

Abstract
Historically accessed through two-electron, anionic chemistry, ketones, alcohols, and amines are of foundational importance to the practice of organic synthesis. After placing this work in proper historical context, this Article reports the development, full scope, and a mechanistic picture for a strikingly different way of forging such functional groups. Thus, carboxylic acids, once converted to redox-active esters (RAEs) can be utilized as formally nucleophilic coupling partners with other carboxylic derivatives (to produce ketones), imines (to produce benzylic amines), or aldehydes (to produce alcohols). The reactions are uniformly mild, operationally simple, and, in the case of ketone synthesis, broad in scope (including several applications to the simplification of synthetic problems and to parallel synthesis). Finally, an extensive mechanistic study of the ketone synthesis is performed to trace the elementary steps of the catalytic cycle and provide the end-user with a clear and understandable rationale for the selectivity, role of additives, and underlying driving forces involved.
Introduction
Roughly 120 years ago, Grignard invented his eponymous reaction that converts alkyl halides into nucleophilic organomagnesium reagents that engage electrophilic C=O bonds.1 This reaction forms one of the foundational reactions of organic synthesis and although the precise mechanism of carbonyl addition is not fully understood, it is generally regarded (and taught to undergraduates) as a two-electron (anionic) process. Grignard reagents and related species are routinely used to generate some of the most useful functional groups such as ketones (via addition to activated ester or amide derivatives), alcohols (via addition to aldehydes), and amines (via addition to imines) as depicted in Figure 1.2 Notwithstanding its indisputable utility and broad scope, it bears with it a restricted window of chemoselectivity – certain functional groups are simply not compatible with the highly basic and nucleophilic properties of the reagents. Additionally, alkyl halides are often inconvenient to prepare and scarcely available compared to other feedstock functional groups such as olefins and carboxylic acids. Thus, the search for Grignard-like reactivity that is milder and more tolerant of air and moisture continues to the present day. Indeed, groundbreaking findings from Lipshutz, Buchwald, Krische, and Miura have opened up a realm of mild nucleophilic additions beginning with olefin precursors.3 We and others have also explored how Mukaiyama-type reactivity can be harnessed on olefins to add to carbonyl groups.4
Figure 1.
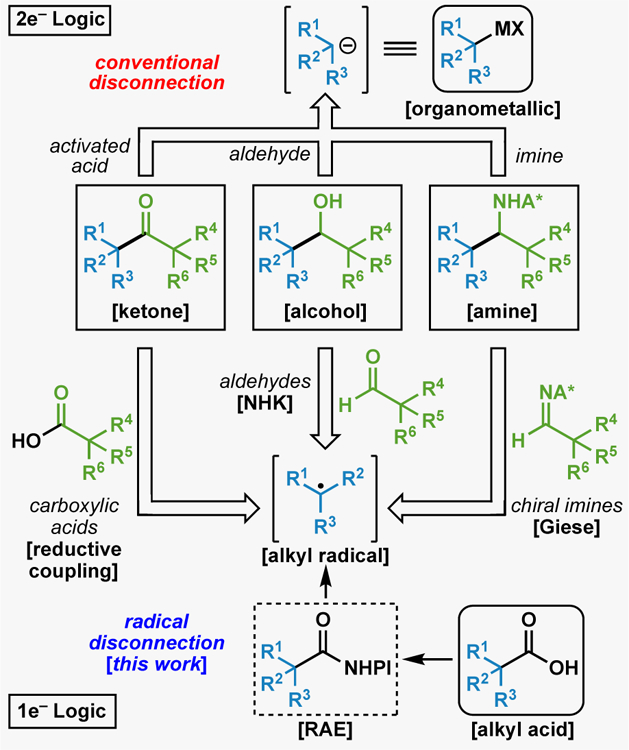
Polar-bond disconnections via conventional 2e− logic vs. newly explored radical processes for ketone, alcohol, and amine synthesis.
In the quest for practicality, alkyl carboxylic acids are perhaps the most ubiquitous functional group from the standpoint of commercial availability.5 They are uniquely modular building blocks in that they can be easily diversified at the alpha-position using anionic chemistry and then disassembled to lose CO2 and afford a carbon-centered radical. Radicals derived in this way have a storied history dating back to Barton’s legendary findings.6 More recently, we and others have explored how such radicals could be intercepted in the same way that alkyl halide-derived radicals have been for decades using transition metals.7 Thus far, most recent studies in this area have been focused on the canonical chemistry of radicals such as their use in cross-coupling and trapping with radicalophiles.
The focus of this work was to see if carboxylate-derived radicals could be engaged in reaction manifolds that have historically been within the purview of anionic (Grignard) chemistry. This Article describes the invention, full scope and limitations, and detailed mechanism of methods for harnessing radicals derived from redox-active esters (RAEs) for addition to electrophilic C=X bonds to generate ketones, alcohols, and amines.
Background and Historical Context
The in situ generation of radicals and subsequent interception with transition metals for the specific purpose of adding to C=X bonds is not new (Figure 2). In fact, this area has a long history dating back to the late 1970’s. Figure 2 outlines the key precedents in this area that have laid the strategic and mechanistic basis for the current studies. Early work in this area focused on the use of electrochemical techniques to generate alkyl-M species (M = Cd and Mg) that could be directly added to acyl halides in situ.8 One of the key breakthroughs in this area can be traced to a report by Périchon who laid some of the groundwork for the cross-electrophile couplings of the modern era.9 In that work, a sacrificial anode such as zinc was utilized in an undivided cell along with catalytic nickel in the presence of a diamine ligand (bpy) to couple simple alkyl and acyl halides to form ketones. In parallel to these electrochemical studies, Mukaiyama and co-workers demonstrated a similar transformation by embedding the ligand for Ni onto the ester (2-pyridyl ester derivatives) rather than using an acyl halide.10 Moving to the modern era, the field of reductive/cross electrophile couplings, specifically those between alkyl halides and acyl halides/anhydrides, has flourished with impactful contributions from the Martin, Gong, Weix, and Reisman groups among many others.11 Finally, the Gong, Weix, and Loren teams have reported single examples of the use of RAEs in place of alkyl halides for ester (Gong) or ketone (Weix and Loren) synthesis.12 This backdrop of precedent places our studies in a proper context.
Figure 2.

Radical methods for accessing ketones: Historical perspective.
