

Abstract
The regioselective Z-isomerization of thermodynamically stable all-trans retinoids remains challenging, and ultimately limits the availability of much needed therapeutics for the treatment of human diseases.1–3 We present here a novel, straightforward approach for the catalytic Z-isomerization of retinoids using conventional heat treatment or microwave irradiation. A screen of 20 transition metal-based catalysts identified an optimal approach for the regioselective production of Z-retinoids. The most effective catalytic system was comprised of a palladium complex with labile ligands. Several mechanistic studies, including isotopic H/D exchange and state-of-the-art quantum chemical calculations using coupled cluster methods indicate that the isomerization is initiated by catalyst dimerization followed by the formation of a cyclic, six-membered chloropalladate catalyst-substrate adduct, which eventually opens to produce the desired Z-isomer. The synthetic development described here, combined with thorough mechanistic analysis of the underlying chemistry, highlights the use of readily available transition metal-based catalysts in straightforward formats for gram-scale drug synthesis.
Keywords: transition metal catalyst, retinoids, catalytic isomerization, microwave reaction, local coupled cluster calculations
Introduction
Retinoids are an essential class of biomolecules4 involved in vertebrate growth and development, cell differentiation, embryonic development5, vision6, 7, immunological mechanisms8, and reproduction.9 The ability to gain straightforward synthetic access to the various cis-isomers of the polyene chain is of utmost translational importance. One example of a clinically important cis-isomer is 13-cis-retinoic acid, which is used clinically to treat a broad number of disorders, including oral leukoplakia and severe acne.10–12 While efficient synthetic access to 13-cis-retinoids has been established for some time, the catalytic production of 9-cis-retinoids from their all-trans counterparts remains elusive. 9-cis-retiniods are of critical importance due to the clinical utility of 9-cis-retinoids in treating a subset of Leber congenital amaurosis (LCA), an inherited retinal degenerative disease.1, 13 The molecular etiology of LCA is the inability to generate the 11-cis-retinoid chromophore required for vision via genetic abnormalities in the enzymes required for either all-trans-retinyl ester formation (lecithin:retinol acyl transferase or LRAT) or isomerization of these esters by a retinoid isomerase, RPE65.14 Although pharmacologic delivery of 11-cis-retioids is not possible due to their metabolic instability, 9-cis-retinoids have been delivered effectively to treat this disease.1, 13 In addition to their use in the treatment of LCA, several retinoids have shown promising activity as antitumor and chemopreventive agents against cancer by inhibiting carcinogenesis at the initiation, promotion, and progression stages.3 The anticancer activity of retinoids is mainly due to their binding to nuclear receptors that act as hormone receptors activating target genes.15, 16
Several methods for the production of 9-cis-retinoids have been reported, including total synthesis,17–20 however these methods are complicated and require multi-step preparations with low yield and contamination by several geometric isomers. In one example, the synthesis of 9-cis-retinyl acetate is comprised of several sequential steps: (i) the Reformatsky reaction between β-cyclocitral and ethyl 4-bromo-3-methylbut-2-enoate, (ii) reduction and acidification steps, (iii) the Horner-Emmons reaction, (iv) ester reduction to 9-cis-retinol and (v) esterification to obtain 9-cis-retinyl acetate with an overall yield < 8%.21
Inspired by the need for more efficient routes to the thermodynamically less stable Z-retinoid, our current investigation examines the effect of the catalytic Z-isomerization of seven representative retinoids. In this study, we examine the ability of 20 commercially available transition metal-based catalysts to induce Z-isomerization and describe a method of repetitive crystallization to increase the yield to 100%. We subsequently perform extensive mechanistic studies to rationalize our catalytic yields and Z-isomer product distribution (Scheme 1)
Scheme 1.
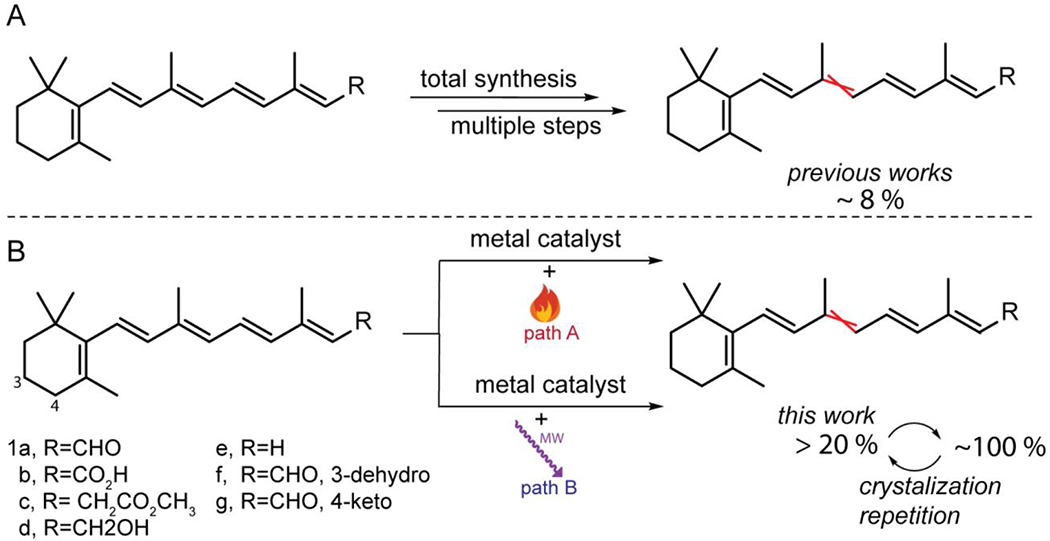
Synthetic strategies for cis-retinoids. A) The E to Z isomerization of retinoids from previous work using multistep total synthesis. B) Our approach illustrating the catalytic synthesis of 9-cis-retinoids using conventional heat and microwave irradiation, followed by a repetitive crystallization method.
Materials and Methods
Reagents
All reagents, catalysts, and standard retinoids were obtained from commercial suppliers (Sigma Aldrich, St. Louis, MO and Toronto Research Chemical, Toronto, Canada) and used without any further purification, unless otherwise noted.
Instrumentation
Nuclear Magnetic Resonance (NMR) Spectroscopy
The NMR spectra were recorded on a Bruker 500 MHz and 800 MHz apparatus at 25 °C, and referenced relative to the residual proton resonances of C6D6 (δ = 7.16 ppm). 1H NMR spectra of the compounds were collected with their corresponding 2D nuclear Overhauser effect spectroscopy, and heteronuclear single quantum coherence (HSQC) spectroscopy. Spectra were processed using MestReNova software (Mestrelab Research). The coupling constants (J) are reported in Hertz (Hz).
Mass Spectroscopy (MS)
MS-based detection was performed with an LTQ linear ion trap mass spectrometer (ThermoFisher Scientific, Waltham, MA) equipped with an electrospray ionization interface operated in the positive ionization mode. Parameters of ionization and detection were tuned with standards.
High Performance Liquid Chromatography (HPLC) Analysis
Procedures were performed under a dim red light. Retinoids were stored in N,N-dimethylformamide under argon at −80 °C. Retinoid analysis was performed on an Agilent 1200 series HPLC equipped with a diode array detector and Agilent Chemstation A.10.01 software (Agilent, Palo Alto, CA). A normal phase column (Beckman Ultrasphere Si 5μ, 4.6 × 250 mm [Beckman Instruments, Fullerton, CA]) and an isocratic solvent system of 2-10% ethyl acetate in hexane (v/v) were used at 20 °C and a flow rate of 1.4 ml/min.
A reverse phase column (C18 4.6 x 250 mm, 5 μm, Phenomenex, Torrance, CA) and an isocratic solvent system of 40% (v/v) CH3CN in aqueous 0.1% trifluoroacetic acid (TFA) were used. For retinal, retinal derivatives, retinol and retinyl acetate, the analyses were performed at 325 nm. Retinoic acid analysis was performed at a flow rate of 1 ml/min at 20 °C with detection at 355 nm.
All the HPLC measurements were calibrated using standards of all-trans-retinoids, 9-cis-retinoids, 13-cis-retinods and 4-keto-9,13-dicis-retinal.
Conventional Reactions
Reactions were carried out by dissolving retinoids (0.1 mmol), triethylamine (TEA, 1 mol %), catalyst (2 mol %) in different solvents for 2-20 h at 65 °C under argon atmosphere and dark conditions. After the treatment, samples were dissolved in hexane or CH3CN and analyzed by reverse phase or normal phase HPLC.
For mechanistic studies, the reaction of all-trans-retinal 0.1 M with 2 mol % catalyst 1, (CH3CN)2PdCl2 in CH3CN at 65 °C was performed in the presence of TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxy), 1, 5 and 10 mol % and monitored by HPLC for 2 h.
Microwave Reactions
Reactions were performed for 5-20 min under dark conditions. Retinoids (0.1 mmol) and selected catalysts (2 mol %) were dissolved in DMF or CH3CN. The samples were then transferred to 10-ml screw-capped glass vials, and the headspace was purged with nitrogen gas, and vials were immediately closed to minimize oxygen exposure. Subsequently, the samples were microwave-treated at 60-150 °C and 70-150 W power (Biotage LLC, NC, USA). After the treatment, samples were extracted with 0.4 mL hexane and analyzed by HPLC.
Kinetic studies
A stock solution of all-trans-retinal 1a, 0.1 M and catalyst I, 0.01 M were prepared. The reactions were performed in four conditions with 1a and dilution of catalyst I (0.95-4 mol %; 0.004-0.00095 M. The reactions were initiated by addition of catalyst I, (CH3CN)2PdCl2 (final concentration of 0.954, 1.53, 2.44 and 3.91 mM), in CH3CN at 65 °C for 2h. All manipulations were done under dim light and the reactions were performed in amber glass vials. At various times a small aliquot of 10 μL was removed and added to a mixture of 100 μL H2O and 50 μL of Hexane. After extraction, 10 μL of Hexane layer were injected into the HPLC to monitor the change in all-trans-retinal absorbance at λ=360 nm. The procedure was repeated three times.
Synthesis of 9-cis-retinyl acetate (2c)
All-trans-retinyl acetate 1c (300 g, 0.91 mol) was dissolved in a solution of hexane (600 ml) and TEA (0.11 ml, 0.83 mmol). After addition of 4 g (0.018 mol) of (CH3CN)2PdCl2 I, the mixture was stirred overnight in the dark at 65 °C under nitrogen. The resulting solution was cooled to room temperature for 1 h and then further cooled to −80 °C. The all-trans-retinyl acetate that crystallized was filtered by suction using a filter precooled with dry ice. The filtrate was concentrated and further crystallized. This process was repeated to obtain a 70:30 mixture of 9-cis-/all-trans-retinyl acetate, which mostly was used for the next steps without further purification (70%, 80 g).
