level genetic data. Therefore, proper adjustments for relatedness could be done before the integration analysis. As demonstrated by previous studies,9,45,46 the integration of multi-omics data is expected to capture additional information that cannot be captured by GWAS of smaller sample size. Both ways (increasing sample size and multi-omics integration) increase statistical power and lead to new findings for GWAS.
Our method has several advantages. Our method does not rely on permutation for collapsing44 and thus is time efficient especially for genes with many variants. Second, we did not filter genetic variants, which would keep all potentially important genetic variants for the analysis. Moreover, we included tissue-specific network interactions that are important to understand potential biological mechanisms within the most relevant tissues.
We acknowledge several limitations of this study. We did not consider the complex interactions between omics data. For example, DNA methylation is one of most common epigenetic mechanisms that regulate gene expression. Similarly, genetic variations could affect both gene expression and DNA methylation. Therefore, gene-based tests of GWAS, EWAS and TWAS could be correlated and varied across different genes. Ignoring such correlations may introduce biased variance estimation for the meta-analysis. Second, the effects of multiple genetic variants and CpG sites were collapsed into gene-level associations solely based on their proximity to genes, which limited our capability to study the heterogeneity effects. Some regulatory variants could exert their effects far from their genomic locations that cannot be captured by our method. Another limitation is the TWAS and EWAS data were limited to a single cohort and without replication, which might result in both false positive and false negative findings. It is worth noting that TWAS and EWAS had overlapping samples from the Offspring cohort of the Framingham Heart Study. However, only weak correlation was observed between TWAS and EWAS (Online Table I), suggesting that two datasets interrogate different stages of biological pathways with marginal effect caused by overlapping samples. In addition, the TWAS and EWAS data were both based on microarray platforms, which have limited transcriptome/epigenome coverage and dynamic ranges. With more samples profiled by RNA-sequencing or bisulfite sequencing with AF status information, we could potentially identify additional new AF-related genes than the microarray data. Most of multi-tissue studies (such as GTEx) do not have AF information in their samples. We therefore used whole blood as a proxy of heart to investigate the association of gene expression and DNA methylation with AF. Similar strategies were previously taken to study the association of gene expression and DNA methylation with heart failure,47 myocardial infarction,48 or coronary artery disease.49,50 Most of our study samples were from participants of European ancestry. Therefore our findings may not be generalizable to samples from other ethnicity groups. It is essential to note that the current approach is hypothesis-generating; computational approaches alone cannot establish causal relations. There is still a long way to translate AF-related genes into clinical practice. Whereas nearly ~2,000 genes were identified as potential AF-related genes, further experimental validation is necessary to understand how these genes interact with each other and affect AF susceptibility.
In conclusion, we developed an analysis strategy to integrate multi-omics data to study AF and identified additional AF-related genes. Our integrative approach could be further extended to the study of other complex diseases with multi-omics data.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Genome-wide association studies (GWAS) have identified more than one hundred genetic loci associated with atrial fibrillation (AF). However, those loci could explain a small proportion of AF heritability.
Epigenome-wide association studies (EWAS) and transcriptome-wide association studies (TWAS) have identified multiple additional genes associated with AF.
What New Information Does This Article Contribute?
We developed a computational strategy to integrate summary statistics from GWAS, EWAS, and TWAS of AF.
The integrative omics strategy showed improved power of identifying AF-related genes than using GWAS alone.
A much larger proportion of AF heritability could be explained by AF-related genes identified by the integrative omics analysis
We developed an approach to integrate summary statistics from GWAS, EWAS and TWAS of AF. The results were incorporated with tissue-specific gene interaction networks to further prioritize AF-related genes. A much larger proportion of AF heritability could be explained by AF-related genes identified by the integrative omics analysis. Many of AF-related genes might be involved in the development of cardiac muscle structure. Our integrative approach might be applicable to the study of other complex diseases using multi-omics data.
SOURCES OF FUNDING
The study was supported by NIH grants 1R01 HL128914; 2R01 HL092577; and American Heart Association 18SFRN341100825, 18SFRN34150007, and 18SFRN34110082. Dr. Lubitz is supported by NIH grant 1R01HL139731 and American Heart Association 18SFRN34250007. Framingham Heart Study was supported by NIH Contracts HHSN268201500001I and N01-HC 25195.
