

Abstract
Normal cardiac function requires that intracellular Ca2+ concentration be reduced to low levels in diastole so that the ventricle can relax and refill with blood. Heart failure is often associated with impaired cardiac relaxation. Little, however, is known about how diastolic intracellular Ca2+ concentration is regulated. This article first discusses the reasons for this ignorance before reviewing the basic mechanisms that control diastolic intracellular Ca2+ concentration. It then considers how the control of systolic and diastolic intracellular Ca2+ concentration is intimately connected. Finally, it discusses the changes that occur in heart failure and how these may result in heart failure with preserved versus reduced ejection fraction.
Keywords: calcium, diastole, heart failure, myofibrils, stroke volume
So then I could tell them
Where the wind goes…
But where the wind comes from
Nobody knows.
—AA Milne, “Wind on the Hill”
In keeping with the above quotation from the collection of poems for children by A.A. Milne,1 the focus of this article is not on the extensively studied mechanisms that deliver calcium ions to the myofilaments and thereby produce systole. Rather, we review the much less well understood removal of Ca2+. Specifically, we will consider how diastolic intracellular Ca2+ concentration ([Ca2+]i) is controlled and how it changes in disease.
Why Is It Important to Control Diastolic [Ca2+]i?
Mechanical Relaxation
An upper limit for diastolic [Ca2+]i results from the need for the myofilaments to be deactivated to allow ventricular filling. There may, however, be reasons for ensuring that diastolic [Ca2+]i is not too low as, the lower it is, to reach a given systolic level, more Ca2+ must be added to and removed from the cytoplasm on each beat. This will increase energy expenditure, and since Ca2+ cycling accounts for about 30% of the energy consumption of the myocardium,2 this may be a significant factor in requiring that diastolic [Ca2+]i is not too low.
Diastolic Influences Systolic [Ca2+]i and Force
There are 2 factors. (1) The lower the diastolic [Ca2+]i, the more Ca2+ must be added to produce a given increase in [Ca2+]i. This is because, at low [Ca2+]i, the cytoplasmic Ca2+ buffers become less saturated and their ability to absorb Ca2+ increases. Conversely, as [Ca2+]i increases, buffering power will decrease so a given increase in [Ca2+]i will require a smaller increase in total Ca2+ (for review, see the article by Smith and Eisner3). In other words, by altering the level of saturation of buffers, diastolic [Ca2+]i determines the amplitude of the systolic transient produced by a given rise of total Ca2+ and therefore alterations of diastolic [Ca2+]i change the inotropic response. (2) A further consideration is that force depends steeply on [Ca2+]i so that, starting from an elevated diastolic [Ca2+]i, a smaller increase in [Ca2+]i will be required to produce the same change of force compared with at a normal diastolic [Ca2+]i.4 Therefore, an increase in diastolic [Ca2+]i will increase the level of developed force produced by a given systolic rise of [Ca2+]i.
Basic Mechanisms Underlying the Ca Transient
The pathways that underlie cardiac calcium cycling are well understood5,6 (Figure 1); the individual mechanisms and their roles in the control of diastolic [Ca2+]i are described in more detail in subsequent sections. Briefly, Ca2+ enters via the L-type Ca channel, and there may also be entry on reverse sodium-calcium exchange (NCX) at the start of the action potential. This Ca2+ entry triggers the release of a larger amount of Ca2+ from the sarcoplasmic reticulum (SR) through the ryanodine receptor (RyR)—a process known as calcium-induced calcium release. Ca2+ is then returned to the SR by the SR Ca-ATPase (SERCA), regulated by the accessory protein PLN (phospholamban). At the surface membrane, Ca2+ is removed from the cell by a combination of NCX and PMCA (plasma membrane Ca-ATPase). Finally, the mitochondria can uptake Ca2+ via the MCU (mitochondrial calcium uniporter). The amplitude of the systolic rise of [Ca2+]i is increased by increasing the size of the L-type Ca current7,8 or the amount of Ca stored in the SR.9,10 The latter is determined by the balance of cellular Ca2+ fluxes. For example, increasing SERCA activity or decreasing Ca efflux on NCX will increase SR Ca2+ content. The decay of the Ca transient is largely due to SERCA-mediated reuptake into the SR with, particularly in larger species, significant contributions from NCX.4 The rate of this decay would be expected to affect end-diastolic [Ca2+]i since, all other things being equal, a faster decay will mean that [Ca2+]i is reduced to a lower level by the time of the next beat, resulting in a lower end-diastolic [Ca2+]i.
Figure 1.
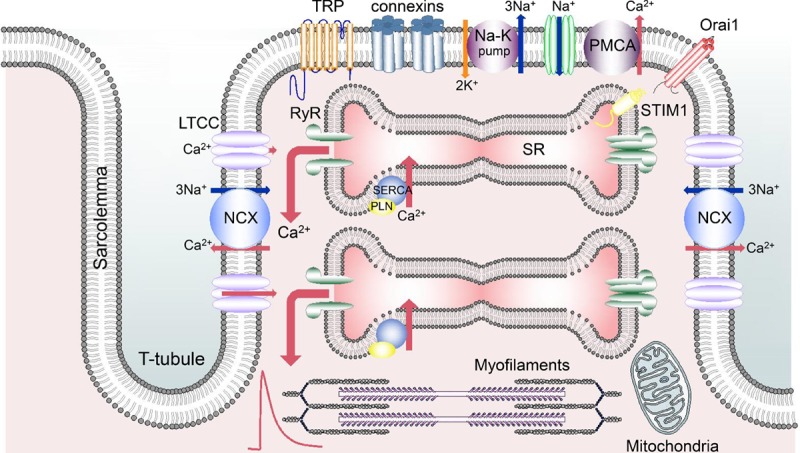
Pathways involved in cardiac cellular calcium cycling. The schematic shows part of a ventricular myocyte including transverse (T) tubules. From left to right, the sarcolemma contains sodium-calcium exchange (NCX); L-type Ca channel (LTCC); transient receptor potential (TRP) channels; connexin hemichannels; Na-K pump; Na channel; PMCA (plasma membrane Ca-ATPase); Orai. The sarcoplasmic reticulum (SR) contains ryanodine receptor (RyR); SR Ca-ATPase (SERCA) and its regulatory protein, PLN (phospholamban); STIM1 (stromal interaction molecule 1).
Why Is So Little Known About Diastolic [Ca2+]i?
There are several reasons for the paucity of data concerning diastolic [Ca2+]i. (1) Problems of indicator calibration make it much easier to measure changes than absolute levels of [Ca2+]i. This is a particular issue when comparing measurements between cells or animals. (2) When nonratiometric, Ca2+-sensitive, fluorescent indicators are used, the records are often normalized to the diastolic or resting fluorescence,11 making it difficult to measure diastolic [Ca2+]i. (3) In experiments using the whole cell version of the patch clamp, diffusion of Ca2+ and Ca2+ buffers into or out of the pipette may contribute to regulation of [Ca2+]i. Indeed, one of the uses of the whole-cell technique is to control the cytoplasmic ionic concentrations. (4) The major issue may be that, particularly in smaller animals, most experimental work studying Ca2+ cycling in cardiac tissues has used rates of stimulation considerably below normal heart rates. While the fact that ion currents and [Ca2+]i have reached steady state values helps dissect the fluxes responsible for the systolic Ca transient, it establishes an artificial situation. As discussed below, end-diastolic [Ca2+]i represents a balance between many Ca2+-handling mechanisms. In contrast, in a quiescent myocyte, the resting level of [Ca2+]i is determined entirely by the fluxes of Ca2+ across the sarcolemma12,13 because, in the steady state, there can be no net flux into or out of organelles. Such a net flux would result in a continuous change of organelle Ca2+ content—a situation incompatible with a steady state. At low rates of stimulation, [Ca2+]i will be identical to the resting level seen in the unstimulated case. These frequency-dependent effects are illustrated by making the RyR leaky with caffeine (Figure 2A and 2B). This has no effect on diastolic [Ca2+]i at a stimulation rate of 0.5 Hz but a marked one at 3 Hz.14 Thus, it is important not to confuse diastolic and resting [Ca2+]i. Finally, as discussed below, physiological changes of heart rate result from those of autonomic tone—a factor that is not examined in studies that simply alter pacing rate.
Figure 2.
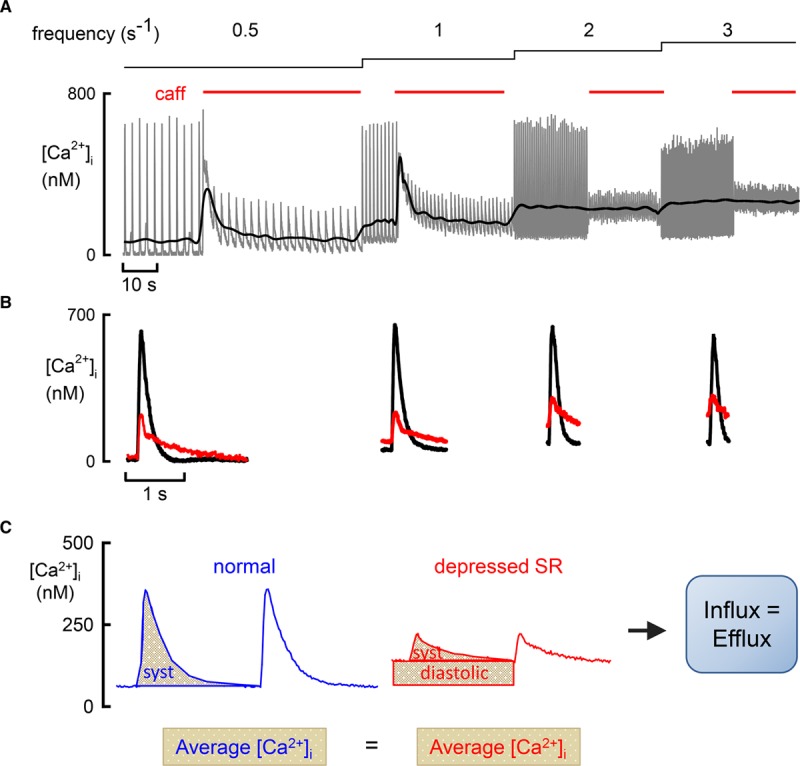
The importance of average intracellular Ca2+ concentration ([Ca2+]i) in the control of systolic (syst) and diastolic [Ca2+]i. A, The effects of application of caffeine (caff) and stimulation rate on [Ca2+]i. A rat ventricular myocyte was stimulated at the frequencies shown above and caff (1 mmol/L) applied for the periods denoted by the red bars. The gray trace is the original data and the black denotes average [Ca2+]i. B, Specimen, averaged traces of [Ca2+]i from the frequencies shown above. For each frequency, the control (black) and caff (red) traces are superimposed. Data reproduced from Sankaranarayanan et al.14 C, Illustration of flux balance in control and with depressed sarcoplasmic reticulum (SR) function. The total Ca2+ efflux via sodium-calcium exchange (NCX) above control diastolic levels is represented by the area under the [Ca2+]i trace. In the depressed SR case, this is separated into 2 components: (1) activated by the syst Ca2+ transient and (2) activated by increased diastolic [Ca2+]i. Average [Ca2+]i is identical with normal and depressed SR (A), and, therefore, Ca2+ efflux is unchanged and equal to influx.
Interaction of Control of Diastolic and Systolic [Ca2+]i
It is tempting to think of the control of diastolic and systolic [Ca2+]i as being separate. From this viewpoint, diastolic [Ca2+]i is controlled at a certain level, and the mechanisms discussed above determine the magnitude of the systolic rise. We think that this is incorrect; the regulation of diastolic and systolic [Ca2+]i is inextricably linked.14,15 This has been demonstrated recently by investigating the effects of interfering with SR function on diastolic and systolic [Ca2+]i. Consistent with previous data,16 making the RyR leaky with caffeine decreased the amplitude of the systolic Ca2+ transient by decreasing SR Ca2+ content. This was accompanied by an increase in diastolic [Ca2+]i such that the average level of [Ca2+]i over the cycle was unaffected (Figure 2A).14 Similar results were found when SERCA activity was decreased with thapsigargin and were accounted for by considerations of cellular Ca2+ flux balance. In the steady state, the Ca2+ influx over the cardiac cycle must equal efflux. Interfering with SR function will have no direct effect on influx, and so efflux must also be unaltered. Since Ca2+ efflux on NCX is proportional to [Ca2+]i,17 constant efflux requires that average [Ca2+]i be unaffected, explaining why the decrease of systolic is accompanied by an increase in diastolic [Ca2+]i (Figure 2C). One caveat is required here; interfering with SR function and thereby decreasing the amplitude of the systolic Ca transient can decrease the degree of Ca-dependent inactivation of the L-type Ca current and thereby increase Ca2+ influx.18,19 In this case, average [Ca2+]i would be elevated, potentially elevating diastolic [Ca2+]i. This does not appear to be an issue in experiments where the RyR was made leaky with caffeine as the L-type Ca2+ influx was unaffected.14
Importance of Average [Ca2+]i
The average [Ca2+]i is determined by Ca2+ entry and efflux across the surface membrane. An increase in rate will increase Ca2+ influx per unit time and thence the average [Ca2+]i (Figure 2A).14 Similar effects would be expected for an increase in the amplitude of the L-type Ca current. Conversely, a decrease in the ability of NCX to pump Ca2+ out of the cell will increase average [Ca2+]i to a level sufficient to maintain Ca2+ efflux. This may arise due to either decreased expression of NCX or an increase in the intracellular Na+ concentration ([Na+]i) and, therefore, a decrease in the energy to pump Ca2+ out of the cell (Figure 3). There is an infinite number of combinations of systolic and diastolic [Ca2+]i that can establish a given average [Ca2+]i. The properties of the SR will be an important factor in determining which occurs. Compromising SR function, by decreasing SERCA activity or increasing Ca2+ (leak) efflux through the RyR, will increase diastolic and decrease systolic [Ca2+]i. For example, in the presence of a normal SR, β-adrenergic stimulation increases systolic but has no effect on diastolic [Ca2+]i. In contrast, when the RyR is leaky, β-stimulation increases diastolic [Ca2+]i.14
Figure 3.
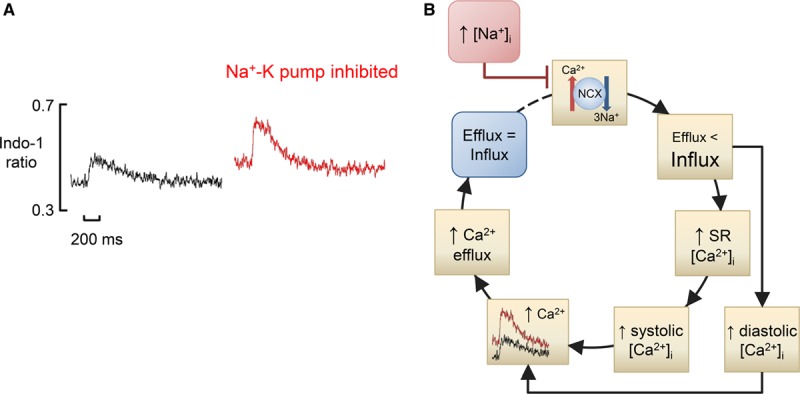
Mechanism of the effects of intracellular Na+ concentration ([Na+]i) on diastolic intracellular Ca2+ concentration ([Ca2+]i). A, Original data. Ca transients recorded using indo-1 in a rat ventricular myocyte. Records show the following: left, control; right, in the presence of the Na-K pump inhibitor, strophanthidin. Data reproduced from Bennett et al.20 B, Flowchart. Initially (blue rectangle), Ca2+ efflux equals influx. The increase in [Na+]i decreases sodium-calcium exchange (NCX) activity making Ca2+ efflux less than influx, leading to an increase in diastolic [Ca2+]i. There will also be an increase in sarcoplasmic reticulum (SR) Ca2+ content and thence an increase in systolic [Ca2+]i. The increases of diastolic and systolic [Ca2+]i will increase Ca2+ efflux until efflux again equals influx.
