

Abstract
FMS‐like tyrosine kinase 3 (FLT3) is a type III receptor tyrosine kinase that plays an important role in hematopoietic cell survival, proliferation and differentiation. The most clinically important point is that mutation of the FLT3 gene is the most frequent genetic alteration and a poor prognostic factor in acute myeloid leukemia (AML) patients. There are two major types of FLT3 mutations: internal tandem duplication mutations in the juxtamembrane domain (FLT3‐ITD) and point mutations or deletion in the tyrosine kinase domain (FLT3‐TKD). Both mutant FLT3 molecules are activated through ligand‐independent dimerization and trans‐phosphorylation. Mutant FLT3 induces the activation of multiple intracellular signaling pathways, mainly STAT5, MAPK and AKT signals, leading to cell proliferation and anti–apoptosis. Because high‐dose chemotherapy and allogeneic hematopoietic stem cell transplantation cannot sufficiently improve the prognosis, clinical development of FLT3 kinase inhibitors expected. Although several FLT3 inhibitors have been developed, it takes more than 20 years from the first identification of FLT3 mutations until FLT3 inhibitors become clinically available for AML patients with FLT3 mutations. To date, three FLT3 inhibitors have been clinically approved as monotherapy or combination therapy with conventional chemotherapeutic agents in Japan and/or Europe and United states. However, several mechanisms of resistance to FLT3 inhibitors have already become apparent during their clinical trials. The resistance mechanisms are complex and emerging resistant clones are heterogenous. Further basic and clinical studies are required to establish the best therapeutic strategy for AML patients with FLT3 mutations.
Keywords: acute myeloid leukemia, FMS‐like tyrosine kinase, inhibitor, resistance, tyrosine kinase
Several mechanisms of resistance to FLT3 inhibitors have already become apparent during their clinical trials. The resistance mechanisms are complex and emerging resistant clones are heterogenous.
1. INTRODUCTION
FMS‐like tyrosine kinase 3 (FLT3) is a type III receptor tyrosine kinase (RTK) together with KIT, FMS and PDGFR.1, 2, 3 FLT3 consists of five immunoglobulin‐like domains in the extracellular region, a juxtamembrane (JM) domain, a tyrosine kinase (TK) domain separated by a kinase insert domain, and a C‐terminal domain in the intracellular region. FLT3 is expressed on normal hematopoietic stem/progenitor cells, and its ligand (FL) is expressed as a membrane‐bound or soluble form by bone marrow stroma cells.4, 5 FLT3 is dimerized by the binding of FL to its extracellular domain, and tyrosine residues in the activation‐loop (A‐loop) are subsequently trans‐phosphorylated. Activated FLT3 induces multiple intracellular signaling pathways, leading to hematopoietic cell survival, proliferation and differentiation.6 FLT3 is also expressed on most acute leukemia cells, and FL stimulation enhances proliferation and reduces apoptosis.7, 8 In 1996, an internal tandem duplication in the JM domain‐coding sequence of the FLT3 gene (FLT3‐ITD) was first identified in acute myeloid leukemia (AML) cells.9 Subsequently, we found a missense point mutation at the D835 residue and point mutations, deletions and insertions in the codons surrounding D835 within a TK domain of FLT3 (FLT3‐TKD).10 FLT3‐ITD and FLT3‐TKD occur in approximately 20 and 10% of AML, respectively.11, 12, 13 Because FLT3 mutation is the most frequent gene mutation in the protein‐coding regions and is associated with a poor prognosis, mutant FLT3 serves as a promising molecular target for the treatment of AML.14, 15 More than 20 years after the discovery of the FLT3 gene mutation, FLT3 inhibitors have been approved for clinical use, leading to therapeutic paradigms for AML with FLT3 mutations (Figure 1). In this review, we summarize the clinical and biological significance of FLT3 mutations, and discuss future therapeutic strategies involving FLT3 inhibitors.
Figure 1.