DISCLOSURES
B. Wang: None
K.L. Lunetta: None
J. Dupuis: None
S.A. Lubitz: Dr. Lubitz receives sponsored research support from Bristol Myers Squibb / Pfizer, Bayer HealthCare, and Boehringer Ingelheim, and has consulted for Abbott, Quest Diagnostics, Bristol Myers Squibb / Pfizer
L. Trinquart: None
L. Yao: None
P.T. Ellinor: Dr. Ellinor is the PI on a grant from Bayer to the Broad Institute focused on the genetics and therapeutics of AF.
E.J. Benjamin: None
H. Lin: None
Nonstandard Abbreviations and Acronyms:
- AF
Atrial fibrillation
- GWAS
Genome-wide association analysis
- AFGen
Atrial Fibrillation Genetics Consortium
- MTAG
multi-trait analysis of GWAS
- TWAS
Transcriptome-wide association study
- EWAS
Epigenome-wide association study
- FHS
Framingham Heart Study
- FastBAT
Fast set-based association analysis
- SKAT
SNP-set Kernel Association Test
- NetWAS
Network-wide association study
- FDR
False discovery rate
- LD
Linkage disequilibrium
- UKBB
United Kingdom Biobank
REFERENCES
- 1.Benjamin EJ, Muntner P, Alonso A, et al. Heart Disease and Stroke Statistics-2019 Update: A Report From the American Heart Association. Circulation. 2019;CIR0000000000000659. [DOI] [PubMed] [Google Scholar]
- 2.Weng LC, Choi SH, Klarin D, Smith JG, Loh PR, Chaffin M, Roselli C, Hulme OL, Lunetta KL, Dupuis J, Benjamin EJ, Newton-Cheh C, Kathiresan S, Ellinor PT, Lubitz SA. Heritability of Atrial Fibrillation. Circ Cardiovasc Genet. 2017;10:1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Roselli C, Chaffin MD, Weng LC, et al. Multi-ethnic genome-wide association study for atrial fibrillation. Nat Genet. 2018;1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nielsen JB, Thorolfsdottir RB, Fritsche LG, et al. Biobank-driven genomic discovery yields new insight into atrial fibrillation biology. Nat Genet. 2018;50:1234–1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nielsen JB, Fritsche LG, Zhou W, et al. Genome-wide Study of Atrial Fibrillation Identifies Seven Risk Loci and Highlights Biological Pathways and Regulatory Elements Involved in Cardiac Development. Am J Hum Genet. 2018;102:103–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ellinor PT, Lunetta KL, Albert CM, et al. Meta-analysis identifies six new susceptibility loci for atrial fibrillation. Nat Genet. 2012;44:670–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Christophersen IE, Rienstra M, Roselli C, et al. Large-scale analyses of common and rare variants identify 12 new loci associated with atrial fibrillation. Nat Genet. 2017;49:946–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Turley P, Walters RK, Maghzian O, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet. 2018;50:229–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ritchie MD, Holzinger ER, Li R, Pendergrass SA, Kim D. Methods of integrating data to uncover genotype-phenotype interactions. Nat Rev Genet. 2015;16:85–97. [DOI] [PubMed] [Google Scholar]
- 10.Lin H, Yin X, Lunetta KL, Dupuis J, McManus DD, Lubitz SA, Magnani JW, Joehanes R, Munson PJ, Larson MG, Levy D, Ellinor PT, Benjamin EJ. Whole blood gene expression and atrial fibrillation: The Framingham Heart Study. PLoS One. 2014;9:e96794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lin H, Yin X, Xie Z, Lunetta KL, Lubitz SA, Larson MG, Ko D, Magnani JW, Mendelson MM, Liu C, McManus DD, Levy D, Ellinor PT, Benjamin EJ. Methylome-wide Association Study of Atrial Fibrillation in Framingham Heart Study. Sci Rep. 2017;7:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281–290. [DOI] [PubMed] [Google Scholar]
- 13.Savage DD, Levy D, Dannenberg AL, et al. Association of echocardiographic left ventricular mass with body size, blood pressure and physical activity (the Framingham Study). Am J Cardiol. 1990;65:371–6. [DOI] [PubMed] [Google Scholar]
