

SUMMARY
Low protein synthesis is a feature of somatic stem cells that promotes regeneration in multiple tissues. Modest increases in protein synthesis impair stem cell function, but the mechanisms by which this occurs are largely unknown. We determine that low protein synthesis within hematopoietic stem cells (HSCs) is associated with elevated proteome quality in vivo. HSCs contain less misfolded and unfolded proteins than myeloid progenitors. Increases in protein synthesis cause HSCs to accumulate misfolded and unfolded proteins. To test how proteome quality affects HSCs, we examine Aarssti/sti mice that harbor a tRNA editing defect that increases amino acid misincorporation. Aarssti/sti mice exhibit reduced HSC numbers, increased proliferation, and diminished serial reconstituting activity. Misfolded proteins overwhelm the proteasome within Aarssti/sti HSCs, which is associated with increased c-Myc abundance. Deletion of one Myc allele partially rescues serial reconstitution defects in Aarssti/sti HSCs. Thus, HSCs are dependent on low protein synthesis to maintain proteostasis, which promotes their self-renewal.
In Brief
Stem cells in multiple tissues depend on low protein synthesis to promote regeneration. Hidalgo San Jose et al. demonstrate that low protein synthesis promotes hematopoietic stem cell function by restricting the biogenesis of misfolded proteins. Misfolded proteins impair stem cell quiescence and self-renewal by overwhelming the proteasome and disrupting proteostasis.
Graphical Abstract
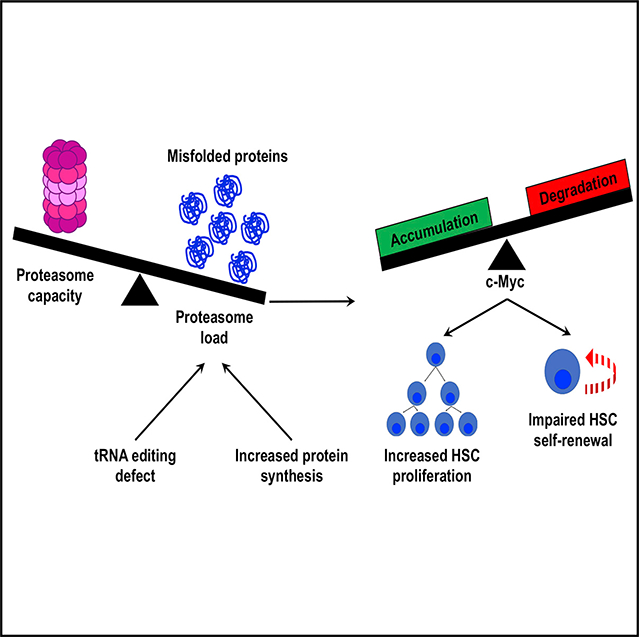
INTRODUCTION
Adult hematopoietic stem cells (HSCs) synthesize less protein per hour than most other hematopoietic progenitors. This does not simply reflect HSC quiescence because dividing HSCs also have lower rates of protein synthesis than dividing restricted progenitors (Signer et al., 2014). Low protein synthesis is necessary for HSC maintenance, as genetic changes that modestly increase protein synthesis impair HSC function (Goncalves et al., 2016; Signer et al., 2014, 2016). Somatic stem cells in multiple other adult tissues also have relatively low rates of protein synthesis, and this is often necessary for stem cell maintenance (Blanco et al., 2016; Llorens-Bobadilla et al., 2015; Sanchez et al., 2016; Zismanov et al., 2016). This raises a fundamental question of why low protein synthesis is necessary for stem cell maintenance and tissue regeneration.
Translation is the most error-prone step in gene expression. Translational errors can lead to protein misfolding and the formation of toxic aggregates. High rates of protein synthesis can increase amino acid misincorporation (Drummond and Wilke, 2009). Reducing translation decreases the synthesis of defective translational products and promotes the clearance of misfolded proteins (Sherman and Qian, 2013). Based on these observations, we hypothesized that low protein synthesis could promote HSC maintenance and function by enhancing proteome quality, which we define as limiting the abundance of mistranslated, unfolded, and/or misfolded proteins. In this study, we set out to examine the effect of protein synthesis rates on proteome quality within HSCs in vivo and to test the consequences of reducing protein quality on HSC function.
RESULTS
HSCs Contain Less Ubiquitylated and Unfolded Protein Than Myeloid Progenitors
To assess proteome quality within hematopoietic stem and progenitor cells, we measured the abundance of ubiquitylated protein by western blot. Ubiquitylated protein includes an abundance of mistranslated or damaged proteins that misfold and are targeted for degradation (Duttler et al., 2013; Kim et al., 2011; Wang et al., 2013). As a consequence, ubiquitylated protein is widely used to assess the abundance of misfolded and unfolded proteins (Kim et al., 2011; Lee et al., 2006; Schubert et al., 2000). To obtain enough cells for western blot analyses, we typically pooled CD150+CD48−Lineage−Sca-1+c-kit+ (CD150+CD48−LSK) HSCs (Kiel et al., 2005) and CD150−CD48−LSK multipotent progenitors (MPPs) (Oguro et al., 2013). HSCs and this population of MPPs (which are sometimes referred to as short-term HSCs) are very similar in terms of their gene expression profile (Signer et al., 2016), cell cycle status (Oguro et al., 2013), protein synthesis rate (Signer et al., 2014), and metabolism (Agathocleous et al., 2017). CD48−LSK HSCs/ MPPs contained considerably less ubiquitylated protein and less LysK48-linkage specific polyubiquitylated protein (which preferentially targets substrates for degradation) than equal numbers of common myeloid progenitors (CMPs), granulocyte macrophage progenitors (GMPs) and megakaryocyte erythroid progenitors (MEPs) (Akashi et al., 2000) isolated from the bone marrow of young adult mice (Figures 1A and S1A).
Figure 1. HSCs Depend upon Low Protein Synthesis to Maintain Proteome Quality.

(A) Western blot examining ubiquitylated protein in 3 × 104 HSCs/MPPs, CMPs, GMPs, and MEPs (one of >5 blots).
(B) Flow cytometry analysis showing ubiquitylated protein content relative to HSCs (n = 11 mice).
(C) Representative histograms of ubiquitylated protein content in HSCs, CMPs, GMPs, and MEPs.
(D) Cell volume of HSCs, CMPs, GMPs, and MEPs (n ≥ 34 cells/population).
(E) Representative gel showing total protein content following SYPRO Ruby staining in HSCs/MPPs, CMPs, GMPs, and MEPs (one of 4 blots).
(F) Total protein content relative to HSCs (n = 4 experiments).
(G) Ubiquitylated protein relative to total protein content in HSCs, CMPs, GMPs, and MEPs (from B and E).
(H) Diagram showing that TMI fluoresces when it binds to free cysteine thiols in unfolded proteins.
(I) Relative TMI fluorescence in bone marrow cells after a 4-h incubation at 37°C or 42°C (n = 8 mice).
(J) Total protein content in bone marrow cells after a 4-h incubation at 37°C or 42°C (n = 3 mice).
(K) TMI fluorescence in bone marrow cells from mice treated 18 h earlier with bortezomib (BZ) or vehicle (DMSO) (n = 6 mice/treatment).
(L) Relative TMI fluorescence in HSCs and progenitors (n = 11 mice).
(M) OP-Puro incorporation by HSCs, CMPs, GMPs, and MEPs in vivo (n = 4 mice).
(N) Diagram representing effects on HSC protein synthesis in wild-type (Ptenfl/fl;Rpl24+/+), Pten-deficient (Mx1-Cre+Ptenfl/fl;Rpl24+/+), and Mx1-Cre+;Ptenfl/fl;Rpl24Bst/+ mice.
(O) Western blot examining ubiquitylated protein in 8.5 × 103 HSCs/MPPs from Ptenfl/fl;Rpl24+/+, Mx1-Cre+Ptenfl/flRpl24+/+, and Mx1-Cre+;Ptenfl/fl; Rpl24Bst/+ mice (one of 3 blots).
(P) TMI fluorescence in wild-type, Mx1-Cre+;Ptenfl/fl;Rpl24+/+, and Mx1-Cre+;Ptenfl/fl;Rpl24Bst/+ mice (n = 3–4 mice/genotype).
Data represent mean ± standard deviation. Statistical significance was assessed using a Student’s t test (I-K) or an ANOVA followed by Dunnett’s multiple comparisons test relative to HSCs (B, D, F, G, L, M, and P). *p < 0.05; **p < 0.01; ***p < 0.001.
Unfortunately, it is not possible to develop western blots with all ubiquitylated protein within the linear range of exposure, which precludes accurate quantification. To circumvent this limitation, we quantified ubiquitylated protein by intracellular flow cytometry using an antibody that preferentially recognizes polyubiquitin chains and exhibits no immunoreactivity with free ubiquitin (Markmiller et al., 2019). HSCs contained significantly less ubiquitylated protein than CMPs (47%; p < 0.01), GMPs (157%; p < 0.001), and MEPs (57%; p < 0.001) (Figures 1B and 1C).
Differences in ubiquitylated protein could not be explained by differences in cell volume, total protein abundance, or proteasome activity. Cell volume was similar between HSCs and CMPs (Figure 1D) despite HSCs having significantly less ubiquitylated protein (Figure 1B). Restricted progenitors also contained similar amounts of total protein as HSCs (Figure 1E and 1F), and HSCs had significantly less ubiquitylated protein than restricted progenitors even when normalized to total protein abundance (Figure 1G). HSCs also exhibited much less proteasome activity than CMPs (10-fold; p < 0.001), GMPs (132-fold; p < 0.001) (Signer et al., 2016), and MEPs (6.5-fold; p < 0.01) (Figure 3F).
Figure 3. Accumulation of Ubiquitylated Protein Overwhelms the Proteasome in Aarssti/sti HSCs, Leading to c-Myc Stabilization.

(A) Heatmap showing 94 differentially expressed transcripts (≥ 2-fold change; padj < 0.1) up- (red) or downregulated (blue) between wild-type (+/+) and Aarssti/sti (sti/sti) HSCs (n = 3 experiments/genotype).
(B) Gene set enrichment analysis demonstrating no significant activation of the UPRer in Aarssti/sti HSCs.