Ketone Synthesis via Cross-Carboxy Coupling: Development and Scope
Retrosynthetic considerations for the synthesis of complex ketones inspired our explorations in this area. For example, as illustrated in Figure 3A, ketone fragment 1 was enlisted as a key intermediate in a total synthesis of anamarine 2. Prior routes to this ketone, which conveniently maps onto readily available tartaric acid, relied on 2e− logic and thus could not easily take advantage of this inexpensive building block because anionic chemistry of the type shown in Figure 3A is not workable.13 Therefore, an olefin-based route was pursued requiring two asymmetric dihydroxylations with Os along with extraneous redox fluctuations and PGs (8 steps overall). In contrast, 1e− logic pointed to a simple (3 step) means of utilizing feedstock chiral pool materials: tartaric and lactic acid. The development of the necessary chemistry to achieve this simplification was accomplished using model substrates 3 and 4 as outlined in Figure 3B. In its fully optimized manifestation, a 73% isolated yield of ketone 5 was obtained from RAE 4 and acid 3 using a carefully engineered set of reagents in concert with a Ni-catalyst: (1) benzoyl anhydride as activating agent for the carboxylic acid, (2) Zn powder as reducing agent, and (3) magnesium and lithium based Lewis-acids (MgCl2 and LiBr). The experimental simplicity is worth pointing out: all components are simply added to a flask followed by addition of solvent. After 3 h of stirring the reaction is quenched. Deviations from the conditions prescribed above are certainly tolerated. For instance, the following set of modifications resulted in only 5–15% decrease in yield: running the reaction under air (entry 2), generating either the RAE (entry 3) or catalyst (entry 4) in situ, reducing the equivalents of acid and anhydride (entry 5), or lowering the catalyst loading (entry 6). Key parameters for the success of this reaction include the specific identity of the RAE (entry 7), Ni-bound ligand (entry 8), anhydride (entries 9–10), reductant (entry 11), and Lewis-acids employed (entries 12–14). Unsurprisingly, inclusion of the anhydride was found to be required for product formation (entry 15). In addition, the use of an acyl chloride in place of the in situ combination of carboxylic acid and anhydride led to even higher conversion (entry 16). For the sake of experimental simplicity and to enable parallel library synthesis (vide infra), the in situ protocol was adopted as the standard protocol. An extensive list of parameters screened can be found in the SI and the role of each component will be discussed in the mechanism section (vide infra).
Figure 3.

(A) Inspiration for a mild ketone and chemoselective ketone synthesis and (B) development and optimization of CCC.
With a set of optimized conditions in hand the scope and versatility of ketone synthesis was systematically explored employing a range of different carboxylic acids (Table 1). This “cross-carboxy coupling” (CCC) tolerates a range of 1°, 2°, 3°, and aromatic carboxylic acids to deliver unsymmetrical ketones in an operationally simple, chemoselective, and scalable way. 1° RAEs were coupled to 1° (6, 7, 9, 11 and 13), 2° (5, 8, 10, 12, 14–17, 19, 20, 22 and 23), and bridged 3° (21) carboxylic acids in good yields. A large variety of functional groups including esters (6), ketones (7–9, 11 and 12), N-Boc (9, 14, 16 and 23) or N-Tosyl (5, 8, 10, 12, 14–17 and 20) protected amines, amides (22 and 23), ethers (7 and 17), and halogens (10, 13, 15, 20, 22 and 23) were tolerated under the reaction conditions. In addition, successful functionalization of fenbufen and indomethacin was achieved to afford compounds 19, 22, and 23 in 75%, 62% and 53% yield, respectively. 2° and 3° RAEs were also coupled to a broad range of 1° (21, 24, 38, 40, 42 and 46) and 2° (25–37, 39, 41, 43–45 and 47) carboxylic acids with similarly exceptional levels of chemoselectivity, including a diol, an epoxide, and a Michael acceptor found on mupirocin (38). Although bridged 3° carboxylic acids and RAEs were well tolerated, lower yields were observed in the case of exceedingly sterically encumbered moieties (18). It is worth noting that the diversification of biotin, a highly polar and oft installed structure in chemical biology was also successful (40).14 Strikingly, almost 90% of the compounds illustrated in Table 1 have never been prepared before, thereby illustrating the capability CCC to access new chemical space. For example, the successful coupling of bridged compounds such as cubane 41 and 2-oxabicyclo[2.1.1]hexane 46 provides a versatile entry into these useful phenyl ring bioisosteres.15 Pleasingly, the developed conditions were easily translated to gram scale experiments affording compounds 5 and 36 in 68% and 69%, respectively.
Table 1.
Scope of the CCC ketone synthesis. Yields of isolated products are indicated in each case. Reaction conditions: RAE (1.0 equiv), carboxylic acid (2.0 equiv), Ni(BPhen)Cl2•2DMF (20 mol%), Zn (3.0 equiv), (PhCO)2O (2.2 equiv), MgCl2 (1.5 equiv), LiBr (1.0 equiv), MeCN/THF (2:3, 0.1 M), rt, 3 h.
 |
using benzoyl chlorides (2.0 equiv) instead of [acid + (PhCO)2O]. DIC = N, N’-diisopropylcarbodiiminde.
When attempting CCC on aryl carboxylic acids, minimal product formation was observed (see SI for optimization details). In this instance, modification of the coupling partner to acyl chlorides, as pioneered by Weix and Loren,12 afforded the desired products in suitable yields (48–52, 54 and 55). Tolerance of protected indole, amine, and ester functionality was observed (49, 50 and 51 for example), although an ortho-methoxy group was required in the case of a pyridine-based substrate (53 and 54).
The range of conceivable coupling partners in CCC is vast and thus from a practitioner’s perspective it may not be intuitive as to which acids serve the role of electrophile/nucleophile. For instance, substrate 21 can be prepared in either fashion (see SI for additional examples). Thus, Table 2 presents a convenient user guide for CCC based on the 16 possible combinations of cross coupling based on empirical findings. For example, in the case of 1° and 2° couplings, the starting acids can be used in either form (free acid or RAE) whereas benzoyl chlorides are best employed as the free carboxylic acid component in all couplings.
Table 2.
An empirically derived user-guide for CCC.
 |
Returning to the original inspiration for the development of this method, Figure 4 outlines a series of applications that serve to simplify synthesis. As depicted in Figure 4A, building block 1, previously prepared through an 8-step sequence relying on olefin functionalization,13 could now be accessed in 3 steps commencing from tartrate-derived acid 56. Subjecting this acid to CCC using commercial lactate-derived acid 57 delivered 1 in 52% yield as a single stereoisomer. In a similar vein, muscone 58, a widely employed fragrance additive, has previously been prepared through RCM/hydrogenation of diene 59.16 The preparation of this simple ketone commences from a relatively expensive alcohol 60 (by fragrance industry standards) and employs three reactions to generate one new C–C bond with the correct ketone oxidation state. In contrast, 10-undecenoic acid 61 is roughly an order of magnitude less expensive and can be smoothly converted to the same building block 59 in 68% yield upon CCC with citronellic acid 62. This one-step process avoids halide waste, water sensitive Grignard reagents, and excess chromium waste (employed solely for the purpose of alcohol oxidation). The experimental simplicity of this method makes it perfectly suited to a parallel library synthesis workflow. Thus, a curated set of diverse building blocks from the Pfizer collection were chosen and subjected to ketone synthesis using 1-cyclopentane carboxylic acid-derived NHPI ester 63. All reagents, with the exception of zinc, were added as stock solutions to facilitate parallel execution in plate format. HPLC analysis indicated that 19 of the 20 substrates (64–83) successfully underwent CCC, while 17 provided sufficient material after isolation (> 1.0 mg) for submission to standard bioactivity assays. The resulting ketones possess drug-like properties, conforming with Lipinski’s “rule of five” (mean molecular weight 371, mean ClogP 4.4, mean hydrogen bond acceptor count 2.5) and include a range of common heterocycles.17 This protocol is particularly well-suited for fragment-based drug discovery (FBDD),18 or as a diversity incorporating event in multi-step parallel library construction (e.g. with subsequent reductive amination).