One hundred milligrams of the above mixture were purified as a control by HPLC with 2% ethyl acetate:hexane, producing the desired compound, 9-cis-retinyl acetate 2c. (65%, 55 mg). 1H NMR (500 MHz, C6D6) δ (ppm) 7.02-6.84 (m, 2H), 6.31(d, J= 15.9 Hz, 1H), 6.20 (d, J= 15.1 Hz, 1H), 6.03 (d, J= 11.4 Hz, 1H), 5.61 (t, J= 7.3 Hz, 1H), 4.61(d, J= 7.3 Hz, 2H), 1.96-1.91 (m, 5H), 1.79 (s, 3H), 1.66 (s, 3H), 1.57-1.53 (m, 5H), 1.46- 1.43 (m, 2H), 1.10 (s, 6H). 13C NMR (126 MHz, C6D6) δ 170.08, 138.94, 138.54, 136.10, 134.97, 130.71, 129.64, 129.61, 128.90, 125.67, 124.75, 61.14, 39.81, 34.53, 33.26, 29.19, 22.07, 20.53, 19.67, 12.53.
NMR assignment was previously reported.22
Synthesis of 9-cis-retinol (2d)
To a mixture of 70:30 (9-cis-retinyl acetate/ All-trans-retinyl acetate) obtained above (50 ml) in ethanol (180 ml), a solution of NaOH (18 g, 0.45 mol) in water (80 ml) was added dropwise at 40 °C under nitrogen in the dark. After stirring for 30 min, the mixture was cooled to 0 °C and then extracted with hexanes (3 × 200 ml). The combined organic layers were washed with ice water twice, dried over anhydrous MgSO4, filtered, and the filtrate was concentrated under vacuum. The resultant yellow oil was dissolved in a solution of pyridine (0.18 ml) in methyl formate (90 ml), stirred at 0 °C for 2 h, and then cooled to −20 °C for overnight crystallization to obtain compound 2d. (90%, 40 g). 1H NMR (500 MHz, C6D6) δ 7.01 (d, J = 16.1 Hz, 1H), 6.91 (dd, J = 15.1, 11.4 Hz, 1H), 6.33 (d, J = 16.0 Hz, 1H), 6.25 (d, J = 15.1 Hz, 1H), 6.09 (d, J = 11.3 Hz, 1H), 5.58 (t, J = 6.8 Hz, 1H), 3.96 (d, J = 6.7 Hz, 2H), 1.94 (s, 5H), 1.87 (d, J = 15.5 Hz, 1H), 1.81 (s, 3H), 1.57 (dd, J = 7.6, 4.3 Hz, 2H), 1.54 (s, 4H), 1.49 – 1.42 (m, 2H), 1.11 (s, 6H).
Complete NMR assignment was previously reported.23
Synthesis of 9-cis-retinal (2a)
To a solution of 2d (20 g, 0.07 mol) in dry methylene chloride (200 ml), manganese dioxide (170 g, 1.9 mol) was added in the dark and the mixture was stirred overnight at room temperature. The mixture was then filtered, and the filtrate was evaporated; both steps were conducted under vacuum. The resulting oil was dissolved in hexane (80 ml) and cooled to −80 °C. Crystals from the oil were filtered by suction using a filter precooled with dry ice to obtain compound 2a. (80%, 15 g). 1H NMR (400 MHz, CDCl3) δ 10.09 (d, J = 8.2 Hz, 1H), 7.22 (dd, J = 15, 11.6 Hz, 1H), 6.66 (d, J = 15.8 Hz, 1H), 6.33 (d, J = 15.8 Hz, 1H), 6.30 (d, J = 15 Hz, 1H), 6.09 (d, J = 11.6 Hz, 1H), 5.97 (d, J = 8.2 Hz, 1H), 2.31 (s, 3H), 2.05 (m, 2H), 2.02 (s, 3H), 1.75 (s, 3H), 1.64 (m, 2H), 1.48 (m, 2H), 1.04 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 191.4, 155.2, 140.4, 138.2, 134.0, 131.5, 131.3, 130.6, 129.5, 129.2, 128.1, 39.64, 34.45 33.28, 29.20, 22.09, 21.21, 19.38, 13.40.
NMR assignment was previously reported.24
Synthesis of 4-keto-retinal (1g)
The compounds were prepared according to the procedure described in the literature.24 Briefly, to the all-trans-retinal (500 mg, 1.76 mmol) in a mixture of CH3CN (34.9 mL) solution, CH2Cl2 (3.5 mL) and H2O (3.5 mL), NBS (622.4 mg, 3.49 mmol) was added at −15 °C under a red dim light. After 5 min, N,N-diethylaniline (349.6 μL) was added. The reaction mixture was stirred an additional 5 min, poured into 5% aqueous Na2S2O3 solution and extracted with Diethylether (x3). The combined organic solution was washed with 1 M aqueous HCl solution, H2O, and brine successively. After drying with MgSO4, the extract was concentrated under reduced pressure and purified by HPLC with 10% ethyl acetate:hexane, producing the desired compound, 4-keto-all-trans-retinal 1g (80%, 350 mg). 1H NMR (800 MHz, C6D6) δ (ppm) 10.00 (d, J= 7.8 Hz, 1H), 6.73 (dd, J= 15.1, 11.4 Hz, 1H), 6.21 (d, J= 16.2 Hz, 1H), 6.14 (d, J= 16.2 Hz, 1H), 6.03 (d, J= 15.1 Hz, 1H), 5.95 (dd, J= 21.5, 9.6 Hz, 2H), 2.39 (m, 2H), 2.10 (s, 3H), 1.69 (s, 3H), 1.65 (s, 3H), 1.47 (m, 2H,), 0.91 (s, 6H). 13C NMR (126 MHz, C6D6) δ (ppm) 196.82, 189.48, 158.59, 152.16, 139.91, 138.42, 136.80, 132.56, 130.50, 130.40, 130.03, 127.98, 127.78, 127.59, 126.44, 37.20, 35.19, 34.19, 27.15, 13.85, 12.15.
NMR assignment was previously reported.25
Synthesis of 4-keto-retinal isomers (2g-4g)
4-keto-all-trans-retinal (400 mg, 1.34 mmol) was dissolved in a solution of CH3CN (80 ml). After addition of 8 mg (0.03 mmol) of bis(benzonitrile)palladium chloride, the mixture was stirred for 2 h in the dark at 65 °C under nitrogen. The resulting solution was cooled to room temperature, concentrated under reduced pressure and purified by HPLC with 10% ethyl acetate:hexane, producing the desired compounds, 4-keto-9-cis-retinal 2g (20%, 100 mg), 4-keto-13-cis-retinal 3g (30%, 120 mg), and 4-keto-9,13-dicis-retinal 4g (10%, 40 mg).
4-keto-9-cis-retinal 2g:
1H NMR (800 MHz, C6D6) δ (ppm) 9.94 (d, J= 7.9 Hz, 1H), 6.95-6.85 ( m, 2H), 6.15 (d, J= 16.0 Hz, 1H), 5.97-5.91 (m, 2H), 5.89 (d, J= 11.6 Hz, 1H), 2.37-2.33 (m, 2H), 2.10 (s, 3H),1.75 (s, 3H), 1.59 (s, 3H), 1.49-1.43 (m, 2H), 0.88 (s, 6H).
4-keto-13-cis-retinal 3g:
1H NMR (800 MHz, C6D6) δ (ppm) 10.13 (d, J= 7.2 Hz, 1H), 7.11 (d, J= 14.9 Hz, 2H), 6.66 (dd, J= 15.0, 11.4 Hz, 1H), 6.24 (d, J= 16.2 Hz, 1H), 6.15 (d, J= 16.2 Hz, 1H), 5.97 (d, J= 11.4 Hz, 1H), 5.75 (d, J= 7.3 Hz, 1H), 2.39 (t, J= 6.9 Hz, 2H), 2.12 (s, 3H), 1.65 (s, 3H), 1.56 (s, 3H), 1.49-1.45 (m, 2H), 0.92 (s, 6H).
4-keto-9,13-dicis-retinal 4g:
1H NMR (800 MHz, C6D6) δ (ppm) 10.08 (d, J= 7.3 Hz, 1H),7.05 (d, J= 14.2 Hz, 1H), 6.90-6.82 (m, 2H), 6.15 (dd, J= 16.0, 8.4 Hz, 1H), 5.92 (d, J= 11.5 Hz, 1H), 5.70 (d, J= 7.3 Hz, 1H), 2.37-2.33 (m, 2H), 2.10 (s, 3H), 1.77 (s, 3H), 1.47 (s, 3H), 1.45-1.42 (m, 2H), 0.88 (s, 6H).
Computational Study
Geometry optimizations were carried out within the density functional theory (DFT) framework using BP86 functional26, 27and def2-TZVP basis set28 along with the corresponding def2-ECP effective core potential assigned for metals (Pd - 28 electrons replaced, Pt – 60 electrons replaced). The description of dispersion interactions was improved with the Grimme’s D3BJ correction.29, 30 This DFT set-up is abbreviated as BP86+D3. The obtained structures were subjected to numerical second derivative calculations – energy minima had only positive normal modes, while transition states had exactly one imaginary mode. To check which minima were connected to a given transition state, we performed steepest descent geometry minimizations in forward and backward directions of the transition mode (this procedure is known as intrinsic-reaction coordinate scan, IRC). All calculations were performed with the ORCA 4.1 suite program.31
To avoid uncertainties associated with common density functional theory approximations, we re-calculated all single-point energies using a high-level wave-function-based domain-based local pair natural orbital-coupled cluster method with a single, double and perturbative triple excitations method32 at the complete basis set limit (DLPNO-CCSD(T)/CBS). The method was extensively benchmarked recently in various applications and showed excellent accuracy with respect to experimental data.33–36
The (complete basis set) CBS limit was estimated in the following way: the DLPNO-CCSD(T) calculations were carried out with the def2-TZVPP basis set to obtain coupled cluster correlation energy values, E(corr)CCdef2-TZVPP. Subsequently, more economic MP2 calculations37, 38 were performed using def2-TZVPP and def2-QZVPP basis sets. The two calculations were used to obtain a Hartree-Fock reference and MP2 correlation energies at the CBS limit using a well-known two-point extrapolation scheme (this yields EHFCBS, E(corr)MP2def2-TZVPP and E(corr)MP2CBS).32 The final DLPNO-CCSD(T)/CBS energy is estimated as: E(DLPNO-CCSD(T)/CBS) ≈ EHFCBS + E(corr)CCdef2-TZVPP + (E(corr)MP2CBS - E(corr)MP2def2-TZVPP). 39 Thermodynamic corrections calculated at the BP86+D3 level were added to the final E(DLPNO-CCSD(T)/CBS) energies to obtain Gibbs free energies. If not stated otherwise, the final relative G(DLPNO-CCSD(T)/CBS) values are reported in the body of the manuscript.