- 14.Splansky GL, Corey D, Yang Q, Atwood LD, Cupples LA, Benjamin EJ, D’Agostino RB, Fox CS, Larson MG, Murabito JM, O’Donnell CJ, Vasan RS, Wolf PA, Levy D. The Third Generation Cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: Design, recruitment, and initial examination. Am J Epidemiol. 2007;165:1328–1335. [DOI] [PubMed] [Google Scholar]
- 15.Bakshi A, Zhu Z, Vinkhuyzen AA, Hill WD, McRae AF, Visscher PM, Yang J. Fast set-based association analysis using summary data from GWAS identifies novel gene loci for human complex traits. Sci Rep. 2016;6:32894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wu MC, Lee S, Cai T, Li Y, Boehnke M, Lin X. Rare-variant association testing for sequencing data with the sequence kernel association test. Am J Hum Genet. 2011;89:82–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhang Q, Zhao Y, Zhang R, Wei Y, Yi H, Shao F, Chen F. A Comparative Study of Five Association Tests Based on CpG Set for Epigenome-Wide Association Studies. PLoS One. 2016;11:e0156895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang B, DeStefano AL, Lin H. Integrative methylation score to identify epigenetic modifications associated with lipid changes resulting from fenofibrate treatment in families. BMC Proc. 2018;12:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Voorman AA, Brody J, Chen H, Lumley T, Davis B. seqMeta: Meta-Analysis of Region-Based Tests of Rare DNA Variants. R package version 1.6.6. 2016. https://CRAN.R-project.org/package=seqMeta [Google Scholar]
- 20.Conneely KN, Boehnke M. So Many Correlated Tests, So Little Time! Rapid Adjustment of P Values for Multiple Correlated Tests. Am J Hum Genet. 2007;81:1158–1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Province MA, Borecki IB. A correlated meta-analysis strategy for data mining “OMIC” scans. Pac Symp Biocomput. 2013;236–246. [PMC free article] [PubMed] [Google Scholar]
- 22.Greene CS, Krishnan A, Wong AK, Ricciotti E, Zelaya RA, Himmelstein DS, Zhang R, Hartmann BM, Zaslavsky E, Sealfon SC, Chasman DI, Fitzgerald GA, Dolinski K, Grosser T, Troyanskaya OG. Understanding multicellular function and disease with human tissue-specific networks. Nat Genet. 2015;47:569–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Turck N, Vutskits L, Sanchez-Pena P, Robin X, Hainard A, Gex-Fabry M, Fouda C, Bassem H, Mueller M, Lisacek F, Puybasset L, Sanchez J-C. pROC: an open-source package for R and S+ to analyze and compare ROC curves. BMC Bioinformatics. 2011;8:12–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B. WebGestalt 2017: A more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Res. 2017;45:W130–W137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Benjamini Y and Hochberg Y. Controlling the False Discovery Rate: a Practical and Powerful Approach to Multiple Testing. J R Stat Soc Series B Stat Methodol. 1995;57:289–300. [Google Scholar]
- 26.Aguet F, Brown AA, Castel SE, et al. Genetic effects on gene expression across human tissues. Nature. 2017;550:204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conover WJ. Practical Nonparametric Statistics, 3rd Edition 1999. [Google Scholar]
- 28.Ferreira MA, Vonk JM, Baurecht H, et al. Shared genetic origin of asthma, hay fever and eczema elucidates allergic disease biology. Nat Genet. 2017;49:1752–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wells A, Kopp N, Xu X, O’Brien DR, Yang W, Nehorai A, Adair-Kirk TL, Kopan R, Dougherty JD. The anatomical distribution of genetic associations. Nucleic Acids Res. 2015;43:10804–10820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shi H, Kichaev G, Pasaniuc B. Contrasting the Genetic Architecture of 30 Complex Traits from Summary Association Data. Am J Hum Genet. 2016;99:139–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berisa T, Pickrell JK. Approximately independent linkage disequilibrium blocks in human populations. Bioinformatics. 2015;32:283–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Neale BM. UK BIOBANK GWAS [Internet]. 2018; Available from: http://www.nealelab.is/uk-biobank/