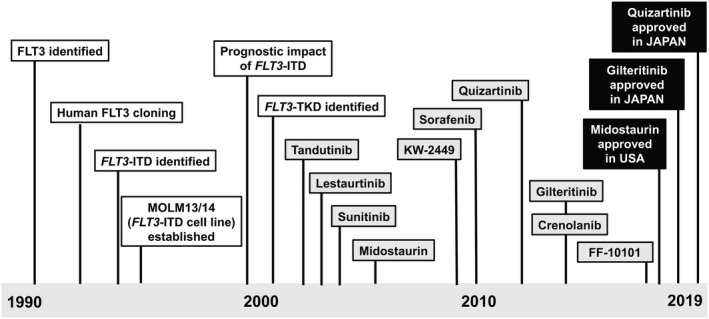
History to practical use of FLT3 inhibitors. The main historical events up to the practical use of FLT3 inhibitors are demonstrated. Indicated points of FLT3 inhibitors are the start times of clinical trials
2. CLINICAL SIGNIFICANCE OF FLT3 MUTATIONS
To date, genetic alterations in AML have been almost completely identified by the next generation sequencing. Several comprehensive genetic studies have revealed that mutations in FLT3, NPM1 and DNMT3A genes are frequently identified in AML patients. Although their frequencies varied slightly between analyzed cohorts, FLT3 mutation was identified in approximately 30% of patients with AML.13, 16, 17 In Japanese adult AML patients registered to the Japan Adult Leukemia Study Group (JALSG) AML201 study, FLT3 was the most frequently (25.4%) identified mutation (Figure 2A).12 Many clinical studies have revealed clinical characteristics of AML with FLT3 mutations.11, 18, 19, 20 FLT3 mutations are mainly found in myeloid neoplasms such as AML and myelodysplastic syndromes (MDS). In MDS, both FLT3‐ITD and FLT3‐TKD are infrequent at a level of approximately 3%, while the frequency increases in advanced‐stage MDS; approximately 15% of patients with AML developing from MDS harbor FLT3 mutations. FLT3‐ITD is far less common in patients with acute lymphocytic leukemia (ALL), while FLT3‐TKD is recurrently found in patients with a KMT2A gene rearrangement or hyperdiploidy.21, 22 FLT3 mutations may be associated with the age of patients with AML. FLT3‐ITD is found in approximately 25% of adult patients but in more than 30% of patients over 55 years of age.23, 24 In contrast, it is found in only approximately 10% of pediatric patients and in less than 5% of infant AML patients under 1 year of age.25
Figure 2.
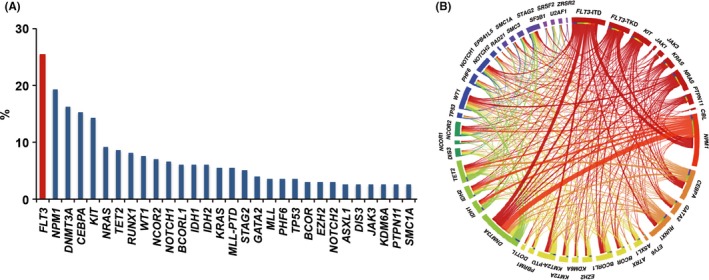
The frequency of FLT3 mutations and co–occurring mutations in acute myeloid leukemia (AML) patients. The frequency of FLT3 mutations and co–occurring mutations in 199 AML patients who were registered in the Japan Adult Leukemia Study Group (JALSG) AML201 study. FLT3 mutation is the most frequently identified in AML patients (A), and frequently co–occurs with NPM1, DNMT3A, IDH1/2, TET2, GATA2 and KMT2A‐partial tandem duplication mutations (B). Figures are adopted from the reference 12
FLT3 mutations are associated with specific cytogenetics or other genetic mutations. FLT3 mutations are frequently found in cytogenetically normal AML (CN‐AML) but are infrequent in AML with altered karyotypes; however, AML with DEK‐NUP214 and PML‐RARA (acute promyelocytic leukemia, APL) frequently harbors FLT3 mutations.11 FLT3‐ITD is particularly associated with a variant morphology of APL and the presence of the short breakpoint cluster region 3 PML‐RARA isoforms.26, 27 In addition, FLT3 mutations are infrequent in core‐binding factor AML (CBF‐AML) consisting of AML with RUNX1‐RUNX1T1 and CBFB‐MYH11, although FLT3‐TKD is frequently found in AML with CBFB‐MYH11.28, 29 FLT3 mutations