The resolution of identity (RI)40, 41 and chain of spheres (COSX)42 approximations were employed to efficiently evaluate Coulomb and exchange integrals, respectively. The DFT integration grid was enlarged to 4 (grid4 in ORCA nomenclature) while for numerical exchange calculations gridx5 was used. DLPNO-CCSD(T) calculations were performed with the NormalPNO settings that were demonstrated to provide excellent balance between calculation time and accuracy.36
Structures of key intermediates, absolute energies used to compute relative energies and number of imaginary frequencies for all species reported in the manuscript are provided in the Supplementary Information.
Results and Discussion
Condition optimization and screening of inorganic catalysts
In the search for an optimal catalyst, we performed the above reactions with twenty transition metal catalyst complexes (all including ruthenium, rhodium, palladium, iridium, platinum, or gold) and investigated their effects on the Z-isomerization of seven retinoid molecules. The rationale behind the utilization of these metals as catalyst complexes is their ability to catalyze reactions under milder conditions with higher selectivity as compared to other metals.43–45
Our list of transition metal catalysts consists of Pd metal-based catalysts with small/labile and bulky/rigid ligands, Pt metal-based catalysts with small and bulky ligands, and Ru, Ir, Au and Rh metal-based catalysts. (Figure 1).
Figure 1.
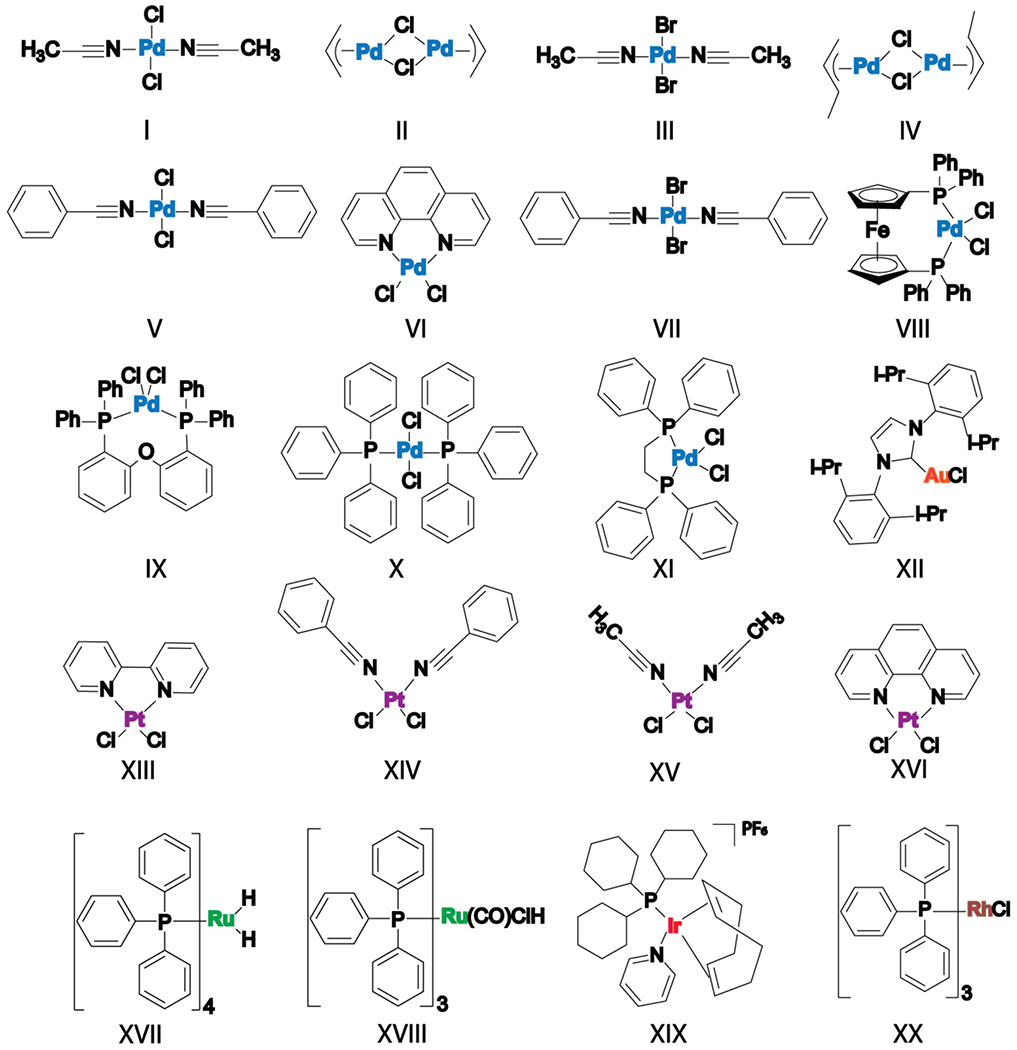
The structures of transition metal-based complexes. Pd metal-based catalysts with small/labile and bulky/rigid ligands are represented in blue for the central metal. Pt metal catalysts with small and bulky ligands are represented in purple for the central metal. Ru metal catalysts are represented in green for the central metal and Ir, Au and Rh metal catalysts are represented in red, orange and brown respectively for the central metal.
First, we performed the catalysis reaction on seven biologically relevant retinoid molecules, extensively optimizing the reaction conditions across different solvents and reaction times (Supporting information, SI Table S5). We found that replacement of non-polar solvent (hexane) with polar, aprotic solvents (CH3CN or acetone) reduced the reaction time from 20 h to 2 h while maintaining a favorable ratio between the cis isomers.
For example, the optimal conditions for heat-induced isomerization of all-trans-retinyl acetate 1c with (CH3CN)2PdCl2 I in non-polar solvent (hexane- this hydrophobic solvent was used because of the propensity of retinoids to convert to anhydro-vitamin A in nucleophilic or acidic solvents) and a mildly basic environment (triethylamine, TEA), at 65 °C for 20 h (standard conditions), produced a 17% conversion of 9-cis isomer 2c (Figure 2 and SI Table S5). While the same reaction in CH3CN produced a 20% conversion of 9-cis after 2 h. This observation might be explained by the greater solubility of the compounds and the catalysts in this solvent. For all-trans-retinal 1a with the palladium catalyst I in CH3CN solvent containing TEA at 65 °C, this yielded 38% conversion to cis isomers 13-cis: 9-cis (3a:2a) ratio of 1.3:1, respectively, in 2 hours, as determined by HPLC (SI Table S5).
Figure 2.
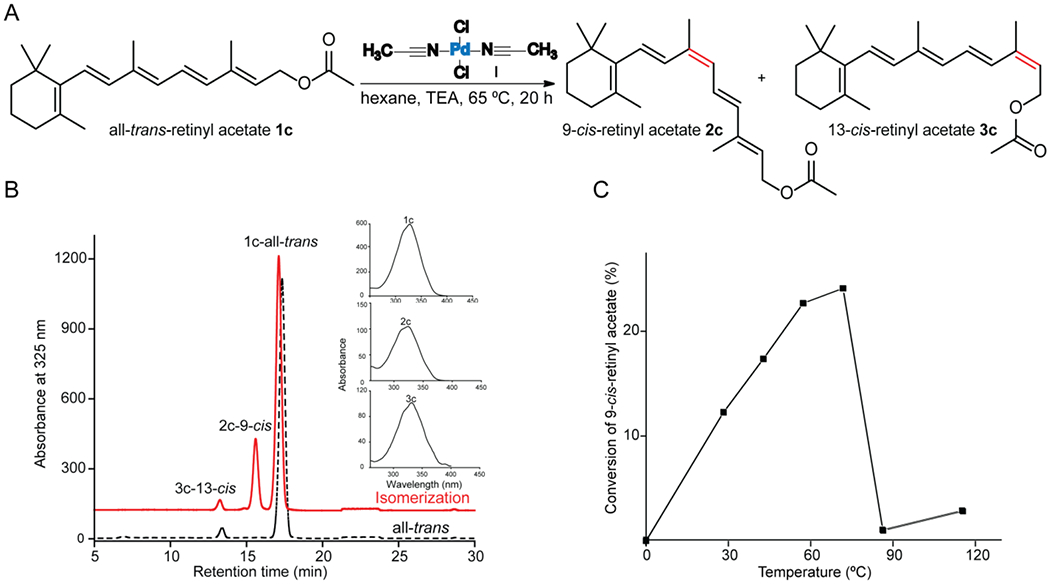
Z-isomerization of all-trans-retinyl acetate. A) Palladium chloride diacetonitrile complex-catalyzed isomerization reaction of all-trans-retinyl acetate 1c, producing 9-cis 2c and 13-cis 3c isomers. B) HPLC chromatogram of a mixture of retinyl acetate isomers obtained from traditional heat treatment. C) Temperature effects on isomerization of 1c to 2c.
We also observed that as the reaction temperature increased, yields of 1c increased, reaching a maximum between 65-75 °C before the retinoid levels decreased (Figure 2C). Further experiments using heat as an energy source were conducted at 65 °C.
Screening using conventional heat-induced isomerization
We first tested heat-induced isomerization with four representative retinoids (all-trans-retinal 1a, all-trans-retinoic acid 1b, all-trans-retinyl acetate 1c and all-trans-retinol 1d) and the catalysts I-XX under standard conditions. As shown in Table 1, the Z-isomerization mostly occurred in the presence of Pd-based catalysts. Reaction of all-trans-retinal 1a yielded a range of 33-39% conversion to cis isomers with the effective catalysts I-V, and VII with a ratio of 1.2-1.6:1 3a:2a (13-cis: 9-cis). In addition, the distribution of the cis-isomers 2a and 3a was monitored over time (SI Figure S3), and showed that 13-cis 3a is the kinetic product as the concentration of this isomer increases quickly in the first 10 min of the reaction followed by a slow decrease in concentration. On the other hand, the 9-cis 2a isomer is most probably the thermodynamic product, with a quasi-linear increase in concentration over time. (SI, Figure S3).
Table 1.