Similar considerations also apply to conditions of calcium overload where waves of calcium release from the SR occur. Much attention has been directed to the detrimental effects of these increases in [Ca2+]i which activate NCX21 and thereby produce arrhythmogenic delayed afterdepolarizations.22 However, the resulting Ca2+ efflux will help maintain Ca2+ flux balance and thereby keep diastolic [Ca2+]i lower than would otherwise be the case. Under these conditions, addition of caffeine to empty the SR and thereby remove waves and their associated efflux results in a steady level of [Ca2+]i, which is greater than the minimum seen in the presence of Ca2+ waves.23
These consequences of flux balance are a generalization of those previously described for changes of systolic [Ca2+]i alone.24–26 That work showed that potentiation of RyR opening had no effect on systolic [Ca2+]i in the steady state. In those earlier experiments, the decrease of SR Ca content was exactly compensated for by an increase in fractional release from the SR so the amplitude of the Ca transient and the accompanying Ca2+ efflux were unaltered. Ca2+ flux balance could, therefore, be maintained at constant diastolic [Ca2+]i. In the more recent work,14 the degree of potentiation of the RyR was greater (higher concentrations of caffeine used), and, therefore, the SR Ca content fell to such a low level that even if it is all released, the systolic Ca transient is smaller than control. The consequent decrease of systolic Ca2+ efflux results in systolic efflux being less than influx, thereby loading the cell with Ca2+ and increasing diastolic [Ca2+]i until the increase of diastolic efflux returns the cell to Ca2+ flux balance.
In the context of the above general considerations, we will review important aspects of the underlying Ca2+ fluxes before discussing how their integration leads to control of diastolic and systolic [Ca2+]i.
Fluxes Regulating Diastolic [Ca2+]i
Ca2+ Buffering
The changes of [Ca2+]i potentially depend as much on the Ca2+ buffering properties of the cell as on the fluxes of total Ca2+.3 In quiescent cells (or at low pacing rates), an increase in buffering is not expected to change diastolic [Ca2+]i, since free [Ca2+]i (and not the Ca2+ bound to buffers) determines efflux, and this must balance influx, which is constant. At higher pacing rates, because an increase in buffering slows the rate of change of [Ca2+]i, the Ca transient cannot decay back to baseline and end-diastolic [Ca2+]i will rise. Accordingly, experimentally increasing the cytoplasmic buffering power slows the rate of decay of [Ca2+]i27 and elevates [Ca2+]i and force in diastole.28 An increase in diastolic [Ca2+]i in hypertrophic cardiomyopathy resulting from some troponin T mutations has been attributed to this mechanism29 and may contribute to contractile impairment at increased heart rates in this condition.
Sarcoplasmic Reticulum Ca-ATPase
The greater the activity of SERCA, the faster systolic [Ca2+]i will decay, and, all other things being equal, the further diastolic [Ca2+]i will fall before the next beat and, therefore, the lower will be end-diastolic [Ca2+]i. Experimentally decreasing SERCA activity can (see above) increase diastolic [Ca2+]i14,16,30 and pressure30 as a consequence of the slowing of the decay of the Ca2+ transient. The increased diastolic [Ca2+]i will compensate for the decreased systolic efflux resulting from the smaller Ca2+ transient thereby maintaining Ca2+ flux balance. It should, however, be noted that acute inhibition of SERCA has been reported to increase [Na+]i,31 and this can elevate diastolic [Ca2+]i via NCX. The origin of this increase in [Na+]i is unclear. One possibility is that the decreased amplitude of the Ca2+ transient will have decreased inactivation of the L-type Ca current, thereby increasing Ca2+ entry and thence efflux on NCX, leading to loading of the cell with Na+. Given that Na+ entry on NCX is a major component of Na+ entry into the cell,32 this will elevate [Na+]i. In another study, knockout of SERCA also elevated [Na+]i.33 These knockout mice have increased L-type Ca current, possibly to compensate for the lack of SERCA. This increased Ca2+ influx will need to be balanced by increased efflux on NCX. The consequent increase in Na+ influx may, therefore, account for the elevation of [Na+]i.
Ryanodine Receptor
As mentioned in a previous section, making the RyR leaky can decrease SR Ca content and thence the amplitude of the systolic Ca transient and systolic Ca2+ efflux. The decrease of efflux means that Ca2+ will accumulate in the cell, increasing diastolic [Ca2+]i until the increase in diastolic Ca2+ efflux restores total efflux to equal influx. Leaky RyRs also slow the rate constant of decay of the systolic Ca transient.34,35 Under normal conditions, Ca release from the SR occurs more or less synchronously, a few milliseconds after the start of depolarization, in response to the rise of [Ca2+]i produced by the L-type current. Release from clusters of RyRs can be seen as calcium sparks.11 In contrast, after myocardial infarction, Ca sparks are observed on the falling phase of the systolic Ca transient.15 Increasing RyR phosphorylation and opening by overexpression of CaMKII (Ca2+/calmodulin-dependent protein kinase II)-δC also leads to the appearance of delayed calcium sparks, which will interfere with the decay of [Ca2+]i and relaxation.35 Such late sparks have also been suggested to be a more general phenomenon particularly when the initial release of Ca2+ from the SR is depressed.36 A study in hypothyroid mice has linked the appearance of late sparks to impaired systolic and diastolic function.37
Sodium-Calcium Exchange
NCX uses the energy provided by 3 Na+ entering to pump 1 Ca2+ out of the cell. This stoichiometry generates an electric current,21,38 and NCX activity is sensitive not only to the Na+ and Ca2+ concentration gradients but also to membrane potential; hyperpolarization increases and depolarization decreases net Ca2+ efflux. Depending on the ionic gradients and membrane potential, NCX can reverse direction with net Ca2+ influx coupled to Na+ efflux (reverse mode). At a normal resting potential, NCX works in the forward direction with Ca2+ efflux roughly proportional to [Ca2+]i.17 It should, however, also be noted that NCX is allosterically regulated by [Ca2+]i, thus limiting Ca2+ efflux at low [Ca2+]i.39 For an extensive review of NCX, see the article by Blaustein and Lederer.40
Intracellular Sodium
An increase in [Na+]i decreases the driving force available for NCX to remove Ca2+ from the cell and thereby increases developed force and the underlying systolic Ca transient. In rabbit ventricular myocytes, inhibition of the Na-K pump increases both [Na+]i and diastolic [Ca2+]i.41 However, at least with moderate increases in [Na+]i, there is often no increase in diastolic [Ca2+]i42,43 or developed force/cell length.44–46 While this may result partly from the low sensitivity of force and some Ca2+ indicators to [Ca2+]i, it may also be explained as follows (Figure 3). In the steady state, the reduction of NCX activity will require an increase in average [Ca2+]i (see above). At first, this will be largely provided by an increase in systolic [Ca2+]i as a result of the increase in SR Ca content. Only with further reduction of NCX, perhaps because there is a limit to how much SR Ca content and thence systolic [Ca2+]i can increase, will diastolic [Ca2+]i increase appreciably.
Plasma Membrane Ca-ATPase
In addition to NCX, the myocyte also expresses a PMCA whose contribution to Ca2+ efflux is less well established.47 It has been argued that the PMCA is irrelevant to the control of bulk cytoplasmic [Ca2+]i and, instead, has a signaling function by controlling [Ca2+]i in microdomains near caveolae.48 Work from the Bers Laboratory finds that the contribution of the PMCA to Ca2+ removal in a variety of species is typically <10% of that of NCX.49 We find a larger contribution; inhibiting NCX with Ni2+ leaves 25% to 33% of the Ca2+ removal from the cell functional in rat.16,50 A concern with the use of Ni2+ is that it may not completely inhibit NCX, but similar results are seen when NCX is stopped by removal of Na+ ions.51,52 The NCX-independent Ca2+ efflux is abolished by the nonspecific PMCA inhibitor carboxyeosin.53,54 A substantial role for PMCA is also suggested by work on myocytes isolated from NCX knockout mice. These animals live normally, and their ventricular myocytes have normal Ca2+ transients. There is no change of PMCA expression, and the myocytes maintain Ca2+ flux balance by decreasing Ca influx through the L-type Ca current to 20%—a level at which PMCA alone can presumably balance it.55,56 This suggests that PMCA makes a contribution equivalent to 25% of that of NCX in the wild type. One caveat is that, as in other studies, the rate of Ca2+ removal from the cell was assessed from the rate of fall of the caffeine-evoked rise of [Ca2+]i. The available data do not provide caffeine exposures of sufficient duration to obtain accurate measurements,56 and further work is required to establish the role of PMCA in the regulation of diastolic [Ca2+]i.
Mitochondrial Ca2+ Handling
In principle, Ca2+ uptake and release from mitochondria could affect diastolic [Ca2+]i. As we have recently reviewed,6 there are conflicting reports in the literature with only some studies finding evidence in favor of beat-to-beat movements of Ca2+ into and out of mitochondria. On balance, at least in adult ventricular myocytes, while changes of mitochondrial [Ca2+] can be observed at slow rates of stimulation,57 they disappear at higher rates questioning their importance in regulating diastolic [Ca2+]i.57
Ca2+ Influx Pathways During the Action Potential
The major route for Ca2+ entry during the action potential is the L-type Ca current.58 In some regions of the heart, particularly in nodal tissues, there are also contributions from the T-type Ca channel.59 The stoichiometry of NCX means that it can also contribute to Ca2+ influx during depolarization, but, under normal conditions, this is much smaller than that through the L-type channel.60 In heart failure, the increase in [Na+]i will increase influx through NCX,61 and it is possible that the magnitude of Ca2+ influx through NCX may have been underestimated due to making measurements at slow rates where [Na+]i is decreased.
Many studies have investigated the effects on systolic [Ca2+]i of maneuvers that alter the L-type Ca current. Inspection of most data shows little effect on diastolic levels,62,63 but the majority of experiments were performed at slow rates or used the whole-cell patch clamp technique. We found that decreasing the L-type Ca current with cadmium in cells where diastolic [Ca2+]i was elevated reduced diastolic [Ca2+]i.14 From first principles, one would expect 2 opposing effects.64 (1) Increased L-type Ca current will increase Ca2+ influx per unit time thereby requiring an increased average [Ca2+]i to balance it. Depending on the conditions, this may be achieved by increased systolic or diastolic [Ca2+]i. (2) The increase in L-type current will increase Ca2+ release from the SR, increasing systolic Ca, thereby contributing to the elevated average without the need to increase diastolic. This latter effect, however, is limited as it is impossible to release >100% of SR content. It should also be noted that, at least under some conditions, increasing L-type Ca current does not increase SR content.64 Additionally, the increase in systolic [Ca2+]i will increase the time taken for [Ca2+]i to decay back to baseline, increasing the tendency for diastolic [Ca2+]i to rise at shorter pacing intervals (higher heart rates). Quantitative considerations will determine whether the increase in systolic efflux is sufficient to balance the increase in influx or, alternatively, whether elevated diastolic [Ca2+]i occurs.
Background Ca2+ Entry Mechanisms
In the absence of stimulation, resting [Ca2+]i is of the order of 100 nmol/L indicating that some kind of background Ca2+ entry pathway must exist to balance Ca2+ efflux on NCX. Such a pathway accounts for the fact that, even in a quiescent cell, after being emptied with caffeine, the SR can be refilled by a mechanism that requires extracellular Ca2+.65 As mentioned above, Ca2+ waves can occur in cells held at a fixed membrane potential,23 again indicating an influx pathway to balance efflux on NCX during the waves. The magnitude of this influx is roughly proportional to external Ca2+ concentration in the range ≤5 mmol/L.23 Subsequent work, examining the effects on resting [Ca2+]i of abruptly removing external Ca2+, provided an estimate for the background Ca2+ influx of the order of 2 to 6 µmol/L per s in rat ventricular myocytes.66 A recent study estimated Ca2+ influx from measurements of average [Ca2+]i (see above) and found a value of about 4 µmol/L per s.14 These values compare to an entry on each action potential via the L-type Ca current of the order of 5 to 10 µmol/L. Therefore, at normal heart rates (in a rat) of 5 s−1, the background influx will be of the order of 10% of that carried by the L-type current. As regards the mechanism of this influx, one study identified a Ca2+ entry mechanism that increased on hyperpolarization of the surface membrane and was blocked by the relatively nonspecific agent gadolinium (Gd3+).67 It is, therefore, important to consider the identity of this flux.
Connexin Hemichannels
One Ca2+ flux inhibited by Gd3+ is that carried by connexins.68 The majority of connexins are found as pairs, made up of 2 hemichannels, one in each of the 2 cell membranes at the intercalated discs. These allow current to flow between cells. However, some connexins are present as hemichannels in the surface membrane of a single cell69,70 and may, therefore, provide a route for Ca2+ entry. Recent work has suggested that this entry may be increased in experimental cardiomyopathy induced by plakophilin-2 deficiency.71
Transient Receptor Potential Channels
Transient receptor potential (TRP) channels are also sensitive to Gd3+, and considerable work has investigated their role in the heart. Knockout of TRPV2 decreases the amplitude of the systolic Ca transient and contraction.72 The compound probenecid, which activates TRPV2, was also shown to increase contractility,73 and a small trial has shown that this compound improves cardiac function in patients with heart failure.74 It should, however, be noted that probenecid has other actions including inhibiting organic anion transporters.75 Furthermore, inhibition of TRPV4 decreases SR Ca release.76 The cardiomyopathy found in the mdx mouse model of muscular dystrophy is associated with elevated diastolic [Ca2+]i, which can be blocked by Gd3+, and has been attributed to Ca2+ entry via TRPC channels.77 Similar results were found for the experimental myopathy produced by infusion of isoproterenol.78 Further evidence suggesting a role for TRP channels in contributing to setting diastolic [Ca2+]i comes from the observation that knocking out both TRPC1 and TRPC4 in mice decreased diastolic [Ca2+]i.79 The recent synthesis of specific antagonists of TRPC channels80 and an agonist81 should make it possible to study the role of these channels more precisely.
TRP channels have also been implicated in the influx of Ca2+ into the cell activated by emptying the SR, so called store-operated Ca2+ entry, and in the HL-1 cell line, this has been suggested to contribute to resting [Ca2+]i.82 One issue is that much of the evidence for a role of store-operated channels in cardiac tissue comes from work on cultured or neonatal cells,83,84 and these may not be representative of adult myocytes. Some recent articles have, however, reported store-operated Ca2+ entry into adult mouse ventricular myocytes85 with the fluxes being inhibited by Gd3+.86,87 In many tissues, store-operated calcium entry is produced by a combination of the endoplasmic reticulum Ca2+ sensor STIM1 (stromal interaction molecule 1) and the surface membrane channel Orai1 (see the article by Qiu and Lewis88 for review). Overexpression of STIM1 in mouse heart increases diastolic [Ca2+]i, as a result of increased Ca2+ entry into the cell and increased leak from the SR.89 It should, however, be noted that STIM1 has also been reported to interact with PLN and thereby control SERCA.90
Finally, some TRP channels and connexins transport Na+ in addition to Ca2+ and, by altering [Na+]i, could affect [Ca2+]i indirectly via NCX. All in all, it is clear that more work is required to characterize the contribution of TRPs, connexins, and as yet unidentified mechanisms to the background Ca2+ influx
Physiological Factors Affecting Diastolic [Ca2+]i
Heart Rate
Increasing the rate of stimulation increases diastolic [Ca2+]i in ventricular trabeculae91 and isolated myocytes.14,92–95 It is important to note that the Ca2+ indicators used to measure [Ca2+]i buffer [Ca2+]i to some degree and potentially exaggerate the effects of increased frequency. It would be useful to repeat these experiments using as low concentrations of Ca indicators as possible. Of course the fact that, even in the absence of indicators, increasing stimulation rate increases diastolic force96 and decreases cell length92 means that excessive buffering cannot account for all the effects.