frequently overlap with NPM1, DNMT3A and KMT2A partial tandem duplication (KMT2A‐PTD) mutations but are mutually exclusive with KIT, K/NRAS and CEBPA‐double (CEBPA‐D) mutations (Figure 2B).12, 13 The co–occurrences of FLT3 mutations and other cytogenetic and genetic alterations reflect the concept that AML is the consequence of two broad complementation classes of mutations: those that confer a proliferative and/or survival advantage to hematopoietic progenitors including activating mutations in tyrosine kinases, such as KIT and FLT3 or their downstream effectors such as RAS, and those that impair hematopoietic differentiation and confer properties of self‐renewal, including PML‐RARA, RUNX1‐RUNX1T1 and CBFB‐MYH11 or NPM1 and DNMT3A mutations.30, 31, 32
FLT3 mutation is strongly associated with leukocytosis and an increased percentage of blast cells in the peripheral blood and bone marrow of AML patients.10, 33 Several large‐scale studies demonstrated that FLT3‐ITD is an independent poor prognostic factor for overall survival (OS), relapse‐free survival (RFS) and event‐free survival (EFS) in patients with AML excluding APL. Although the clinical impact of FLT3‐TKD on the long‐term outcome is controversial, our meta‐analysis of four published studies including 1160 adult patients with AML indicated its adverse effects.34
Because FLT3 mutations are closely associated with a poor prognosis in patients with AML, the WHO classification and the guidelines of the European LeukemiaNet (ELN) and National Comprehensive Cancer Network (NCCN) recommend that FLT3 mutations should be analyzed for stratifying patients into distinct risk groups at the diagnosis of AML.15 The ELN first recommended a risk classification system based on the cytogenetic and genetic status in 2010 (ELN‐2010).35 In this system, CN‐AML patients with mutated NPM1 and wild‐type FLT3 were categorized into a favorable risk group, and those with FLT3‐ITD were classified into an intermediate‐II group. Recently, ELN revised the risk stratification system for AML (ELN‐2017), in which the allelic ratio (AR) of FLT3‐ITD is considered for risk stratification because several groups demonstrated that high FLT3‐ITD AR (≥0.5) (FLT3‐ITDhigh) is associated with a poor prognosis but not low FLT3‐ITD AR (<0.5) (FLT3‐ITDlow).15 Therefore, the ELN‐2017 system includes CN‐AML patients with mutated NPM1 and FLT3‐ITDlow in addition to those with mutated NPM1 and wild‐type FLT3. However, we and others reported that FLT3‐ITDlow has an impact on the long‐term prognosis, as for FLT3‐ITDhigh, in Japanese AML patients.36, 37
Although it has been reported that high‐dose daunorubicin and gemtuzumab ozogamicin might reduce the adverse effect of FLT3 mutations, a retrospective analysis showed that allogeneic hematopoietic stem cell transplantation (allo‐HSCT) did not improve the poor prognosis of patients with FLT3 mutations.38, 39 Therefore, novel treatment paradigms including FLT3 inhibitors for patients with FLT3 mutations should be further evaluated.