Transition metal-catalyzed Z-isomerization of all-trans-retinoid using traditional heat treatment.a,b
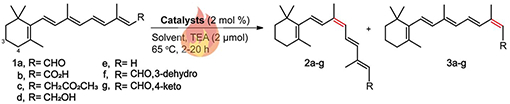 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Entry | Sub | Solvent | Time | Conversionc (%) | Selectivity of 9-cisd (%) | Product (3:2) | Ratio range | Effective Cat |
| 1 | 1a | C6H14 | 20 h | 33-39 | 37-44 | 13-cis: 9-cis | 1.2- 1.6: 1 | I-V, VII |
| 2 | 1b | C6H14 | 20 h | 15-34 | 21-98 | 13-cis: 9-cis | 0- 3.9: 1 | I, III-V, VII |
| 3 | 1c | C6H14 | 20 h | 18-24 | 85-90 | 13-cis: 9-cis | 0.1- 5.6: 1 | I-V, VII |
| 4 | 1d | C6H14 | 20 h | 16-21 | 77-94 | 13-cis: 9-cis | 0.1- 0.6: 1 | II, IV, VII |
| 5 | 1e | CH3CN | 2 h | 24 | 83 | 12-cis: 8-cis | 0.2: 1 | I |
| 6 | 1f | CH3CN | 2 h | 43-44 | 39-45 | 13-cis: 9-cis | 0- 3: 1 | I-V, XIX |
| 7 | 1g | CH3CN | 2 h | 33-46e | 45-55 | 13-cis: 9-cis | 0- 1.05: 1 | I-XI |
Reactions were carried out with all-trans-retinoid (0.1 mmol), catalyst (2 mol %), and TEA (0.2 μmol) at 65 °C under N2.
E/Z ratios reported are the average of two experiments and were determined by HPLC analysis.
Conversion to Z-isomer of the most effective catalysts.
Yields of 9-cis isomer for the most effective catalysts. 4.4 % of 9,13-dicis was detected. No reaction occurred without a catalyst for all substrates.
Retinoid 1b isomerization yielded a range of 15-34% cis isomers when catalysts I, III-V and VII were used, and the best result was obtained in the presence of catalyst III with 34% conversion to 9-cis isomer. Reaction of all-trans-retinyl acetate 1c provided yields ranging from 18-24% in the presence of I-V, and VII with the best ratio 0.1:1 for 3c:2c (13-cis: 9-cis). All-trans-retinol 1d was mostly reactive with catalysts II and IV, with 19% conversion. This analogue is unstable and has a high degradation rate (SI Tables S1–S4).
We further investigated the reaction of three additional retinoid derivatives (3-dehydro-retinal 1f, 4-keto-retinal 1g and anhydroretinol 1e) in CH3CN for 2 h at 65 °C. The results showed that both 1f and 1g displayed the highest conversion to 9-cis isomer in the presence of catalysts II and V with 19% and 22% of 2f and 2g, respectively. 1f displayed reactivity also with catalyst IX (17%). Moreover, slightly lower conversion values were observed for 1g with XIII-XX catalysts ranging from 10-16% (SI Tables S7 and S8).
We also examined the possibility of synergistic catalysis effects but did not detect higher yields as a result of these combinations (SI Table S9).
Overall, the conventional heating method for the Z-isomerization of retinoids showed that six palladium metal-based complexes (I-V and VII), all share relatively small and labile ligands, were more active than the rest of the metal catalysts. Palladium complexes with bulky ligands were less reactive perhaps due to steric hindrances which prevented them from forming dimers (vide infra). Also, catalysts with other central metals (e.g. Pt or Ru) were dramatically less reactive.
Screening using microwave irradiation-induced isomerization
We subsequently investigated the effect of microwave irradiation on the formation of Z-isomers based on the previously reported advantage of combining homogeneous catalysts with microwaves. Microwave irradiation produces efficient internal heating and dramatically reduces reaction time.46, 47 Three representative retinoids, (all-trans-retinal 1a, all-trans-retinoic acid 1b and all-trans-retinyl acetate 1c) were chosen for studies of microwave irradiation.
We investigated the effects of representative catalysts (I, V, XI, XV, and XVIII-XX) on the Z-isomerization of these retinoids. We also evaluated the effects of solvent, temperature and reaction time on Z-isomerization. Time points were taken after 5-20 min to investigate the reactions in two different solvents (DMF and CH3CN). The optimization indicated a superior 13-cis: 9-cis (3: 2) ratio in DMF. The efficiency of different temperatures and reaction times varied for individual retinoid derivatives (SI Table S11).
The results of the catalyst screen showed that only catalysts I and V were effective for the Z-isomerization (Table 2 and SI Table S12–S14). Retinoid 1a Z-isomerization with the active catalysts showed that the average of the 9-cis isomer 2a reached 15% in 5 min of microwave irradiation, when the temperature reached 80 °C (SI Table S1). 1b isomerization showed an average yield of 22% 2b with catalyst I and V, and the reaction of 1c yielded an average of 18% 2c in 15 min. (SI Table S14)
Table 2.
 | |||||||
|---|---|---|---|---|---|---|---|
| Entry | Sub | Time | Temp (°C) | Conversionc (%) | Selectivity of 9-cisd (%) | Ratio range (13-cis: 9-cis) | Effective Cat |
| 1 | 1a | 5 min | 80 | 27 | 58, 60 | 0.67: 1 | I, V |
| 2 | 1b | 10 min | 100 | 24 | 93 | 0.07- 0.09: 1 | I, V |
| 3 | 1c | 15 min | 80 | 23-27 | 67, 85 | 0.2- 0.5: 1 | I, V |
Reaction conditions: all-trans-retinoid (0.1 mmol) and catalyst (2 mol %) under N2 as determined by HPLC.
Each yield was averaged from n ≥ 2 reactions.
Conversion to Z-isomer of the most effective catalysts.
Yields of 9-cis-isomer for the most effective catalysts. No reaction occurred without a catalyst for all substrates.
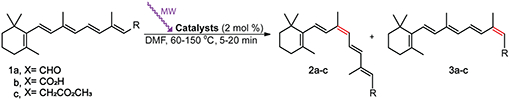
Additionally, we performed a time course comparison between conventional heating and microwave irradiation treatment. As shown in Figure 3, when the conventional heat-induced isomerization was conducted at 65 °C for 2 h, the 9-cis isomer of retinoid 1c reached a 15% yield for the effective catalyst V. By microwave treatment, the 9-cis isomer content reached 13% after 10 min of irradiation.
Figure 3.
Z-isomerization of all-trans-retinyl acetate 1c (0.1 mmol) with catalyst V (2 mol %) in CH3CN. 9-cis-retinyl acetate 2c yields under conventional heating and microwave irradiation.
Our findings demonstrate that microwave-induced Z-isomerization of retinoids is more efficient as compared to conventional heat-induced isomerization, particularly due to dramatically decreased reaction time. The most efficient catalysts were found to be Pd-based metal catalysts in both methods, suggesting that internal and external heat sources may retain similar mechanisms.
Mechanistic study
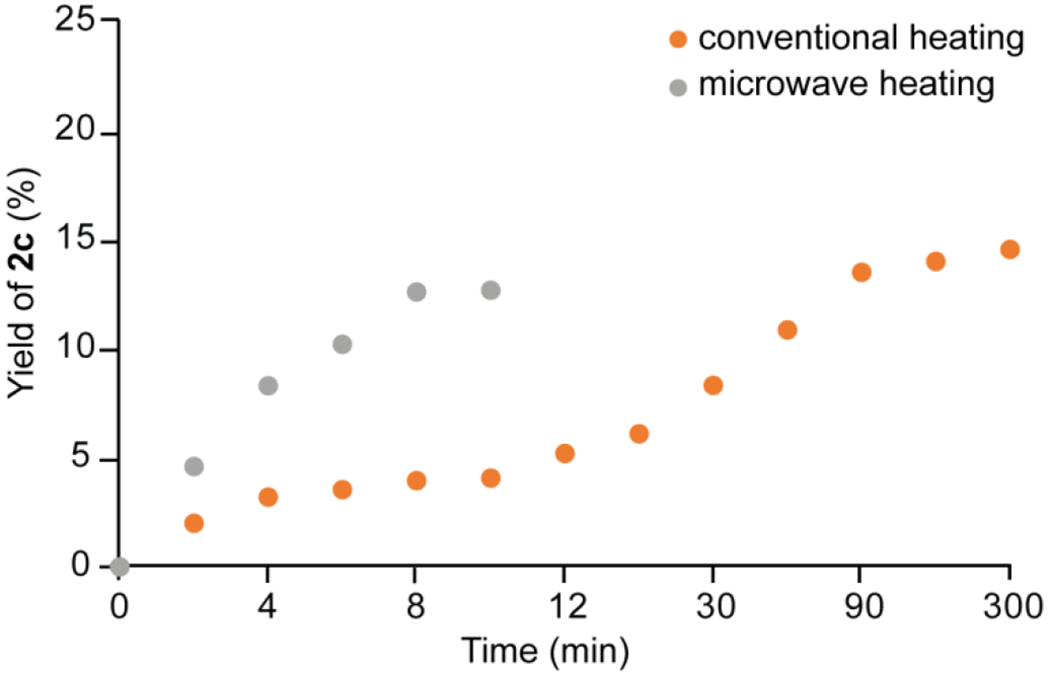
To further elucidate the mechanistic details related to formation of the Z-isomer using the effective palladium metal-based catalysts, we carried out several experiments. The literature suggests that an alkene-transition metal catalyst dependent isomerization can go through two main reaction pathways — isomerization via hydride and non hydride mechanisms.48–51 Hydrogen-deuterium exchange (HDX) experiments showed no deuterium incorporation by the Z-retinyl products in the presence of CD3CN or CD3OD, indicating that there is no solvent-dependent behavior necessary for hydride-based mechanisms (Figure 4A, B). Moreover, reaction of all-trans-retinal 1a in the presence of a Pd-H trap, TEMPO, at several concentrations and time points, indicated that there is no inhibition of Z-isomerization in the presence of the Pd-H trap, thereby confirming that the Z-isomerization mechanism does not involve a Pd-H intermediate (Figure 4C). In light of these findings, the reaction most likely proceeds through the non-hydride mechanism first suggested by Tan et al.50
Figure 4.
Mechanistic study of Pd-catalyzed retinyl acetate Z-isomerization reaction. A) Reaction of all-trans-retinyl acetate 1c with 2 mol % (CH3CN)2PdCl2 I in CD3CN or CD3OD (1) or in the presence of TEMPO (2) at several concentrations. B) LC-MS of retinyl acetate isomers obtained after heat treatment with catalyst I and characteristic MS/MS fragmentation patterns of the parent ion m/z = 269 [M + H]+. C) Comparison of 1c yields in the absence and presence of TEMPO, monitored by HPLC.