There are at least 2 possible explanations for the frequency-dependent increase in diastolic [Ca2+]i (Figure 4). One is that increasing frequency increases [Na+]i41,97,98 and, as discussed above, decreases NCX activity, requiring an increase in average [Ca2+]i to maintain flux balance. This explanation is consistent with the parallel increase in [Na+]i and diastolic [Ca2+]i.99 Two arguments, however, suggest that Na-independent mechanisms may also be involved. (1) The increase in diastolic [Ca2+]i can occur abruptly on increase in rate,94 much faster than the presumed increase in at least global [Na+]i. (2) In the NCX knockout mouse (where changes of [Na+]i would not be expected to increase [Ca2+]i), the effects of rate on diastolic [Ca2+]i are similar to those in wild type.55 As discussed above, and in Figure 4, this Na-independent factor is likely to be the need for the increase in Ca2+ influx to be balanced by increased efflux and, therefore, elevated average [Ca2+]i. Why does diastolic [Ca2+]i increase at higher rates? It might be thought that balance could occur simply by the increased frequency of Ca transients resulting in more systolic efflux. This, however, ignores that (1) the Ca transient cannot decay back to equilibrium before the next beat, resulting in end-diastolic [Ca2+]i rising and (2) there is less time for NCX Ca removal from the cell per beat. Finally, in species with a negative force-frequency relationship (and humans with heart failure100), the systolic Ca transient decreases at higher rates presumably reducing the systolic efflux per beat. Both Na-dependent and independent mechanisms may contribute. The increase in Ca2+ entry at higher rates demands increased Ca2+ efflux for flux balance. This will require an increase in average [Ca2+]i. The elevated [Na+]i will decrease NCX activity thereby requiring a greater increase in average [Ca2+]i.
Figure 4.
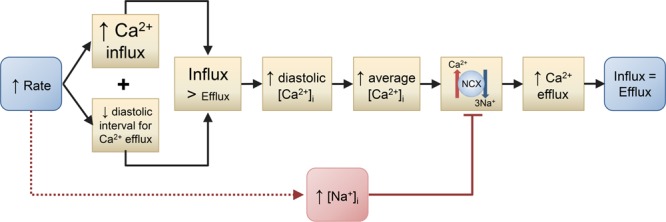
Possible mechanisms for the increase in diastolic intracellular Ca2+ concentration ([Ca2+]i) with increased stimulation rate. Two mechanisms are shown. (1) The increase in rate increases Ca2+ influx per unit time and decreases the time over which efflux can occur resulting in influx becoming greater than efflux. This increases diastolic [Ca2+]i and thence average [Ca2+]i. Via sodium-calcium exchange (NCX), this elevation of average [Ca2+]i increases Ca2+ efflux to equal the influx so that influx and efflux are again equal. (2) Increased rate elevates intracellular Na+ concentration ([Na+]i), which decreases Ca2+ efflux on NCX. Therefore, a greater increase in average [Ca2+]i is required to maintain Ca2+ efflux equal to influx.
β-Adrenergic Stimulation
Physiologically, changes of β-adrenergic stimulation are the main cause of changes of heart rates. The effects of β-adrenergic stimulation will, therefore, be a combination of those of rate, discussed above, as well as direct effects. The latter include an increase in both Ca entry through the L-type Ca current and of SERCA activity (see above—via phosphorylation of PLN). The expected effects of these have been discussed earlier. In brief, the level of diastolic [Ca2+]i will be determined by the net effects of β-adrenergic stimulation on Ca entry via the L-type current and on the shape and size of the Ca transient, which determine NCX removal. β-Adrenergic stimulation increases influx via ICa-L, which is partly balanced by a larger Ca transient amplitude and, therefore, greater systolic efflux via NCX. This latter effect is curtailed, however, by the accelerated decay of the Ca transient resulting from greater SERCA activity.101 Accordingly, in rat ventricular myocytes, studied at constant rate, β-adrenergic stimulation increases diastolic [Ca2+]i.94 It should also be noted that β-adrenergic stimulation phosphorylates phospholemman, increasing Na-K pump activity, decreasing [Na+]i, and thereby increasing the driving force for NCX-mediated Ca2+ efflux. This will decrease the level of average [Ca2+]i required to balance the increased Ca2+ influx.
The Ca transient and accompanying NCX removal could also be affected by changes in Ca2+ buffering arising from PKA-dependent phosphorylation of cardiac troponin I and PLN during β-adrenergic stimulation. The effect on cardiac troponin I will lower the affinity for Ca2+ binding and, except at high levels of [Ca2+]i, would be expected to decrease Ca2+ buffering and accelerate the decay of the Ca transient. Conversely, phosphorylation of PLN will increase affinity and increase buffering and should slow the decay. Experiments on mouse ventricular myocytes found that these effects were balanced such that there was no net effect on Ca2+ buffering.101 Further work is required to investigate the effects of β-adrenergic stimulation in a wider range of conditions.
Clinical Aspects of Abnormal Diastolic Function
The previous sections have reviewed the fundamental mechanisms that regulate diastolic [Ca2+]i. Before turning to the changes of calcium cycling that occur in heart failure, it is important to set these in a clinical context.
Diastolic Dysfunction
Ventricular filling in diastole relies on both a compliant ventricle and a pressure gradient between the left atrium and left ventricle (LV). In the early phase of diastole, active ventricular relaxation helps to generate this gradient by actively sucking blood into the ventricle via elastic recoil. This active phase is myocyte dependent and relies on the rapid decline in [Ca2+]i at the beginning of diastole, leading to dissociation of the thick and thin filaments. In the subsequent, passive phases of diastole, the pressure gradient distends the ventricle.102 While this phase depends heavily on passive properties of the myocardium including wall thickness and fibrosis, it is also determined by diastolic [Ca2+]i by setting the baseline myofilament activation and thus tension. Both active and passive processes require the heart to be sufficiently relaxed and compliant to fill with blood. During exercise, heart rate rises and diastolic interval decreases. Here, LV filling is maintained by increasing transmitral flow via an increase in pressure gradient. In the healthy heart, this gradient is generated by enhanced elastic recoil to reduce LV pressure in early diastole, without significantly changing left atrium pressure.103
Slowing of relaxation leads to diastolic dysfunction, and this is particularly pronounced during dynamic exercise with exercise intolerance—a frequent presenting feature of heart failure. Consequently, LV diastolic pressure increases, and filling can only be achieved by an increase in left atrium pressure, resulting in pulmonary congestion, breathlessness, and effort intolerance.104 More advanced stages of diastolic dysfunction display elevated filling pressures at rest.
Diastolic dysfunction is frequently observed alongside systolic dysfunction in heart failure with reduced ejection fraction (HFrEF). There are multiple mechanisms for slowed relaxation in HFrEF, including abnormalities in Ca2+ cycling (including reduced Ca uptake via SERCA)105 and changes in diastolic [Ca2+]i (see below), increased extracellular collagen,106 increased myofilament crossbridge interactions due to metabolic changes independent of Ca2+,107,108 and loss of elastic recoil (due to failure of elastic compression in systole).109
Importantly, however, about half of patients with heart failure have diastolic dysfunction but a normal ejection fraction or heart failure with preserved ejection fraction (HFpEF; for review, see the article by Pfeffer et al110). The increase in chamber stiffness and slowed relaxation observed in HFpEF causes a rise in LV filling pressures, which, when sufficiently high, results in the heart failure syndrome.111 It is increasingly clear that HFpEF is not a condition of diastolic dysfunction alone and some impairment in systolic function is present at rest and becomes more prominent during exercise.112 This systolic impairment may further exacerbate diastolic dysfunction because contractile impairment modifies the restoring forces that drive early diastolic recoil.113 Additionally, HFpEF is associated with a constellation of comorbidities such as diabetes mellitus, obesity, hypertension, aging, and kidney disease. Consequently, HFpEF is accompanied by systemic changes, including inflammation and endothelial dysfunction, tissue fibrosis, microvascular dysfunction and ischemia, and multiorgan impairment such as renal failure and sarcopenia. These contribute both to the diastolic impairment and the overall clinical phenotype.110 In spite of its complexity, the inherent defects underlying diastolic dysfunction can be broadly grouped into 2 classes: (1) external to the cardiac myocyte and (2) resulting from impaired myocyte function.
Myocyte-Independent Mechanisms
The extracellular matrix is a major determinant of myocardial stiffness, and increases in interstitial fibrosis and collagen are observed in HFpEF,114 as well as being part of the aging process.115 In addition to increasing stiffness,116 expansion of the extracellular matrix in HFpEF is associated with increased mortality and rates of hospitalization.117 It has also been proposed that elevations in LV filling pressure may result from increased extrinsic restraint on the heart,118 for example, in the obese phenotype of HFpEF where epicardial fat may cause mechanical compression of the heart, as well as exerting paracrine effects.119,120 Finally, it is worth noting that LV geometry itself may impact on diastolic function. Concentric hypertrophy is commonly observed clinically in HFpEF,121 particularly in patients with systemic arterial hypertension, and results from both expansion of the interstitium and myocyte hypertrophy.106,122 Here, an increase in wall thickness elevates stiffness and contributes to the diastolic impairment.123,124
Myocyte-Dependent Mechanisms
Dysfunctional relaxation and higher passive stiffness in HFpEF is present at the level of the cardiac myocyte.125 Traditionally, diastolic dysfunction has been attributed to increased stiffness secondary to gross concentric hypertrophy (typically caused by hypertension),126 which is also present in isolated myocytes.127 However, a significant proportion of HFpEF patients do not have LV hypertrophy, and severity of hypertrophy does not closely correlate with diastolic dysfunction.128 Instead, the bulk of this increase in resting tension can be explained at the sarcomere.
The giant molecular spring titin, which spans the Z disk to M band, is a major determinant of passive tension by providing recoil in early diastole and resistance to stretch in late diastole.129,130 Its properties can be directly modified by phosphorylation (by protein kinases A and G, and CaMKII, which reduce tension)129–131 and oxidative modification via disulphide bonds132 and S-glutathionylation.133 As such, in addition to changes in its expression, posttranslational modifications in titin allow for dynamic changes in cellular and diastolic stiffness, which are implicated in the pathophysiology of HFpEF. Finally and intriguingly, the titin N2BA isoform exhibits a small [Ca2+]i-dependent increase in stiffness.134,135 Although this may add further importance to the role of diastolic Ca2+ in diastolic dysfunction, the significance of this finding in vivo has not yet been established.
At the sarcomere level, there is also evidence implicating the actin-myosin filaments in HFpEF. Relaxation of these depends on both diastolic [Ca2+]i (see subsequent sections) and their sensitivity to Ca2+. Increased myofilament Ca2+ sensitivity secondary to hypophosphorylation of cardiac troponin I has been reported in HFpEF.136 Furthermore, abnormally high myofilament Ca sensitivity also contributes to the diastolic dysfunction observed in hypertrophic cardiomyopathy caused by sarcomeric gene mutations.137,138 Accordingly, the increase in resting tension in HFpEF myocytes has been linked with low PKG (protein kinase G) levels, which may impair relaxation by reducing phosphorylation of titin, cardiac troponin I, and PLN.139,140 A role for defective CaMKII phosphorylation of titin has also been proposed.131 In conclusion, although other factors may contribute to impaired diastolic function, it is important to consider the role of abnormalities in Ca2+ signaling.
Diastolic [Ca2+]i in Heart Failure
Does diastolic [Ca2+]i change in heart failure? We will first consider data from animal and human studies where systolic function is also impaired before moving on to HFpEF.
Heart Failure With Reduced Ejection Fraction
The decreased systolic Ca transient in heart failure may result in large part from a decrease in SR Ca2+ content caused by one or more of decreased SERCA activity, leaky RyRs, or increased NCX activity (see the article by Bers105 for review). As far as diastolic [Ca2+]i is concerned, measurements on ventricular strips from patients with heart failure found increases in diastolic force and [Ca2+]i, which were most obvious at higher stimulation frequencies.141 Ca transients in cells isolated from patients had a smaller amplitude and also slowed decay,42 which would be expected to increase diastolic [Ca2+]i. A subsequent study found little elevation of diastolic [Ca2+]i or force96 but pointed out that the lack of sensitivity of the Ca2+ indicator used may have made it hard to resolve changes of diastolic [Ca2+]i. Experiments on myocytes from patients with heart failure, using more sensitive fluorescent indicators (fluo-3 and fura-red), demonstrated an increase in diastolic [Ca2+]i with increasing rate,142 but no control data were available. Increasing the rate of stimulation increased diastolic force in ventricular muscle strips from patients with heart failure but not controls.143 In a rabbit model of aortic insufficiency/restriction, the amplitude of the systolic Ca transient decreased to about 70% of control with no change of diastolic [Ca2+]i.144 It should, however, be noted that the experiments were performed at a slow rate (0.5 Hz). Another study on rabbit myocytes found that pressure and volume overload–induced heart failure increased diastolic [Ca2+]i,145 and, in contrast to much of the other work discussed here, this was unaffected by stimulation frequency. Finally, a study of right side heart failure (induced in rats with monocrotaline) showed a tendency to increased diastolic [Ca2+]i, particularly at elevated stimulation frequencies.146 Further complication is added by reports of a decrease in diastolic [Ca2+]i in a sheep tachypacing model of heart failure, albeit studied at low stimulation rates.147 Interestingly, this was accompanied by a decrease in the L-type Ca current, which would be expected to decrease average [Ca2+]i, perhaps contributing to the decrease in diastolic [Ca2+]i. Unchanged diastolic [Ca2+]i was found in ventricular myocytes from tachypaced dogs, but this also used low stimulation rates and whole-cell patch clamp.148 Decreased diastolic [Ca2+]i has also been found in a ferret aortic banding model of hypertrophy, again at low stimulation rates.149
An important clinical situation that produces heart failure and depressed myocardial contractility is sepsis. Cecal ligation and puncture in the rat slowed the decay of the Ca transient; this was attributed to increased frequency of Ca sparks and accompanied by decreased systolic and increased diastolic [Ca2+]i.150 In another study on rats, using lipopolysaccharide administration, septic cardiomyopathy slowed the decay of [Ca2+]i.151 This was suggested to result from decreased activity of NCX and PMCA. This is surprising because these sarcolemmal transporters make only a small contribution (compared with SERCA) to the decay of the systolic Ca transient in small animals. In contrast, lipopolysaccharide administration in mice also slowed the decrease in the Ca transient, but this was associated with decreased SERCA activity due to sulphonylation.152 This was accompanied by a small decrease in diastolic [Ca2+]i over the range of 1 to 6 Hz, the explanation for which is not clear.
Although it is not easy to draw conclusions from the above work on patients and animal models with HFrEF, it does appear that in the majority of studies where physiological rates have been studied, there is a frequency-dependent increase in diastolic [Ca2+]i and force. More work is needed at physiological rates to characterize this.