3. BIOLOGICAL EFFECTS OF FLT3 MUTATIONS ON ACUTE MYELOID LEUKEMIA
Wild‐type (Wt)‐FLT3 is activated through ligand‐dependent dimerization and trans‐phosphorylation. Activated Wt‐FLT3 induces activations of multiple intracellular signaling pathways, mainly MAPK and AKT signals, leading to cell proliferation and anti–apoptosis. Mutant FLT3 ligand independently forms a dimer, resulting in constitutive activation.40, 41 Notably, mutant FLT3 activates STAT5 in addition to MAPK and AKT signals.40, 42 In vitro, it was demonstrated that the constitutively active mutant FLT3 kinase induces autonomous cell proliferation to cytokine‐dependent cell lines, such as Ba/F3, FDCP1 and 32D cells.23 Importantly, mice transplanted with mutant FLT3‐transfected hematopoietic stem cells develop oligoclonal myeloproliferative disorder (MPD) but not clonal AML.43 However, when mutant FLT3‐transfected PML‐RARA‐expressing or dnmt3a‐null hematopoietic stem cells are transplanted, the mice rapidly develop AML.31, 32 These results collectively indicate that mutant FLT3 is sufficient to induce MPD, but additional mutations that impair hematopoietic differentiation and/or proliferation are necessary for the development of clonal AML.
4. FIRST‐GENERATION FLT3 INHIBITORS
Because FLT3 mutation is the most frequent genetic alteration associated with a poor prognosis in AML patients, mutant FLT3 serves as a promising molecular target for the treatment of AML patients with FLT3 mutations. At first, tyrosine kinase inhibitors (TKI), which have a potency to inhibit FLT3 kinase, were subjected to clinical trials for evaluating efficacy and safety; these TKI are called first‐generation FLT3 inhibitors, including tandutinib, sunitinib, midostaurin, lestaurtinib and sorafenib (Table 1).44, 45, 46, 47, 48 However, the clinical efficacy as a monotherapy of the first‐generation FLT3 inhibitors for AML was unimpressive in early phase studies because of low clinical efficacy and adverse events.14 Furthermore, these studies revealed that maintaining an effective plasma concentration of inhibitors is essential for achieving clinical efficacy.
Table 1.
FLT3 inhibitors in clinical development
| Tandutinib | Lestaurtinib | Sorafenib | Midostaurin | Quizartinib | Gilteritinib | Crenolanib | FF‐10101 | |
|---|---|---|---|---|---|---|---|---|
| FLT3 kinase inhibition IC50 (nmol/L) | 220 | 3 | 58 | 6.3 | 1.6 | 0.29 | 1.3 | 0.2 |
| Plasma/Medium ratios of GI50 values in FLT3‐ITD‐expressing‐cells | Unknown | 350 | 88.3 | 300 | 18 | 22.8 | 20.5 | 26.2 |
| Growth inhibition to Wt‐FLT3 and ITD‐FLT3‐co–expressing‐32D GI50 (nmol/L) | ||||||||
| FL (0 ng/mL) | Unknown | 9.7 | 5.3 | 20.3 | 1.4 | 8.9 | 23.4 | 1.4 |
| FL (10 ng/mL) | Unknown | 9.2 | 16.0 | 22.7 | 6.5 | 36.4 | 43.5 | 7.5 |
| Inhibitory type | Type II | Type I | Type II | Type I | Type II | Type I | Type I | Type I (Covalent) |
| Other targets |
PDGFR KIT |
TRKA PKC KDR PDGFR |
RAF VEGFR PDGFR KIT |
PKC KDR KIT PDGFR |
KIT |
AXL LTK ALK |
KIT PDGFR RET |
FMS |
| Development stage for AML | Withdrawn | Phase 2 | Phase 3 | Approved as combination with chemo for AML | Approved as single agent for R/R AML | Approved as single agent for R/R AML | Phase 3 | Phase 1/2a |
Blood concentration is widely used as a target indicator of the pharmacological effects of drugs. However, it does not reflect the clinical efficacy of FLT3 inhibitors because most of the inhibitors are bound to plasma proteins, resulting in a decrease of free compounds that actually exert a pharmacological action. As shown in the Table 1, GI50 values of FLT3 inhibitors in human plasma are higher than in culture medium. In particular, GI50 values of lestaurtinib and midostaurin in human plasma were more than 300 times higher than those in culture medium.