Figure 4.

Applications of CCC to the formal synthesis of anamarine (A), muscone (B), and parallel library synthesis (C).
Asymmetric Synthesis of Benzylic Amines
Compelled by ongoing medicinal chemistry projects at Pfizer, attention then turned to how the putatively nucleophilic species generated from RAEs under Ni-catalysis could be engaged by other electrophilic species such as imines. Prior studies from our labs (Figure 5A) focused on the synthesis of enantiopure amino acids using the glyoxyl-derived chiral sulfinyl imine reagent 84.7n That chemistry exhibits remarkable substrate scope and utilizes a TCNHPI-based RAE in concert with an inexpensive reducing agent (Zn) and catalyst [Ni(OAc)2•4H2O]. Translation of those exact conditions using the less electrophilic aryl sulfinyl imines in order to generate enantiopure benzylic amines furnished only trace product (Figure 5B, entry 1). Thus, a thorough re-examination of conditions was pursued culminating in the identification of a mild and robust set of conditions (entry 3). Three key modifications relative to prior studies were an increased concentration (0.2 M vs. 1.0 M, entry 10), use of NHPI instead of TCNHPI as the RAE (entry 6) and a more reactive reducing agent (standard zinc powder vs. zinc nanopowder, entry 8). This unique form of zinc, available from Sigma-Aldrich,19 was also found to be critical in radical couplings in DNA-encoded library synthesis.7k Similar to ketone synthesis via CCC, the reaction tolerates air/moisture (entry 4) and the RAE can be made in situ to enable parallel synthesis endeavors (entry 5). Attempts to utilize the corresponding Ellman imine (R = tBu) failed due to competitive reduction of the sulfinimine (entry 2). Interestingly, unlike previous studies where low yields of product were observed in the absence of Ni-catalyst, the current reaction completely shuts down without it (with near quantitative imine recovery, entry 7).
Figure 5.

(A) Synthesis of amines from RAEs previously limited to amino acid synthesis and (B) an alternative protocol to access benzylic amines.
With this new set of conditions in hand a range of 1°, 2°, and 3° carboxylic acids could be coupled with imine 85 via the corresponding RAE to rapidly provide a range of chiral benzylic amines in good yield and high dr (Table 3). Ethers (88 and 97), esters (91–93 and 96), Boc- and Ts-protected amines (87, 89, 94, 95, 98 and 99), pyridines (96–98), and aryl halides (94 and 95) were all tolerated. Of the substrates screened, the most rapid and high yielding reactions involved bridgehead RAEs (90–93) presumably due to the increased stability and nucleophilicity of the putative radical intermediates. A notable limitation of this work is that alkyl sulfinyl imines are not viable coupling partners. The majority of products in Table 3 are novel (including their deprotected analogs). In the case of substrate 86, the deprotected racemic amine is expensive (from Sigma-Aldrich, ca. $1/mg).20 The deprotected analog of 87 has been prepared in racemic form during the search for new kinase inhibitors by way of pipecolic acid through a classical three step sequence (Weinreb amide, Grignard, reductive amination).21
Table 3.
Scope of the radical addition into chiral imines. Reaction conditions: RAE (2.0 equiv), sulfinimine (1.0 equiv), NiCl2•glyme (25 mol%), Zn nanopowder (6.0 equiv), DMF (1.0 M), rt, 2 d.
 |
See SI for reaction conditions.
Extending the Scope of Nucleophilic RAE Chemistry: Alkyl NHK reactivity
The addition of standard 2e− based nucleophiles to aldehydes is often beleaguered with competing enolization/polymerization problems (Figure 6A). The alkyl NHK reaction provides a viable solution to this issue but still requires an alkyl halide precursor.22 Shenvi and co-workers recently reported an impressive departure from this requirement by utilizing olefin-derived radicals in a HAT/Cr system to deliver branched products from terminal olefins.23 At this juncture it is now self-evident that RAEs represent viable surrogates for alkyl halides in a myriad of different reactions under Ni-, Fe-, Co-, Cu-, Ru-, Pd- and Ir-based catalysis.24 The use of alkyl halides in the classic Nozaki-Hiyama-Kishi (NHK) reaction is rare, and within the theme of utilizing RAEs to add to C=X bonds this seemed like a useful reaction to explore (Figure 6A).
Figure 6.

(A) Alcohol synthesis through conventional and radical means and (B) the development of an alkyl-NHK reaction employing RAEs.
Access to such reactivity via RAEs versus olefins could provide a complementary solution to give linear rather than branched products.
The execution of a RAE-based alkyl NHK was straightforward and optimized conditions simply involved the addition of RAE to aldehyde in the presence of CrCl2 and TMSCl (Figure 6B). This is exemplified using model RAE 4 and aldehyde 100 to deliver TMS-protected alcohol 101 in 79% isolated yield. Although the RAE could be generated in situ to give a similar yield (entry 2), the reaction is sensitive to air (entry 3) and should be conducted using an Ar or N2 balloon. NHPI is the preferred RAE (entry 4) and Cr is clearly essential for the reaction (entry 5). The free alcohol could also be obtained in the absence of TMSCl, albeit in diminished yield (entry 6). Unlike the original alkyl NHK using alkyl halides, low valent transition metal mediators such as CoPc or Ni have no effect on conversion (entries 7 and 8). However, excess Cr is still required (2.0 equiv leads to diminished yield, entry 9). Although reversing the stoichiometry of substrates had no effect, a lower yield was observed with one equivalent of RAE (entries 10 and 11). The RAE-based alkyl NHK has admirable scope with regards to functional group compatibility tolerating N-Boc protected amines (102, 104 and 105), halogens (103 and 109–111), and alkenes (106 and 114). However only primary RAEs provide synthetically useful yields thus mirroring the historic limitations of alkyl-halide based NHK. In addition, pyridine-containing building blocks did not afford synthetically useful yields (113).
Mechanistic Inquiry
The synthetic utility of RAEs to function as nucleophilic (Grignard-like) reagents for the addition to C=X bonds presents an exciting opportunity to interrogate this unique chemical reactivity. For this purpose, the CCC described above was chosen for in-depth study.25 Figure 7A outlines the complete mechanistic picture of this transformation supported by kinetics, radical-clock studies, UV spectroscopy, isotopic labeling, and byproduct analysis.26
Figure 7.

CCC: The complete mechanistic picture and supporting experiments. Phth = phthalimide.