Heart Failure With Preserved Ejection Fraction
A major issue with studying HFpEF in animals has been the difficulty of producing an appropriate model.153,154 Work in 2 articles has developed potential models of HFpEF by banding the aorta in rats. In one, ventricular myocytes displayed an increase in both diastolic [Ca2+]i and the amplitude of the Ca transient. These effects were attributed, at least in part, to increased Ca2+ leak through the RyR (seen as increased Ca spark frequency) and decreased NCX activity.155 In the other, although the animals and isolated ventricular trabeculae had impaired diastolic function, isolated myocytes, taken from the same hearts, showed lower diastolic [Ca2+]i and shortening, suggesting that the main cause of mechanical dysfunction involved passive mechanisms rather than Ca2+ handling.156 Modeling studies have pointed out that the maintained ejection fraction in HFpEF could be achieved despite a decrease in systolic [Ca2+]i due to the compensatory effect of concentric ventricular hypertrophy.157 Another, recently developed, model of HFpEF is that of an inbred rat with a hypertrophic heart. This has increased diastolic and systolic [Ca2+]i accompanied by an increase in the L-type Ca current.127 It is, therefore, possible that the increase in both diastolic and systolic [Ca2+]i augments Ca2+ efflux to compensate for the increased influx. In the absence of measurements, however, it is impossible to exclude a contribution from effects of [Na+]i mediated via NCX. Interestingly, the rate of decay of the Ca transient was accelerated arguing against decreased SERCA activity.
Kidney disease is a risk factor for HFpEF, and this has been modeled experimentally by removing 80% of renal tissue resulting in prolonged ventricular relaxation and elevated end-diastolic pressure. Early work found elevated diastolic [Ca2+]i, attributed to altered NCX possibly due to increased [Na+]i.158 A subsequent study159 showed a slowing of the rate of decay of both cell shortening and the systolic Ca transient but no effect on the level of either systolic or diastolic [Ca2+]i. The experiments were, however, performed at a slow rate. This study also found that acute administration of the NCX inhibitor SEA0400 accelerated the decay of the systolic Ca transient, but the mechanism was unclear. Another study from this group found that in vivo administration of another NCX inhibitor (ORM-11035) also accelerated relaxation160—a result consistent with studies on the Dahl salt-sensitive rat where the improved relaxation produced by NCX inhibition was attributed to an effect on fibroblasts, decreasing fibrosis.161
As mentioned above, diastolic dysfunction is clinically observed in diabetes mellitus. Work on a streptozotocin rat model, with normal systolic and impaired diastolic function, found a decrease in the rate constant of decay of the systolic Ca transient due to decreased SERCA activity.162 Another study using the same model observed a slowing of decay, but this was accompanied by a fall not only of systolic but also diastolic [Ca2+]i during stimulation at 1 Hz.163 It is unclear why diastolic [Ca2+]i should decrease. Some studies have found decreased L-type Ca current,164 which may decrease average [Ca2+]i. In addition, the slowing of decay of the Ca transient will increase average [Ca2+]i thereby allowing a lower diastolic [Ca2+]i as long as there is sufficient time for [Ca2+]i to fall in diastole. A similar study found a decrease in resting [Ca2+]i in unstimulated cells.165 This argues for alterations of background Ca2+ influx or NCX/PMCA. It should, however, be noted that there is evidence that the depression of contractility in the streptozotocin rat may be independent of changes of [Ca2+]i.166 Metabolic dysregulation is also associated with development of HFpEF. A recent study found that the ZSF-1 obese rat had elevated diastolic but similar systolic [Ca2+]i compared with controls.167 Mitochondrial [Ca2+] was also elevated and suggested to partly compensate by stimulating metabolism but also to result in adverse consequences of mitochondrial overload. This article also reported a decrease in diastolic [Ca2+]i with increasing stimulation rate—a result that differs from other studies reviewed here. Atria from the ZSF-1 obese rat also show impaired function but no effect on either the rate constant of decay of the Ca transient or diastolic [Ca2+]i was observed.168 Finally, a recent publication has introduced a mouse model of HFpEF using a combination of high-fat diet and nitrosative stress, which seems to mimic many of the features of the human condition.169 It would be useful to use this model to study [Ca2+]i.
Work on ventricular strips from patients with cardiac hypertrophy but normal ejection fraction has found interesting results. When the stimulation rate was increased, preparations from hearts showing LV hypertrophy developed increased diastolic force, and this was abolished by the contractile uncoupler butanedione monoxime suggesting that it resulted from myofilament activation. When SR function was inhibited by the SERCA inhibitor cyclopiazonic acid plus ryanodine, there was an elevation of diastolic force, which was much greater in those preparations that had previously developed significant diastolic force with raised frequency.170
Finally, hypertrophic cardiomyopathy can also result in a heart failure syndrome. While this is a separate disease entity to HFpEF, it also leads to diastolic dysfunction. Hypertrophic cardiomyopathy is often an inherited condition that can result from mutations in the sarcomeric proteins, which make up the thick and thin filaments, (reviewed in171). Many of these mutations increase the sensitivity of the myofilaments for [Ca2+]i. This, alone, would increase diastolic force, but, in addition, the increase in Ca2+ buffering slows the decay of [Ca2+]i, elevating end-diastolic [Ca2+]i,29 and, therefore, diastolic force/pressure.
How Does Diastolic [Ca2+]i Increase in Heart Failure?
Although, as discussed above, there is considerable variation between studies, the consensus appear to be that diastolic [Ca2+]i increases in heart failure. There are at least 2 (nonexclusive) explanations for this. (1) As discussed in an earlier section, any decrease in the systolic Ca transient will decrease Ca2+ efflux during systole, requiring a compensatory increase in diastolic [Ca2+]i. (2) Another explanation is provided by the increase in [Na+]i commonly observed in heart failure,41,42,143,172 which will decrease Ca2+ efflux on NCX, thereby requiring an increase in average [Ca2+]i, which may, in part, be provided by increased diastolic [Ca2+]i. Consistent with this, elevation of [Na+]i by inhibition of the sodium pump increased diastolic force at elevated stimulation rates.143 Further evidence linking NCX to diastolic function came from work on ventricular strips from failing human hearts showing that the greater the expression of NCX, the better the diastolic function.173 The increase in diastolic force and [Ca2+]i can also be attenuated by the drug ranolazine—a blocker of the late sodium current that decreases [Na+]i.174 Similarly, work on rats found that ranolazine reversed the diastolic impairment produced by the anticancer drug doxyrubicin.175 In canine myocytes, experimental ischemic heart failure increased diastolic [Ca2+]i at elevated rates, and this was normalized by ranolazine or tetrodotoxin.176 Work on mice found that overexpressing CaMKIIδC decreased systolic and increased diastolic force.35 The decrease of systolic [Ca2+]i has been attributed to excessive phosphorylation of RyRs leading to diastolic Ca2+ leak177 as evidenced by increased Ca2+ spark frequency. Again, these effects were reversed by ranolazine thereby linking them to changes of [Na+]i.178 In contrast, a recent study found that ranolazine had no effect on diastolic force in ventricular muscle taken from patients with HFpEF, arguing against a role for changes of [Na+]i.179
As mentioned in an earlier section, a different explanation of elevated diastolic [Ca2+]i has been suggested in the cardiomyopathy observed in the mdx mouse—a model of Duchenne muscular dystrophy. Here, the elevated diastolic [Ca2+]i is normalized by Gd3+ suggesting that it originates from Ca2+ entry through TRP channels.77
Differences of Ca2+ Handling in HFrEF and HFpEF: a Role for NCX and [Na+]i?
An unresolved question concerns the cellular mechanisms responsible for the difference in systolic function between HFrEF and HFpEF. Figure 5 shows a speculative hypothesis. For the sake of argument, we will assume that it results from differences in Ca2+ signaling and that, in both cases, there is a combination of increased NCX, leaky RyR, and decreased SERCA activity resulting in decreased SR Ca content and thence the amplitude of the systolic Ca transient and systolic function. The decreased systolic efflux will require an increase in diastolic efflux so increasing diastolic [Ca2+]i. These changes could, therefore, account for HFrEF (Figure 5A). The rise of [Na+]i often seen in heart failure will slow NCX and, if sufficient, will overcome the effects of the other changes thereby maintaining SR Ca content and systolic [Ca2+]i at control levels. Diastolic [Ca2+]i will be increased to maintain Ca2+ efflux despite the inhibited NCX. The combination would, therefore, produce an HFpEF phenotype (Figure 5B). It is, therefore, possible that the changes of Ca2+ cycling that underlie HFrEF and HFpEF are qualitatively identical but that, in HFpEF, the increase in [Na+]i dominates over the other changes. Clearly, experimental studies are required to see whether this simplistic hypothesis has any validity.
Figure 5.
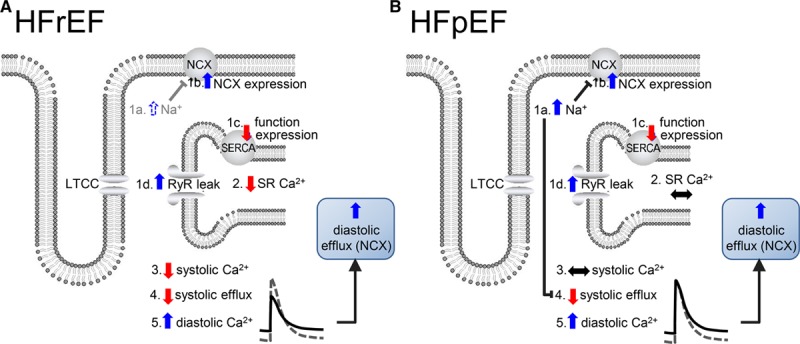
Speculative hypothesis to account for the difference in Ca2+ handling between heart failure with reduced ejection fraction (HFrEF) and heart failure with preserved ejection fraction (HFpEF). A, HFrEF. B, HFpEF. In both panels, heart failure results in an increase in intracellular Na+ concentration ([Na+]i), 1a; increase in sodium-calcium exchange (NCX) expression, 1b; decrease in sarcoplasmic reticulum Ca-ATPase (SERCA) function or expression, 1c; increase in Ca2+ leak through ryanodine receptor (RyR), 1d. We assume that in A (HFrEF) 1b–1d dominate over 1a with the net result being a decrease of sarcoplasmic reticulum (SR) Ca2+ content (2) and systolic intracellular Ca2+ concentration ([Ca2+]I; 3), systolic efflux (4) and a consequent increase in diastolic [Ca2+]i (5), which (via NCX) raises diastolic efflux. In B (HFpEF), the increase in [Na+]i (1a) dominates over 1b-d so that SR Ca2+ content (2) and systolic [Ca2+]i (3) are unchanged. The increase in [Na+]i decreases NCX activity so that diastolic [Ca2+]i (5) has to increase to maintain systolic efflux (4) and flux balance. LTCC indicates L-type Ca channel.
Conclusions
Control of diastolic calcium concentration is essential for normal cardiac function. As we have discussed, this regulation depends on precise balance between influx and efflux. However, there are still major uncertainties about how this is achieved. In particular, more work is required to investigate the role of the PMCA, as well as the nature of the background Ca2+ influx. It is essential that studies are performed at physiological heart rates. It is also important to characterize the alterations of Ca2+ signaling that occur in heart failure and how they may differ in failure with preserved compared with reduced ejection fraction.
Sources of Funding
This work is supported by grants from the British Heart Foundation (CH/2000004/12801 to D.A. Eisner; FS/12/57/29717 to A.W. Trafford). D.C. Hutchings is an academic clinical lecturer supported by the National Institute of Health Research.
Disclosures
None.
Footnotes
Nonstandard Abbreviations and Acronyms
- [Ca2+]i
- intracellular Ca2+ concentration
- [Na+]i
- intracellular Na+ concentration
- CaMKII
- Ca2+/calmodulin-dependent protein kinase II
- Gd3+
- gadolinium
- HFpEF
- heart failure with preserved ejection fraction
- HFrEF
- heart failure with reduced ejection fraction
- LV
- left ventricle
- MCU
- mitochondrial calcium uniporter
- NCX
- sodium-calcium exchange
- PKG
- protein kinase G
- PLN
- phospholamban
- PMCA
- plasma membrane Ca-ATPase
- RyR
- ryanodine receptor
- SERCA
- sarcoplasmic reticulum Ca-ATPase
- SR
- sarcoplasmic reticulum
- STIM1
- stromal interaction molecule 1
- TRP
- transient receptor potential
For Sources of Funding and Disclosures, see page 407.