(C) Western blot examining phosphorylated (Ser51)-Eif2α in 2 × 104 wild-type (+/+) and Aarssti/sti (sti/sti) HSCs/MPPs.
(D) Frequency of Annexin V+ HSCs in wild-type (+/+) and Aarssti/sti (sti/sti) (n = 3 mice/genotype).
(E) Diagram of the proteostasis network.
(F) Proteasome activity in 5 × 103 HSCs/MPPs, CMPs, GMPs, and MEPs (n = 5–9 replicates in 4 experiments). Data are shown in relative luminescence units (RLUs).
(G) Representative histogram showing GFP expression in ubG76V-GFP HSCs/MPPs in vitro treated for 18 h with (gray) or without (black) BZ.
(H) Frequency of HSCs that are GFP+ in UbG76V-GFP (+/+) and UbG76V-GFP;Aarssti/sti (sti/sti) bone marrow (n = 4–5 mice/genotype).
(I) Relative proteasome activity in wild-type (+/+) and Aarssti/sti (sti/sti) HSCs/MPPs (n = 9 replicates in 3 experiments).
(J) Number of HSCs in the bone marrow (1 femur and 1 tibia) in mice treated with BZ or vehicle (DMSO) (n = 5–6 mice/treatment).
(K) Frequency of wild-type (+/+) and Aarssti/sti (sti/sti) HSCs that incorporated BrdU after a 72-h pulse in vivo (n = 6–8 mice/genotype).
(L and M) Western blot examining c-Myc protein in 104 HSCs/MPPs (L) or 1.8 × 104 CMPs, GMPs and MEPs (M) isolated from wild-type (+/+) and Aarssti/sti (sti/sti) mice (one of 2 (L) or 3 (M) blots).
(N) Myc expression normalized to β-Actin in wild-type (+/+) and Aarssti/sti (sti/sti) HSCs (n = 3).
(O) Fbxw7 expression in wild-type (+/+) and Aarssti/sti (sti/sti) HSCs (n = 3/genotype).
Data represent mean ± standard deviation. Statistical significance was assessed using a Student’s t test (D, H, I, J, K, N, and O) or an ANOVA followed by Dunnett’s test relative to HSCs/MPPs (F). *p < 0.05; **p < 0.01; ***p < 0.001.
Although ubiquitylated protein is used as a surrogate to quantify misfolded and unfolded proteins (Lee et al., 2006; Sontag et al., 2014; Vilchez et al., 2012), not all ubiquitylated protein arises from misfolding events. We thus sought an orthogonal assay to measure protein quality. Tetraphenylethene maleimide (TMI) is a cell-permeable dye that fluoresces upon binding to cysteine thiols (Chen et al., 2017). The free thiol side chains of non-disulphide-bonded hydrophobic cysteines are typically buried within the core of globular proteins (Marino and Gladyshev, 2010) but can be exposed in unfolded proteins (Figure 1H). We adapted TMI to quantify unfolded proteins within single hematopoietic cells by flow cytometry. Hematopoietic cells treated with TMI exhibited a clear increase in fluorescence relative to untreated controls (Figure S1B). Although glutathione represents a major pool of non-protein thiols within cells, it does not induce TMI fluorescence (Chen et al., 2017). Furthermore, glutathione levels do not significantly differ between HSCs and restricted progenitors (Agathocleous et al., 2017).
To validate TMI fluorescence as a reporter of unfolded protein accumulation in hematopoietic cells, we performed several independent experiments. First, we assessed the effect of heat shock, which induces protein unfolding (Morimoto, 2011), on TMI fluorescence in bone marrow cells. Bone marrow cells incubated at 42°C for 4 h exhibited a 22% increase in TMI fluorescence relative to an aliquot of the same cells incubated at 37°C (Figures 1I and S1B; p < 0.05). Heat-shocked bone marrow cells had statistically equivalent amounts of total protein compared to bone marrow cells incubated at 37°C (Figure 1J), indicating that differences in TMI fluorescence do not simply reflect differences in total protein abundance. Next, because unfolded proteins are primarily degraded by the proteasome, we assessed the effects of proteasome inhibition on TMI fluorescence in vivo. Bone marrow cells isolated from mice treated with the proteasome inhibitor bortezomib exhibited a ~30% increase in TMI fluorescence compared to cells from vehicle-treated controls (Figures 1K and S1C; p < 0.05). Finally, we compared levels of ubiquitylated protein within TMIlow (lowest quartile of TMI fluorescence), TMIhigh (highest quartile of TMI fluorescence), and unfractionated bone marrow cells by western blot. TMIlow bone marrow cells contained less ubiquitylated protein than unfractionated bone marrow cells, which in turn contained less ubiquitylated protein than TMIhigh bone marrow cells (Figure S1D). These data suggest that TMI fluorescence accurately reflects the amount of unfolded proteins within primary hematopoietic cells.
The mean fluorescence intensity of TMI was significantly lower in HSCs than CMPs (32%; p < 0.01), GMPs (25%; p < 0.05), and MEPs (48%; p < 0.01) (Figures 1L and S1E–S1K). Although these differences appear rather modest, the increase in TMI fluorescence in restricted progenitors compared to HSCs was similar or greater in magnitude to that observed in bone marrow cells following either heat shock (Figure 1I) or bortezomib administration (Figure 1K), which are both treatments that significantly disrupt proteostasis. Overall, these data suggest that HSCs contain significantly elevated proteome quality compared to restricted myeloid progenitors in vivo.
Increasing Protein Synthesis Reduces Protein Quality within HSCs In Vivo
Adult HSCs exhibit unusually low protein synthesis compared to other hematopoietic cells (Figure 1M; Hidalgo San Jose and Signer, 2019; Signer et al., 2014). Translation is the most error-prone step in gene expression, and 10%−30% of all newly synthesized proteins are ubiquitylated and targeted for degradation (Lykke-Andersen and Bennett, 2014; Schubert et al., 2000). This raises the question of whether low protein synthesis in HSCs contributes to their reduced abundance of ubiquitylated and unfolded protein in vivo.
To test if increasing protein synthesis reduces proteome quality within HSCs, we took advantage of two genetic mouse models that modestly alter protein synthesis rates within HSCs in vivo. Conditional deletion of Pten from adult hematopoietic cells in Mx1-Cre;Ptenfl/fl mice leads to hyperactivation of the PI3K and mTor signaling pathways, which increases protein synthesis by ~30% in HSCs (Signer et al., 2014). Introduction of a ribosomal mutation (Rpl24Bst/+) (Oliver et al., 2004) functionally blocks this increase in protein synthesis within Pten-deficient HSCs without reducing hyperactivation of the downstream signaling pathways (Figure 1N; Signer et al., 2014). Increased protein synthesis within Pten-deficient HSCs was accompanied by an accumulation of ubiquitylated (Figure 1O) and unfolded protein (Figure 1P; ~30%; p < 0.05) relative to controls. Rpl24Bst/+, which blocks the increase in protein synthesis (Signer et al., 2014), also blocked the increase in ubiquitylated and unfolded protein in Pten-deficient HSCs (Figures 1O and 1P). These data indicate that increased protein synthesis reduces protein quality within HSCs in vivo and suggests that HSCs depend upon low protein synthesis to maintain the integrity of their proteome.
Defects in tRNA Editing Preferentially Impair HSC Self-Renewal
We previously demonstrated that conditional deletion of Pten increases protein synthesis and severely impairs HSC function. Blocking the increase in protein synthesis with the Rpl24Bst/+ mutation largely rescues the function of Pten-deficient HSCs (Signer et al., 2014). These data indicate that increased protein synthesis impairs HSC function. Consistent with these observations, other genetic interventions that modestly increase protein synthesis within HSCs, such as deletion of Eif4ebp1 and Eif4ebp2 (Signer et al., 2016) or deletion of Angiogenin (Goncalves et al., 2016), also impair HSC function.
Given that increased protein synthesis impairs HSC function and reduces proteome quality (Figures 1O and 1P), it raised the question of whether the accumulation of misfolded and unfolded proteins contributes to HSC dysfunction. To test this, we sought a mouse model that could decouple increased protein synthesis and reduced protein quality. Aarssti/sti mice harbor a mutation in the alanyl-tRNA synthetase that causes a tRNA editing defect that increases amino acid misincorporation at alanine residues, which can lead to an accumulation of mistranslated and ubiquitylated proteins within slowly and non-dividing cells (Lee et al., 2006). Aarssti/sti mice exhibit some skin and hair texture defects as well as age-related neurological defects (Lee et al., 2006). Despite these defects, Aarssti/sti mice are otherwise grossly normal and survive into adulthood. Aarssti/sti HSCs had an 18% increase in ubiquitylated protein compared to littermate control HSCs (Figures 2A and 2B; p < 0.01). Notably, the accumulation of ubiquitylated protein in Aarssti/sti HSCs occurred without significant changes in overall protein synthesis (Figure 2C), cell size (Figure S2A) or proteasome activity (Figure 3I). Furthermore, the increase in ubiquitylated protein within Aarssti/sti HSCs was within the physiological range observed within restricted progenitors (Figure S3A), making Aarssti/sti HSCs more similar to other hematopoietic cells. These data establish Aarssti/sti mice as a model to study the effects of a physiological decline in protein quality in vivo that is independent of changes in protein synthesis rate.
Figure 2. A tRNA Editing Defect that Reduces Proteome Quality Impairs HSC Self-Renewal.

(A) Western blot examining ubiquitylated protein in 2.5 × 104 wild-type (+/+) and Aarssti/sti (sti/sti) HSCs/MPPs, CMPs, GMPs, and MEPs (one of 2 blots).
(B) Flow cytometry analysis showing ubiquitylated protein content in Aarssti/sti (sti/sti) relative to wild-type (+/+) HSCs (n = 4 mice/genotype).
(C) OP-Puro incorporation by wild-type (+/+) and Aarssti/sti (sti/sti) HSCs and progenitor cells in vivo (n = 3 mice/genotype).
(D) Bone marrow cellularity (BM; 1 femur and 1 tibia) in wild-type (+/+) and Aarssti/sti (sti/sti) mice (n = 6 mice/genotype).
(E) Frequency of HSCs in wild-type (+/+) and Aarssti/sti (sti/sti) bone marrow (n = 6 mice/genotype).
(F and G) Absolute number of HSCs in the bone marrow (1 femur and 1 tibia; F) and spleen (G) of wild-type (+/+) and Aarssti/sti (sti/sti) mice (n = 6 mice/genotype).