A catalytic cycle that is fully consistent with the data (Figure 7A, see high level summary) consists of initial oxidative addition by the electrophilically activated (anhydride or acyl chloride) carboxy group (R1-CO2H) to a tBubpy-ligated Ni(0)-species I. Here, MgCl2 serves to facilitate the formation of a mixed anhydride species as verified in control studies outlined in inset A.27 The critical oxidative addition step occurs rapidly to furnish acyl-bound Ni(II)-carboxylate species II. Indeed, zero-order kinetics in acid/anhydride were measured, as evidenced in the kinetic orders, Figure 7. This step was confirmed through the discrete preparation of Ni(0)-complex I (see inset B and C) and exposure to either a symmetrical anhydride or acyl chloride which both resulted in near instantaneous oxidative addition to complexes II and III, respectively.28 Subsequent ligand exchange (carboxylate for chloride) then occurs as supported by UV-vis spectroscopy wherein the addition of MgCl2 appears to convert II to III.29 It is also important to note that control studies with complex I and the RAE 120 lead to radical-based decomposition pathways rather than OA products (see SI). Competition experiments between RAE 120 and acyl chloride in the presence of complex I show complete consumption of the acyl chloride to the OA complex III (see SI). As aided by the persistent radical effect,30 complex III captures a radical derived from the RAE (R2-CONHPI) to deliver Ni(III) intermediate IV that undergoes rapid reductive elimination to provide the ketone product and Ni(I)-complex V.31 The observation of small amounts of dimerized and decarboxylated byproducts from the RAE indicates that transient radical R2• is captured by persistent metalloradical complex III (see byproduct analysis, Figure 7A and SI). Support for these two steps stems from the direct reaction of either complex II or III with RAE 120 (insets D and E). Presumably due to the differences in disproportionation rates for II and III, carboxylate complex II does not require the presence of a reductant whereas chloride complex III does (see initiation, Figure 7A).32 Thus, either complex may exist in the reaction mixture and both of them can participate in the catalytic cycle. The Ni(I)-complex V produced can then engage another molecule of RAE to perpetuate the cycle and generate Ni(II)-species VI.33 Alternative pathways to RAE decomposition (to produce R2•) can be rationalized through disproportionation or via a Ni/Zn-mediated process (see initiation, Figure 7A).34 To deconvolute the role of Ni and Zn in the reductive cleavage of RAEs, the kinetics of this step were studied (inset F). Strikingly, if a RAE is exposed to standard reaction conditions in the absence of Ni, it undergoes rapid decomposition to a mixture of dimerized and decarboxylated products without any ketone product (see byproduct analysis, Figure 7A and the SI for the kinetic profile). The chart in inset F illustrates the “buffering” effect of Ni to slow down Zn-induced RAE consumption and increase product formation as the catalyst loading is increased. This creates a complex kinetic picture with slight positive orders in Ni at low catalyst loading and saturation kinetics observed at higher loadings (>10%) indicative of off-cycle pathways. This data, combined with that mentioned in insets D and E, support a Ni-based pathway for radical formation from the RAE. A positive kinetic order of 0.3 in the concentration of RAE is observed, suggesting it is involved in one of multiple rate-determining in this complex mechanistic pathway (see evaluation of kinetic orders, Figure 7).35
The key evidence for a radical chain pathway came from analysis of the extent of cyclization upon exposure of RAE 122 and acid 123 to the standard reaction conditions (see inset G).36 The direct linear relationship between concentration of nickel catalyst and the ratio of 124 to 13 forges a mechanistic picture that is consistent with radical formation from complex V, diffusion of R2• out of the solvent cage (wherein cyclization is proposed to occur) before capture by complex III.37 The addition of more Ni catalyst to the reaction mixture shortens the lifetime of the radical in solution resulting in diminished cyclization. Ni(II)-species VI is then reduced by Zn to afford complex I and complete the cycle. The observation of positive kinetic orders in Zn, MgCl2, LiBr indicate this step may be partially rate determining and supports the crucial role that is experimentally observed for the additives (see SI for complete kinetic analysis). Indeed, a complete shutdown of reactivity occurs in the absence of Zn or both MgCl2 and LiBr (see byproduct analysis, Figure 7A and SI). To evaluate the effects of each additive on the reduction from complex VI to complex I, a solution of NiCl2(tBubpy) (inset H, orange line) was stirred in the presence of Zn for an hour. The UV spectra (inset H, blue line) clearly demonstrate formation of Ni(0)(tBubpy) complex I (inset H, purple line). However, addition of MgCl2 (inset H, grey line) or LiBr (inset H, green line) accelerated the reduction from Ni(II) to Ni(0), LiBr to a greater extent. Thus, MgCl2 serves a triple role in facilitating anhydride formation (as a Lewis acid), ligand exchange (via salt metathesis), and Ni-reduction.
To summarize, although the mechanistic picture outlined above is complex, each elementary step is supported, and the role of each essential additive is justified. The studies outlined above help in rationalizing the empirically generated user guide in Table 2 (vide supra) and should aid in the troubleshooting of difficult couplings or the large-scale implementation of CCC.
Conclusion
Barton’s pioneering studies taught the community that there is much value in using carboxylic acids as precursors to a realm of new chemical space via C–C breaking radical fragmentations rather than simple dehydrations (to make amides or esters). Barton esters, Okada’s NHPI esters, and other RAEs (e.g. TCNHPI, -OAt, or -OBt) as well as redox active pyridiniums and sulfones all provide a useful way of breaking bonds in order to modularly install new ones in a versatile way. This study centered on the use of RAEs to access products that have heretofore resided within the scope of two-electron retrosynthetic disconnections. In accessing common functional groups like ketones, alcohols, and amines, students are generally taught that polar bond analysis should lead to nucleophilic and electrophilic starting materials. Implementing radical retrosynthetic logic to the same targets results in the use of RAEs in an unusual way. Thus, these three functional groups can now be accessed commencing from readily available carboxylic acids via RAEs (which become the “nucleophilic” component) with mixed anhydrides, acyl halides, imines, or aldehydes (the canonical electrophilic component). Although certain limitations were encountered (Figure 8), these mild methods offer enhanced scope and orthogonal access to the same functionality previously accessed through two-electron chemistry and are amenable to parallel synthesis. In some cases, the enabled reactivity permits access to disconnections completely unavailable to the two-electron world (i.e. Figure 4). The underlying mechanistic pathways for these transformations are clearly more complex than their classical surrogates however the studies documented herein within the context of CCC help to bring clarity and should instill a sense of confidence and predictability in their implementation. It is therefore expected that these techniques for ketone (via CCC), amine (via RAE couplings with imines), and alcohol (via alkyl NHK reactivity) construction will find utility in a variety of known and unanticipated avenues of synthetic exploration.
Figure 8.