References
- 1.Milne AA. Now We Are Six. London: Methuen; 1927. [Google Scholar]
- 2.Gibbs CL, Loiselle DS, Wendt IR. Activation heat in rabbit cardiac muscle. J Physiol. 1988;395:115–130. doi: 10.1113/jphysiol.1988.sp016911. doi: 10.1113/jphysiol.1988.sp016911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Smith GL, Eisner DA. Calcium buffering in the heart in health and disease. Circulation. 2019;139:2358–2371. doi: 10.1161/CIRCULATIONAHA.118.039329. doi: 10.1161/CIRCULATIONAHA.118.039329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bers DM. Excitation-Contraction Coupling and Cardiac Contractile Force. 2nd ed. Dordrecht, London: Kluwer Academic; 2001. [Google Scholar]
- 5.Bers DM. Cardiac excitation-contraction coupling. Nature. 2002;415:198–205. doi: 10.1038/415198a. doi: 10.1038/415198a. [DOI] [PubMed] [Google Scholar]
- 6.Eisner DA, Caldwell JL, Kistamás K, Trafford AW. Calcium and excitation-contraction coupling in the heart. Circ Res. 2017;121:181–195. doi: 10.1161/CIRCRESAHA.117.310230. doi: 10.1161/CIRCRESAHA.117.310230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cannell MB, Berlin JR, Lederer WJ. Effect of membrane potential changes on the calcium transient in single rat cardiac muscle cells. Science. 1987;238:1419–1423. doi: 10.1126/science.2446391. doi: 10.1126/science.2446391. [DOI] [PubMed] [Google Scholar]
- 8.Barcenas-Ruiz L, Wier WG. Voltage dependence of intracellular [Ca2+]i transients in guinea pig ventricular myocytes. Circ Res. 1987;61:148–154. doi: 10.1161/01.res.61.1.148. doi: 10.1161/01.res.61.1.148. [DOI] [PubMed] [Google Scholar]
- 9.Bassani JW, Yuan W, Bers DM. Fractional SR Ca release is regulated by trigger Ca and SR Ca content in cardiac myocytes. Am J Physiol. 1995;268:C1313–C1319. doi: 10.1152/ajpcell.1995.268.5.C1313. doi: 10.1152/ajpcell.1995.268.5.C1313. [DOI] [PubMed] [Google Scholar]
- 10.Trafford AW, Díaz ME, Eisner DA. Stimulation of Ca-induced Ca release only transiently increases the systolic Ca transient: measurements of Ca fluxes and sarcoplasmic reticulum Ca. Cardiovasc Res. 1998;37:710–717. doi: 10.1016/s0008-6363(97)00266-6. doi: 10.1016/s0008-6363(97)00266-6. [DOI] [PubMed] [Google Scholar]
- 11.Cheng H, Lederer WJ, Cannell MB. Calcium sparks: elementary events underlying excitation-contraction coupling in heart muscle. Science. 1993;262:740–744. doi: 10.1126/science.8235594. doi: 10.1126/science.8235594. [DOI] [PubMed] [Google Scholar]
- 12.Allen DG, Eisner DA, Orchard CH. Characterization of oscillations of intracellular calcium concentration in ferret ventricular muscle. J Physiol. 1984;352:113–128. doi: 10.1113/jphysiol.1984.sp015281. doi: 10.1113/jphysiol.1984.sp015281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ríos E. The cell boundary theorem: a simple law of the control of cytosolic calcium concentration. J Physiol Sci. 2010;60:81–84. doi: 10.1007/s12576-009-0069-z. doi: 10.1007/s12576-009-0069-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sankaranarayanan R, Kistamas K, Greensmith DJ, Venetucci LA, Eisner DA. Systolic [Ca2+]i regulates diastolic levels in rat ventricular myocytes. J Physiol. 2017;595:5545–5555. doi: 10.1113/JP274366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Litwin SE, Zhang D, Bridge JH. Dyssynchronous Ca2+ sparks in myocytes from infarcted hearts. Circ Res. 2000;87:1040–1047. doi: 10.1161/01.res.87.11.1040. doi: 10.1161/01.res.87.11.1040. [DOI] [PubMed] [Google Scholar]
- 16.Negretti N, O’Neill SC, Eisner DA. The effects of inhibitors of sarcoplasmic reticulum function on the systolic Ca2+ transient in rat ventricular myocytes. J Physiol. 1993;468:35–52. doi: 10.1113/jphysiol.1993.sp019758. doi: 10.1113/jphysiol.1993.sp019758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Barcenas-Ruiz L, Beuckelmann DJ, Wier WG. Sodium-calcium exchange in heart: membrane currents and changes in [Ca2+]i. Science. 1987;238:1720–1722. doi: 10.1126/science.3686010. doi: 10.1126/science.3686010. [DOI] [PubMed] [Google Scholar]
- 18.Trafford AW, Díaz ME, Negretti N, Eisner DA. Enhanced Ca2+ current and decreased Ca2+ efflux restore sarcoplasmic reticulum Ca2+ content after depletion. Circ Res. 1997;81:477–484. doi: 10.1161/01.res.81.4.477. doi: 10.1161/01.res.81.4.477. [DOI] [PubMed] [Google Scholar]
- 19.Shannon TR, Wang F, Puglisi J, Weber C, Bers DM. A mathematical treatment of integrated Ca dynamics within the ventricular myocyte. Biophys J. 2004;87:3351–3371. doi: 10.1529/biophysj.104.047449. doi: 10.1529/biophysj.104.047449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bennett DL, O’Neill SC, Eisner DA. Strophanthidin-induced gain of Ca2+ occurs during diastole and not systole in guinea-pig ventricular myocytes. Pflugers Arch. 1999;437:731–736. doi: 10.1007/s004240050839. doi: 10.1007/s004240050839. [DOI] [PubMed] [Google Scholar]
- 21.Mechmann S, Pott L. Identification of Na-Ca exchange current in single cardiac myocytes. Nature. 1986;319:597–599. doi: 10.1038/319597a0. doi: 10.1038/319597a0. [DOI] [PubMed] [Google Scholar]
- 22.Kass RS, Lederer WJ, Tsien RW, Weingart R. Role of calcium ions in transient inward currents and aftercontractions induced by strophanthidin in cardiac Purkinje fibres. J Physiol. 1978;281:187–208. doi: 10.1113/jphysiol.1978.sp012416. doi: 10.1113/jphysiol.1978.sp012416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Díaz ME, Trafford AW, O’Neill SC, Eisner DA. Measurement of sarcoplasmic reticulum Ca2+ content and sarcolemmal Ca2+ fluxes in isolated rat ventricular myocytes during spontaneous Ca2+ release. J Physiol. 1997;501(pt 1):3–16. doi: 10.1111/j.1469-7793.1997.003bo.x. doi: 10.1111/j.1469-7793.1997.003bo.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Trafford AW, Díaz ME, Sibbring GC, Eisner DA. Modulation of CICR has no maintained effect on systolic Ca2+: simultaneous measurements of sarcoplasmic reticulum and sarcolemmal Ca2+ fluxes in rat ventricular myocytes. J Physiol. 2000;522((pt 2)):259–270. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. doi: 10.1111/j.1469-7793.2000.t01-2-00259.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Greensmith DJ, Galli GL, Trafford AW, Eisner DA. Direct measurements of SR free Ca reveal the mechanism underlying the transient effects of RyR potentiation under physiological conditions. Cardiovasc Res. 2014;103:554–563. doi: 10.1093/cvr/cvu158. doi: 10.1093/cvr/cvu158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eisner D, Bode E, Venetucci L, Trafford A. Calcium flux balance in the heart. J Mol Cell Cardiol. 2013;58:110–117. doi: 10.1016/j.yjmcc.2012.11.017. doi: 10.1016/j.yjmcc.2012.11.017. [DOI] [PubMed] [Google Scholar]
- 27.Díaz ME, Trafford AW, Eisner DA. The effects of exogenous calcium buffers on the systolic calcium transient in rat ventricular myocytes. Biophys J. 2001;80:1915–1925. doi: 10.1016/S0006-3495(01)76161-9. doi: 10.1016/S0006-3495(01)76161-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Steenbergen C, Murphy E, Levy L, London RE. Elevation in cytosolic free calcium concentration early in myocardial ischemia in perfused rat heart. Circ Res. 1987;60:700–707. doi: 10.1161/01.res.60.5.700. doi: 10.1161/01.res.60.5.700. [DOI] [PubMed] [Google Scholar]
- 29.Schober T, Huke S, Venkataraman R, Gryshchenko O, Kryshtal D, Hwang HS, Baudenbacher FJ, Knollmann BC. Myofilament Ca sensitization increases cytosolic Ca binding affinity, alters intracellular Ca homeostasis, and causes pause-dependent Ca-triggered arrhythmia. Circ Res. 2012;111:170–179. doi: 10.1161/CIRCRESAHA.112.270041. doi: 10.1161/CIRCRESAHA.112.270041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elliott EB, Kelly A, Smith GL, Loughrey CM. Isolated rabbit working heart function during progressive inhibition of myocardial SERCA activity. Circ Res. 2012;110:1618–1627. doi: 10.1161/CIRCRESAHA.111.262337. doi: 10.1161/CIRCRESAHA.111.262337. [DOI] [PubMed] [Google Scholar]
- 31.Maier LS, Pieske B, Allen DG. Influence of stimulation frequency on [Na+]i and contractile function in Langendorff-perfused rat heart. Am J Physiol. 1997;273:H1246–H1254. doi: 10.1152/ajpheart.1997.273.3.H1246. doi: 10.1152/ajpheart.1997.273.3.H1246. [DOI] [PubMed] [Google Scholar]
- 32.Despa S, Bers DM. Na+ transport in the normal and failing heart - remember the balance. J Mol Cell Cardiol. 2013;61:2–10. doi: 10.1016/j.yjmcc.2013.04.011. doi: 10.1016/j.yjmcc.2013.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Louch WE, Hougen K, Mørk HK, Swift F, Aronsen JM, Sjaastad I, Reims HM, Roald B, Andersson KB, Christensen G, et al. Sodium accumulation promotes diastolic dysfunction in end-stage heart failure following Serca2 knockout. J Physiol. 2010;588:465–478. doi: 10.1113/jphysiol.2009.183517. doi: 10.1113/jphysiol.2009.183517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sankaranarayanan R, Li Y, Greensmith DJ, Eisner DA, Venetucci L. Biphasic decay of the Ca transient results from increased sarcoplasmic reticulum Ca leak. J Physiol. 2016;594:611–623. doi: 10.1113/JP271473. doi: 10.1113/JP271473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guo T, Zhang T, Ginsburg KS, Mishra S, Brown JH, Bers DM. CaMKIIδC slows [Ca]i decline in cardiac myocytes by promoting Ca sparks. Biophys J. 2012;102:2461–2470. doi: 10.1016/j.bpj.2012.04.015. doi: 10.1016/j.bpj.2012.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fowler ED, Kong CHT, Hancox JC, Cannell MB. Late Ca2+ sparks and ripples during the systolic Ca2+ transient in heart muscle cells. Circ Res. 2018;122:473–478. doi: 10.1161/CIRCRESAHA.117.312257. doi: 10.1161/CIRCRESAHA.117.312257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Montalvo D, Pérez-Treviño P, Madrazo-Aguirre K, González-Mondellini FA, Miranda-Roblero HO, Ramonfaur-Gracia D, Jacobo-Antonio M, Mayorga-Luna M, Gómez-Víquez NL, García N, et al. Underlying mechanism of the contractile dysfunction in atrophied ventricular myocytes from a murine model of hypothyroidism. Cell Calcium. 2018;72:26–38. doi: 10.1016/j.ceca.2018.01.005. doi: 10.1016/j.ceca.2018.01.005. [DOI] [PubMed] [Google Scholar]
- 38.Kimura J, Noma A, Irisawa H. Na-Ca exchange current in mammalian heart cells. Nature. 1986;319:596–597. doi: 10.1038/319596a0. doi: 10.1038/319596a0. [DOI] [PubMed] [Google Scholar]
- 39.Ginsburg KS, Weber CR, Bers DM. Cardiac Na+-Ca2+ exchanger: dynamics of Ca2+-dependent activation and deactivation in intact myocytes. J Physiol. 2013;591:2067–2086. doi: 10.1113/jphysiol.2013.252080. doi: 10.1113/jphysiol.2013.252080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Blaustein MP, Lederer WJ. Sodium/calcium exchange: its physiological implications. Physiol Rev. 1999;79:763–854. doi: 10.1152/physrev.1999.79.3.763. doi: 10.1152/physrev.1999.79.3.763. [DOI] [PubMed] [Google Scholar]
- 41.Baartscheer A, Schumacher CA, van Borren MM, Belterman CN, Coronel R, Fiolet JW. Increased Na+/H+-exchange activity is the cause of increased [Na+]i and underlies disturbed calcium handling in the rabbit pressure and volume overload heart failure model. Cardiovasc Res. 2003;57:1015–1024. doi: 10.1016/s0008-6363(02)00809-x. doi: 10.1016/s0008-6363(02)00809-x. [DOI] [PubMed] [Google Scholar]
- 42.Beuckelmann DJ, Näbauer M, Erdmann E. Intracellular calcium handling in isolated ventricular myocytes from patients with terminal heart failure. Circulation. 1992;85:1046–1055. doi: 10.1161/01.cir.85.3.1046. doi: 10.1161/01.cir.85.3.1046. [DOI] [PubMed] [Google Scholar]
- 43.Wier WG, Hess P. Excitation-contraction coupling in cardiac Purkinje fibers. Effects of cardiotonic steroids on the intracellular [Ca2+] transient, membrane potential, and contraction. J Gen Physiol. 1984;83:395–415. doi: 10.1085/jgp.83.3.395. doi: 10.1085/jgp.83.3.395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Eisner DA, Lederer WJ, Vaughan-Jones RD. The quantitative relationship between twitch tension and intracellular sodium activity in sheep cardiac Purkinje fibres. J Physiol. 1984;355:251–266. doi: 10.1113/jphysiol.1984.sp015417. doi: 10.1113/jphysiol.1984.sp015417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Eisner DA, Lederer WJ. Inotropic and arrhythmogenic effects of potassium-depleted solutions on mammalian cardiac muscle. J Physiol. 1979;294:255–277. doi: 10.1113/jphysiol.1979.sp012929. doi: 10.1113/jphysiol.1979.sp012929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Harrison SM, McCall E, Boyett MR. The relationship between contraction and intracellular sodium in rat and guinea-pig ventricular myocytes. J Physiol. 1992;449:517–550. doi: 10.1113/jphysiol.1992.sp019100. doi: 10.1113/jphysiol.1992.sp019100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Stafford N, Wilson C, Oceandy D, Neyses L, Cartwright EJ. The plasma membrane calcium atpases and their role as major new players in human disease. Physiol Rev. 2017;97:1089–1125. doi: 10.1152/physrev.00028.2016. doi: 10.1152/physrev.00028.2016. [DOI] [PubMed] [Google Scholar]
- 48.Mohamed TM, Oceandy D, Zi M, Prehar S, Alatwi N, Wang Y, Shaheen MA, Abou-Leisa R, Schelcher C, Hegab Z, et al. Plasma membrane calcium pump (PMCA4)-neuronal nitric-oxide synthase complex regulates cardiac contractility through modulation of a compartmentalized cyclic nucleotide microdomain. J Biol Chem. 2011;286:41520–41529. doi: 10.1074/jbc.M111.290411. doi: 10.1074/jbc.M111.290411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bassani RA, Bassani JW, Bers DM. Mitochondrial and sarcolemmal Ca2+ transport reduce [Ca2+]i during caffeine contractures in rabbit cardiac myocytes. J Physiol. 1992;453:591–608. doi: 10.1113/jphysiol.1992.sp019246. doi: 10.1113/jphysiol.1992.sp019246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Varro A, Negretti N, Hester SB, Eisner DA. An estimate of the calcium content of the sarcoplasmic reticulum in rat ventricular myocytes. Pflugers Arch. 1993;423:158–160. doi: 10.1007/BF00374975. doi: 10.1007/bf00374975. [DOI] [PubMed] [Google Scholar]