The plasma inhibitory activity (PIA) assay has been established as a novel index for evaluating the clinical efficacy of FLT3 inhibitors, replacing the blood concentration, and has been incorporated into the evaluation criteria in clinical trials.49 In the PIA assay, mutant FLT3‐expressing cells are cultured with plasma obtained from a drug‐administered patient, and then the phosphorylation status of the mutant FLT3 is evaluated by western blot analysis. In general, the phosphorylation statuses are compared between the culture with the plasma before drug administration and the trough plasma after administration. Previous clinical trials showed that clinical efficacies of FLT3 inhibitors were achieved when the phosphorylation of mutant FLT3 was completely suppressed even in the trough plasma by the PIA assay. The PIA assay clearly showed that the first‐generation FLT3 inhibitors did not sufficiently suppress the phosphorylation of mutant FLT3 even at the target blood concentration. In particular, because lestaurtinib and midostaurin consist of an indolocarbazole molecule, which is known to be tightly bound to acid‐‐glycoprotein (AGP) in human plasma, the PIA assay demonstrated that the plasma concentration of these compounds could not reach a biologically effective level.49, 50
Due to the pharmacological and clinical disadvantages, the clinical studies of first‐generation FLT3 inhibitors as monotherapy for AML were discontinued. Midostaurin, lestaurtinib and sorafenib were, therefore, evaluated in combination with chemotherapies for AML with FLT3 mutations. Although the addition of lestaurtinib or sorafenib to conventional chemotherapy did not show benefits in clinical trials, a randomized phase 3 study (RATIFY study) demonstrated the superiority of midostaurin in addition to the conventional induction and consolidation chemotherapies for overall survival (OS).50, 51, 52 Based on the results of this study, midostaurin was approved as a combination agent with standard chemotherapy by the US Food and Drug Administration (FDA) in 2017. Of interest is the fact that sorafenib maintenance therapy significantly improved RFS and OS after allo‐HSCT in AML patients with FLT3‐ITD in CR (SORMAIN study).53 Notably, this result is supported by a report that sorafenib may induce graft versus host disease (GVHD) as a result of increased IL‐15 and donor cytotoxic T‐cell activity.54
5. SECOND‐GENERATION FLT3 INHIBITORS
Because first‐generation FLT3 inhibitors were not originally screened for sensitivity and selectivity against activated FLT3 kinase, the development of second‐generation FLT3 inhibitors, which have more selective and potent inhibitory activities, continues (Table 1).
Gilteritinib is a highly selective and potent FLT3 inhibitor, and it also shows inhibitory activity against the receptor tyrosine kinase AXL.55, 56 AXL was shown to be upregulated and activated in malignant cells, including AML, and its high expression was reported to be associated with a poor prognosis.57 Furthermore, it was reported that FLT3 inhibitors induce AXL activation, reducing their growth inhibitory effects in vitro.58 In a phase 1/2 trial of 252 patients with relapsed or refractory (R/R) AML over 18 years of age, safety and efficacy were examined at doses from 20 to 450 mg/day.59 The PIA assay revealed that >90% suppression of FLT3 phosphorylation was achieved at >80mg/day. In an interim analysis of a phase 3 trial for R/R AML with FLT3 mutations (ADMIRAL study), gilteritinib showed superior complete remission (CR) and CR with partial recovery of blood cells (CRh) than conventional chemotherapy. Furthermore, the subsequent analysis for the primary endpoint showed that the median OS of gilteritinib‐treated patients (9.3 months) was significantly longer than that of conventional chemotherapy‐treated patients (5.6 months) (P < .001).60 Gilteritinib was approved for R/R AML patients with FLT3 mutations as the first single agent FLT3 inhibitor in Japan in 2018.