- 33.Ashburner M, Ball CA, Blake JA, et al. Gene ontology: tool for the unification of biology. Nat Genet. 2000;25(1):25–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carbon S, Douglass E, Dunn N, et al. The Gene Ontology Resource: 20 years and still GOing strong. Nucleic Acids Res. 2019;47(D1):D330–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jia G, Jia Y, Sowers JR. Role of mineralocorticoid receptor activation in cardiac diastolic dysfunction. Biochim Biophys Acta - Mol Basis Dis. 2017;1863:2012–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.H GL C GC, Yozamp N, Wang M, Vaidya A. Incidence of atrial fibrillation and mineralocorticoid receptor activity in patients with medically and surgically treated primary aldosteronism. JAMA Cardiol. 2018;3:768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lubitz SA, Brody JA, Bihlmeyer NA, et al. Whole Exome Sequencing in Atrial Fibrillation. PLoS Genet. 2016;12:e1006284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Feghaly J, Zakka P, London B, MacRae CA, Refaat MM. Genetics of Atrial Fibrillation. J Am Heart Assoc. 2018;7:e009884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kayvanpour E, Sedaghat-Hamedani F, Amr A, Lai A, Haas J, Holzer DB, Frese KS, Keller A, Jensen K, Katus HA, Meder B. Genotype-phenotype associations in dilated cardiomyopathy: meta-analysis on more than 8000 individuals. Clin Res Cardiol. 2017;106:127–139. [DOI] [PubMed] [Google Scholar]
- 41.NIH Office of Extramural Research. Key Elements to Consider in Preparing a Data Sharing Plan Under NIH Extramural Support [Internet]. 2012; Available from: https://www.nlm.nih.gov/NIHbmic/data_sharing_plan.html
- 42.Libraries MIT. Data management, Share your data [Internet]. 2019; Available from: https://libraries.mit.edu/data-management/share/journal-requirements/
- 43.Hoff K, Lemme M, Kahlert A-K, et al. DNA methylation profiling allows for characterization of atrial and ventricular cardiac tissues and hiPSC-CMs. Clin Epigenetics. 2019;11:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shu L, Zhao Y, Kurt Z, Byars SG, Tukiainen T, Kettunen J, Orozco LD, Pellegrini M, Lusis AJ, Ripatti S, Zhang B, Inouye M, Mäkinen VP, Yang X. Mergeomics: Multidimensional data integration to identify pathogenic perturbations to biological systems. BMC Genomics. 2016;17:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang S, Chaudhary K, Garmire LX. More Is Better: Recent Progress in Multi-Omics Data Integration Methods. Front Genet. 2017;8:84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hasin Y, Seldin M, Lusis A. Multi-omics approaches to disease. Genome Biol. 2017;18:83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Meder B, Haas J, Sedaghat-Hamedani F, et al. Epigenome-Wide Association Study Identifies Cardiac Gene Patterning and a Novel Class of Biomarkers for Heart Failure. Circulation. 2017;136:1528–1544. [DOI] [PubMed] [Google Scholar]
- 48.Nakatochi M, Ichihara S, Yamamoto K, Naruse K, Yokota S, Asano H, Matsubara T, Yokota M. Epigenome-wide association of myocardial infarction with DNA methylation sites at loci related to cardiovascular disease. Clin Epigenetics. 2017;9:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wingrove JA, Daniels SE, Sehnert AJ, Tingley W, Elashoff MR, Rosenberg S, Buellesfeld L, Grube E, Newby LK, Ginsburg GS, Kraus WE. Correlation of peripheral-blood gene expression with the extent of coronary artery stenosis. Circ Cardiovasc Genet. 2008;1:31–38. [DOI] [PubMed] [Google Scholar]
- 50.Thomas GS, Voros S, McPherson JA, Lansky AJ, Winn ME, Bateman TM, Elashoff MR, Lieu HD, Johnson AM, Daniels SE, Ladapo JA, Phelps CE, Douglas PS, Rosenberg S. A blood-based gene expression test for obstructive coronary artery disease tested in symptomatic nondiabetic patients referred for myocardial perfusion imaging the COMPASS study. Circ Cardiovasc Genet. 2013;6:154–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.