- 51.O’Neill SC, Valdeolmillos M, Lamont C, Donoso P, Eisner DA. The contribution of Na-Ca exchange to relaxation in mammalian cardiac muscle. Ann N Y Acad Sci. 1991;639:444–452. doi: 10.1111/j.1749-6632.1991.tb17331.x. doi: 10.1111/j.1749-6632.1991.tb17331.x. [DOI] [PubMed] [Google Scholar]
- 52.Lamont C, Eisner DA. The sarcolemmal mechanisms involved in the control of diastolic intracellular calcium in isolated rat cardiac trabeculae. Pflugers Arch. 1996;432:961–969. doi: 10.1007/s004240050223. doi: 10.1007/s004240050223. [DOI] [PubMed] [Google Scholar]
- 53.Bassani RA, Bassani JW, Bers DM. Relaxation in ferret ventricular myocytes: role of the sarcolemmal Ca ATPase. Pflugers Arch. 1995;430:573–578. doi: 10.1007/BF00373894. doi: 10.1007/bf00373894. [DOI] [PubMed] [Google Scholar]
- 54.Choi HS, Eisner DA. The effects of inhibition of the sarcolemmal Ca-ATPase on systolic calcium fluxes and intracellular calcium concentration in rat ventricular myocytes. Pflugers Arch. 1999;437:966–971. doi: 10.1007/s004240050868. doi: 10.1007/s004240050868. [DOI] [PubMed] [Google Scholar]
- 55.Henderson SA, Goldhaber JI, So JM, Han T, Motter C, Ngo A, Chantawansri C, Ritter MR, Friedlander M, Nicoll DA, et al. Functional adult myocardium in the absence of Na+-Ca2+ exchange: cardiac-specific knockout of NCX1. Circ Res. 2004;95:604–611. doi: 10.1161/01.RES.0000142316.08250.68. doi: 10.1161/01.RES.0000142316.08250.68. [DOI] [PubMed] [Google Scholar]
- 56.Pott C, Philipson KD, Goldhaber JI. Excitation-contraction coupling in Na+-Ca2+ exchanger knockout mice: reduced transsarcolemmal Ca2+ flux. Circ Res. 2005;97:1288–1295. doi: 10.1161/01.RES.0000196563.84231.21. doi: 10.1161/01.RES.0000196563.84231.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lu X, Ginsburg KS, Kettlewell S, Bossuyt J, Smith GL, Bers DM. Measuring local gradients of intramitochondrial [Ca2+] in cardiac myocytes during sarcoplasmic reticulum Ca2+ release. Circ Res. 2013;112:424–431. doi: 10.1161/CIRCRESAHA.111.300501. doi: 10.1161/CIRCRESAHA.111.300501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sipido KR, Wier WG. Flux of Ca2+ across the sarcoplasmic reticulum of guinea-pig cardiac cells during excitation-contraction coupling. J Physiol. 1991;435:605–630. doi: 10.1113/jphysiol.1991.sp018528. doi: 10.1113/jphysiol.1991.sp018528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mesirca P, Torrente AG, Mangoni ME. T-type channels in the sino-atrial and atrioventricular pacemaker mechanism. Pflugers Arch. 2014;466:791–799. doi: 10.1007/s00424-014-1482-6. doi: 10.1007/s00424-014-1482-6. [DOI] [PubMed] [Google Scholar]
- 60.Sipido KR, Maes M, Van de Werf F. Low efficiency of Ca2+ entry through the Na+-Ca2+ exchanger as trigger for Ca2+ release from the sarcoplasmic reticulum. A comparison between L-type Ca2+ current and reverse-mode Na+-Ca2+ exchange. Circ Res. 1997;81:1034–1044. doi: 10.1161/01.res.81.6.1034. doi: 10.1161/01.res.81.6.1034. [DOI] [PubMed] [Google Scholar]
- 61.Dipla K, Mattiello JA, Margulies KB, Jeevanandam V, Houser SR. The sarcoplasmic reticulum and the Na+/Ca2+ exchanger both contribute to the Ca2+ transient of failing human ventricular myocytes. Circ Res. 1999;84:435–444. doi: 10.1161/01.res.84.4.435. doi: 10.1161/01.res.84.4.435. [DOI] [PubMed] [Google Scholar]
- 62.Sipido KR, Carmeliet E, Pappano A. Na+ current and Ca2+ release from the sarcoplasmic reticulum during action potentials in guinea-pig ventricular myocytes. J Physiol. 1995;489(pt 1):1–17. doi: 10.1113/jphysiol.1995.sp021025. doi: 10.1113/jphysiol.1995.sp021025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Huang MH, Knight PR, III, Izzo JL., Jr. Ca2+-induced Ca2+ release involved in positive inotropic effect mediated by CGRP in ventricular myocytes. Am J Physiol. 1999;276:R259–R264. doi: 10.1152/ajpregu.1999.276.1.R259. doi: 10.1152/ajpregu.1999.276.1.R259. [DOI] [PubMed] [Google Scholar]
- 64.Díaz ME, Trafford AW, Eisner DA. The role of intracellular Ca buffers in determining the shape of the systolic Ca transient in cardiac ventricular myocytes. Pflugers Arch. 2001;442:96–100. doi: 10.1007/s004240000509. doi: 10.1007/s004240000509. [DOI] [PubMed] [Google Scholar]
- 65.Terracciano CM, MacLeod KT. Reloading of Ca2+-depleted sarcoplasmic reticulum during rest in guinea pig ventricular myocytes. Am J Physiol. 1996;271:H1814–H1822. doi: 10.1152/ajpheart.1996.271.5.H1814. doi: 10.1152/ajpheart.1996.271.5.H1814. [DOI] [PubMed] [Google Scholar]
- 66.Choi HS, Trafford AW, Eisner DA. Measurement of calcium entry and exit in quiescent rat ventricular myocytes. Pflugers Arch. 2000;440:600–608. doi: 10.1007/s004240000295. doi: 10.1007/s004240000295. [DOI] [PubMed] [Google Scholar]
- 67.Kupittayanant P, Trafford AW, Díaz ME, Eisner DA. A mechanism distinct from the L-type Ca current or Na-Ca exchange contributes to Ca entry in rat ventricular myocytes. Cell Calcium. 2006;39:417–423. doi: 10.1016/j.ceca.2006.01.011. doi: 10.1016/j.ceca.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 68.Stout CE, Costantin JL, Naus CC, Charles AC. Intercellular calcium signaling in astrocytes via ATP release through connexin hemichannels. J Biol Chem. 2002;277:10482–10488. doi: 10.1074/jbc.M109902200. doi: 10.1074/jbc.M109902200. [DOI] [PubMed] [Google Scholar]
- 69.Leybaert L, Lampe PD, Dhein S, Kwak BR, Ferdinandy P, Beyer EC, Laird DW, Naus CC, Green CR, Schulz R. Connexins in cardiovascular and neurovascular health and disease: pharmacological implications. Pharmacol Rev. 2017;69:396–478. doi: 10.1124/pr.115.012062. doi: 10.1124/pr.115.012062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wang N, De Bock M, Antoons G, Gadicherla AK, Bol M, Decrock E, Evans WH, Sipido KR, Bukauskas FF, Leybaert L. Connexin mimetic peptides inhibit Cx43 hemichannel opening triggered by voltage and intracellular Ca2+ elevation. Basic Res Cardiol. 2012;107:304. doi: 10.1007/s00395-012-0304-2. doi: 10.1007/s00395-012-0304-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kim JC, Pérez-Hernández M, Alvarado FJ, Maurya SR, Montnach J, Yin Y, Zhang M, Lin X, Vasquez C, Heguy A, et al. Disruption of Ca2+i homeostasis and connexin 43 hemichannel function in the right ventricle precedes overt arrhythmogenic cardiomyopathy in plakophilin-2-deficient mice. Circulation. 2019;140:1015–1030. doi: 10.1161/CIRCULATIONAHA.119.039710. doi: 10.1161/CIRCULATIONAHA.119.039710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Rubinstein J, Lasko VM, Koch SE, Singh VP, Carreira V, Robbins N, Patel AR, Jiang M, Bidwell P, Kranias EG, et al. Novel role of transient receptor potential vanilloid 2 in the regulation of cardiac performance. Am J Physiol Heart Circ Physiol. 2014;306:H574–H584. doi: 10.1152/ajpheart.00854.2013. doi: 10.1152/ajpheart.00854.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Koch SE, Gao X, Haar L, Jiang M, Lasko VM, Robbins N, Cai W, Brokamp C, Varma P, Tranter M, et al. Probenecid: novel use as a non-injurious positive inotrope acting via cardiac TRPV2 stimulation. J Mol Cell Cardiol. 2012;53:134–144. doi: 10.1016/j.yjmcc.2012.04.011. doi: 10.1016/j.yjmcc.2012.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Robbins N, Gilbert M, Kumar M, McNamara JW, Daly P, Koch SE, Conway G, Effat M, Woo JG, Sadayappan S, et al. Probenecid improves cardiac function in patients with heart failure with reduced ejection fraction in vivo and cardiomyocyte calcium sensitivity in vitro. J Am Heart Assoc. 2018;7:e007148. doi: 10.1161/JAHA.117.007148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Di Virgilio F, Steinberg TH, Silverstein SC. Inhibition of Fura-2 sequestration and secretion with organic anion transport blockers. Cell Calcium. 1990;11:57–62. doi: 10.1016/0143-4160(90)90059-4. doi: 10.1016/0143-4160(90)90059-4. [DOI] [PubMed] [Google Scholar]
- 76.Jones JL, Peana D, Veteto AB, Lambert MD, Nourian Z, Karasseva NG, Hill MA, Lindman BR, Baines CP, Krenz M, et al. TRPV4 increases cardiomyocyte calcium cycling and contractility yet contributes to damage in the aged heart following hypoosmotic stress. Cardiovasc Res. 2019;115:cvy156–cvy156. doi: 10.1093/cvr/cvy156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Mijares A, Altamirano F, Kolster J, Adams JA, López JR. Age-dependent changes in diastolic Ca2+ and Na+ concentrations in dystrophic cardiomyopathy: role of Ca2+ entry and IP3. Biochem Biophys Res Commun. 2014;452:1054–1059. doi: 10.1016/j.bbrc.2014.09.045. doi: 10.1016/j.bbrc.2014.09.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Chen MS, Xiao JH, Wang Y, Xu BM, Gao L, Wang JL. Upregulation of TRPC1 contributes to contractile function in isoproterenol-induced hypertrophic myocardium of rat. Cell Physiol Biochem. 2013;32:951–959. doi: 10.1159/000354498. doi: 10.1159/000354498. [DOI] [PubMed] [Google Scholar]
- 79.Camacho Londoño JE, Tian Q, Hammer K, Schröder L, Camacho Londoño J, Reil JC, He T, Oberhofer M, Mannebach S, Mathar I, et al. A background Ca2+ entry pathway mediated by TRPC1/TRPC4 is critical for development of pathological cardiac remodelling. Eur Heart J. 2015;36:2257–2266. doi: 10.1093/eurheartj/ehv250. doi: 10.1093/eurheartj/ehv250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Rubaiy HN, Ludlow MJ, Henrot M, Gaunt HJ, Miteva K, Cheung SY, Tanahashi Y, Hamzah N, Musialowski KE, Blythe NM, et al. Picomolar, selective, and subtype-specific small-molecule inhibition of TRPC1/4/5 channels. J Biol Chem. 2017;292:8158–8173. doi: 10.1074/jbc.M116.773556. doi: 10.1074/jbc.M116.773556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akbulut Y, Gaunt HJ, Muraki K, Ludlow MJ, Amer MS, Bruns A, Vasudev NS, Radtke L, Willot M, Hahn S, et al. (-)-Englerin A is a potent and selective activator of TRPC4 and TRPC5 calcium channels. Angew Chem Int Ed Engl. 2015;54:3787–3791. doi: 10.1002/anie.201411511. doi: 10.1002/anie.201411511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Touchberry CD, Elmore CJ, Nguyen TM, Andresen JJ, Zhao X, Orange M, Weisleder N, Brotto M, Claycomb WC, Wacker MJ. Store-operated calcium entry is present in HL-1 cardiomyocytes and contributes to resting calcium. Biochem Biophys Res Commun. 2011;416:45–50. doi: 10.1016/j.bbrc.2011.10.133. doi: 10.1016/j.bbrc.2011.10.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hunton DL, Lucchesi PA, Pang Y, Cheng X, Dell’Italia LJ, Marchase RB. Capacitative calcium entry contributes to nuclear factor of activated T-cells nuclear translocation and hypertrophy in cardiomyocytes. J Biol Chem. 2002;277:14266–14273. doi: 10.1074/jbc.M107167200. doi: 10.1074/jbc.M107167200. [DOI] [PubMed] [Google Scholar]
- 84.Uehara A, Yasukochi M, Imanaga I, Nishi M, Takeshima H. Store-operated Ca2+ entry uncoupled with ryanodine receptor and junctional membrane complex in heart muscle cells. Cell Calcium. 2002;31:89–96. doi: 10.1054/ceca.2001.0257. doi: 10.1054/ceca.2001.0257. [DOI] [PubMed] [Google Scholar]
- 85.Kojima A, Kitagawa H, Omatsu-Kanbe M, Matsuura H, Nosaka S. Presence of store-operated Ca2+ entry in C57BL/6J mouse ventricular myocytes and its suppression by sevoflurane. Br J Anaesth. 2012;109:352–360. doi: 10.1093/bja/aes212. doi: 10.1093/bja/aes212. [DOI] [PubMed] [Google Scholar]
- 86.Hunton DL, Zou L, Pang Y, Marchase RB. Adult rat cardiomyocytes exhibit capacitative calcium entry. Am J Physiol Heart Circ Physiol. 2004;286:H1124–H1132. doi: 10.1152/ajpheart.00162.2003. doi: 10.1152/ajpheart.00162.2003. [DOI] [PubMed] [Google Scholar]
- 87.Wen H, Zhao Z, Fefelova N, Xie LH. Potential arrhythmogenic role of trpc channels and store-operated calcium entry mechanism in mouse ventricular Myocytes. Front Physiol. 2018;9:1785. doi: 10.3389/fphys.2018.01785. doi: 10.3389/fphys.2018.01785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Qiu R, Lewis RS. Structural features of STIM and orai underlying store-operated calcium entry. Curr Opin Cell Biol. 2019;57:90–98. doi: 10.1016/j.ceb.2018.12.012. doi: 10.1016/j.ceb.2018.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Correll RN, Goonasekera SA, van Berlo JH, Burr AR, Accornero F, Zhang H, Makarewich CA, York AJ, Sargent MA, Chen X, et al. STIM1 elevation in the heart results in aberrant Ca2+ handling and cardiomyopathy. J Mol Cell Cardiol. 2015;87:38–47. doi: 10.1016/j.yjmcc.2015.07.032. doi: 10.1016/j.yjmcc.2015.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Zhao G, Li T, Brochet DX, Rosenberg PB, Lederer WJ. STIM1 enhances SR Ca2+ content through binding phospholamban in rat ventricular myocytes. Proc Natl Acad Sci USA. 2015;112:E4792–E4801. doi: 10.1073/pnas.1423295112. doi: 10.1073/pnas.1423295112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Layland J, Kentish JC. Positive force- and [Ca2+]i-frequency relationships in rat ventricular trabeculae at physiological frequencies. Am J Physiol. 1999;276:H9–H18. doi: 10.1152/ajpheart.1999.276.1.H9. doi: 10.1152/ajpheart.1999.276.1.H9. [DOI] [PubMed] [Google Scholar]
- 92.Antoons G, Mubagwa K, Nevelsteen I, Sipido KR. Mechanisms underlying the frequency dependence of contraction and [Ca2+]i transients in mouse ventricular myocytes. J Physiol. 2002;543:889–898. doi: 10.1113/jphysiol.2002.025619. doi: 10.1113/jphysiol.2002.025619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Frampton JE, Orchard CH, Boyett MR. Diastolic, systolic and sarcoplasmic reticulum [Ca2+] during inotropic interventions in isolated rat myocytes. J Physiol. 1991;437:351–375. doi: 10.1113/jphysiol.1991.sp018600. doi: 10.1113/jphysiol.1991.sp018600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Dibb KM, Eisner DA, Trafford AW. Regulation of systolic [Ca2+]i and cellular Ca2+ flux balance in rat ventricular myocytes by SR Ca2+, L-type Ca2+ current and diastolic [Ca2+]i. J Physiol. 2007;585:579–592. doi: 10.1113/jphysiol.2007.141473. doi: 10.1113/jphysiol.2007.141473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Horváth B, Szentandrássy N, Veress R, Almássy J, Magyar J, Bányász T, Tóth A, Papp Z, Nánási PP. Frequency-dependent effects of omecamtiv mecarbil on cell shortening of isolated canine ventricular cardiomyocytes. Naunyn Schmiedebergs Arch Pharmacol. 2017;390:1239–1246. doi: 10.1007/s00210-017-1422-z. doi: 10.1007/s00210-017-1422-z. [DOI] [PubMed] [Google Scholar]