(H-J) Frequency of CMPs (H), GMPs (I), and MEPs (J) in wild-type (+/+) and Aarssti/sti (sti/sti) bone marrow (n = 6 mice/genotype).
(K) Frequency of colony-forming unit (CFU) progenitors in wild-type (+/+) and Aarssti/sti (sti/sti) bone marrow (n = 4 mice/genotype).
(L) Diagram showing HSC transplantation strategy.
(M) Donor cell engraftment when 10 HSCs from wild-type (+/+) and Aarssti/sti (sti/sti) mice were transplanted with 2 × 105 recipient-type bone marrow cells into irradiated mice. Total hematopoietic cell, B cell, T cell, and myeloid cell engraftment is shown (n = 4 donors and 14–21 total recipients/genotype).
(N) Donor cell engraftment in secondary recipients (n = 2 donors and 8–9 total recipients/genotype).
(O) Frequency of secondary recipients in (N) that exhibited long-term (16-week) multilineage reconstitution (≥0.5% donor derived peripheral blood B, T, and myeloid cells).
Data represent mean ± standard deviation (B-K) or standard error of the mean (M and N). Statistical significance was assessed using a Student’s t test or a Fisher’s exact test (O). *p < 0.05; **p < 0.01; ***p < 0.001.
To assess the effects of reduced protein quality on hematopoiesis, we analyzed 8-week-old Aarssti/sti and littermate control mice. Aarssti/sti mice had normal bone marrow cellularity (Figure 2D) but significantly reduced spleen cellularity (Figure S2I) compared to controls. The frequency and absolute numbers of HSCs were significantly reduced by ~40% in the bone marrow of Aarssti/sti mice (Figures 2E and 2F; p < 0.01). The decline in HSCs was not due to mobilization to the spleen, as Aarssti/sti mice did not exhibit an increase in spleen HSCs (Figure 2G). In contrast to HSCs, the frequencies of CMPs, GMPs, MEPs, CD71+Ter119+ erythroid progenitors, CD11b+Gr1+ myeloid cells, pro-B cells, pre-B cells, IgM+ B cells, CD4+ T cells, and CD8+ T cells were unchanged in the bone marrow and spleen of Aarssti/sti mice compared to controls (Figures 2H–2J and S2B–S2P). The frequency of colony-forming progenitors was also unchanged in Aarssti/sti mice (Figure 2K). These data indicate that reduced protein quality in Aarssti/sti mice impairs the maintenance of HSCs but does not significantly impact the maintenance of restricted hematopoietic progenitors.
To test if reduced protein quality impairs HSC function, we performed serial long-term reconstitution assays. We competitively transplanted 10 CD150+CD48−LSK HSCs from Aarssti/sti or littermate control mice (CD45.2+) together with 2 × 105 wild-type congenic (CD45.1+) bone marrow cells into lethally irradiated mice (CD45.1+) (Figure 2L). Overall, long-term hematopoietic reconstitution from Aarssti/sti HSCs was statistically indistinguishable from controls in primary recipients (Figure 2M). After 16 weeks, 3 × 106 bone marrow cells from primary recipient mice were serially transplanted into lethally irradiated mice. Secondary recipients of Aarssti/sti HSCs exhibited significantly less donor cell reconstitution after transplantation (Figure 2N). None (0/8) of the secondary recipients of Aarssti/sti HSCs exhibited long-term multilineage reconstitution compared to 67% (6/9) of controls (Figure 2O; p < 0.01). These data indicate that Aarssti/sti HSCs have impaired self-renewal potential in vivo.
We next sought to determine whether reduced proteome quality preferentially impaired HSC function or whether it also impaired the function of restricted progenitors. In contrast to HSCs, we observed no significant increase in ubiquitylated protein among Aarssti/sti myeloid progenitors (Figures S3A–S3D). This observation is consistent with previous reports that rapidly dividing cells efficiently dilute out misfolded proteins (Lee et al., 2006). Because Aarssti/sti myeloid progenitors do not accumulate more ubiquitylated proteins than controls, we are precluded from making direct conclusions on the effect of reduced protein quality within those populations. We thus tested whether CD150−CD48−LSK MPPs (short-term HSCs), a progenitor cell population with similar cycling kinetics as HSCs (Oguro et al., 2013), would be susceptible to an accumulation of ubiquitylated proteins. Indeed, similar to HSCs, Aarssti/sti MPPs exhibited a significant ~16% increase in ubiquitylated protein compared to controls (Figure S3E; p < 0.05). However, in contrast to HSCs, we observed no significant change in MPP frequency (Figure S3F). To test MPP function, we competitively transplanted 50 Aarssti/sti or control MPPs into irradiated mice (Figure S3G). Levels of reconstitution by Aarssti/sti MPPs were indistinguishable from controls (Figure S3H). Although HSC defects largely appeared during serial transplantation, it is not possible to perform secondary transplants with MPPs because they lack serial reconstituting activity. Nevertheless, these data suggest that HSCs are particularly sensitive to disruptions in proteostasis, as modest declines in protein quality preferentially impair HSC self-renewal.
Accumulation of Misfolded Proteins Overwhelms and/or Impairs the Proteasome in HSCs
An accumulation of misfolded and unfolded proteins can activate proteotoxic stress response pathways, including the endoplasmic reticulum unfolded protein response (UPRER) (Walter and Ron, 2011). Basal activity of the UPRER can promote HSC survival and engraftment (Chapple et al., 2018; Liu et al., 2019; van Galen et al., 2018). Under more severe stress conditions, activation of the UPRER can induce apoptosis within HSCs (van Galen et al., 2014) and fetal HSC self-renewal depends upon bile acids to suppress UPRER activation (Sigurdsson et al., 2016). This raised the question of whether the reduced HSC number in Aarssti/sti mice was a consequence of activation of the UPRER and increased cell death. To test this, we performed RNA sequencing on Aarssti/sti and control HSCs (Figure 3A) and looked for changes in the expression of gene sets related to the UPRER. We observed no significant changes in the expression of UPRER-related gene sets in Aarssti/sti compared to control HSCs (Figure 3B). Consistent with this observation, Aarssti/sti HSCs did not exhibit increased abundance of phosphorylated (Ser51) Eif2α (Figure 3C), which is one marker of UPRER activation (Harding et al., 1999). In addition, there was no significant difference in the frequency of Annexin V+ HSCs between Aarssti/sti and control mice (Figure 3D), indicating that reduced HSC numbers in Aarssti/sti mice are not due to increased cell death.
In the absence of detectable UPRER activation, we sought to uncover other potential mechanisms contributing to Aarssti/sti HSC dysfunction. Proteostasis, in addition to protein biogenesis, is maintained by an integrated network of physiological mechanisms that promote protein folding, repair, and degradation (Labbadia and Morimoto, 2015; Figure 3E). Previous work has demonstrated that overexpression of aggregation-prone proteins can overwhelm and/or impair the ubiquitin proteasome system in HEK293 cells (Bence et al., 2001). Because HSCs exhibited less proteasome activity than restricted progenitors (Figure 3F), we hypothesized that an accumulation of misfolded proteins might overwhelm the capacity of the proteasome within HSCs to efficiently degrade all ubiquitylated substrates.
To test this, we used UbG76V-GFP proteasome reporter mice (Lindsten et al., 2003). UbG76V-GFP mice ubiquitously express GFP that is fused to a constitutive degradation signal. GFP transcripts can be detected in various tissues, but no GFP fluorescence is detected because the protein is rapidly degraded by the proteasome. However, administration of a proteasome inhibitor leads to robust GFP fluorescence (Figure 3G; Lindsten et al., 2003). Similar to other cell types, UbG76V-GFP HSCs did not express significant GFP fluorescence in vivo (Figure 3H). We crossed UbG76V-GFP mice to Aarssti/sti mice and assessed GFP expression within HSCs. Strikingly, we observed a significant accumulation of GFP+ HSCs within UbG76V-GFP;Aarssti/sti mice (Figure 3H; p < 0.01), and this was not due to a reduction in proteasome activity (Figure 3I). These data indicate that the proteasome can become overwhelmed and/or impaired when misfolded proteins accumulate within HSCs.
To test if insufficient proteasome activity impairs HSC maintenance, we treated mice with bortezomib for 3 consecutive days and assessed HSC numbers in the bone marrow on day 4. Mice treated with bortezomib exhibited a ~50% reduction in HSCs compared to vehicle (DMSO)-treated controls (Figure 3J; p < 0.01). The bortezomib-mediated decline in HSCs was similar to the decline in HSCs observed within Aarssti/sti mice (Figure 2F). These data indicate that impaired proteasome activity impairs HSC maintenance and suggest that the decline in Aarssti/sti HSCs may be caused by their overwhelmed/impaired proteasome.
Accumulation of Misfolded Proteins Is Associated with c-Myc Accumulation in HSCs
In addition to regulating proteostasis, the ubiquitin proteasome system post-translationally regulates the abundance of multiple proteins that affect diverse cellular functions, including HSC fate (Moran-Crusio et al., 2012). As a consequence, we wondered if overwhelming the proteasome might lead to stabilization of some of these proteasome targets within HSCs. Our RNA sequencing data revealed a significant increase in the expression of cell cycle genes within Aarssti/sti HSCs (Figure 3B). In accordance with these gene expression studies, we determined that Aarssti/sti HSCs exhibited increased proliferation in vivo compared to controls. After a 72-h pulse of bromodeoxyuridine (BrdU) in vivo, 41.5% of Aarssti/sti HSCs incorporated BrdU compared to 23.8% of control HSCs (Figure 3K; p < 0.05).
Because Aarssti/sti HSCs exhibited increased proliferation (Figure 3K) and diminished self-renewal activity (Figures 2N and 2O), we sought to identify known proteasome-regulated targets that could contribute to these phenotypes. The target that we focused on was c-Myc. c-Myc has an established role in regulating HSC quiescence, cell cycle entry, and self-renewal. Although Myc is transcribed at similar levels in HSCs and progenitors, the c-Myc protein is mostly absent from HSCs but accumulates in more differentiated progenitors (Reavie et al., 2010). Conditional deletion of the E3 ubiquitin ligase Fbxw7 leads to an accumulation of c-Myc protein within HSCs (Reavie et al., 2010; Thompson et al., 2008), suggesting that Fbxw7 targets c-Myc for degradation. Fbxw7−/− HSCs exhibit increased cycling and premature exhaustion (Matsuoka et al., 2008; Thompson et al., 2008), which is similar to HSCs with enforced expression of c-Myc (Wilson et al., 2004). Genetic inactivation of one allele of Myc is sufficient to partially rescue Fbxw7−/− HSCs (Reavie et al., 2010). These studies indicate that proteasome-mediated degradation of c-Myc is essential for normal HSC quiescence and self-renewal.