Quizartinib was screened for selectivity and affinity against FLT3 kinase using the KinomeScan technique.61 Quizartinib shows high selectivity for and strong inhibitory activity against FLT3‐ITD but not FLT3‐TKD.62 In phase 1 and 2 clinical trials involving R/R AML patients, quizartinib showed a >50% response rate for FLT3‐ITD‐positive AML patients; however, many CR with incomplete hematologic recovery (CRi) cases were observed in the patients who responded, and QTc prolongation was demonstrated to have a marked toxicity.63, 64 It was suggested that poor blood cell recovery after treatment with quizartinib is caused by its inhibitory activity against KIT.65 However, in a phase 3 trial for R/R FLT3‐ITD‐positive AML (QuANTUM‐R study), in which an amendment of the administration dose reduced the frequency of QTc prolongation, the median OS of quizartinib‐treated patients (6.2 months) was significantly longer than that of conventional chemotherapy‐treated patients (4.7 months) (P = .02).66 Quizartinib was approved for R/R AML patients with FLT3‐ITD in Japan in 2019.
Crenolanib has inhibitory activity both against FLT3‐ITD and FLT3‐TKD. It also has inhibitory activity against PDGFR.67, 68 A single agent phase 2 trial involving FLT3 mutation‐positive R/R AML patients showed a response rate of 47%, but digestive toxicity was revealed.69 A phase 3 study of crenolanib versus midostaurin administered following induction chemotherapy and consolidation therapy in newly diagnosed AML patients with FLT3 mutation is currently ongoing.
6. PRIMARY RESISTANCE MECHANISMS OF FLT3 INHIBITORS
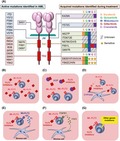
Although three FLT3 inhibitors have been approved for clinical use in Japan and/or Europe and United states, several resistance mechanisms of FLT3 inhibitors have become apparent in clinical studies. These resistance mechanisms are classified into primary and secondary resistance mechanisms. Primary resistance includes different inhibitory activity against types of FLT3 mutations (FLT3‐ITD and FLT3‐TKD), FL‐dependent impedance, FGF2‐promoting resistance, bone marrow stromal CYP3A‐mediated resistance and other activating signals (Figure 3).70, 71, 72, 73, 74
Figure 3.

Resistant mechanism of FLT3 inhibitors. A, On–target resistance of FLT3 inhibitors. To date, several activating FLT3 mutations have been identified in acute myeloid leukemia (AML) cells. During the treatment with FLT3 inhibitors, additional mutations in the FLT3 gene are acquired. Although the potencies of FLT3 inhibitors against each acquired mutation are different, those against the gatekeeper mutation, F691L, are low. B, In the culture medium, most FLT3 inhibitors exist as free‐forms, and inhibit the proliferation of mutant FLT3‐expressing cells. C, Binding to plasma proteins, such as AGP, reduce the free‐inhibitor concentration in blood. The bone marrow microenvironment is associated with a primary resistant mechanism. D, FL reduces the inhibitory activity of FLT3 inhibitors through the activation of Wt‐FLT3. E, FGF2 reduces the inhibitory activity of FLT3 inhibitors through the activation of FGFR1. F, CYP3A4 expressed in bone marrow stromal cells metabolizes FLT3 inhibitors. G, Other gene mutations, particularly RAS/MAPK pathway gene mutations, confer resistance during treatment with FLT3 inhibitors. AGP, acid‐α‐glycoprotein; BM, bone marrow; EC, extra‐cellular; FL, FLT3 ligand; JM, juxtamembrane; PB, peripheral blood; TK, tyrosine kinase
6.1. Type I and type II inhibitors
Different inhibitory activities between FLT3‐ITD and FLT3‐TKD are caused by the binding affinity of FLT3 inhibitors. FLT3 has an active or inactive conformation according to the orientation of three amino acid residues, Asp‐Phe‐Gly, in the activation loop (A‐loop). FLT3 inhibitors can be divided into two major types (type I and type II) according to the mode of binding to FLT3. The type I inhibitor binds to both active and inactive conformations of FLT3, but the type II inhibitor can bind to only an inactive conformation (Figure 4).75 Midostaurin, gilteritinib and crenolanib are type I inhibitors, and are designed to have a high affinity for the ATP‐binding region of the active conformation of FLT3. Because the protein structure of the ATP‐binding region is similar between kinases, many type I inhibitors show potency against multiple kinases. Quizartinib and sorafenib are type II inhibitors. They fit into the back pocket of the ATP‐binding region maintained in the inactive conformation and interact with the side chains of related amino acid residues, improving inhibitory activity and selectivity. However, they lose binding affinity to an active conformation of FLT3 because of the use of the back pocket in addition to the ATP‐binding region for the interaction with FLT3.