- 96.Pieske B, Kretschmann B, Meyer M, Holubarsch C, Weirich J, Posival H, Minami K, Just H, Hasenfuss G. Alterations in intracellular calcium handling associated with the inverse force-frequency relation in human dilated cardiomyopathy. Circulation. 1995;92:1169–1178. doi: 10.1161/01.cir.92.5.1169. doi: 10.1161/01.cir.92.5.1169. [DOI] [PubMed] [Google Scholar]
- 97.Bountra C, Kaila K, Vaughan-Jones RD. Effect of repetitive activity upon intracellular pH, sodium and contraction in sheep cardiac Purkinje fibres. J Physiol. 1988;398:341–360. doi: 10.1113/jphysiol.1988.sp017046. doi: 10.1113/jphysiol.1988.sp017046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Cohen CJ, Fozzard HA, Sheu SS. Increase in intracellular sodium ion activity during stimulation in mammalian cardiac muscle. Circ Res. 1982;50:651–662. doi: 10.1161/01.res.50.5.651. doi: 10.1161/01.res.50.5.651. [DOI] [PubMed] [Google Scholar]
- 99.Frampton JE, Harrison SM, Boyett MR, Orchard CH. Ca2+ and Na+ in rat myocytes showing different force-frequency relationships. Am J Physiol. 1991;261:C739–C750. doi: 10.1152/ajpcell.1991.261.5.C739. doi: 10.1152/ajpcell.1991.261.5.C739. [DOI] [PubMed] [Google Scholar]
- 100.Mulieri LA, Hasenfuss G, Leavitt B, Allen PD, Alpert NR. Altered myocardial force-frequency relation in human heart failure. Circulation. 1992;85:1743–1750. doi: 10.1161/01.cir.85.5.1743. doi: 10.1161/01.cir.85.5.1743. [DOI] [PubMed] [Google Scholar]
- 101.Briston SJ, Dibb KM, Solaro RJ, Eisner DA, Trafford AW. Balanced changes in Ca buffering by SERCA and troponin contribute to Ca handling during β-adrenergic stimulation in cardiac myocytes. Cardiovasc Res. 2014;104:347–354. doi: 10.1093/cvr/cvu201. doi: 10.1093/cvr/cvu201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yellin EL, Nikolic S, Frater RW. Left ventricular filling dynamics and diastolic function. Prog Cardiovasc Dis. 1990;32:247–271. doi: 10.1016/0033-0620(90)90016-u. doi: 10.1016/0033-0620(90)90016-u. [DOI] [PubMed] [Google Scholar]
- 103.Cheng CP, Igarashi Y, Little WC. Mechanism of augmented rate of left ventricular filling during exercise. Circ Res. 1992;70:9–19. doi: 10.1161/01.res.70.1.9. doi: 10.1161/01.res.70.1.9. [DOI] [PubMed] [Google Scholar]
- 104.Borlaug BA, Jaber WA, Ommen SR, Lam CS, Redfield MM, Nishimura RA. Diastolic relaxation and compliance reserve during dynamic exercise in heart failure with preserved ejection fraction. Heart. 2011;97:964–969. doi: 10.1136/hrt.2010.212787. doi: 10.1136/hrt.2010.212787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bers DM. Altered cardiac myocyte Ca regulation in heart failure. Physiology (Bethesda) 2006;21:380–387. doi: 10.1152/physiol.00019.2006. doi: 10.1152/physiol.00019.2006. [DOI] [PubMed] [Google Scholar]
- 106.van Heerebeek L, Borbély A, Niessen HW, Bronzwaer JG, van der Velden J, Stienen GJ, Linke WA, Laarman GJ, Paulus WJ. Myocardial structure and function differ in systolic and diastolic heart failure. Circulation. 2006;113:1966–1973. doi: 10.1161/CIRCULATIONAHA.105.587519. doi: 10.1161/CIRCULATIONAHA.105.587519. [DOI] [PubMed] [Google Scholar]
- 107.Fowler ED, Benoist D, Drinkhill MJ, Stones R, Helmes M, Wüst RC, Stienen GJ, Steele DS, White E. Decreased creatine kinase is linked to diastolic dysfunction in rats with right heart failure induced by pulmonary artery hypertension. J Mol Cell Cardiol. 2015;86:1–8. doi: 10.1016/j.yjmcc.2015.06.016. doi: 10.1016/j.yjmcc.2015.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sequeira V, Najafi A, McConnell M, Fowler ED, Bollen IA, Wüst RC, dos Remedios C, Helmes M, White E, Stienen GJ, et al. Synergistic role of ADP and Ca2+ in diastolic myocardial stiffness. J Physiol. 2015;593:3899–3916. doi: 10.1113/JP270354. doi: 10.1113/JP270354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Bell SP, Nyland L, Tischler MD, McNabb M, Granzier H, LeWinter MM. Alterations in the determinants of diastolic suction during pacing tachycardia. Circ Res. 2000;87:235–240. doi: 10.1161/01.res.87.3.235. doi: 10.1161/01.res.87.3.235. [DOI] [PubMed] [Google Scholar]
- 110.Pfeffer MA, Shah AM, Borlaug BA. Heart failure with preserved ejection fraction in perspective. Circ Res. 2019;124:1598–1617. doi: 10.1161/CIRCRESAHA.119.313572. doi: 10.1161/CIRCRESAHA.119.313572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Westermann D, Kasner M, Steendijk P, Spillmann F, Riad A, Weitmann K, Hoffmann W, Poller W, Pauschinger M, Schultheiss HP, et al. Role of left ventricular stiffness in heart failure with normal ejection fraction. Circulation. 2008;117:2051–2060. doi: 10.1161/CIRCULATIONAHA.107.716886. doi: 10.1161/CIRCULATIONAHA.107.716886. [DOI] [PubMed] [Google Scholar]
- 112.Tan YT, Wenzelburger F, Lee E, Heatlie G, Leyva F, Patel K, Frenneaux M, Sanderson JE. The pathophysiology of heart failure with normal ejection fraction: exercise echocardiography reveals complex abnormalities of both systolic and diastolic ventricular function involving torsion, untwist, and longitudinal motion. J Am Coll Cardiol. 2009;54:36–46. doi: 10.1016/j.jacc.2009.03.037. doi: 10.1016/j.jacc.2009.03.037. [DOI] [PubMed] [Google Scholar]
- 113.Opdahl A, Remme EW, Helle-Valle T, Lyseggen E, Vartdal T, Pettersen E, Edvardsen T, Smiseth OA. Determinants of left ventricular early-diastolic lengthening velocity: independent contributions from left ventricular relaxation, restoring forces, and lengthening load. Circulation. 2009;119:2578–2586. doi: 10.1161/CIRCULATIONAHA.108.791681. doi: 10.1161/CIRCULATIONAHA.108.791681. [DOI] [PubMed] [Google Scholar]
- 114.Mohammed SF, Hussain S, Mirzoyev SA, Edwards WD, Maleszewski JJ, Redfield MM. Coronary microvascular rarefaction and myocardial fibrosis in heart failure with preserved ejection fraction. Circulation. 2015;131:550–559. doi: 10.1161/CIRCULATIONAHA.114.009625. doi: 10.1161/CIRCULATIONAHA.114.009625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Biernacka A, Frangogiannis NG. Aging and cardiac fibrosis. Aging Dis. 2011;2:158–173. [PMC free article] [PubMed] [Google Scholar]
- 116.Zile MR, Baicu CF, Ikonomidis JS, Stroud RE, Nietert PJ, Bradshaw AD, Slater R, Palmer BM, Van Buren P, Meyer M, et al. Myocardial stiffness in patients with heart failure and a preserved ejection fraction: contributions of collagen and titin. Circulation. 2015;131:1247–1259. doi: 10.1161/CIRCULATIONAHA.114.013215. doi: 10.1161/CIRCULATIONAHA.114.013215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Kato S, Saito N, Kirigaya H, Gyotoku D, Iinuma N, Kusakawa Y, Iguchi K, Nakachi T, Fukui K, Futaki M, et al. Prognostic significance of quantitative assessment of focal myocardial fibrosis in patients with heart failure with preserved ejection fraction. Int J Cardiol. 2015;191:314–319. doi: 10.1016/j.ijcard.2015.05.048. doi: 10.1016/j.ijcard.2015.05.048. [DOI] [PubMed] [Google Scholar]
- 118.Obokata M, Reddy YNV, Pislaru SV, Melenovsky V, Borlaug BA. Evidence supporting the existence of a distinct obese phenotype of heart failure with preserved ejection fraction. Circulation. 2017;136:6–19. doi: 10.1161/CIRCULATIONAHA.116.026807. doi: 10.1161/CIRCULATIONAHA.116.026807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.van Woerden G, Gorter TM, Westenbrink BD, Willems TP, van Veldhuisen DJ, Rienstra M. Epicardial fat in heart failure patients with mid-range and preserved ejection fraction. Eur J Heart Fail. 2018;20:1559–1566. doi: 10.1002/ejhf.1283. doi: 10.1002/ejhf.1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Iacobellis G, Corradi D, Sharma AM. Epicardial adipose tissue: anatomic, biomolecular and clinical relationships with the heart. Nat Clin Pract Cardiovasc Med. 2005;2:536–543. doi: 10.1038/ncpcardio0319. doi: 10.1038/ncpcardio0319. [DOI] [PubMed] [Google Scholar]
- 121.Redfield MM, Chen HH, Borlaug BA, Semigran MJ, Lee KL, Lewis G, LeWinter MM, Rouleau JL, Bull DA, Mann DL, et al. RELAX Trial. Effect of phosphodiesterase-5 inhibition on exercise capacity and clinical status in heart failure with preserved ejection fraction: a randomized clinical trial. JAMA. 2013;309:1268–1277. doi: 10.1001/jama.2013.2024. doi: 10.1001/jama.2013.2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Mirsky I, Laks MM. Time course of changes in the mechanical properties of the canine right and left ventricles during hypertrophy caused by pressure overload. Circ Res. 1980;46:530–542. doi: 10.1161/01.res.46.4.530. doi: 10.1161/01.res.46.4.530. [DOI] [PubMed] [Google Scholar]
- 123.Grossman W, McLaurin LP, Moos SP, Stefadouros M, Young DT. Wall thickness and diastolic properties of the left ventricle. Circulation. 1974;49:129–135. doi: 10.1161/01.cir.49.1.129. doi: 10.1161/01.cir.49.1.129. [DOI] [PubMed] [Google Scholar]
- 124.Fouad FM, Slominski JM, Tarazi RC. Left ventricular diastolic function in hypertension: relation to left ventricular mass and systolic function. J Am Coll Cardiol. 1984;3:1500–1506. doi: 10.1016/s0735-1097(84)80289-2. doi: 10.1016/s0735-1097(84)80289-2. [DOI] [PubMed] [Google Scholar]
- 125.Borbély A, van der Velden J, Papp Z, Bronzwaer JG, Edes I, Stienen GJ, Paulus WJ. Cardiomyocyte stiffness in diastolic heart failure. Circulation. 2005;111:774–781. doi: 10.1161/01.CIR.0000155257.33485.6D. doi: 10.1161/01.CIR.0000155257.33485.6D. [DOI] [PubMed] [Google Scholar]
- 126.Lam CS, Roger VL, Rodeheffer RJ, Bursi F, Borlaug BA, Ommen SR, Kass DA, Redfield MM. Cardiac structure and ventricular-vascular function in persons with heart failure and preserved ejection fraction from Olmsted County, Minnesota. Circulation. 2007;115:1982–1990. doi: 10.1161/CIRCULATIONAHA.106.659763. doi: 10.1161/CIRCULATIONAHA.106.659763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Curl CL, Danes VR, Bell JR, Raaijmakers AJA, Ip WTK, Chandramouli C, Harding TW, Porrello ER, Erickson JR, Charchar FJ, et al. Cardiomyocyte functional etiology in heart failure with preserved ejection fraction is distinctive-a new preclinical model. J Am Heart Assoc. 2018;7:e007451. doi: 10.1161/JAHA.117.007451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Solomon SD, Janardhanan R, Verma A, Bourgoun M, Daley WL, Purkayastha D, Lacourcière Y, Hippler SE, Fields H, Naqvi TZ, et al. Valsartan In Diastolic Dysfunction (VALIDD) Investigators. Effect of angiotensin receptor blockade and antihypertensive drugs on diastolic function in patients with hypertension and diastolic dysfunction: a randomised trial. Lancet. 2007;369:2079–2087. doi: 10.1016/S0140-6736(07)60980-5. doi: 10.1016/S0140-6736(07)60980-5. [DOI] [PubMed] [Google Scholar]
- 129.Krüger M, Kötter S, Grützner A, Lang P, Andresen C, Redfield MM, Butt E, dos Remedios CG, Linke WA. Protein kinase G modulates human myocardial passive stiffness by phosphorylation of the titin springs. Circ Res. 2009;104:87–94. doi: 10.1161/CIRCRESAHA.108.184408. doi: 10.1161/CIRCRESAHA.108.184408. [DOI] [PubMed] [Google Scholar]
- 130.Hidalgo C, Hudson B, Bogomolovas J, Zhu Y, Anderson B, Greaser M, Labeit S, Granzier H. PKC phosphorylation of titin’s PEVK element: a novel and conserved pathway for modulating myocardial stiffness. Circ Res. 2009;105:631–638, 617 p following 638. doi: 10.1161/CIRCRESAHA.109.198465. doi: 10.1161/CIRCRESAHA.109.198465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hamdani N, Krysiak J, Kreusser MM, Neef S, Dos Remedios CG, Maier LS, Krüger M, Backs J, Linke WA. Crucial role for Ca2+/calmodulin-dependent protein kinase-II in regulating diastolic stress of normal and failing hearts via titin phosphorylation. Circ Res. 2013;112:664–674. doi: 10.1161/CIRCRESAHA.111.300105. doi: 10.1161/CIRCRESAHA.111.300105. [DOI] [PubMed] [Google Scholar]
- 132.Grützner A, Garcia-Manyes S, Kötter S, Badilla CL, Fernandez JM, Linke WA. Modulation of titin-based stiffness by disulfide bonding in the cardiac titin N2-B unique sequence. Biophys J. 2009;97:825–834. doi: 10.1016/j.bpj.2009.05.037. doi: 10.1016/j.bpj.2009.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernández JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell. 2014;156:1235–1246. doi: 10.1016/j.cell.2014.01.056. doi: 10.1016/j.cell.2014.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Labeit D, Watanabe K, Witt C, Fujita H, Wu Y, Lahmers S, Funck T, Labeit S, Granzier H. Calcium-dependent molecular spring elements in the giant protein titin. Proc Natl Acad Sci USA. 2003;100:13716–13721. doi: 10.1073/pnas.2235652100. doi: 10.1073/pnas.2235652100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fujita H, Labeit D, Gerull B, Labeit S, Granzier HL. Titin isoform-dependent effect of calcium on passive myocardial tension. Am J Physiol Heart Circ Physiol. 2004;287:H2528–H2534. doi: 10.1152/ajpheart.00553.2004. doi: 10.1152/ajpheart.00553.2004. [DOI] [PubMed] [Google Scholar]
- 136.Hamdani N, Bishu KG, von Frieling-Salewsky M, Redfield MM, Linke WA. Deranged myofilament phosphorylation and function in experimental heart failure with preserved ejection fraction. Cardiovasc Res. 2013;97:464–471. doi: 10.1093/cvr/cvs353. doi: 10.1093/cvr/cvs353. [DOI] [PubMed] [Google Scholar]
- 137.Robinson P, Griffiths PJ, Watkins H, Redwood CS. Dilated and hypertrophic cardiomyopathy mutations in troponin and alpha-tropomyosin have opposing effects on the calcium affinity of cardiac thin filaments. Circ Res. 2007;101:1266–1273. doi: 10.1161/CIRCRESAHA.107.156380. doi: 10.1161/CIRCRESAHA.107.156380. [DOI] [PubMed] [Google Scholar]
- 138.Braunwald E, Lambrew CT, Rockoff SD, Ross J, Jr, Morrow AG. Idiopathic hypertrophic subaortic stenosis. I. A description of the disease based upon an analysis of 64 patients. Circulation. 1964;30(suppl 4):3–119. doi: 10.1161/01.cir.29.5s4.iv-3. [DOI] [PubMed] [Google Scholar]
- 139.van Heerebeek L, Hamdani N, Falcão-Pires I, Leite-Moreira AF, Begieneman MP, Bronzwaer JG, van der Velden J, Stienen GJ, Laarman GJ, Somsen A, et al. Low myocardial protein kinase G activity in heart failure with preserved ejection fraction. Circulation. 2012;126:830–839. doi: 10.1161/CIRCULATIONAHA.111.076075. doi: 10.1161/CIRCULATIONAHA.111.076075. [DOI] [PubMed] [Google Scholar]