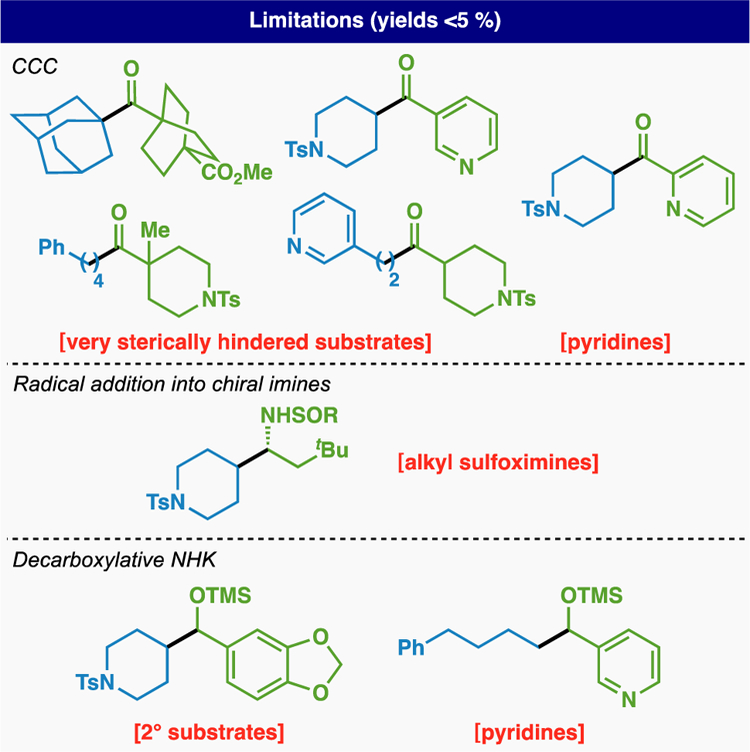
Limitations of the described methodologies.
Supplementary Material
Table 4.
Scope of the alkyl NHK reactions with RAEs. Reaction conditions: RAE (2.0 equiv), aldehydes (1.0 equiv), CrCl2 (4.0 equiv), TMSCl (2.0 equiv), THF/DMF (3:5, 0.06 M), rt, 1 h. TMS = trimethylsilane.
 |
ACKNOWLEDGMENT
Financial support for this work was provided by NIH (GM-118176), the China Scholarship Council (CSC, S.N.), the Marie Skłodowska-Curie Global Fellowships (749359-EnanSET, N.M.P) within the European Union research and innovation framework programme (2014–2020), and the fulbright Scholar Program (P.K.M.). We would like to thank Enamine Ltd for providing samples of several acids. We are grateful to Dr. Dee-Hua Huang and Dr. Laura Pasternack (Scripps Research) for assistance with nuclear magnetic resonance (NMR) spectroscopy and Mr. Andrew Badwal (Pfizer) for high throughput purification. We also thank Stephen Harwood (Scripps Research) for helpful advice and David Hill and Prof. Donna Blackmond (both Scripps Research) for assistance with the mechanistic studies.
Footnotes
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Detailed experimental procedures and analytical data (PDF)
REFERENCES
- 1).Kagan HB. Victor Grignard and Paul Sabatier: Two Showcase Laureates of the Nobel Prize for Chemistry. Angew. Chem. Int. Ed 2012, 51, 7376–7382. [DOI] [PubMed] [Google Scholar]
- 2).Seyferth D. The Grignard Reagents. Organometallics 2009, 28, 1598–1605 and references therein. [Google Scholar]
- 3).For reviews and selected examples, see: (a) Liu RY; Zhou Y; Yang Y; Buchwald SL. Enantioselective Allylation Using Allene, a Petroleum Cracking Byproduct. J. Am. Chem. Soc 2019, 141, 2251–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang Y; Perry IB; Buchwald SL. Copper-Catalyzed Enantioselective Addition of Styrene-Derived Nucleophiles to Imines Enabled by Ligand-Controlled Chemoselective Hydrocupration. J. Am. Chem. Soc 2016, 138, 9787–9790. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang Y-M; Bruno NC; Placeres ÁL; Zhu S; Buchwald SL. Enantioselective Synthesis of Carbo- and Heterocycles through a CuH-Catalyzed Hydroalkylation Approach. J. Am. Chem. Soc 2015, 137, 10524–10527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Zhou Y; Bandar JS; Buchwald SL. Enantioselective CuH-Catalyzed Hydroacylation Employing Unsaturated Carboxylic Acids as Aldehyde Surrogates. J. Am. Chem. Soc 2017, 139, 8126–8129. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Obora Y; Hatanaka S; Ishii Y. Iridium-Catalyzed Coupling Reaction of Primary Alcohols with 1-Aryl-1-propynes Leading to Secondary Homoallylic Alcohols. Org. Lett 2009, 11, 3510–3513. [DOI] [PubMed] [Google Scholar]; (f) Bower JF; Kim IS; Patman RL; Krische MJ. Catalytic Carbonyl Addition through Transfer Hydrogenation: A Departure from Preformed Organometallic Reagents. Angew. Chem. Int. Ed 2008, 48, 34–46. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Kokubo K; Miura M; Nomura M. Rhodium-Catalyzed Reaction of Benzoic Anhydride with Styrene under Molecular Hydrogen. Organometallics 1995, 14, 4521–4524. [Google Scholar]; (h) Kim SW; Zhang W; Krische MJ. Catalytic Enantioselective Carbonyl Allylation and Propargylation via Alcohol-Mediated Hydrogen Transfer: Merging the Chemistry of Grignard and Sabatier. Acc. Chem. Res 2017, 50, 2371–2380. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Nguyen KD; Park BY; Luong T; Sato H; Garza VJ; Krische MJ. Metal-catalyzed reductive coupling of olefin-derived nucleophiles: Reinventing carbonyl addition. Science 2016, 354, aah5133. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Deutsch C; Krause N; Lipshutz BH. CuH-Catalyzed Reactions. Chem. Rev 2008, 108, 2916–2927. [DOI] [PubMed] [Google Scholar]; (k) Mohr J; Oestreich M. Balancing C=C Functionalization and C=O Reduction in Cu−H Catalysis. Angew. Chem. Int. Ed 2016, 55, 12148–12149. [DOI] [PubMed] [Google Scholar]
- 4).(a) Crossley SWM; Obradors C; Martinez RM; Shenvi RA. Mn-, Fe-, and Co-Catalyzed Radical Hydrofunctionalizations of Olefins. Chemical Reviews 2016, 116, 8912–9000. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Green SA; Crossley SWM; Matos JLM; Vásquez-Céspedes S; Shevick SL; Shenvi RA. The High Chemofidelity of Metal-Catalyzed Hydrogen Atom Transfer. Acc. Chem. Res 2018, 51 (11), 2628–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lo JC; Kim D; Pan C-M; Edwards JT; Yabe Y; Gui J; Qin T; Gutiérrez S; Giacoboni J; Smith MW; Holland PL; Baran PS. Fe-Catalyzed C–C Bond Construction from Olefins via Radicals. J. Am. Chem. Soc 2017, 139, 2484–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Shevick SL; Obradors C; Shenvi RA. Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Gooßen LJ; Rodríguez N; Gooßen K. Carboxylic Acids as Substrates in Homogeneous Catalysis. Angew. Chem., Int. Ed 2008, 47, 3100–3120. [DOI] [PubMed] [Google Scholar]
- 6).Barton DHR; Zard SZ. Invention of New Reactions Useful in the Chemistry of Natural Products. Pure Appl. Chem 1986, 58, 675–684. [Google Scholar]