Given that c-Myc is heavily regulated by the proteasome, promotes HSC cell cycle entry, and influences HSC self-renewal, we investigated whether c-Myc was dysregulated in Aarssti/sti mice. We determined that Aarssti/sti HSCs/MPPs expressed elevated amounts of c-Myc protein relative to controls (Figure 3L), but no such changes were evident within restricted progenitors (Figure 3M). The increase in the c-Myc protein could not be explained by increased Myc mRNA expression, which was unchanged within Aarssti/sti HSCs (Figure 3N). Increased c-Myc expression also could neither be explained by reduced expression of Fbxw7 nor any other E3 ubiquitin ligases implicated in c-Myc ubiquitylation (Figures 3O and S4H–S4S). Although we cannot rule out all possible mechanisms, our data suggest that c-Myc accumulates within Aarssti/sti HSCs at least in part because of impaired degradation as a consequence of overwhelmed and/or impaired proteasome capacity.
To further support our findings in Aarssti/sti HSCs, we tested whether an accumulation of misfolded and unfolded proteins had similar effects on proteasome capacity and c-Myc expression in Pten-deficient HSCs that accumulate ubiquitylated proteins as a consequence of increased protein synthesis (Figure 1O). Consistent with our observations in Aarssti/sti HSCs, Pten-deficient HSCs exhibited a robust increase in c-Myc protein expression (Figure S4B) without any increase in Myc mRNA expression (Figure S4C) or decline in Fbxw7 expression (Figure S4D). In addition, we observed a significant increase in GFP+ HSCs within Mx1-Cre+;Ptenfl/fl;UBG76V-GFP mice (Figure S4E), indicating that Pten-deficient HSCs also experience an overwhelming or impairment of their proteasome without any significant change in overall proteasome capacity (Figure S4F). Notably, the Rpl24Bst/+ mutation reduced the abundance of c-Myc in Pten-deficient HSCs/MPPs to near wild-type levels (Figure S4G). Taken together, these data strongly suggest that a modest accumulation of misfolded proteins is sufficient to overwhelm the capacity of the proteasome within HSCs and is associated with accumulation of c-Myc.
Reducing c-Myc Expression Partially Rescues Self-Renewal Defects in Aarssti/sti HSCs
Because c-Myc overexpression can impair HSC self-renewal, we hypothesized that c-Myc accumulation within Aarssti/sti HSCs was contributing to defects in serial reconstituting activity. To test this, we sought to reduce c-Myc expression within Aarssti/sti HSCs. We could not completely delete Myc because Myc deficiency causes severe hematopoietic defects (Wilson et al., 2004). We, thus, sought to reduce c-Myc expression by conditionally deleting a single allele of Myc in Aarssti/sti mice by breeding Mx1-Cre+;Mycfl/+;Aarssti/sti (Aarssti/sti;Myc+/−) mice. Remarkably, c-Myc expression in Aarssti/sti HSCs was reduced to near wild-type levels in Aarssti/sti;Myc+/− HSCs (Figure 4A), whereas levels of ubiquitylated protein were still elevated (data not shown).
Figure 4. Reducing c-Myc Expression Largely Rescues Serial Reconstituting Activity of Aarssti/sti HSCs.

(A) Western blot examining c-Myc in 104 HSCs/MPPs isolated from wild-type (+/+), Mx1-Cre+;Aarssti/sti;Mycfl/+ (sti/sti;Myc+/−) and Aarssti/sti (sti/sti;Myc+/+) mice.
(B) Diagram showing bone marrow transplantation strategy.
(C) Donor cell engraftment 16 weeks after 5 × 105 bone marrow cells from wild-type (+/+), Aarssti/sti (sti/sti;Myc+/+) and Mx1-Cre+;Aarssti/sti;Mycfl/+ (sti/sti;Myc+/−) mice were transplanted with 5 × 105 recipient-type bone marrow cells into irradiated mice. Total hematopoietic cells, B cell, T cell, and myeloid cell engraftment is shown (n = 4–8 donors and 26–44 total recipients/genotype).
(D) Donor cell engraftment in secondary recipients (n = 4–10 donors and 17–44 recipients/genotype).
(E) Frequency of secondary recipients in (D) that exhibited long-term multilineage reconstitution.
Data represent mean ± standard error of the mean. Statistical significance was assessed using a Student’s t test (C and D) or Fisher’s exact test (E). *p < 0.05; **p < 0.01; ***p < 0.001.
To test if reducing c-Myc expression could partially rescue Aarssti/sti HSC self-renewal, we performed serial long-term reconstitution assays. We competitively transplanted 5×105 Mx1-Cre+;Mycfl/+;Aarssti/sti, Aarssti/sti or wild-type control bone marrow cells (CD45.2+) together with 5 × 105 congenic bone marrow cells (CD45.1+) into lethally irradiated mice (CD45.1+) (Figure 4B). Similar to our HSC transplants (Figure 2M), long-term hematopoietic reconstitution from Aarssti/sti bone marrow cells was statistically indistinguishable from controls in primary recipients (Figure 4C). Aarssti/sti;Myc+/− bone marrow cells also gave similar levels of reconstitution in primary transplants, although they did exhibit a slight, but significant reduction in B cell reconstitution. After 16 weeks, 3 × 106 bone marrow cells from primary recipient mice were serially transplanted into lethally irradiated mice. Secondary recipients of Aarssti/sti bone marrow cells exhibited significantly less donor cell reconstitution in all lineages after transplantation (Figure 4D). Strikingly, reducing c-Myc expression in Aarssti/sti;Myc+/− HSCs partially prevented these defects (Figure 4D). Aarssti/sti;Myc+/− cells gave similar levels of reconstitution as wild-type cells, except in the B cell lineage where reconstitution was still reduced (Figure 4D). Overall, long-term multilineage reconstitution in secondary transplants was reduced from 95% (35/37) in recipients of wild-type cells to 39% (17/44) in recipients of Aarssti/sti cells (p < 0.001), but was increased back to 94% (16/17) in recipients of Aarssti/sti;Myc+/− cells (Figure 4E; p < 0.001). These data indicate that reducing c-Myc expression partially prevents self-renewal defects within Aarssti/sti HSC, and suggest that c-Myc accumulation contributes to impaired HSC function when proteostasis is perturbed.
DISCUSSION
Low protein synthesis is a broadly conserved feature of somatic stem cells (Blanco et al., 2016; Llorens-Bobadilla et al., 2015; Sanchez et al., 2016; Signer et al., 2014; Zismanov et al., 2016) that promotes regeneration in multiple tissues. However, little is known about why stem cells depend upon low protein synthesis and why modest increases in protein synthesis impair stem cell function. In this study, we set out to address this question by examining the influence of protein synthesis on protein quality and homeostasis. We found that HSCs contain less ubiquitylated and unfolded protein than restricted myeloid progenitors in vivo (Figure 1) and that modest increases in protein synthesis reduce protein quality within HSCs. To investigate the consequences of reduced protein quality on HSCs independent of changes in protein synthesis rate, we examined Aarssti/sti mice that have a defect in tRNA editing activity. The accumulation of defective translational products within Aarssti/sti HSCs impaired their serial long-term multilineage reconstituting activity (Figure 2). Surprisingly, we found that the accumulation of misfolded proteins overwhelmed and/or impaired the proteasome and led to increased c-Myc abundance in both Aarssti/sti and Pten-deficient HSCs (Figures 3 and S4). Reducing c-Myc expression by deleting a single allele of Myc partially prevented the self-renewal defects observed in Aarssti/sti HSCs (Figure 4). Overall, our studies indicate that stem cells depend upon low protein synthesis to maintain proteome quality and that modest accumulation of misfolded proteins preferentially impairs HSC self-renewal.
Our finding that the proteasome gets overloaded and impaired in HSCs raises the possibility that stem cells may depend on alternative mechanisms to detoxify and eliminate misfolded proteins. One possibility is that HSCs preferentially depend upon autophagy to degrade misfolded proteins. In support of this possibility, two recent studies reported that autophagy promotes proteostasis maintenance within aging neural stem cells (Leeman et al., 2018) and muscle satellite cells (García-Prat et al., 2016). Similar to HSCs, quiescent neural stem cells also contain less proteasome activity than restricted progenitors and neural stem cells preferentially store misfolded proteins as aggregates in lysosomes (Leeman et al., 2018). Several studies have demonstrated an essential role for autophagy in HSCs (Ho et al., 2017; Liu et al., 2010; Mortensen et al., 2011; Warr et al., 2013), but to our knowledge, whether autophagy promotes proteostasis maintenance within HSCs is unstudied.
In addition to degradation pathways, HSCs could eliminate misfolded proteins through proliferation. Cell division enables cells to dilute out misfolded proteins by distributing them to daughter cells. We determined that a buildup of misfolded proteins led to an accumulation of c-Myc and increased proliferation in HSCs but not restricted progenitors (Figure 3). These data reveal a stem-cell-specific molecular mechanism connecting the proteostasis network to cell cycle control. An intriguing possibility is that HSCs sense changes in protein synthesis and homeostasis by proteasome-mediated turnover of c-Myc, and c-Myc accumulation could be an adaptive mechanism used by individual HSCs to restore proteostasis within daughter stem cells generated by self-renewing divisions. Furthermore, this mechanism could also protect the HSC pool because HSCs unable to restore proteostasis would have sustained c-Myc expression, which promotes HSC differentiation (Wilson et al., 2004).
c-Myc is not the only protein in HSCs whose expression is regulated by the proteasome (Moran-Crusio et al., 2012). Interestingly, a previous report determined that expression of p53, which is also heavily regulated by the proteasome in HSCs (Abbas et al., 2010), is elevated within Pten-deficient HSCs and contributes to self-renewal defects in that background (Lee et al., 2010). Deleting a single allele of Myc also modestly restores long-term multilineage, reconstituting activity within Pten-deficient HSCs (data not shown). Overwhelming of the proteasome due to an accumulation of misfolded and unfolded proteins is likely to have more widespread effects on HSC proteome content. However, detailed proteomic analyses needed to broadly investigate this possibility remain technically difficult to perform within rare HSCs.