Kinetic study
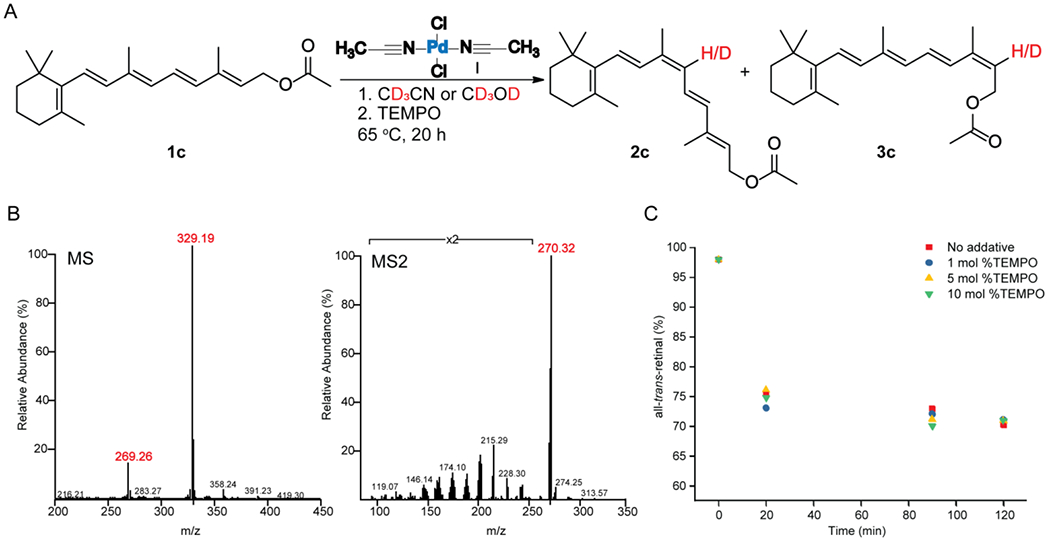
The kinetics of E/Z interconversion of all-trans-retinal 1a was studied by variation of the concentration of added (CH3CN)2PdCl2 I, 0.95 ≤ [catalyst I] ≤ 4 mM. Complete dataset and fits are shown in Figure 5A. As illustrated here, addition of catalyst I to the all-trans-retinal gives rise to kinetic traces that are described by single-exponential functions. Linear regression of pseudo first-order afforded kobs values that displayed a positive non-linear deviation for catalyst I.
Figure 5.
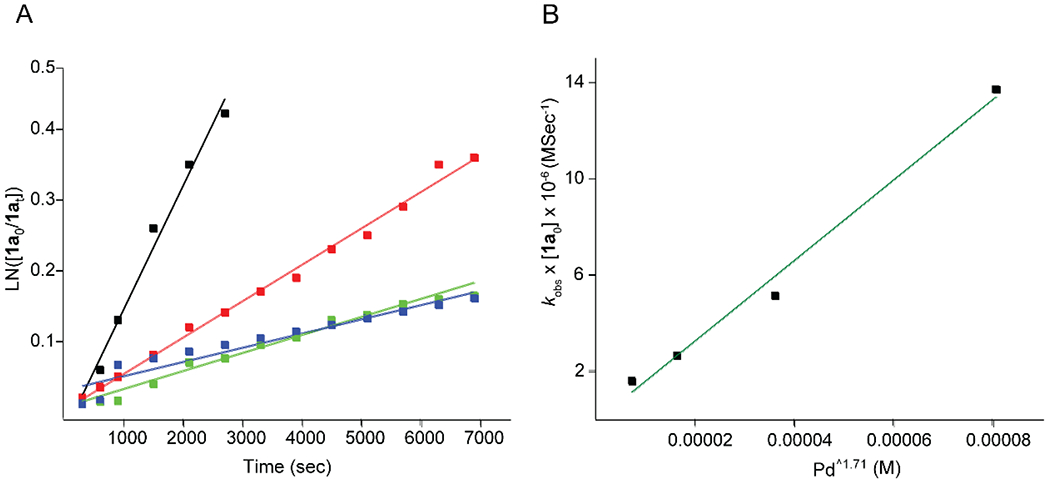
Z-isomerization of all-trans-retinal 1a catalyzed by (CH3CN)2PdCl2 I in CH3CN at 65 °C. A) Concentration change of 1a (0.1 M) as a function of time in different concentrations of catalyst I, 0.95 mM represented by blue, 1.53 mM represented by green, 2.44 mM represented by red and 3.91 mM represented by black. B) Observed pseudo-first-order rate constants (kobs)*[1a0] plotted as a function of Pd1.71. kobs= 0.24[Pd]1.71[1a0]−1.
Moreover, when the observed rate constants are plotted as a function of concentration of the added catalyst I, a non-linear dependence is observed. Non-linear regression of kobs×[all-trans-retinal0] versus [Pd] yielded the relationship kobs×[all-trans-retinal0] = 0.24(±0.16)×[Pd]1.71(±0.14) (Figure 5B). Consequently, the reaction corresponded to pseudo first order conversion with dependency on Pd catalyst which is larger than 1 that implies that palladium may form a dimer in order to react with all-trans-retinal 1a.
Computational study
To gain insights into the mechanism of Z-isomerization using metal-based catalysis, we further investigated the reaction using quantum chemical methods. The E to Z isomerization of an alkene involves rotation around an otherwise rigid carbon-carbon double bond. Figure 6 presents the relaxed potential energy surface scans for rotations around three selected double bonds of retinal 1a. As expected, in all cases a distinct maximum is found at the dihedral angle α=90°. The lowest electronic barrier of about 26 kcal/mol is associated with trans to 13-cis isomerization while other rotations have barriers of more than 30 kcal/mol.
Figure 6.
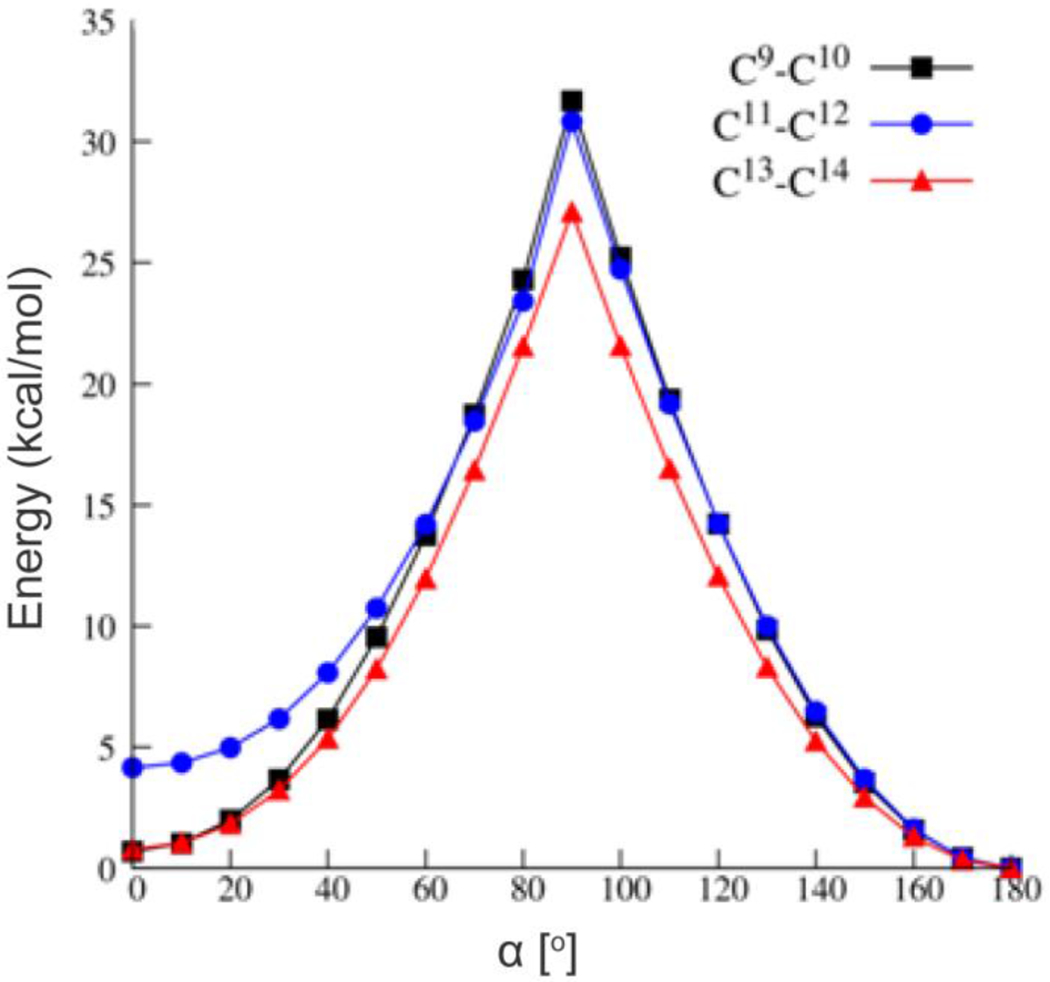
Relaxed potential energy surface scans along the dihedral angle (α) that defines the rotations around three key double bonds of 1a. α=0° corresponds to the cis isomer, while α=180° corresponds to the trans conformation. Calculations were performed at the BP86+D3 level of theory.
To obtain a better estimate for barrier heights we took efforts to compute true transition states (TS) for these processes. Hessian calculations carried out with the highest energy structures always yielded a single imaginary mode that was very low (about 30i cm−1). The TS optimizations along these modes consistently revealed the all-trans minimum structure. Thus, we were unable to locate true transition states and decided to accept the highest energy structures from the relaxed surface scans as the approximate TS. The changes in Gibbs free energy (ΔGE/Z) values as well as Gibbs free activation barrier (ΔGǂE/Z) values for the E to Z isomerization of 1a are presented in Table 3. BP86+D3 and reference DLPNO-CCSD(T) methods provide a consistent picture for 1a isomerization with barriers calculated using the latter approach of 33.3, 32.0 and 28.0 kcal/mol for all-trans conversion into 9-cis, 11-cis and 13-cis isomers, respectively. Interestingly, the 9-cis and 13-cis isomers are very close in energy to all-trans 1a irrespective of the method used in the calculations (ΔGE/Z < 0.5 kcal/mol). These calculations demonstrate that 11-cis formation is a rather thermodynamically unfavorable process with ΔGE/Z = 5.0 kcal/mol.
Table 3.