- 7).For seminal studies on the use of RAEs, see: (a) Okada K; Okamoto K; Oda M. A New and Practical Method of Decarboxylation: Photosensitized Decarboxylation of N-Acyloxyphthalimides via Electron-Transfer Mechanism. J. Am. Chem. Soc 1988, 110, 8736. [Google Scholar]; (b) Okada K; Okamoto K; Morita N; Okubo K; Oda M. Photosensitized Decarboxylative Michael Addition Through N-(Acyloxy)phthalimides via an Electron-Transfer Mechanism. J. Am. Chem. Soc 1991, 113, 9401. [Google Scholar]; For recent investigations, see: (c) Cornella J; Edwards JT; Qin T; Kawamura S; Wang J; Pan C-M; Gianatassio R; Schmidt M; Eastgate MD; Baran PS. Practical Ni-Catalyzed Aryl–Alkyl Cross-Coupling of Secondary Redox-Active Esters. J. Am. Chem. Soc 2016, 138, 2174. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Qin T; Cornella J; Li C; Malins LR; Edwards JT; Kawamura S; Maxwell BD; Eastgate MD; Baran PS. A General Alkyl-Alkyl Cross-Coupling Enabled by Redox-Active Esters and Alkylzinc Reagents. Science 2016, 352, 801. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Wang J; Qin T; Chen T-G; Wimmer L; Edwards JT; Cornella J; Vokits B; Shaw SA; Baran PS. Nickel-Catalyzed Cross-Coupling of Redox-Active Esters with Boronic Acids. Angew. Chem. Int. Ed 2016, 55, 9676. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Toriyama F; Cornella J; Wimmer L; Chen T-G; Dixon DD; Creech G; Baran PS. Redox-Active Esters in Fe-Catalyzed C–C Coupling. J. Am. Chem. Soc 2016, 138, 11132. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Qin T; Malins LR; Edwards JT; Merchant RR; Novak AJE; Zhong JZ; Mills RB; Yan M; Yuan C; Eastgate MD; Baran PS. Nickel-Catalyzed Barton Decarboxylation and Giese Reactions: A Practical Take on Classic Transforms. Angew. Chem. Int. Ed 2017, 56, 260. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Sandfort F; O’Neill MJ; Cornella J; Wimmer L; Baran PS. Alkyl–(Hetero)aryl Bond Formation via Decarboxylative Cross-Coupling: A Systematic Analysis. Angew. Chem. Int. Ed 2017, 56, 3319. [DOI] [PMC free article] [PubMed] [Google Scholar]; (i) Edwards JT; Merchant RR; McClymont KS; Knouse KW; Qin T; Malins LR; Vokits B; Shaw SA; Bao D-H; Wei F-L; Zhou T; Eastgate MD; Baran PS. Decarboxylative Alkenylation. Nature 2017, 545, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]; (j) Smith JM; Qin T; Merchant RR; Edwards JT; Malins LR; Liu Z; Che G; Shen Z; Shaw SA; Eastgate MD; Baran PS. Decarboxylative Alkynylation. Angew. Chem. Int. Ed 2017, 56, 11906. [DOI] [PMC free article] [PubMed] [Google Scholar]; (k) Wang J; Lundberg H; Asai S; Martin-Acosta P; Chen JC; Brown S; Farrell W; Dushin R; O’Donnell CJ; Ratnayake AS; Richardson P; Liu Z; Qin T; Blackmond DG; Baran PS. Kinetically Guided Radical-Based Synthesis of C(sp3)−C(sp3) Linkages on DNA. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E6404. [DOI] [PMC free article] [PubMed] [Google Scholar]; (l) Chen T-G; Barton LM; Lin Y; Tsien J; Kossler D; Bastida I; Asai S; Bi C; Chen JS; Shan M; Fang H; Fang FG; Choi H; Hawkins L; Qin T; Baran PS. Building C(sp3)-Rich Complexity by Combining Cycloaddition and C-C Cross-Coupling Reactions. Nature 2018, 560, 350. [DOI] [PMC free article] [PubMed] [Google Scholar]; (m) Wang J; Shang M; Lundberg H; Feu KS; Hecker SJ; Qin T; Blackmond DG; Baran PS. Cu-Catalyzed Decarboxylative Borylation. ACS Catal 2018, 8, 9537. [DOI] [PMC free article] [PubMed] [Google Scholar]; (n) Ni S; Garrido-Castro A; Merchant R; deGruyter J; Schmitt DC; Mousseau JJ; Gallego GM; Yang S; Collins MR; Qiao JX; Yeung K-S; Langley DR; Poss MA; Scola PM; Qin T; Baran PS. A General Amino Acid Synthesis Enabled by Innate Radical Cross-Coupling. Angew. Chem. Int. Ed 2018, 57, 14560. [DOI] [PMC free article] [PubMed] [Google Scholar]; (o) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ. Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Jamison CR; Overman LE. Fragment Coupling with Tertiary Radicals Generated by Visible-Light Photocatalysis. Acc. Chem. Res 2016, 49, 1578. [DOI] [PubMed] [Google Scholar]; (q) Fawcett A; Pradeilles J; Wang Y; Mutsuga T; Myers EL; Aggarwal VK. Photoinduced Decarboxylative Borylation of Carboxylic Acids. Science 2017, 357, 283. [DOI] [PubMed] [Google Scholar]; (r) Zhao W; Wurz RP; Peters JC; Fu GC. Photoinduced, Copper-Catalyzed Decarboxylative C–N Coupling to Generate Protected Amines: An Alternative to the Curtius Rearrangement. J. Am. Chem. Soc 2017, 139, 12153. [DOI] [PMC free article] [PubMed] [Google Scholar]; (s) Candish L; Teders M; Glorius F. Transition-Metal-Free, Visible-Light-Enabled Decarboxylative Borylation of Aryl N-Hydroxyphthalimide Esters. J. Am. Chem. Soc 2017, 139, 7440. [DOI] [PubMed] [Google Scholar]; (t) Li H; Breen CP; Seo H; Jamison TF; Fang Y-Q; Bio MM. Ni-Catalyzed Electrochemical Decarboxylative C–C Couplings in Batch and Continuous Flow. Org. Lett 2018, 20, 1338. [DOI] [PMC free article] [PubMed] [Google Scholar]; (u) Mao R; Balon J; Hu X. Decarboxylative C(sp3)−O Cross-Coupling. Angew. Chem. Int. Ed 2018, 57, 13624. [DOI] [PubMed] [Google Scholar]; (v) Liu