Figure 4.

Binding modes of FLT3 inhibitors to FLT3 molecule. Schematic diagrams of FLT3 kinase inhibition are shown. FLT3 inhibitors are classified into Type I and Type II inhibitors according to the mode of binding to FLT3. The Type I inhibitors bind to only the ATP‐binding site, enabling binding to both active and inactive conformations of FLT3. In contrast, because the type II inhibitors are designed to favorably bind to the back pocket of the inactive conformation of FLT3 to increase the affinity for the ATP‐binding site, they can bind to only the inactive conformation. FF‐10101 is designed to form a covalent binding between the C695 residue of FLT3. This covalent bond formation of FF‐10101 maintains the ability to bind to both the active and inactive conformations of FLT3
6.2. FL‐dependent resistance
FL‐dependent impedance of an FLT3 inhibitor is a clinically important resistance mechanism. It was reported that an increased plasma FL concentration after chemotherapy impeded the efficacies of FLT3 inhibitors.50, 71 We focused on evidence that most FLT3‐mutated AML cells co–express Wt‐FLT3, and demonstrated that FL‐dependent Wt‐FLT3 activation reduced inhibitory effects of FLT3 inhibitors.76 We evaluated the growth‐inhibitory effects of FLT3 inhibitors in the presence or absence of exogenous FL using sole FLT3‐ITD‐FLT3‐expressing and Wt‐FLT3 and ITD‐FLT3 co–expressing 32D cells. FL stimulation significantly increased GI50 values of all FLT3 inhibitors in Wt‐FLT3 and ITD‐FLT3 co–expressing cells compared to sole ITD‐FLT3 cells. In sole ITD‐FLT3 cells, quizartinib inhibited the phosphorylation of ITD‐FLT3 and its downstream molecules, STAT5, AKT and MAPK. Although ITD‐FLT3 was re–phosphorylated by the addition of FL, downstream molecules were not affected by FL. In contrast, FL activated Wt‐FLT3, but not ITD‐FLT3, in Wt‐FLT3 and ITD‐FLT3 co–expressing cells in the presence of quizartinib. In parallel with the activation of Wt‐FLT3, AKT and MAPK, but not STAT5, were re–phosphorylated by FL. These results indicate that FL activated Wt‐FLT3, but not ITD‐FLT3, in the co–expressing cells. In kinase‐dead (KD) FLT3 (K644A) and ITD‐FLT3 co–expressing cells, FL did not activate MAPK nor affect the growth‐inhibitory effect of FLT3 inhibitors. These results indicate that FL activated Wt‐FLT3, but not ITD‐FLT3, in the co–expressing cells. Furthermore, the addition of a MEK inhibitor blocked the reduced effect of FL on the growth inhibitory effect of FLT3 inhibitors, indicating that FL‐dependent activation of the Wt‐FLT3‐MAPK axis is a key mechanism in FL‐induced resistance of FLT3 inhibitors. Interestingly, the addition of FL more strongly reduces inhibitory activities of gilteritinib, quizartinib, and sorafenib on Wt‐ and ITD‐FLT3 co–expressing cells than those of midostaurin and lestaurtinib (Table 1). Although midostaurin and lestaurtinib have similar inhibitory activities against both ITD‐FLT3 and Wt‐FLT3, gilteritinib, quizartinib and sorafenib show a lower potency against Wt‐FLT3 than ITD‐FLT3. These results indicate that different potencies against ITD‐FLT3 and Wt‐FLT3 are associated with the FL‐dependent impendence of growth‐inhibitory effects on Wt‐FLT3 and ITD‐FLT3 co–expressing cells.