Comparison of Gibbs free energies (ΔGE/Z) and approximate activation Gibbs free energies (ΔGǂE/Z) calculated with BP86+D3 and DLPNO-CCSD(T) methods for the E to Z isomerization process that takes place around three key double bonds of 1a. All data in kcal/mol.
| C9-C10 | C11-C12 | C13-C14 | ||||
|---|---|---|---|---|---|---|
| ΔGE/Z | ΔGǂE/Z | ΔGE/Z | ΔGǂE/Z | ΔGE/Z | ΔGǂE/Z | |
| BP86+D3 | 0.4 | 31.0 | 4.1 | 30.1 | 0.6 | 26.4 |
| DLPNO-CCSD(T) | 0.0 | 33.3 | 5.0 | 32.0 | 0.5 | 28.0 |
Monomer mechanism
A good catalyst for E to Z isomerization should impact two key elements of the transformation: (1) weaken the C=C bond and (2) stabilize the TS structure. One can expect an alkene to replace a labile ligand of the catalyst. We selected two similar catalysts from our screening list (Figure 1), PdCl2(CH3CN) I and PtCl2(CH3CN) XV, to explore the Z-isomerization of retinoid 1a. These two catalysts differ only by the central atom, however they display dramatically different catalytic behavior with catalyst I the most effective catalyst in the set while catalyst XV is almost completely inactive. Table 4 shows the calculated Gibbs free energies (ΔGmono-complex) for the reaction of catalyst (I or XV) with retinoid 1a. The DLPNO-CCSD(T) numbers suggest that the complex formation is either mildly exothermic (complexation via C9-C10 and C11-C12 double bonds) or mildly endothermic (complexation via C13-C14). According to our results, the differences between catalysts I and XV are negligible (ΔGmono-complex differences up to 0.4 kcal/mol). The BP86+D3 method shows significant deviations from the DLPNO-CCSD(T) values of up to 5.1 kcal/mol in the case of catalyst I interacting via the C13-C14 double bond. Such differences highlight the complexity of electron correlation effects in the examined species.
Table 4.
Comparison of Gibbs free energies (ΔGmono-complex) calculated with BP86+D3 and DLPNO-CCSD(T) methods for the catalyst + 1a → adduct + CH3CN reaction. The catalyst was chosen as species I or XV (see Figure 1). All values in kcal/mol.
| C9-C10 | C11-C12 | C13-C14 | ||||
|---|---|---|---|---|---|---|
| Pd(I) | Pt(XV) | Pd(I) | Pt(XV) | Pd(I) | Pt(XV) | |
| BP86+D3 | −6.3 | −2.1 | −0.4 | −2.9 | −2.2 | 1.6 |
| DLPNO-CCSD(T) | −1.8 | −1.4 | −2.4 | −2.7 | 3.3 | 2.5 |
Based on Table 4, we first considered the reaction mechanism that involves simple rotation around selected C=C bonds in the complex of the catalyst with substrate 1a. The C=C bond lengths in the isolated 1a are all equal at 1.38 Å. They become elongated upon complexation to 1.44 Å (C9-C10 and C13-C14) or 1.42 Å (C11-C12) irrespective of the catalyst used. Thus, we expect the double bonds to be weakened when compared to the isolated species. Any attempt to locate the TS structures for rotations around C=C bonds in the complex of 1a with catalysts I and XV failed. This finding is consistent with the results obtained by Tan and co-workers.50 Thus, we decided to accept the highest energy structures from the relaxed potential energy surface scans along dihedral angles that correspond to rotations around three key double bonds in the complexes of 1a with the catalysts. These approximate activation Gibbs free energies (ΔGǂrot) calculated with BP86+D3 and DLPNO-CCSD(T) methods are reported in Table 5. It is clear that all barriers in the complex are larger than for the rotations in the free molecule 1a. For example, in the case of rotation around the C11-C12 double bond catalyzed by XV, the barrier increased by 20.1 kcal/mol. This observation could be explained by the formation of a Pd/Pt-C bond in the complex that is accompanied by a positive charge accumulation on the proximal carbon atom, which in turn is stabilized by the presence of the transition metal ion in close proximity. In the TS, such stabilization is lost and the quasi-carbocation has high relative energy with respect to the substrate. Therefore, catalysts I and XV do indeed offer C=C bond weakening (postulate 1 for a good catalyst) but do not provide TS stabilization (postulate 2). High energy barriers as well as the absence of pronounced experimental solvent effect (carbocation stabilization), rule out the possibility of the reaction occurring through a simple complexation-and-rotation mechanism.
Table 5.
Comparison of approximate activation Gibbs free energies (ΔGǂrot) calculated with BP86+D3 and DLPNO-CCSD(T) methods for the E to Z rotations around C9-C10, C11-C12 and C13-C14 double bonds that lead to 9-cis, 11-cis and 13-cis 1a isomers, respectively, catalyzed by complexes I or XV (see Figure 1). All values in kcal/mol.
| C9-C10 | C11-C12 | C13-C14 | ||||
|---|---|---|---|---|---|---|
| Pd(I) | Pt(XV) | Pd(I) | Pt(XV) | Pd(I) | Pt(XV) | |
| BP86+D3 | 34.7 | 31.7 | 32.5 | 37.7 | 26.5 | 23.8 |
| DLPNO-CCSD(T) | 46.2 | 45.2 | 49.2 | 52.1 | 32.5 | 41.7 |
Dimer mechanism
Based on the results for monomeric catalysts I and XV we postulate that the reaction follows the non-hydride binuclear addition-elimination pathway.50 This pathway involves formation of a chloropalladate intermediate in which the excessive positive charge of one of the carbon atoms is stabilized by the chlorine atom. This mechanism requires the catalyst to dimerize prior to complexation with alkene and is consistent with the presented kinetic data.
To test our hypothesis regarding the formation of a dimeric complex as a critical step in the overall reaction, we calculated the dimerization energy of catalyst I and its platinum analogue, catalyst XV, according to the general reaction 2 x catalyst I/XV → dimer + 2CH3CN.
We found that the monomeric catalyst I exists in a thermodynamic equilibrium with its dimer (ΔGdimerization= −0.02 kcal/mol with the dimer favored). Catalyst XV was used in a cis form but we expect that at reaction temperatures it will be present in a thermodynamically more stable trans form, especially when considering an isomerization mechanism via CH3CN loss.52 For the trans form of catalyst XV we found that dimerization is highly unfavorable with ΔGdimerization= +12.00 kcal/mol. We associate such a large difference in monomer-dimer preference with the difference in a labile ligand (CH3CN) dissociation free energy, which increases by +5.3 kcal/mol when going from palladium to platinum (the dimerization process requires two CH3CN molecules to dissociate). While it is known that ligands become less labile when going down the nickel group,53, 54 this effect was not explored in the present case. Platinum is expected to show significant relativistic effects that manifest particularly in low 5d-6s/p atomic levels splitting as compared to 4d-5s/p splitting found in palladium.55 Thus, in the former case we expect large 5d-6p hybridization (indirectly also with 6s) that should enhance binding of σ-donating/π-accepting molecules such as acetonitrile. According to Löwdin population (BP86+D3),56, 57 the effective electron configuration of Pd in catalyst I is [Kr]4d9.055s0.435p0.86 and Pt in catalyst XV is [Xe]4f145d8.935f0.056s0.556p0.61. At the same level of theory the metal-nitrogen Mayer bond orders58 were found to be 0.56 and 0.79 for catalyst I and XV, respectively. Thus, the increased population of 6s in catalyst XV as compared to 5s in catalyst I reflects a stronger σ-bond in the case of the platinum complex.
These results were obtained at the BP86+D3 level with a quasi-relativistic effective core potential (ECP) coupled to specially optimized def2-TZVP basis set applied for Pd and Pt atoms (see above). The interaction energy of a CH3CN fragment with MCl2(CH3CN), where M = Pd or Pt, calculated in this way yields interaction energies of −39.3 and −52.1 kcal/mol, respectively. An alternative way to cover relativistic effects59 is to use a scalar-relativistic Hamiltonian such as Douglas-Kroll-Hess at second-order (DKH2)60 along with a specially designed all-electron def2-TZVP basis set (all-electron analogue of ECP-based def2-TZVP).61 We performed such calculations and obtained interaction energies of −37.8 and −53.4 kcal/mol for Pd and Pt complexes, respectively. The agreement between quasi-relativistic and scalar-relativistic approaches is very close and encouraging owing to the simplicity of the latter method. However, the key step is to turn off relativistic effects entirely, i.e. adapt all-electron def2-TZVP basis set for non-relativistic calculations. In this case the interaction energies were found to be −31.6 and −32.3 kcal/mol for catalysts I and XV, respectively. For both systems studied, relativistic effects are pronounced – they make the interaction with CH3CN stronger but to different degrees. In the case of catalyst I, relativistic effects account for 6.2 kcal/mol of the total DKH2 interaction energy (16%). This number increases to 21.1 kcal/mol for catalyst XV where relativistic effects are responsible for 40% of the interaction energy.
Additionally, we examined another palladium catalyst with bulky ligands and calculated the dimerization energy. Catalyst X, PdCl2(PPh3), is closely related to catalyst I but like catalyst XV does not show significant activity in the E to Z isomerization reactions. The calculated dimerization energy for this catalyst was 27.1 kcal/mol, thus it exists mainly in the monomeric form. Phosphorus-based ligands are not as labile as, for example, CH3CN or PhCN and thus do not facilitate dimerization of the parent complexes.
Based on these results, catalysts X and XV do not form a dimer, which explains their lack of reactivity. This also explains the lack of reactivity of most of the catalysts which contain palladium complexes with bulky ligands that prevent their dimerization.
In the following, we decided to focus solely on (CH3CN)2PdCl2 catalyst I that can form a dimer, a prerequisite for the non-hydride binuclear addition-elimination pathway. The calculations were performed for all-trans-retinal 1a as a representative retinoid to study the Z-isomerization reaction pathway. The free energy profile and key intermediates involved in the proposed mechanisms are shown in Figure 7. Catalyst I dimer forms a reversible adduct with the all-trans substrate (calculated Pd-C bond formation values are ΔG = 4.7, 2.0 and 6.4 kcal/mol for C9, C11 and C13, respectively). The reaction proceeds via a six-membered cyclic chloropalladate intermediate. In contrast to the mechanism described by Tan et al., we were able to find only full chair-like chloropalladate conformations. For trans to 9-cis and 13-cis isomerizations, the intermediates are stable minima at their respective potential energy surfaces, while for trans to 11-cis conversion the intermediate has a transition state character (TSchloro). The stability of the intermediate is dictated by the partial positive charge stabilization that is largest for tertiary carbon atoms C9 and C13. Thus, in our model, rotations around the C9=C10 and C13=C14 double bonds are two-step processes, while rotation around the C11=C12 bond is a one-step process. We found that trans to 13-cis isomerization has the lowest energy barriers of ΔGǂ= +20.3 kcal/mol for chloropalladate intermediate formation and ΔGǂ= +3.1 kcal/mol for the chloropalladate ring-opening resulting in 13-cis product. These barriers for trans to 9-cis isomerization were 2.2 kcal/mol and 0.8 kcal/mol higher in energy, respectively. Thus, the catalytic effect of the palladium complex is to decrease the barrier for the rate-determining step of the reaction by about 10 kcal/mol in these cases. The trans to 11-cis one-step isomerization had a barrier of ΔGǂ= +25.2 kcal/mol, which is only 6 kcal/mol below the calculated barrier for uncatalyzed rotation around C11=C12 bond (Table 3). Moreover, the 11-cis adduct with the catalyst had 9.4 kcal/mol higher energy as compared to the all-trans adduct due to a steric encumbrance (Figure 7). Therefore, we conclude that trans to 11-cis isomerization is both kinetically and thermodynamically less favorable when compared to trans to 9- and 13-cis isomerization.