Disrupted proteostasis is a hallmark of aging (López-Otín et al., 2013), but whether it contributes to HSC aging is still untested. An intriguing possibility is that disrupted proteostasis in aging HSCs could contribute to HSC dysfunction and the increased incidence of blood cancer in the elderly by preventing the degradation of c-Myc. A potential role for c-Myc in stem cell aging could extend a recent study that demonstrated that reducing Myc expression enhances longevity in mice (Hofmann et al., 2015).
Low protein synthesis is a shared feature of many types of somatic stem cells (Blanco et al., 2016; Llorens-Bobadilla et al., 2015; Sanchez et al., 2016; Signer et al., 2014; Zismanov et al., 2016). Our findings raise the possibility that the maintenance of proteostasis is similarly required by stem cells in diverse tissues. Interestingly, interventions that reduce protein synthesis and enhance proteostasis extend organismal lifespan in an evolutionary conserved manner (Taylor and Dillin, 2011). Our current findings suggest that mechanisms that promote organismal longevity are conserved at the cellular level to maintain long-lived stem cells in vivo.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Robert Signer (rsigner@ucsd.edu). TMI was synthesized by Dr. Yuning Hong and is available from her upon request (Y.Hong@latrobe.edu.au).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
C57BL/6J, C57BL6.SJL, Ptenfl/fl, Mx1-Cre, Rpl24Bst/+, Aarssti/sti, UbG76V-GFP, and Mycfl mice were housed in pathogen-free conditions.
Ptenfl/fl (Groszer et al., 2006), and Mycfl mice (de Alboran et al., 2001) were bred to Mx1-Cre (Cre+) (Kühn et al., 1995) mice to generate Mx1-Cre+;Ptenfl/fl and Mx1-Cre+;Mycfl mice, respectively. Mx1-Cre+;Ptenfl/fl mice were further crossed to Rpl24Bst/+ mice (Oliver et al., 2004) to yield Mx1-Cre+;Ptenfl/fl;Rpl24Bst/+ mice. Aarssti/sti mice (Lee et al., 2006) were bred to Mx1-Cre+;Mycfl mice to produce Mx1-Cre+;Mycfl/+;Aarssti/sti mice. Mx1-Cre+;Ptenfl/fl and Aarssti/sti were bred to UbG76V-GFP mice (Lindsten et al., 2003) to generate Mx1-Cre+;Ptenfl/fl;UBG76V-GFP and UbG76V-GFP;Aarssti/sti mice, respectively. Mutant mice were all backcrossed for at least ten generations onto a C57BL background. C57BL6/J (CD45.2) and C57BL6.SJL (CD45.1) mice were used in transplantation experiments.
Both male and female mice between 6 and 12 weeks of age were used in these studies. All mice were housed in the vivarium at the UC San Diego Moores Cancer Center. All protocols were approved by the UC San Diego Institutional Animal Care and Use Committee.
Cell isolation
Bone marrow cells were isolated by flushing the long bones (femurs and tibias) or by crushing the long bones, vertebrae and pelvic bones with a mortar and pestle in Ca2+- and Mg2+-free Hank’s buffered salt solution (HBSS; Corning) supplemented with 2% (v/v) heat-inactivated bovine serum (GIBCO). Spleens were prepared by crushing tissues between frosted slides. All cells were filtered through a 40 μm cell strainer to obtain single cell suspensions. Cell number and viability were assessed with a hemocytometer based on trypan blue exclusion.
METHOD DETAILS
PIPC treatment
Expression of Mx1-Cre was induced by three or four intraperitoneal injections of 10 mg polyinosinic-polycytidylic acid (pIpC; GE Healthcare) administered every other day, beginning at approximately 6 weeks of age.
Proteasome inhibition
For proteasome inhibition experiments, mice were administered bortezomib (Cell Signaling; 1 mg/kg) diluted in 10% (v/v) DMSO via retro-orbital injection.
Flow cytometry and sorting
For flow cytometric analysis and isolation of specific hematopoietic progenitors, cells were incubated with combinations of antibodies to the following cell-surface markers, conjugated to FITC, PE, PerCP-Cy5.5, APC, PE-Cy7, eFluor 660, Alexa Fluor 700, APC-eFluor 780 or biotin (antibody clones are given in brackets in the following list): CD3ε (17A2), CD4 (GK1.5), CD5 (53–7.3), CD8α (53–6.7), CD11b (M1/70), CD16/32 (FcγRII/III; 93), CD34 (RAM34), CD43 (R2/60), CD45.1 (A20), CD45.2 (104), CD45R (B220; RA3–6B2), CD48 (HM48–1), CD71 (R17217), CD117 (cKit; 2B8), CD127 (IL7Rα; A7R34), CD150 (TC15–12F12.2), Ter119 (TER-119), Sca-1 (D7, E13–161.7), Gr-1 (RB6–8C5) and IgM (II/41). For isolation of HSCs and MPPs, Lineage markers included CD3, CD5, CD8, B220, Gr-1 and Ter119. For isolation of CMPs, GMPs and MEPs, these Lineage markers were supplemented with additional antibodies against CD4 and CD11b. Biotinylated antibodies were visualized by incubation with PE-Cy7 conjugated streptavidin. All reagents were acquired from eBiosciences, or BioLegend. All incubations were for 30–90 min on ice.
HSCs, MPPs, CD34+CD16/32lowCD127−Lineage−Sca-1−c-kit+ CMPs (Akashi et al., 2000), CD34+CD16/32highCD127−Lineage−Sca-1−c-kit+ GMPs (Akashi et al., 2000), and CD34−CD16/32−/lowCD127−Lineage−Sca-1−c-kit+ MEPs (Akashi et al., 2000) were pre-enriched by selecting c-kit+ cells using paramagnetic microbeads and an autoMACS magnetic separator (Miltenyi Biotec) before sorting.
Non-viable cells were excluded from sorts and analyses using 4’,6-diamidino-2-phenylindole (DAPI). Apoptotic cells were identified using APC annexin V (BDBiosciences). Proliferation was assessed by administering 5-bromo-2′-deoxyuridine (BrdU; Sigma) to mice. BrdU dissolved in PBS was administered with an initial intraperitoneal injection (2 mg) and then mice were maintained on drinking water containing 1 mg/ml BrdU for 72 h. BrdU incorporation in HSCs was measured using the APC BrdU Flow Kit (BD Biosciences).
For analysis of ubiquitylated protein following cell surface staining, cells were fixed in 0.5 mL of 1% paraformaldehyde (Affymetrix) in PBS for 10–15 min on ice. Cells were washed in PBS, then permeabilized in 200 mL PBS supplemented with 3% (v/v) fetal bovine serum (Life Technologies) and 0.1% (m/v) saponin (Sigma) for 5 min at room temperature (20–25°C). Cells were then incubated with anti-ubiquitylated protein antibody (clone FK2, Millipore) at a concentration of 1:500 for 30 min at room temperature. This was followed by incubation with anti-mouse Alexa Fluor 488 (Thermo Scientific) at a concentration of 1:500 for 30 min at room temperature. Cells were washed twice in PBS supplemented with 3% fetal bovine serum and 0.1% saponin, then resuspended in PBS supplemented with DAPI (4 mg/ml final concentration) and analyzed by flow cytometry.
Data acquisition and cell sorting were performed on a FACSAria II, LSR II or FACSCanto flow cytometer (BD Biosciences). Sorted fractions were typically double sorted to ensure high purity. Data were analyzed by FlowJo (TreeStar) software.
Competitive repopulation assay
Adult recipient mice (CD45.1) were administered a minimum lethal dose of radiation using a Mark I Cesium source irradiator (J.L. Sheperd) to deliver two doses of 550 rad (1,100 rad in total) at least 4 h apart. Cells were injected into the retro-orbital venous sinus of anaesthetized recipients. For competitive bone marrow transplants 5×105 donor and 5×105 recipient-type cells were transplanted. For HSC transplants 10 donor CD150+CD48−Lineage−Sca-1+c-kit+ HSCs and 2×105 recipient-type bone marrow cells were transplanted. Blood was obtained from the tail veins of recipient mice every 4 weeks for at least 16 weeks after transplantation. Red blood cells were lysed with ammonium chloride potassium buffer. The remaining cells were stained with antibodies against CD45.2, CD45.1, CD45R (B220), CD11b, CD3 and Gr-1 to assess donor-cell engraftment.
For secondary transplants, 3×106 bone marrow cells collected from primary recipients were transplanted non-competitively into irradiated recipient mice. Primary recipients used for secondary transplantation had long-term multilineage reconstitution by donor cells and median levels of donor-cell reconstitution for the treatments from which they originated. Mice that died over the course of transplantation experiments were omitted from the analyses.
For MPP transplants, 50 CD150−CD48−Lineage−Sca-1+c-kit+ MPPs and 2×105 recipient-type bone marrow cells were transplanted. Blood was obtained from the tail veins of recipient mice 3,5 and 7 weeks after transplantation.
Western blot analysis
Equal numbers of cells from each stem or progenitor population were sorted into trichloracetic acid (TCA, Sigma). The final concentration was adjusted to 10% (v/v) TCA. Extracts were incubated on ice for at least 15 min and centrifuged at 15,000 g at 4°C for 15 min. Precipitates were washed in acetone twice and dried. The pellets were solubilized in 9 M urea, 2% (v/v) Triton X-100, and 1% DTT. LDS loading buffer (Life Technologies) was added and the pellet was heated at 70°C for 10 min. Samples were separated on Bis-Trispolyacrylamide gels (Life Technologies) and transferred to PVDF membranes (Bio-Rad). Western blot analyses were performed according to the protocol from Cell Signaling Technologies. Blots were stripped with 1% SDS, 25 mM glycine (pH 2) before re-probing. The following primary and secondary antibodies were used for western blot analyses: Ubiquitin (P4D1; Cell Signaling), K48-Ubiquitin (polyclonal; Cell Signaling 4289), c-Myc (polyclonal; Cell Signaling 9402S), phos-Eif2α (S51; Cell Signaling) β-Actin (AC74; Sigma), Gapdh (14C10; Cell Signaling), HRP-linked anti-rabbit IgG (Cell Signaling) and HRP-linked anti-mouse IgG (Cell Signaling). Blots were developed with the SuperSignal West Femto or Pico PLUS chemiluminescence kit (Thermo Scientific).