- 140.Scotcher J, Prysyazhna O, Boguslavskyi A, Kistamas K, Hadgraft N, Martin ED, Worthington J, Rudyk O, Rodriguez Cutillas P, Cuello F, et al. Disulfide-activated protein kinase G Iα regulates cardiac diastolic relaxation and fine-tunes the Frank-Starling response. Nat Commun. 2016;7:13187. doi: 10.1038/ncomms13187. doi: 10.1038/ncomms13187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gwathmey JK, Slawsky MT, Hajjar RJ, Briggs GM, Morgan JP. Role of intracellular calcium handling in force-interval relationships of human ventricular myocardium. J Clin Invest. 1990;85:1599–1613. doi: 10.1172/JCI114611. doi: 10.1172/JCI114611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Sipido KR, Stankovicova T, Flameng W, Vanhaecke J, Verdonck F. Frequency dependence of Ca2+ release from the sarcoplasmic reticulum in human ventricular myocytes from end-stage heart failure. Cardiovasc Res. 1998;37:478–488. doi: 10.1016/s0008-6363(97)00280-0. doi: 10.1016/s0008-6363(97)00280-0. [DOI] [PubMed] [Google Scholar]
- 143.Pieske B, Maier LS, Piacentino V, III, Weisser J, Hasenfuss G, Houser S. Rate dependence of [Na+]i and contractility in nonfailing and failing human myocardium. Circulation. 2002;106:447–453. doi: 10.1161/01.cir.0000023042.50192.f4. doi: 10.1161/01.cir.0000023042.50192.f4. [DOI] [PubMed] [Google Scholar]
- 144.Pogwizd SM, Qi M, Yuan W, Samarel AM, Bers DM. Upregulation of Na+/Ca2+ exchanger expression and function in an arrhythmogenic rabbit model of heart failure. Circ Res. 1999;85:1009–1019. doi: 10.1161/01.res.85.11.1009. doi: 10.1161/01.res.85.11.1009. [DOI] [PubMed] [Google Scholar]
- 145.Baartscheer A, Schumacher CA, Belterman CN, Coronel R, Fiolet JW. SR calcium handling and calcium after-transients in a rabbit model of heart failure. Cardiovasc Res. 2003;58:99–108. doi: 10.1016/s0008-6363(02)00854-4. doi: 10.1016/s0008-6363(02)00854-4. [DOI] [PubMed] [Google Scholar]
- 146.Lamberts RR, Hamdani N, Soekhoe TW, Boontje NM, Zaremba R, Walker LA, de Tombe PP, van der Velden J, Stienen GJ. Frequency-dependent myofilament Ca2+ desensitization in failing rat myocardium. J Physiol. 2007;582:695–709. doi: 10.1113/jphysiol.2007.134486. doi: 10.1113/jphysiol.2007.134486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Briston SJ, Caldwell JL, Horn MA, Clarke JD, Richards MA, Greensmith DJ, Graham HK, Hall MC, Eisner DA, Dibb KM, et al. Impaired β-adrenergic responsiveness accentuates dysfunctional excitation-contraction coupling in an ovine model of tachypacing-induced heart failure. J Physiol. 2011;589:1367–1382. doi: 10.1113/jphysiol.2010.203984. doi: 10.1113/jphysiol.2010.203984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Hobai IA, Maack C, O’Rourke B. Partial inhibition of sodium/calcium exchange restores cellular calcium handling in canine heart failure. Circ Res. 2004;95:292–299. doi: 10.1161/01.RES.0000136817.28691.2d. doi: 10.1161/01.RES.0000136817.28691.2d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Díaz ME, Graham HK, Trafford AW. Enhanced sarcolemmal Ca2+ efflux reduces sarcoplasmic reticulum Ca2+ content and systolic Ca2+ in cardiac hypertrophy. Cardiovasc Res. 2004;62:538–547. doi: 10.1016/j.cardiores.2004.01.038. doi: 10.1016/j.cardiores.2004.01.038. [DOI] [PubMed] [Google Scholar]
- 150.Zhu X, Bernecker OY, Manohar NS, Hajjar RJ, Hellman J, Ichinose F, Valdivia HH, Schmidt U. Increased leakage of sarcoplasmic reticulum Ca2+ contributes to abnormal myocyte Ca2+ handling and shortening in sepsis. Crit Care Med. 2005;33:598–604. doi: 10.1097/01.ccm.0000152223.27176.a6. doi: 10.1097/01.ccm.0000152223.27176.a6. [DOI] [PubMed] [Google Scholar]
- 151.Wagner S, Schürmann S, Hein S, Schüttler J, Friedrich O. Septic cardiomyopathy in rat LPS-induced endotoxemia: relative contribution of cellular diastolic Ca2+ removal pathways, myofibrillar biomechanics properties and action of the cardiotonic drug levosimendan. Basic Res Cardiol. 2015;110:507. doi: 10.1007/s00395-015-0507-4. doi: 10.1007/s00395-015-0507-4. [DOI] [PubMed] [Google Scholar]
- 152.Hobai IA, Buys ES, Morse JC, Edgecomb J, Weiss EH, Armoundas AA, Hou X, Khandelwal AR, Siwik DA, Brouckaert P, et al. SERCA Cys674 sulphonylation and inhibition of L-type Ca2+ influx contribute to cardiac dysfunction in endotoxemic mice, independent of cGMP synthesis. Am J Physiol Heart Circ Physiol. 2013;305:H1189–H1200. doi: 10.1152/ajpheart.00392.2012. doi: 10.1152/ajpheart.00392.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Valero-Muñoz M, Backman W, Sam F. Murine models of heart failure with preserved ejection fraction: a “Fishing Expedition”. JACC Basic Transl Sci. 2017;2:770–789. doi: 10.1016/j.jacbts.2017.07.013. doi: 10.1016/j.jacbts.2017.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Porrello ER, Delbridge LMD. HFpEF-time to explore the role of genetic heterogeneity in phenotypic variability: new mechanistic insights offer promise for personalized therapies. Circulation. 2019;140:1607–1609. doi: 10.1161/CIRCULATIONAHA.119.042496. doi: 10.1161/CIRCULATIONAHA.119.042496. [DOI] [PubMed] [Google Scholar]
- 155.Rouhana S, Farah C, Roy J, Finan A, Rodrigues de Araujo G, Bideaux P, Scheuermann V, Saliba Y, Reboul C, Cazorla O, et al. Early calcium handling imbalance in pressure overload-induced heart failure with nearly normal left ventricular ejection fraction. Biochim Biophys Acta Mol Basis Dis. 2019;1865:230–242. doi: 10.1016/j.bbadis.2018.08.005. doi: 10.1016/j.bbadis.2018.08.005. [DOI] [PubMed] [Google Scholar]
- 156.Røe ÅT, Aronsen JM, Skårdal K, Hamdani N, Linke WA, Danielsen HE, Sejersted OM, Sjaastad I, Louch WE. Increased passive stiffness promotes diastolic dysfunction despite improved Ca2+ handling during left ventricular concentric hypertrophy. Cardiovasc Res. 2017;113:1161–1172. doi: 10.1093/cvr/cvx087. doi: 10.1093/cvr/cvx087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Adeniran I, MacIver DH, Hancox JC, Zhang H. Abnormal calcium homeostasis in heart failure with preserved ejection fraction is related to both reduced contractile function and incomplete relaxation: an electromechanically detailed biophysical modeling study. Front Physiol. 2015;6:78. doi: 10.3389/fphys.2015.00078. doi: 10.3389/fphys.2015.00078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.McMahon AC, Naqvi RU, Hurst MJ, Raine AE, MacLeod KT. Diastolic dysfunction and abnormality of the Na+/Ca2+ exchanger in single uremic cardiac myocytes. Kidney Int. 2006;69:846–851. doi: 10.1038/sj.ki.5000193. doi: 10.1038/sj.ki.5000193. [DOI] [PubMed] [Google Scholar]
- 159.Primessnig U, Schönleitner P, Höll A, Pfeiffer S, Bracic T, Rau T, Kapl M, Stojakovic T, Glasnov T, Leineweber K, et al. Novel pathomechanisms of cardiomyocyte dysfunction in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. 2016;18:987–997. doi: 10.1002/ejhf.524. doi: 10.1002/ejhf.524. [DOI] [PubMed] [Google Scholar]
- 160.Primessnig U, Bracic T, Levijoki J, Otsomaa L, Pollesello P, Falcke M, Pieske B, Heinzel FR. Long-term effects of Na+/Ca2+ exchanger inhibition with ORM-11035 improves cardiac function and remodelling without lowering blood pressure in a model of heart failure with preserved ejection fraction. Eur J Heart Fail. 2019;21:1543–1552. doi: 10.1002/ejhf.1619. doi: 10.1002/ejhf.1619. [DOI] [PubMed] [Google Scholar]
- 161.Kamimura D, Ohtani T, Sakata Y, Mano T, Takeda Y, Tamaki S, Omori Y, Tsukamoto Y, Furutani K, Komiyama Y, et al. Ca2+ entry mode of Na+/Ca2+ exchanger as a new therapeutic target for heart failure with preserved ejection fraction. Eur Heart J. 2012;33:1408–1416. doi: 10.1093/eurheartj/ehr106. doi: 10.1093/eurheartj/ehr106. [DOI] [PubMed] [Google Scholar]
- 162.Lacombe VA, Viatchenko-Karpinski S, Terentyev D, Sridhar A, Emani S, Bonagura JD, Feldman DS, Györke S, Carnes CA. Mechanisms of impaired calcium handling underlying subclinical diastolic dysfunction in diabetes. Am J Physiol Regul Integr Comp Physiol. 2007;293:R1787–R1797. doi: 10.1152/ajpregu.00059.2007. doi: 10.1152/ajpregu.00059.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Lagadic-Gossmann D, Buckler KJ, Le Prigent K, Feuvray D. Altered Ca2+ handling in ventricular myocytes isolated from diabetic rats. Am J Physiol. 1996;270:H1529–H1537. doi: 10.1152/ajpheart.1996.270.5.H1529. doi: 10.1152/ajpheart.1996.270.5.H1529. [DOI] [PubMed] [Google Scholar]
- 164.Hamouda NN, Sydorenko V, Qureshi MA, Alkaabi JM, Oz M, Howarth FC. Dapagliflozin reduces the amplitude of shortening and Ca2+ transient in ventricular myocytes from streptozotocin-induced diabetic rats. Mol Cell Biochem. 2015;400:57–68. doi: 10.1007/s11010-014-2262-5. doi: 10.1007/s11010-014-2262-5. [DOI] [PubMed] [Google Scholar]
- 165.Noda N, Hayashi H, Miyata H, Suzuki S, Kobayashi A, Yamazaki N. Cytosolic Ca2+ concentration and pH of diabetic rat myocytes during metabolic inhibition. J Mol Cell Cardiol. 1992;24:435–446. doi: 10.1016/0022-2828(92)93197-r. doi: 10.1016/0022-2828(92)93197-r. [DOI] [PubMed] [Google Scholar]
- 166.Zhang L, Ward ML, Phillips AR, Zhang S, Kennedy J, Barry B, Cannell MB, Cooper GJ. Protection of the heart by treatment with a divalent-copper-selective chelator reveals a novel mechanism underlying cardiomyopathy in diabetic rats. Cardiovasc Diabetol. 2013;12:123. doi: 10.1186/1475-2840-12-123. doi: 10.1186/1475-2840-12-123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Miranda-Silva D, Wust RC, Conceicao G, Goncalves-Rodrigues P, Goncalves N, Goncalves A, Kuster DW, Leite-Moreira AF, van der Velden J, de Sousa Beleza JM, et al. Disturbed cardiac mitochondrial and cytosolic calcium handling in a metabolic-risk related rat model of heart failure with preserved ejection fraction [published online September 13, 2019]. Acta Physiol (Oxf) 2019:e13378. doi: 10.1111/apha.13378. doi: 10.1111/apha.13378. https://onlinelibrary.wiley.com/doi/full/10.1111/apha.13378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Hohendanner F, Bode D, Primessnig U, Guthof T, Doerr R, Jeuthe S, Reimers S, Zhang K, Bach D, Wakula P, et al. Cellular mechanisms of metabolic syndrome-related atrial decompensation in a rat model of HFpEF. J Mol Cell Cardiol. 2018;115:10–19. doi: 10.1016/j.yjmcc.2017.12.012. doi: 10.1016/j.yjmcc.2017.12.012. [DOI] [PubMed] [Google Scholar]
- 169.Schiattarella GG, Altamirano F, Tong D, French KM, Villalobos E, Kim SY, Luo X, Jiang N, May HI, Wang ZV, et al. Nitrosative stress drives heart failure with preserved ejection fraction. Nature. 2019;568:351–356. doi: 10.1038/s41586-019-1100-z. doi: 10.1038/s41586-019-1100-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Selby DE, Palmer BM, LeWinter MM, Meyer M. Tachycardia-induced diastolic dysfunction and resting tone in myocardium from patients with a normal ejection fraction. J Am Coll Cardiol. 2011;58:147–154. doi: 10.1016/j.jacc.2010.10.069. doi: 10.1016/j.jacc.2010.10.069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Frey N, Luedde M, Katus HA. Mechanisms of disease: hypertrophic cardiomyopathy. Nat Rev Cardiol. 2011;9:91–100. doi: 10.1038/nrcardio.2011.159. doi: 10.1038/nrcardio.2011.159. [DOI] [PubMed] [Google Scholar]
- 172.Despa S, Islam MA, Weber CR, Pogwizd SM, Bers DM. Intracellular Na+ concentration is elevated in heart failure but Na/K pump function is unchanged. Circulation. 2002;105:2543–2548. doi: 10.1161/01.cir.0000016701.85760.97. doi: 10.1161/01.cir.0000016701.85760.97. [DOI] [PubMed] [Google Scholar]
- 173.Hasenfuss G, Schillinger W, Lehnart SE, Preuss M, Pieske B, Maier LS, Prestle J, Minami K, Just H. Relationship between Na+-Ca2+-exchanger protein levels and diastolic function of failing human myocardium. Circulation. 1999;99:641–648. doi: 10.1161/01.cir.99.5.641. doi: 10.1161/01.cir.99.5.641. [DOI] [PubMed] [Google Scholar]
- 174.Sossalla S, Wagner S, Rasenack EC, Ruff H, Weber SL, Schöndube FA, Tirilomis T, Tenderich G, Hasenfuss G, Belardinelli L, et al. Ranolazine improves diastolic dysfunction in isolated myocardium from failing human hearts–role of late sodium current and intracellular ion accumulation. J Mol Cell Cardiol. 2008;45:32–43. doi: 10.1016/j.yjmcc.2008.03.006. doi: 10.1016/j.yjmcc.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 175.Cappetta D, Esposito G, Coppini R, Piegari E, Russo R, Ciuffreda LP, Rivellino A, Santini L, Rafaniello C, Scavone C, et al. Effects of ranolazine in a model of doxorubicin-induced left ventricle diastolic dysfunction. Br J Pharmacol. 2017;174:3696–3712. doi: 10.1111/bph.13791. doi: 10.1111/bph.13791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Undrovinas NA, Maltsev VA, Belardinelli L, Sabbah HN, Undrovinas A. Late sodium current contributes to diastolic cell Ca2+ accumulation in chronic heart failure. J Physiol Sci. 2010;60:245–257. doi: 10.1007/s12576-010-0092-0. doi: 10.1007/s12576-010-0092-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Zhang T, Maier LS, Dalton ND, Miyamoto S, Ross J, Jr, Bers DM, Brown JH. The deltaC isoform of CaMKII is activated in cardiac hypertrophy and induces dilated cardiomyopathy and heart failure. Circ Res. 2003;92:912–919. doi: 10.1161/01.RES.0000069686.31472.C5. doi: 10.1161/01.RES.0000069686.31472.C5. [DOI] [PubMed] [Google Scholar]
- 178.Sossalla S, Maurer U, Schotola H, Hartmann N, Didié M, Zimmermann WH, Jacobshagen C, Wagner S, Maier LS. Diastolic dysfunction and arrhythmias caused by overexpression of CaMKIIδC can be reversed by inhibition of late Na+ current. Basic Res Cardiol. 2011;106:263–272. doi: 10.1007/s00395-010-0136-x. doi: 10.1007/s00395-010-0136-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Runte KE, Bell SP, Selby DE, Haussler TN, Ashikaga T, LeWinter MM, Palmer BM, Meyer M. Relaxation and the role of calcium in isolated contracting myocardium from patients with hypertensive heart disease and heart failure with preserved ejection fraction. Circ Heart Fail. 2017;10:e004311. doi: 10.1161/CIRCHEARTFAILURE.117.004311. [DOI] [PMC free article] [PubMed] [Google Scholar]