Figure 7.
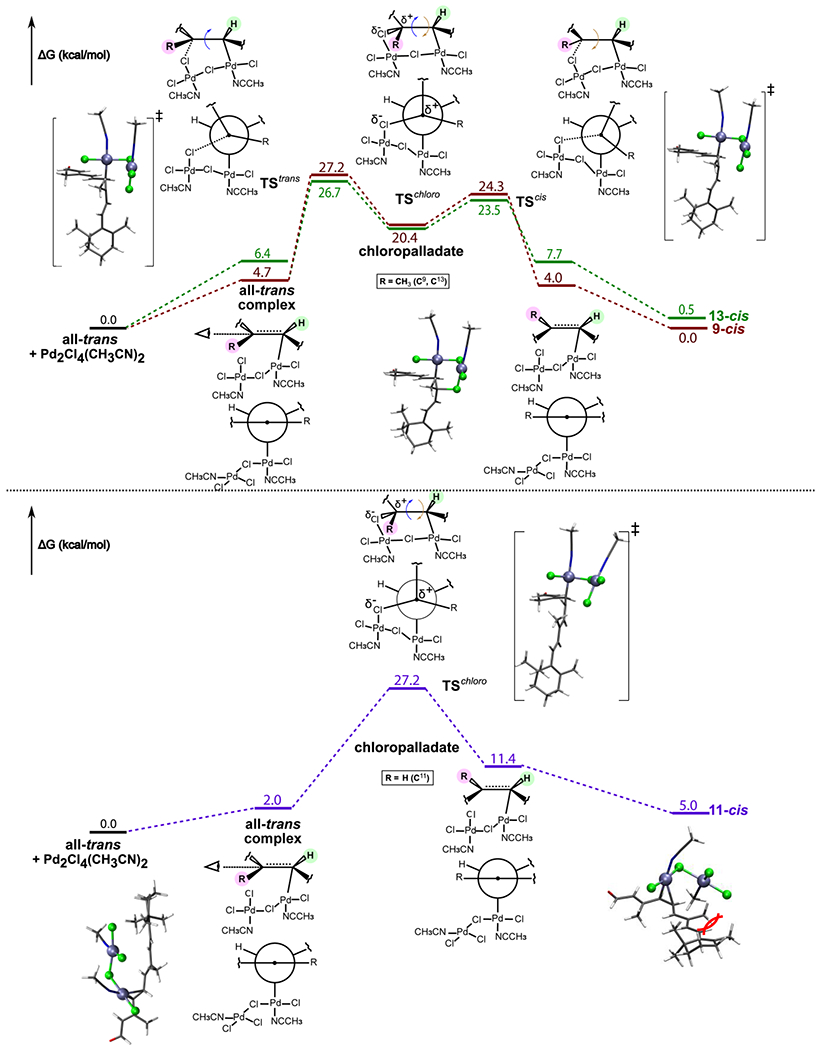
DLPNO-CCSD(T) free energy surfaces for all-trans-retinal 1a Z-isomerization reaction catalyzed by a dimeric form of catalyst I.
All-trans 1a and 9-cis 2a are virtually isoenergetic, and the 13-cis 3a isomer is only 0.5 kcal/mol higher in energy. The high relative stability of 9-cis and 13-cis can be explained by the fact that the trans-to-cis rotation swaps the positions of the -CH3 and –C(H)= groups (R1 and R2, respectively) interacting with the H atom attached to the neighboring carbon atom in the R1(R2)C=C(H)- bonding situation. In the presence of catalyst I, formation of the Pd-C9 bond was favored over Pd-C13 (Figure 7). The 9-cis isomer had the lowest absolute energy at the end; thus, at 65 °C, the reaction is largely thermodynamically controlled.
To further confirm that the reaction is thermodynamically controlled we performed simple kinetic simulations based on the dimeric mechanism presented in Figure 7. The changes of concentrations of various species involved in E/Z isomerization of 1a using catalyst I over time are presented graphically in the SI, Figure S6. Obtained curves resemble the shape of experimental concentration traces (SI, Figure S3): initially, the major product is 13-cis but its concentration decreases slowly in favor of the 9-cis product. Although the latter requires that a slightly larger energetic barrier must be passed, it features the lowest energy at the end of the reaction. Thus, based on the compatibility of experimental and computational results, we conclude that transition metal-based complexes with small, labile ligands capable of forming dimeric complexes are most effective for the Z-isomerization of retinoids.
The application
Here, we successfully developed a method which overcomes the obstacles associated with the total synthesis of Z-retinoids (which result in a relatively low yield of ~ 8%), and demonstrate the application of the current method by synthesizing gram-scale quantities of 9-cis-retinyl acetate, -retinol, and -retinal (2c, 2b, and 2a, respectively) (Scheme 2). Such quantities allow for large-scale tests of their therapeutic capabilities in retinal degeneration models in murine and canine models as a prelude to human investigations.62–65 As we have shown previously, supplementation of 3 mg of 9-cis-retinal 2a in the LCA dog model, which lacks visual cycle function, showed improvement of 25% in photoreceptor function. 62
Scheme 2.
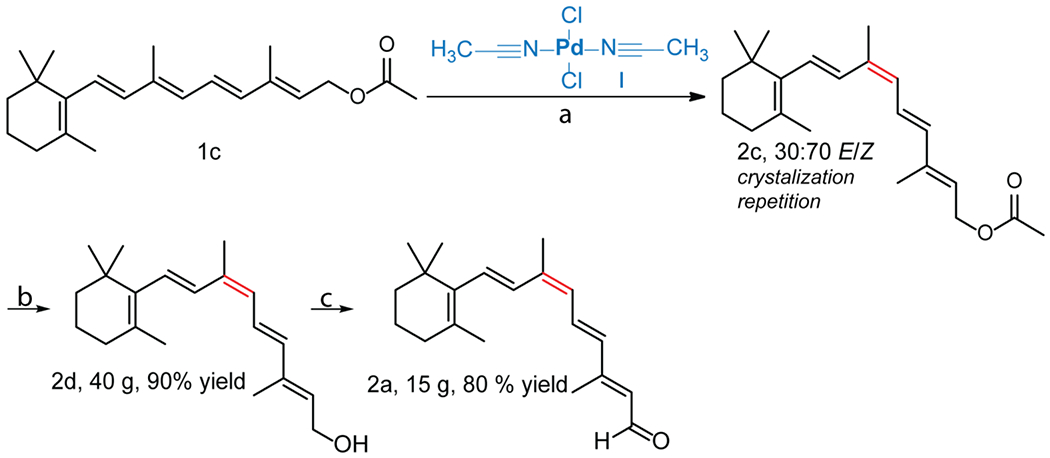
Large-scale synthesis pathway of 9-cis-retinal 2a. a) (CH3CN)2PdCl2, TFA, hexane at 65 °C, 20 h. b) NaOH, ethanol at 40 °C, 30 min. c) MnO2, DCM, 24 h.
The presented reaction of all-trans-retinyl acetate 1c that occurs most favorably in the presence of catalyst I yields an exceptional 20% of the desired 9-cis isomer. Under these conditions, we enriched the solution to a 70:30 mixture of 9-cis/all-trans by recrystallization and re-use of the parent compound 1c (See experimental section). Theoretically, the yield of this reaction could be increased to 100% due to our discovery of optimal conditions for crystallizing the parent compound for further loop reactions with the catalyst.
Conclusions
In this investigation, we developed a novel method for the Z-isomerization of retinoids with readily available transition metal catalysts in mild conditions using conventional heating or microwave irradiation. We were able to produce the stable 9-cis (2a-2g) isomers with a single step in relatively high yields, 16-25%. While it is common for other organic catalytic reactions to reach even higher yields, the method reported here roughly triples the best yield reported to date and also reduces the number of reaction steps as well as the time of the reaction.18, 19 In fact, the yields we report, coupled with the use of a crystallization loop technique, suggest that this method could be extremely useful in gram-scale synthesis of 9-cis isomers for studies on the level of large animals or humans.
The conventional heating method showed that palladium-based catalysts, in particular palladium catalysts capable of dimer formation complexes (I-V, VII), were more catalytically reactive than the rest of the metal catalysts. Moreover, we have shown that microwave-induced Z-isomerization of retinoids is even more efficient, and dramatically alters catalyst efficiency, decreasing the reaction time as compared to conventional heat-induced isomerization over 50 times.
In this report, we also presented a thorough mechanistic study of the catalyst I dimer-catalyzed isomerization of representative retinoids, all-trans-retinal to cis-retinal, by means of state-of-the art quantum chemical calculations and kinetic simulations to further support our hypothesis. The calculations explain the catalytic yields, the distribution of Z-isomers, the rationale behind the reactivity of the catalyst, all of which may help guide future catalyst selection and improvement.
Supplementary Material
Acknowledgements
This work was partially carried out at the Department of Pharmacology, Case Western Reserve University, Cleveland, Ohio, USA, and supported in part by grants from the National Institutes of Health (NIH) (EYR24024864 and R24EY027283 to KP), RPB (Research to Prevent Blindness) to the Department of Ophthalmology at UCI, the Canadian Institute for Advanced Research (CIFAR), and the Alcon Research Institute (ARI). K.P. is the Leopold Chair of Ophthalmology. A.K. acknowledges support from the National Science Centre, Poland, grant number 2018/30/E/ST4/00004. Access to high performance computing resources was provided by the Interdisciplinary Centre for Mathematical and Computational Modelling in Warsaw, Poland, under grants no. G64-9 and GB77-11.
Footnotes
Supporting Information
The Supporting Information is available.
Experimental details and characterization data (PDF)
Conflicts of interest
The authors declare no competing financial interests.