Measurement of unfolded proteins
4×106 bone marrow cells were stained for cell surface markers as described above. After staining, cells were washed twice in Ca2+- and Mg2+-free PBS. Tetraphenylethene maleimide (TMI; stock 2 mM in DMSO) was diluted in PBS (50 μM final concentration), added to each sample and samples were incubated at 37°C for 45 minutes. Samples were washed twice in PBS and analyzed by flow cytometry. DAPI was omitted from these samples because of spectral overlap.
To assess the effects of heat shock, 106 BM cells were incubated Ca2+- and Mg2+-free HBSS (Corning) supplemented with 2% heat-inactivated bovine serum at 37°C or 42°C for 4 h. Cells were then washed twice in Ca2+- and Mg2+-free PBS (Corning), and TMI was added as described above.
In vivo measurement of protein synthesis
OP-Puro (Medchem Source; 50 mgkg−1 body mass; pH 6.4–6.6 in PBS) was injected intraperitoneally. One hour later mice were euthanized. Bone marrow was collected, and 4×106 cells were stained with combinations of antibodies against cell-surface markers as described above. After washing, cells were fixed in 0.5 mL of 1% paraformaldehyde (Affymetrix) in PBS for 10–15 min on ice. Cells were washed in PBS, then permeabilized in 200 mL PBS supplemented with 3% (v/v) fetal bovine serum (Life Technologies) and 0.1% (m/v) saponin (Sigma) for 5 min at room temperature (20–25°C). The azide-alkyne cycloaddition was performed using the Click-iT Cell Reaction Buffer Kit (Life Technologies) and azide conjugated to Alexa Fluor 555 (Life Technologies) at 5 mM final concentration. After the 30-min reaction, the cells were washed twice in PBS supplemented with 3% fetal bovine serum and 0.1% saponin, then resuspended in PBS supplemented with DAPI (4 mgml−1 final concentration) and analyzed by flow cytometry (Hidalgo San Jose and Signer, 2019).
In vitro analysis
Colony forming units were assessed by sorting 500 live bone marrow cells per well of a 96-well plate (24 wells per mouse) containing methylcellulose culture medium (M3434; STEMCELL Technologies). Colonies were counted after 14 days.
Cell volume
Average diameter was measured by sorting cells into flat-bottom 96-well plates and analyzing micrographs with ImageJ software. Cell volume was calculated based on the assumption that cells are spherical (4/3*π*r3).
Protein content
Equal numbers of cells from each stem or progenitor population were sorted into HBSS supplemented with 2% (v/v) heat-inactivated bovine serum (GIBCO). Samples were washed once with PBS and centrifuged at 2500 g at 4°C for 5 min. Supernatant was discarded and protein pellets were directly lysed and solubilized in 9M urea, 2% SDS, 50 mM DTT, and 50 mM Tris pH 7.4. After incubating the samples for 10–15 min at room temperature, LDS loading buffer (Life Technologies) was added and the sample was heated at 70°C for 10 minutes. Samples were separated on Bis-Trispolyacrylamide gels (Life Technologies). The Bis-Trispolyacrylamide gels were then stained with SYPRO Ruby Protein Gel Stain (Bio-Rad) for 3 h and washed in a solution containing 7% acetic acid and 10% methanol for 1 h. Gels were imaged using Gel Doc XR (Bio-Rad) and analyzed with Image Lab 6.0.1 software (Bio-Rad).
For bone marrow cells, incubated at 37°C or 42°C, 105 cells were sorted into TCA. The final concentration was adjusted to 10% TCA. Extracts were incubated on ice for at least 15 min and centrifuged at 15,000 g at 4°C for 15 min. Precipitates were washed in acetone twice and dried. The pellets were solubilized in 6 M urea, 2% (v/v) Triton X-100, and 1% (v/v) 2-mercaptoethanol. The protein content in the supernatant was assessed with the microBCA assay (Pierce).
Proteasome activity
5×103 cells from each cell population were plated into 96-well plates in triplicate. Proteasome activity was measured with the Proteasome-Glo chymotrypsin-like cell based assay (Promega). Luminescence was monitored after a 15 min incubation using a Synergy HTX multi-mode plate reader. HSCs/MPPs incubated with 100 μM MG132 were used as controls.
RNA sequencing
For RNA-seq analysis, total RNA was extracted from ~3000 HSCs using the RNeasy Plus Micro Kit (QIAGEN). Illumina mRNA libraries were prepared using the SMARTseq2 protocol (Picelli et al., 2014). 2.6 μL of total RNA was used in the SMARTSeq2 protocol (70pg to 1.62ng). 18 cycles of PCR were performed for the cDNA preamplification step and 12 cycles were performed for the tagmentation library preparation. The resulting libraries were pooled and deep sequenced in two lanes on the Illumina HiSeq 2500 in high output mode using single-end reads with lengths of 50 nucleotides (25–35M reads per condition). The single-end reads that passed Illumina filters were filtered for reads aligning to tRNA, rRNA, adaptor sequences, and spike-in controls. The reads were then aligned to the mm10 reference genome using TopHat (v 1.4.1) (Trapnell et al., 2009). DUST scores were calculated with PRINSEQ Lite (v 0.20.3) (Schmieder and Edwards, 2011) and low-complexity reads (DUST > 4) were removed from the BAM files. The alignment results were parsed via the SAMtools (Li et al., 2009) to generate SAM files. Read counts to each genomic feature were obtained with the htseq-count program (v 0.7.1) (Anders et al., 2015) using the “union” option. After removing absent features (zero counts in all samples), the raw counts were then imported to Bioconductor package DESeq2 (v 1.6.3) (Love et al., 2014) to identify differentially expressed genes among samples. P values for differential expression are calculated using the Wald test for differences between the base means of two conditions. These P values are then adjusted for multiple test correction using Benjamini Hochberg algorithm. We considered genes differentially expressed between two groups of samples when the DESeq2 analysis resulted in an adjusted P value of < 0.10 and the difference in gene expression was at least 2-fold. Principal Component Analysis (PCA) was performed using the ‘prcomp’ function in R. Heatmaps of differentially expressed genes were generated from log2(RPKM+1) values using the function heatmap.2(package gplots) in R. GSEA with classic scoring scheme was run for the geneset Biological processes (c5.bp.v6.1.symbols.gmt) from MSigDB on pre-ranked gene lists. For genes with log2FoldChange greater than zero, a rank −log10(pValue) was assigned and for genes with log2FoldChange less than zero, a rank of log10(pValue) was assigned.
Quantitative PCR
Cells were sorted directly into RLT plus buffer (QIAGEN) supplemented with 2-mercaptoethanol. RNA was extracted with the RNeasy micro plus kit (QIAGEN) and cDNA was synthesized with the RT2 First Strand Kit (QIAGEN). Reactions were run in 20 μL volumes with SYBR green and a LightCycler 480 (Roche Applied Science). Primer sequences were: Myc F, 5′- CTTCTCTCCTTCCTCGGACTC-3′; Myc R 5′- GGAGATGAGCCCGACTCCGACCTC-3′; β-Actin F, 5′-CGTCGACAACGGCTCCGGCATG-3′; β-Actin R, 5′-GGGCC TCGTCACCCACATAGGAG-3′
QUANTIFICATION AND STATISTICAL ANALYSIS
Group data are represented by mean ± standard deviation, except for transplantation data which are represented as mean ± standard error of the mean. To test statistical significance between two samples, two-tailed Student’s t tests were used. When multiple samples were compared, statistical significance was assessed using a one-way ANOVA or a repeated-measures one-way ANOVA (when comparing multiple time points or populations from the same mouse) followed by Dunnett’s test for multiple comparisons. Statistical significance comparing overall numbers of mice with long-term multilineage reconstitution was assessed by a Fisher’s exact test. Statistical tests were performed using GraphPad Prism software. The specific type of test used for each figure panel is described in the figure legends, except for the RNA sequencing data which is described in the “RNA sequencing” paragraph of the STAR Methods section above.
For normalized data, means were calculated and statistical tests were performed using log10-transformed data and then means were back-transformed to prevent data skewing. No randomization or blinding was used in any experiments. The only mice excluded from any experiment were those that died after transplantation.