X-G; Zhou C-J; Lin E; Han X-L; Zhang S-S; Li Q; Wang H. Decarboxylative Negishi Coupling of Redox-Active Aliphatic Esters by Cobalt Catalysis. Angew. Chem. Int. Ed 2018, 57, 13096. [DOI] [PubMed] [Google Scholar]; (w) Kingston C; Wallace MA; Allentoff AJ; deGruyter JN; Chen JS; Gong SX; Bonacorsi S; Baran PS. Direct Carbon Isotope Exchange through Decarboxylative Carboxylation. J. Am. Chem. Soc 2019, 141, 774–779. [DOI] [PMC free article] [PubMed] [Google Scholar]; (x) Chen TG; Zhang H; Mykhailiuk PK; Merchant RR; Smith CA; Qin T; Baran PS. Quaternary Centers by Nickel-Catalyzed Cross-Coupling of Tertiary Carboxylic Acids and (Hetero)Aryl Zinc Reagents. Angew. Chem. Int. Ed 2019, 58, 2454–2458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8).Habeeb JJ; Tuck DG. Electrochemical synthesis of unsymmetric ketones and some related coupling reactions. J. Chem. Soc., Chem. Commun 1976, 17, 696–697. [Google Scholar]
- 9).(a) Marzouk H; Rollin Y; Folest JC; Nédélec JY; Périchon J. Electrochemical synthesis of ketones from acid chlorides and alkyl and aryl halides catalysed by nickel complexes. J. Organomet. Chem 1989, 369, C47–C50. [Google Scholar]; (b) Amatore C; Jutand A; Périchon J; Rollin Y. Mechanism of the Nickel-Catalyzed Electrosynthesis of Ketones by Heterocoupling of Acyl and Benzyl Halides. Monatsh. Chem 2000, 131, 1293–1304. [Google Scholar]
- 10).Onaka M; Matsuoka Y; Mukaiyama T. A convient method for the direct preparation of ketones from 2-(6-(2-methoxyethyl)pyridyl) carboxylates and alkyl iodides by use of zinc dust and a catalytic amount of nickel dichloride. Chem. Lett 1981, 10, 531–534. [Google Scholar]
- 11).(a) Yin H; Zhao C; You H; Lin K; Gong H. Mild ketone formation via Ni-catalyzed reductive coupling of unactivated alkyl halides with acid anhydrides. Chem. Comm 2012, 48, 7034–7036. [DOI] [PubMed] [Google Scholar]; (b) Wotal AC; Weix DJ. Synthesis of Functionalized Dialkyl Ketones from Carboxylic Acid Derivatives and Alkyl Halides. Org. Lett 2012, 14, 1476–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Cherney AH; Kadunce NT; Reisman SE. Catalytic Asymmetric Reductive Acyl Cross-Coupling: Synthesis of Enantioenriched Acyclic α,α-Disubstituted Ketones. J. Am. Chem. Soc 2013, 135, 7442–7445. [DOI] [PubMed] [Google Scholar]; (d) Jia X; Zhang X; Qian Q; Gong H. Alkyl–aryl ketone synthesis via nickel-catalyzed reductive coupling of alkyl halides with aryl acids and anhydrides. Chem. Comm 2015, 51, 10302–10305. [DOI] [PubMed] [Google Scholar]; For reviews, see: (e)Moragas T; Correa A; Martin R. Metal-Catalyzed Reductive Coupling Reactions of Organic Halides with Carbonyl-Type Compounds. Chem. Eur. J 2014, 20, 8242–8258. [DOI] [PubMed] [Google Scholar]; (f) Everson DA; Weix DJ. Cross-Electrophile Coupling: Principles of Reactivity and Selectivity. J. Org. Chem 2014, 79, 4793–4798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).(a) Zheng M; Xue W; Xue T; Gong H. Ester Formation via Nickel-Catalyzed Reductive Coupling of Alkyl Halides with Chloroformates. Org. Lett 2016, 18, 6152–6155. [DOI] [PubMed] [Google Scholar]; (b) Huihui KMM; Caputo JA; Melchor Z; Olivares AM; Spiewak AM; Johnson KA; DiBenedetto TA; Kim S; Ackerman LKG; Weix DJ. Decarboxylative Cross-Electrophile Coupling of N-Hydroxyphthalimide Esters with Aryl Iodides. J. Am. Chem. Soc 2016, 138, 5016–5019. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Koyanagi T; Herath A; Chong A; Ratnikov M; Valiere A; Chang J; Molteni V; Loren J. One-Pot Electrochemical Nickel-Catalyzed Decarboxylative Sp2–Sp3 Cross-Coupling. Org. Lett 2019, 21, 816–820. [DOI] [PubMed] [Google Scholar]
- 13).Gao D; O’Doherty GA. De Novo Asymmetric Synthesis of Anamarine and Its Analogues. J. Org. Chem 2005, 70, 9932–9939. [DOI] [PubMed] [Google Scholar]
- 14).Prescher JA; Bertozzi CR. Chemistry in living systems. Nat. Chem. Biol 2005, 1, 13–21. [DOI] [PubMed] [Google Scholar]
- 15).(a) Reekie TA; Williams CM; Rendina LM; Kassiou M. Cubanes in Medicinal Chemistry. J. Med. Chem, 2019, 62, 1078–1095; [DOI] [PubMed] [Google Scholar]; (b) Mykhailiuk PK. Saturated bioisosteres of benzene: where to go next? Org. Biomol. Chem 2019, DOI: 10.1039/C8OB02812E. [DOI] [PubMed]; (c) Locke GM; Bernhard SSR; Senge MO. Nonconjugated Hydrocarbons as Rigid-Linear Motifs: Isosteres for Material Sciences and Bioorganic and Medicinal Chemistry. Chem. Eur. J 2019, DOI: 10.1002/chem.201804225. [DOI] [PubMed]
- 16).Narula APS; Arruda EM; Amorelli B; Schiet FT. 3-methyl-6-cyclohexadecen-1-one and its use in perfume compositions. US patent US20130052151A1 (2013).
- 17).Lipinski CA; Lombardo F; Dominy BW; Feeney PJ. Experimental and Computational Approaches to Estimate Solubility and Permeability in Drug Discovery and Development Settings. Adv. Drug Delivery Rev 1997, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 18).Murray CW; Rees DC. The rise of fragment-based drug discovery. Nat. Chem 2009, 1, 187–192. [DOI] [PubMed] [Google Scholar]
- 19).Catalogue number 578002 (40–60 nm particle size).
- 20).Sigma-Aldrich cat no. ENA371463686.