References
- 1.Maeda T, Maeda A, Casadesus G, Palczewski K and Margaron P, Invest. Ophthalmol. Vis. Sci, 2009, 50, 4368–4378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gearhart PM, Gearhart C, Thompson DA and Petersen-Jones SM, Arch. Ophthalmol, 2010, 128, 1442–1448. [DOI] [PubMed] [Google Scholar]
- 3.Altucci L and Gronemeyer H, Nat. Rev. Cancer, 2001, 1, 181. [DOI] [PubMed] [Google Scholar]
- 4.Ross AC and Gardner EM, Adv Exp Med Biol, 1994, 352, 187–200. [DOI] [PubMed] [Google Scholar]
- 5.Canete A, Cano E, Munoz-Chapuli R and Carmona R, Nutrients, 2017, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Palczewski K, J. Biol. Chem, 2012, 287, 1612–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hofmann L and Palczewski K, Prog Retin Eye Res, 2015, 49, 46–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang L, Dong R, Ying M, He Q, Cao J and Yang B, Br. J. Pharmacol, 2018, 175, 4285–4294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Clagett-Dame M and Knutson D, Nutrients, 2011, 3, 385–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Handkiewicz-Junak D, Roskosz J, Turska M, Wygoda Z and Jarzab B, Wiad Lek, 2001, 54, 301–306. [PubMed] [Google Scholar]
- 11.Jones H, Blanc D and Cunliffe WJ, Lancet, 1980, 316, 1048–1049. [DOI] [PubMed] [Google Scholar]
- 12.Hong WK, Endicott J, Itri LM, Doos W, Batsakis JG, Bell R, Fofonoff S, Byers R, Atkinson EN, Vaughan C and et al. , N. Engl. J. Med, 1986, 315, 1501–1505. [DOI] [PubMed] [Google Scholar]
- 13.Van Hooser JP, Aleman TS, He YG, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG and Palczewski K, Proc. Natl. Acad. Sci. U. S. A, 2000, 97, 8623–8628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kiser PD, Golczak M, Maeda A and Palczewski K, Biochim. Biophys. Acta, 2012, 1821, 137–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Okuno M, Kojima S, Matsushima-Nishiwaki R, Tsurumi H, Muto Y, Friedman SL and Moriwaki H, Curr. Cancer Drug Targets, 2004, 4, 285–298. [DOI] [PubMed] [Google Scholar]
- 16.Suzuki T, Makino-Tasaka M and Eguchi E, Vision Res, 1984, 24, 783–787. [DOI] [PubMed] [Google Scholar]
- 17.canada Pat., 02 665 909, 2008.
- 18.Bienayme H and Yezeguelian C, Tetrahedron, 1994, 50, 3389–3396. [Google Scholar]
- 19.Duhamel L, Duhamel P and Lecouvé JP, Tetrahedron, 1987, 43, 4349–4358. [Google Scholar]
- 20.Duhamel L, Duhamel P and Lecouve JP, Tetrahedron, 1987, 43, 4339–4348. [Google Scholar]
- 21.Sher I, Tzameret A, Peri-Chen S, Edelshtain V, Ioffe M, Sayer A, Buzhansky L, Gazit E and Rotenstreich Y, Sci. Rep, 2018, 8, 6130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albert K, Schlotterbeck G, Braumann U, Händel H, Spraul M and Krack G, Angew. Chem. Int. Ed. Engl, 1995, 34, 1014–1016. [Google Scholar]
- 23.Choi YH, Kim HK, Wilson EG, Erkelens C, Trijzelaar B and Verpoorte R, Anal. Chim. Acta, 2004, 512, 141–147. [Google Scholar]
- 24.Wang KW, Wang SW and Du QZ, Magn. Reson. Chem, 2013, 51, 435–438. [DOI] [PubMed] [Google Scholar]
- 25.Hashimoto M and Fujimoto Y, Synth. Commun, 1999, 29, 3793–3797. [Google Scholar]
- 26.Perdew JP, Phys. Rev. B, 1986, 33, 8822–8824. [DOI] [PubMed] [Google Scholar]
- 27.Becke AD, Phy. Rev. A, 1988, 38, 3098–3100. [DOI] [PubMed] [Google Scholar]
- 28.Weigend F and Ahlrichs R, Phys. Chem. Chem. Phys, 2005, 7, 3297–3305. [DOI] [PubMed] [Google Scholar]
- 29.Grimme S, Ehrlich S and Goerigk L, J. Comput. Chem, 2011, 32, 1456–1465. [DOI] [PubMed] [Google Scholar]
- 30.Grimme S, Antony J, Ehrlich S and Krieg H, J. Chem. Phys, 2010, 132, 154104. [DOI] [PubMed] [Google Scholar]
- 31.Neese F, Software update: the ORCA program system, version 4.0, WIREs Comput Mol Sci 2018, 2018:e1327 doi: 2010.1002/wcms.2132. [Google Scholar]
- 32.Riplinger C, Sandhoefer B, Hansen A and Neese F, J. Chem. Phys, 2013, 139, 134101. [DOI] [PubMed] [Google Scholar]
- 33.Minenkov Y, Chermak E and Cavallo L, J. Chem. Theory. Comput, 2015, 11, 4664–4676. [DOI] [PubMed] [Google Scholar]
- 34.Kubas A, Berger D, Oberhofer H, Maganas D, Reuter K and Neese F, J. Phys. Chem. Lett, 2016, 7, 4207–4212. [DOI] [PubMed] [Google Scholar]
- 35.Minenkov Y, Chermak E and Cavallo L, J. Chem. Theory. Comput, 2016, 12, 1542–1560. [DOI] [PubMed] [Google Scholar]
- 36.Paulechka E and Kazakov A, J. Phy. Chem. A, 2017, 121, 4379–4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Møller C and Plesset MS, Phys. Rev, 1934, 46, 618–622. [Google Scholar]
- 38.Attila Szabo NSO, Modern Quantum Chemistry: Introduction to Advanced Electronic Structure Theory, Dover, 1989. [Google Scholar]
- 39.Jurečka P, Šponer J, Černý J and Hobza P, Phys. Chem. Chem. Phys, 2006, 8, 1985–1993. [DOI] [PubMed] [Google Scholar]
- 40.Eichkorn K, Weigend F, Treutler O and Ahlrichs R, Theor. Chem. Acc, 1997, 97, 119–124. [Google Scholar]
- 41.Eichkorn K, Treutler O, Öhm H, Häser M and Ahlrichs R, Chem. Phys. Lett, 1995, 242, 652–660. [Google Scholar]
- 42.Izsak R and Neese F, J. Chem. Phys, 2011, 135, 144105. [DOI] [PubMed] [Google Scholar]
- 43.Renner H, Schlamp G, Kleinwächter I, Drost E, Lüschow HM, Tews P, Panster P, Diehl M, Lang J, Kreuzer T, Knödler A, Starz KA, Dermann K, Rothaut J, Drieselmann R, Peter C, Schiele R, Coombes J, Hosford M and Lupton DF, Platinum Group Metals and Compounds, Ullmann’s Encyclopedia of Industrial Chemistry, 2018 [Google Scholar]
- 44.Ji Y, Jain S and Davis RJ, J. Phy. Chem. B, 2005, 109, 17232–17238. [DOI] [PubMed] [Google Scholar]
- 45.Oro LA and Claver C, Iridium complexes in organic synthesis, Wiley-VCH, Weinheim, 2009. [Google Scholar]
- 46.Oliver Kappe C, Chem. Soc. Rev, 2008, 37, 1127–1139. [DOI] [PubMed] [Google Scholar]
- 47.Larhed M, Moberg C and Hallberg A, Acc. Chem. Res, 2002, 35, 717–727. [DOI] [PubMed] [Google Scholar]
- 48.Trost BM and Kulawiec RJ, J. Am. Chem. Soc, 1993, 115, 2027–2036. [Google Scholar]
- 49.Paulson ER and Grotjahn DB, Isomerization and Hydrogenation of Alkenes, Encyclopedia of Inorganic and Bioinorganic Chemistry, Scott RA, 2017. [Google Scholar]
- 50.Tan EH, Lloyd-Jones GC, Harvey JN, Lennox AJ and Mills BM, Angew Chem Int Ed Engl, 2011, 50, 9602–9606. [DOI] [PubMed] [Google Scholar]
- 51.Pünner F, Schmidt A and Hilt G, Angew. Chem. Int. Ed. Engl, 2012, 51, 1270–1273. [DOI] [PubMed] [Google Scholar]
- 52.Fanizzi FP, Intini FP, Maresca L and Natile G, J. Chem. Soc., Dalton Trans, 1990, 199–202. [Google Scholar]
- 53.Strömberg S, Svensson M and Zetterberg K, Organometallics, 1997, 16, 3165–3168. [Google Scholar]
- 54.Gruene P, Fielicke A, Meijer G and Rayner DM, Phys. Chem. Chem. Phys, 2008, 10, 6144–6149. [DOI] [PubMed] [Google Scholar]
- 55.Desclaux JP, At. Data Nucl. Data Tables, 1973, 12, 311–406. [Google Scholar]
- 56.Löwdin P-O, in Adv. Quantum Chem, ed. Löwdin P-O, Academic Press, 1970, vol. 5, pp. 185–199. [Google Scholar]
- 57.Löwdin PO, J. Chem. Phys, 1950, 18, 365–375. [Google Scholar]
- 58.Mayer I, J. Comput. Chem, 2007, 28, 204–221. [DOI] [PubMed] [Google Scholar]
- 59.Pyykko P, Annu. Rev. Phys. Chem, 2012, 63, 45–64. [DOI] [PubMed] [Google Scholar]
- 60.Nakajima T and Hirao K, Chem. Rev, 2012, 112, 385–402. [DOI] [PubMed] [Google Scholar]
- 61.Pantazis DA and Neese F, Theor. Chem. Acc, 2012, 131, 1292. [Google Scholar]
- 62.Gao S, Kahremany S, Zhang J, Jastrzebska B, Querubin J, Petersen-Jones SM and Palczewski K, Mol. Pharmacol, 2018, 93, 438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Van Hooser JP, Aleman TS, He Y-G, Cideciyan AV, Kuksa V, Pittler SJ, Stone EM, Jacobson SG and Palczewski K, Proc. Natl. Acad. Sci. U.S.A, 2000, 97, 8623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gearhart PM, Gearhart C, Thompson DA and Petersen-Jones SM, Arch. Ophthalmol, 2010, 128, 1442–1448. [DOI] [PubMed] [Google Scholar]
- 65.Maeda A, Golczak M, Palczewski K, Maeda T, Cideciyan AV, Jacobson SG and Aleman TS, Hum. Mol. Gen, 2009, 18, 2277–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.