DATA AND CODE AVAILABILITY
The RNA-sequencing data comparing Aarssti/sti and control HSCs is available at Gene Expression Omnibus: GSE141008.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| APC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100236 |
| FITC Anti-mouse CD3ε (17A2) | BioLegend | Cat #100204 |
| PE Anti-mouse CD3ε (17A2) | BioLegend | Cat #100206 |
| APC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100412 |
| FITC Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100406 |
| PE Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100408 |
| APC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100626 |
| FITC Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100606 |
| PE Anti-mouse CD5 (53–7.3) | BioLegend | Cat #100608 |
| APC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #17–0081–82 |
| FITC Anti-mouse CD8α (53–6.7) | eBioscience | Cat #11–0081–85 |
| PE Anti-mouse CD8α (53–6.7) | eBioscience | Cat #12–0081–83 |
| APC Anti-mouse CD11b (M1/70) | eBioscience | Cat #17–0112–82 |
| APC efluor780 Anti-mouse CD11b (M1/70) | eBioscience | Cat #47–0112–82 |
| FITC Anti-mouse CD11b (M1/70) | eBioscience | Cat #12–0112–83 |
| PE Anti-mouse CD11b (M1/70) | eBioscience | Cat #11–0112–85 |
| PE/Cy7 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101318 |
| PerCP/Cy5.5 Anti-mouse CD16/32 (FcγRII/III; 93) | BioLegend | Cat #101324 |
| Biotin Anti-mouse CD34 (RAM34) | eBioscience | Cat #13–0341–85\ |
| eFluor660 Anti-mouse CD34 (RAM34) | eBioscience | Cat #50–0341–82 |
| Alexa Fluor 700 Anti-mouse CD34 (RAM34) | eBioscience | Cat #56–0341–82 |
| FITC Anti-mouse CD34 (RAM34) | eBioscience | Cat #11–0341–85 |
| FITC Anti-mouse CD43 (R2/60) | eBioscience | Cat #11–0431–85 |
| PE Anti-mouse CD43 (R2/60) | eBioscience | Cat #12–0431–83 |
| APC eFluor 780 Anti-mouse CD45.1 (A20) | eBioscience | Cat #47–0453–82 |
| Alexa Fluor 700 Anti-mouse CD45.2 (104) | BioLegend | Cat #109822 |
| FITC Anti-mouse CD45.2 (104) | BioLegend | Cat #109806 |
| APC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #17–0452–83 |
| FITC Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #11–0452–85 |
| PE Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #12–0452–85 |
| PerCP-Cyanine5 Anti-mouse CD45R (B220) (RA3–6B2) | eBioscience | Cat #45–0452–82 |
| APC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103412 |
| FITC Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103404 |
| PE Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103406 |
| PE/Cy7 Anti-mouse CD48 (HM48–1) | BioLegend | Cat #103424 |
| FITC Anti-mouse CD71 (R17217) | eBioscience | Cat #11–0711–85 |
| APC Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #17–1171–83 |
| APC eFluor 780 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #47–1711–82 |
| PE-Cyanine7 Anti-mouse CD117 (cKit) (2B8) | eBioscience | Cat #25–1711–82 |
| PE Anti-mouse CD127 (IL7Rα; A7R34) | BioLegend | Cat #135010 |
| APC Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115910 |
| PE Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115904 |
| PE-Cyanine7 Anti-mouse CD150 (TC15–12F12.2) | BioLegend | Cat #115914 |
| APC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116212 |
| FITC Anti-mouse Ter119 (TER-119) | Biolegend | Cat #116206 |
| PE Ter119 (TER-119) | Biolegend | Cat #116208 |
| APC Anti-mouse Sca-1 (D7, E13–161.7)\ | eBioscience | Cat #17–5981–82 |
| Alexa Fluor 700 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #56–5981–82 |
| FITC Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #11–5981–85 |
| PerCp-Cyanine5.5 Anti-mouse Sca-1 (D7, E13–161.7) | eBioscience | Cat #45–5981–82 |
| APC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108412 |
| FITC Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108406 |
| PE Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108408 |
| PE/Cy7 Anti-mouse Gr-1 (RB6–8C5) | BioLegend | Cat #108416 |
| APC Anti-mouse IgM (II/41) | eBioscience | Cat #17–5790–82 |
| PE Anti-mouse IgM (II/41) | eBioscience | Cat #12–5790–83 |
| Streptavidin-PECy7 | BioLegend | Cat #405206 |
| Anti-Ubiquitin (P4D1) | Cell Signaling | Cat #3933S |
| Anti-K48-Polyubiquitin (polyclonal) | Cell Signaling | Cat# 4289S |
| Anti-c-Myc (polyclonal) | Cell Signaling | Cat# 9402S |
| Anti-phos-Eif2α (S51) | Cell Signaling | Cat #9721S |
| Anti-β-Actin (AC74) | Sigma | Cat #A2228 |
| Anti-Gapdh (14C10) | Cell Signaling | Cat #2118S |
| HRP-linked anti-rabbit IgG | Cell Signaling | Cat #7074S |
| HRP-linked anti-mouse IgG | Cell Signaling | Cat #7076S |
| Chemicals, Peptides, and Recombinant Proteins | ||
| PIPC | GE Healthcare Biosciences Corp | Cat #27473201 |
| Anti-mouse CD117 microbeads | Miltenyi | Cat #130–091–224 |
| TCA | Sigma | Cat #T6399 |
| Triton X-100 | CalBiochem | Cat #648466 |
| LDS loading buffer | Life Technologies | Cat #NP007 |
| SDS | Life Technologies | Cat #NP0060 |
| Glycine | Sigma | Cat #50046 |
| Tetraphenylethene maleimide (TMI) | Dr. Yuning Hong | Custom Synthesized |
| OP-Puro | Medchem Source | Custom Synthesized |
| SYPRO Ruby Protein Gel Stain | Bio-Rad | Cat #1703125 |
| 2-mercaptoethanol | Sigma | Cat #M3148 |
| SYBR green | Roche | Cat #4707516001 |
| Critical Commercial Assays | ||
| SuperSignal West Femto or Pico PLUS chemiluminescence kit | Thermo Scientific | Cat #34095 |
| Click-iT Cell Reaction Buffer Kit | Life Technologies | Cat #C10269 |
| Alexa Fluor 555-conjugated azide | Life Technologies | Cat #A20012 |
| Proteasome-Glo chymotrypsin-like cell based assay | Promega | Cat #G8660 |
| RNeasy Plus Micro Kit | QIAGEN | Cat #74034 |
| RT2 First Strand Kit | QIAGEN | Cat #330404 |
| Deposited Data | ||
| RNA-sequencing data (Aarssti/sti and Aars+/+ HSCs) | GEO | GSE141008 |
| Experimental Models: Organisms/Strains | ||
| Mouse: B6: C57BL/6J | The Jackson Laboratory | Cat #000664 |
| Mouse: B6SJL: B6.SJL | The Jackson Laboratory | Cat #002014 |
| Mouse: Ptenfl/fl | The Jackson Laboratory | Cat #006440 |
| Mouse: Mx1-Cre | The Jackson Laboratory | Cat #003556 |
| Mouse: Rpl24Bst/+ | The Jackson Laboratory | Cat #000516 |
| Mouse: Aarssti/sti | The Jackson Laboratory | Cat #002560 |
| Mouse: UbG76V-GFP | The Jackson Laboratory | Cat #008111 |
| Mouse: Mycfl | The Jackson Laboratory | Cat #011123 |
| Oligonucleotides | ||
| Myc forward | IDT | 5′- CTTCTCTCCTTCCTCGGACTC-3′ |
| Myc reverse | IDT | 5′- GGAGATGAGCCCGACTCCGACCTC-3′ |
| β-Actin forward | IDT | 5′-CGTCGACAACGGCTCCGGCATG-3′ |
| β-Actin reverse | IDT | 5′-GGGCCTCGTCACCCACATAGGAG-3′ |
| Software and Algorithms | ||
| FlowJo | FlowJo, LLC | N/A |
| FACSDiva | BD Bioscience | N/A |
| Prism 8 | GraphPad | N/A |
| ImageJ | NIH | N/A |
| Image Lab 6.0.1 | Bio-Rad | N/A |
| TopHat 1.4.1 | JHU CCB | N/A |
| PRINSEQ Lite 0.20.3 | N/A | N/A |
| Htseq-count 0.7.1 | N/A | N/A |
| Bioconductor package DESeq2 1.6.3 | Bioconductor | N/A |
Highlights.
HSCs have fewer misfolded and unfolded proteins than restricted myeloid progenitors in vivo
HSCs depend on low protein synthesis to maintain their elevated proteome quality
tRNA editing defect impairs HSC self-renewal by increasing genesis of misfolded proteins
Misfolded proteins overwhelm proteasome in HSCs, leading to c-Myc accumulation
ACKNOWLEDGMENTS
We would like to thank E.J. Bennett, M. Leonard, S.A. Ackerman, J.S. Gutkind, S.J. Morrison, and J.A. Magee, C. Kim and the staff at the LJI Flow Cytometry core facility, J. Day and S. Wlodychak at the LJI NGS core facility, and J. Greenbaum and A. Chawla at the LJI Bioinformatics core facility. This work was supported by the NIH/NIDDK (R01DK116951), Scholar Awards from the V Foundation for Cancer Research (V2017–003) and the American Society of Hematology, the American Cancer Society Institutional Research Grant (IRG-15–172-45-IRG), the Sanford Stem Cell Clinical Center, and the UCSD Moores Cancer Center, which is supported by the NIH/NCI Specialized Cancer Center Support Grant (2P30CA023100–33). The LJI Flow Cytometry core facility is supported by the NIH Shared Instrumentation Grant Program (S10 RR027366). L.H.S.J. was supported by a Chancellor’s Research Excellence Scholarship.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing financial interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.003.