- 21).Caldwell JJ; Davies TG; Donald A; McHardy T; Rowlands MG; Aherne GW; Hunter LK; Taylor K; Ruddle R; Raynaud FI; Verdonk M; Workman P; Garrett MD; Collins I. Identification of 4-(4-Aminopiperidin-1-yl)-7H-pyrrolo[2,3-d]pyrimidines as Selective Inhibitors of Protein Kinase B through Fragment Elaboration. J. Med. Chem 2008, 51, 2147–2157. [DOI] [PubMed] [Google Scholar]
- 22).Fürstner A. Carbon–Carbon Bond Formations Involving Organochromium (III) Reagents. Chem. Rev 1999, 99, 991–1046. [DOI] [PubMed] [Google Scholar]
- 23).Matos JLM; Vásquez-Céspedes S; Gu J; Oguma T; Shenvi RA. Branch-Selective Addition of Unactivated Olefins into Imines and Aldehydes. J. Am. Chem. Soc 2018, 140, 16976–16981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Murarka S. N-(Acyloxy)phthalimides as Redox-Active Esters in Cross-Coupling Reactions. Adv. Synth. Catal 2018, 360, 1735–1753. [Google Scholar]
- 25).For general reviews on the mechanism of Ni-catalyzed cross-coupling reactions, see:(a) Hu X. Nickel-catalyzed cross coupling of non-activated alkyl halides: a mechanistic perspective. Chem. Sci 2011, 2, 1867–1886. [Google Scholar]; (b) Tasker SZ; Standley EA; Jamison TF. Recent advances in homogeneous nickel catalysis. Nature 2014, 509, 299–309. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Lucas EL; Jarvo ER. Keeping Track of the Electrons. Acc. Chem. Res 2018, 51, 567–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Alternative pathways consisting of in situ organometallic formation, oxidative addition of the two substrates to two separate Ni species followed by transmetalation or consecutive double oxidative addition to the same Ni species were ruled out through the mechanistic experiments in Figure 7 and further studies fully detailed in the SI. For information on these mechanistic pathways, see: (a) Biswas S; Weix DJ. Mechanism and Selectivity in Nickel-Catalyzed Cross-Electrophile Coupling of Aryl Halides with Alkyl Halides. J. Am. Chem. Soc 2013, 135, 16192–16197 and [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Everson DA; Jones BA; Weix DJ. Replacing Conventional Carbon Nucleophiles with Electrophiles: Nickel-Catalyzed Reductive Alkylation of Aryl Bromides and Chlorides. J. Am. Chem. Soc 2012, 134, 6146–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) For previous DFT studies on the preference for radical chain or consecutive double oxidative addition pathways in Ni-catalyzed cross-couplings, see: Ren Q; Jiang F; Gong H. DFT study of the single electron transfer mechanisms in Ni-Catalyzed reductive cross-coupling of aryl bromide and alkyl bromide. J. Organomet. Chem 2014, 770, 130–135. [Google Scholar]; (d) Wang X; Ma G; Peng Y; Pitsch CE; Moll BJ; Ly TD; Wang X; Gong H. Ni-Catalyzed Reductive Coupling of Electron-Rich Aryl Iodides with Tertiary Alkyl Halides. J. Am. Chem. Soc 2018, 140, 14490–14497. [DOI] [PubMed] [Google Scholar]
- 27).A mixed anhydride was synthesized, isolated and found to be reactive under the standard conditions, see the SI for full details.
- 28).For UV studies on the oxidative addition of Ni(0) species to cyclic anhydrides, see: Stache EE; Rovis T; Doyle AG. Dual Nickel- and Photoredox-Catalyzed Enantioselective Desymmetrization of Cyclic meso-Anhydrides. Angew. Chem., Int. Ed 2017, 56, 3679–3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).The results are consistent with Gong’s report, see: Zhao C; Jia X; Wang X; Gong H. Ni-Catalyzed Reductive Coupling of Alkyl Acids with Unactivated Tertiary Alkyl and Glycosyl Halides. J. Am. Chem. Soc 2014, 136, 17645–17651. [DOI] [PubMed] [Google Scholar]; MgCl2 may also influence the dielectric constant of the solvent: McCann LC; Organ MG. On the Remarkably Different Role of Salt in the Cross-Coupling of Arylzincs From That Seen with Alkylzincs. Angew. Chem., Int. Ed 2014, 53, 4386–4389, [DOI] [PubMed] [Google Scholar]; while Li salts have been shown to clean the surface of Zn by removal of organometallic species, see: Feng C; Cunningham DW; Easter QT; Blum SA. Role of LiCl in Generating Soluble Organozinc Reagents. J. Am. Chem. Soc 2016, 138, 11156–11159. [DOI] [PubMed] [Google Scholar]
- 30).For reviews, see: (a) Studer A. The Persistent Radical Effect in Organic Synthesis. Chem. Eur. J 2001, 7, 1159–1164. [DOI] [PubMed] [Google Scholar]; (b) Fischer H. The Persistent Radical Effect: A Principle for Selective Radical Reactions and Living Radical Polymerizations. Chem. Rev 2001, 101, 3581–3610. [DOI] [PubMed] [Google Scholar]
- 31).For a study on the reactivity of a Ni(III) species, see: Zheng B; Tang F; Luo J; Schultz JW; Rath NP; Mirica LM. Organometallic Nickel(III) Complexes Relevant to Cross-Coupling and Carbon–Heteroatom Bond Formation Reactions. J. Am. Chem. Soc 2014, 136, 6499–6504. [DOI] [PubMed] [Google Scholar]
- 32).In contrast to the results with complexes II and III, no product formation is observed using Ni(BPhen)Cl2•2DMF without zinc under the standard reaction conditions (see byproduct analysis, Figure 7A), indicating this species does not disproportionate and initiate radical formation.
- 33).For a study on the reactivity of a Ni(I) species, see: Jones GD; Martin JL; McFarland C; Allen OR; Hall RE; Haley AD; Brandon RJ; Konovalova T; Desrochers PJ; Pulay P; Vicic DA. Ligand Redox Effects in the Synthesis, Electronic Structure, and Reactivity of an Alkyl–Alkyl Cross-Coupling Catalyst. J. Am. Chem. Soc 2006, 128, 13175–13183. [DOI] [PubMed] [Google Scholar]
- 34). Such pathways to initiation have also been proposed in cross-electrophile coupling with alkyl halides, see: ref. 26a.
- 35).The precise kinetic order was determined by variable time normalization analysis (VTNA), see: (a) Nielsen CDT; Burés J. Visual kinetic analysis. Chem. Sci 2019, 10, 348–353. The results contrast our previous investigations wherein zero-order in RAE was observed, see Ref. 7k and 7m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36). For selected recent examples of radical chain mechanisms in Ni-catalysis, see: (a) Ref. 26. (b) Ref. 29a.; (c) Breitenfeld J; Ruiz J; Wodrich MD; Hu X. Bimetallic Oxidative Addition Involving Radical Intermediates in Nickel-Catalyzed Alkyl–Alkyl Kumada Coupling Reactions. J. Am. Chem. Soc 2013, 135, 12004–12012. [DOI] [PubMed] [Google Scholar]; (d) Schley ND; Fu GC. Nickel-Catalyzed Negishi Arylations of Propargylic Bromides: A Mechanistic Investigation. J. Am. Chem. Soc 2014, 136, 16588–16593. [DOI] [PMC free article] [PubMed] [Google Scholar]; For general discussion, see: (a) Lucas EL; Jarvo ER. Stereospecific and stereoconvergent cross-couplings between alkyl electrophiles. Nat. Rev. Chem 2017, 1, 0065. [Google Scholar]; (b) Weix DJ. Methods and Mechanisms for Cross-Electrophile Coupling of Csp2 Halides with Alkyl Electrophiles. Acc. Chem. Res 2015, 48, 1767–1775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37).Shevick SL; Obradors C; Shenvi RA. Mechanistic Interrogation of Co/Ni-Dual Catalyzed Hydroarylation. J. Am. Chem. Soc 2018, 140, 12056–12068. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.