REFERENCES
- Abbas HA, Maccio DR, Coskun S, Jackson JG, Hazen AL, Sills TM, You MJ, Hirschi KK, and Lozano G (2010). Mdm2 is required for survival of hematopoietic stem cells/progenitors via dampening of ROS-induced p53 activity. Cell Stem Cell 7, 606–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akashi K, Traver D, Miyamoto T, and Weissman IL (2000). A clonogenic common myeloid progenitor that gives rise to all myeloid lineages. Nature 404, 193–197. [DOI] [PubMed] [Google Scholar]
- Anders S, Pyl PT, and Huber W (2015). HTSeq—a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bence NF, Sampat RM, and Kopito RR (2001). Impairment of the ubiquitin-proteasome system by protein aggregation. Science 292, 1552–1555. [DOI] [PubMed] [Google Scholar]
- Blanco S, Bandiera R, Popis M, Hussain S, Lombard P, Aleksic J, Sajini A, Tanna H, Cortés-Garrido R, Gkatza N, et al. (2016). Stem cell function and stress response are controlled by protein synthesis. Nature 534, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapple RH, Hu T, Tseng YJ, Liu L, Kitano A, Luu V, Hoegenauer KA, Iwawaki T, Li Q, and Nakada D (2018). ERα promotes murine hematopoietic regeneration through the Ire1α-mediated unfolded protein response. eLife 7, e31159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MZ, Moily NS, Bridgford JL, Wood RJ, Radwan M, Smith TA, Song Z, Tang BZ, Tilley L, Xu X, et al. (2017). A thiol probe for measuring unfolded protein load and proteostasis in cells. Nat. Commun. 8, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Alboran IM, O’Hagan RC, Gärtner F, Malynn B, Davidson L, Rickert R, Rajewsky K, DePinho RA, and Alt FW (2001). Analysis of C-MYC function in normal cells via conditional gene-targeted mutation. Immunity 14, 45–55. [DOI] [PubMed] [Google Scholar]
- Drummond DA, and Wilke CO (2009). The evolutionary consequences of erroneous protein synthesis. Nat. Rev. Genet. 10, 715–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duttler S, Pechmann S, and Frydman J (2013). Principles of cotranslational ubiquitination and quality control at the ribosome. Mol. Cell 50, 379–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, et al. (2016). Autophagy maintains stemness by preventing senescence. Nature 529, 37–42. [DOI] [PubMed] [Google Scholar]
- Goncalves KA, Silberstein L, Li S, Severe N, Hu MG, Yang H, Scadden DT, and Hu GF (2016). Angiogenin Promotes Hematopoietic Regeneration by Dichotomously Regulating Quiescence of Stem and Progenitor Cells. Cell 166, 894–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groszer M, Erickson R, Scripture-Adams DD, Dougherty JD, Le Belle J, Zack JA, Geschwind DH, Liu X, Kornblum HI, and Wu H (2006). PTEN negatively regulates neural stem cell self-renewal by modulating G0-G1 cell cycle entry. Proc. Natl. Acad. Sci. USA 103, 111–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harding HP, Zhang Y, and Ron D (1999). Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature 397, 271–274. [DOI] [PubMed] [Google Scholar]
- San Jose Lorena Hidalgo, and Signer RAJ (2019). Cell-type-specific quantification of protein synthesis in vivo. Nat. Protoc. 14, 441–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegué E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hofmann JW, Zhao X, De Cecco M, Peterson AL, Pagliaroli L, Manivannan J, Hubbard GB, Ikeno Y, Zhang Y, Feng B, et al. (2015). Reduced expression of MYC increases longevity and enhances healthspan. Cell 160, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Yilmaz OH, Terhorst C, and Morrison SJ (2005). SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell 121, 1109–1121. [DOI] [PubMed] [Google Scholar]
- Kim W, Bennett EJ, Huttlin EL, Guo A, Li J, Possemato A, Sowa ME, Rad R, Rush J, Comb MJ, et al. (2011). Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kühn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Labbadia J, and Morimoto RI (2015). The biology of proteostasis in aging and disease. Annu. Rev. Biochem. 84, 435–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cook SA, Davisson MT, Sundberg JP, Schimmel P, and Ackerman SL (2006). Editing-defective tRNA synthetase causes protein misfolding and neurodegeneration. Nature 443, 50–55. [DOI] [PubMed] [Google Scholar]
- Lee JY, Nakada D, Yilmaz OH, Tothova Z, Joseph NM, Lim MS, Gilliland DG, and Morrison SJ (2010). mTOR activation induces tumor suppressors that inhibit leukemogenesis and deplete hematopoietic stem cells after Pten deletion. Cell Stem Cell 7, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leeman DS, Hebestreit K, Ruetz T, Webb AE, McKay A, Pollina EA, Dulken BW, Zhao X, Yeo RW, Ho TT, et al. (2018). Lysosome activation clears aggregates and enhances quiescent neural stem cell activation during aging. Science 359, 1277–1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The Sequence Alignment/Map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindsten K, Menéndez-Benito V, Masucci MG, and Dantuma NP (2003). A transgenic mouse model of the ubiquitin/proteasome system. Nat. Biotechnol. 21, 897–902. [DOI] [PubMed] [Google Scholar]
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, and Guan JL (2010). FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 116, 4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L, Zhao M, Jin X, Ney G, Yang KB, Peng F, Cao J, Iwawaki T, Del Valle J, Chen X, and Li Q (2019). Adaptive endoplasmic reticulum stress signalling via IRE1α-XBP1 preserves self-renewal of haematopoietic and pre-leukaemic stem cells. Nat. Cell Biol. 21, 328–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens-Bobadilla E, Zhao S, Baser A, Saiz-Castro G, Zwadlo K, and Martin-Villalba A (2015). Single-Cell Transcriptomics Reveals a Population of Dormant Neural Stem Cells that Become Activated upon Brain Injury. Cell Stem Cell 17, 329–340. [DOI] [PubMed] [Google Scholar]
- López-Otín C, Blasco MA, Partridge L, Serrano M, and Kroemer G (2013). The hallmarks of aging. Cell 153, 1194–1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lykke-Andersen J, and Bennett EJ (2014). Protecting the proteome: Eukaryotic cotranslational quality control pathways. J. Cell Biol. 204, 467–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marino SM, and Gladyshev VN (2010). Cysteine function governs its conservation and degeneration and restricts its utilization on protein surfaces. J. Mol. Biol. 404, 902–916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markmiller S, Fulzele A, Higgins R, Leonard M, Yeo GW, and Bennett EJ (2019). Active Protein Neddylation or Ubiquitylation Is Dispensable for Stress Granule Dynamics. Cell Rep. 27, 1356–1363.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka S, Oike Y, Onoyama I, Iwama A, Arai F, Takubo K, Mashimo Y, Oguro H, Nitta E, Ito K, et al. (2008). Fbxw7 acts as a critical fail-safe against premature loss of hematopoietic stem cells and development of T-ALL. Genes Dev. 22, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moran-Crusio K, Reavie LB, and Aifantis I (2012). Regulation of hematopoietic stem cell fate by the ubiquitin proteasome system. Trends Immunol. 33, 357–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto RI (2011). The heat shock response: systems biology of proteotoxic stress in aging and disease. Cold Spring Harb. Symp. Quant. Biol. 76, 91–99. [DOI] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, Sadighi- Akha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE, et al. (2011). The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med. 208, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguro H, Ding L, and Morrison SJ (2013). SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell 13, 102–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver ER, Saunders TL, Tarlé SA, and Glaser T (2004). Ribosomal protein L24 defect in belly spot and tail (Bst), a mouse Minute. Development 131, 3907–3920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picelli S, Faridani OR, Björklund AK, Winberg G, Sagasser S, and Sandberg R (2014). Full-length RNA-seq from single cells using Smart-seq2. Nat. Protoc. 9, 171–181. [DOI] [PubMed] [Google Scholar]
- Reavie L, Della Gatta G, Crusio K, Aranda-Orgilles B, Buckley SM, Thompson B, Lee E, Gao J, Bredemeyer AL, Helmink BA, et al. (2010). Regulation of hematopoietic stem cell differentiation by a single ubiquitin ligase-substrate complex. Nat. Immunol. 11, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez CG, Teixeira FK, Czech B, Preall JB, Zamparini AL, Seifert JR, Malone CD, Hannon GJ, and Lehmann R (2016). Regulation of Ribosome Biogenesis and Protein Synthesis Controls Germline Stem Cell Differentiation. Cell Stem Cell 18, 276–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmieder R, and Edwards R (2011). Quality control and preprocessing of metagenomic datasets. Bioinformatics 27, 863–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schubert U, Antón LC, Gibbs J, Norbury CC, Yewdell JW, and Bennink JR (2000). Rapid degradation of a large fraction of newly synthesized proteins by proteasomes. Nature 404, 770–774. [DOI] [PubMed] [Google Scholar]
- Sherman MY, and Qian SB (2013). Less is more: improving proteostasis by translation slow down. Trends Biochem. Sci. 38, 585–591. [DOI] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signer RA, Qi L, Zhao Z, Thompson D, Sigova AA, Fan ZP, DeMartino GN, Young RA, Sonenberg N, and Morrison SJ (2016). The rate of protein synthesis in hematopoietic stem cells is limited partly by 4E-BPs. Genes Dev. 30, 1698–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sigurdsson V, Takei H, Soboleva S, Radulovic V, Galeev R, Siva K, Leeb-Lundberg LM, Iida T, Nittono H, and Miharada K (2016). Bile Acids Protect Expanding Hematopoietic Stem Cells from Unfolded Protein Stress in Fetal Liver. Cell Stem Cell 18, 522–532. [DOI] [PubMed] [Google Scholar]
- Sontag EM, Vonk WIM, and Frydman J (2014). Sorting out the trash: the spatial nature of eukaryotic protein quality control. Curr. Opin. Cell Biol. 26, 139–146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor RC, and Dillin A (2011). Aging as an event of proteostasis collapse. Cold Spring Harb. Perspect. Biol. 3, a004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson BJ, Jankovic V, Gao J, Buonamici S, Vest A, Lee JM, Zavadil J, Nimer SD, and Aifantis I (2008). Control of hematopoietic stem cell quiescence by the E3 ubiquitin ligase Fbw7. J. Exp. Med. 205, 1395–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell C, Pachter L, and Salzberg SL (2009). TopHat: discovering splice junctions with RNA-Seq. Bioinformatics 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Galen P, Kreso A, Mbong N, Kent DG, Fitzmaurice T, Chambers JE, Xie S, Laurenti E, Hermans K, Eppert K, et al. (2014). The unfolded protein response governs integrity of the haematopoietic stem-cell pool during stress. Nature 510, 268–272. [DOI] [PubMed] [Google Scholar]
- van Galen P, Mbong N, Kreso A, Schoof EM, Wagenblast E, Ng SWK, Krivdova G, Jin L, Nakauchi H, and Dick JE (2018). Integrated Stress Response Activity Marks Stem Cells in Normal Hematopoiesis and Leukemia. Cell Rep. 25, 1109–1117.e5. [DOI] [PubMed] [Google Scholar]
- Vilchez D, Boyer L, Morantte I, Lutz M, Merkwirth C, Joyce D, Spencer B, Page L, Masliah E, Berggren WT, et al. (2012). Increased proteasome activity in human embryonic stem cells is regulated by PSMD11. Nature 489, 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walter P, and Ron D (2011). The unfolded protein response: from stress pathway to homeostatic regulation. Science 334, 1081–1086. [DOI] [PubMed] [Google Scholar]
- Wang F, Durfee LA, and Huibregtse JM (2013). A cotranslational ubiquitination pathway for quality control of misfolded proteins. Mol. Cell 50, 368–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warr MR, Binnewies M, Flach J, Reynaud D, Garg T, Malhotra R, Debnath J, and Passegué E (2013). FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 494, 323–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Murphy MJ, Oskarsson T, Kaloulis K, Bettess MD, Oser GM, Pasche AC, Knabenhans C, Macdonald HR, and Trumpp A (2004). c-Myc controls the balance between hematopoietic stem cell self-renewal and differentiation. Genes Dev. 18, 2747–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zismanov V, Chichkov V, Colangelo V, Jamet S, Wang S, Syme A, Koromilas AE, and Crist C (2016). Phosphorylation of eIF2α Is a Translational Control Mechanism Regulating Muscle Stem Cell Quiescence and Self-Renewal. Cell Stem Cell 18, 79–90. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The RNA-sequencing data comparing Aarssti/sti and control HSCs is available at Gene Expression Omnibus: GSE141008.