6.3. Another primary resistance mechanisms
In addition to FL, other cytokines, growth factors and soluble proteins from the bone marrow microenvironment were evaluated for their resistance effects against quizartinib. Among them, fibroblast growth factor 2 (FGF2) promoted resistance through activation of FGFR1 and downstream MAPK effectors. Notably, FGF2 expression in marrow stromal cells was increased in FLT3‐ITD‐positive AML patients treated with quizartinib, and the expression level peaked prior to overt clinical relapse and the detection of resistant mutations (Figure 3).72 Furthermore, expression of CYP3A4 in bone marrow stromal cells was reported to inhibit the activity of three different FLT3 inhibitors (sorafenib, quizartinib and gilteritinib) against FLT3‐ITD positive AML (Figure 3).73 It is well known that CYP3A4 inactivates many chemotherapeutic agents for TKI. In particular, hepatic CYP3A4 has been shown to inactivate essentially all TKI, including FLT3 inhibitors. These results collectively suggested that AML cells in BM might escape inhibitor exposure at an effective concentration through a bone marrow microenvironment‐mediated mechanism, resulting in the persistence of AML cells, which will acquire resistance.
7. SECONDARY RESISTANCE MECHANISMS OF FLT3 INHIBITORS
The accumulation of resistant cases in clinical trials of FLT3 inhibitors has revealed their secondary resistance mechanisms. Secondary resistance mechanisms against FLT3 inhibitors were classified into on‐target and off‐target mechanisms, in which leukemia cells become dependent on other signaling pathways.
7.1. On‐target resistance
In on‐target resistance, leukemia cells retain dependency on FLT3 signals but show resistance to FLT3 inhibitors by acquiring mutations in the FLT3 gene (Figure 3). Secondary resistance mutations were first identified in patients with FLT3‐ITD who relapsed following quizartinib treatment.74 In those patients, secondary mutations at the D835 residue, Y842 residue, or the gatekeeper residue F691 in the kinase domain were identified, and FLT3‐ITD + D835Y, +D835V, +Y842C, +Y842H,or +F691L‐expressing Ba/F3 cells showed resistance to growth‐inhibitory and dephosphorylation activities of quizartinib. These resistance mutations in the A‐loop were also identified in patients who were treated with the other type II inhibitor sorafenib. Such acquired resistance reflects the lower binding affinity of type II inhibitors to the activated form of FLT3, which is mediated by TK domain mutations. Notably, the variant allele frequency (VAF) of the acquired mutation on the FLT3‐ITD allele was less than 50% in patients who were clinically resistant to the type II inhibitor quizartinib, suggesting the existence of another resistance mechanism. Subsequent analysis demonstrated that 4 of the 8 quizartinib‐resistant patients harbored more than one resistance mutation in the TK domain.77 Furthermore, 7 of the 8 resistant patients harbored the TK domain mutation in the native FLT3 allele in addition to the FLT3‐ITD allele. Importantly, there were patients in whom the mutation type and mutation frequency were different between the native and ITD FLT3 alleles. In this study, it was also identified that AML cells from 1 quizartinib‐resistant patient acquired no resistance mutation either in native or ITD FLT3 alleles. Although no gene mutation other than FLT3 was identified, an off‐target resistance mechanism might be involved in this patient. These findings collectively indicate that polyclonal mechanisms occurred in most AML patients who relapsed following quizartinib treatment.
7.2. Off‐target resistance
Because quizartinib and sorafenib are type II inhibitors, it is understandable that resistant clones acquired mutations in the TK domain of the FLT3 gene. In contrast, resistant clones after treatment with the type I inhibitors gilteritinib and crenolanib, which have a potency against FLT3‐TKD, showed different resistance features from those after type II inhibitor treatment. Comparable genetic analysis between baseline and progression samples from patients treated with gilteritinib demonstrated several distinct patterns of clonal selection during the gilteritinib treatment.78 In 5 of the 41 gilteritinib‐resistant patients (12.2%), FLT3 mutations were not identified in AML cells after gilteritinib treatment; however, RAS/MAPK pathway mutations were identified in all of them, indicating that FLT3 mutation‐negative clones newly acquired RAS/MAPK pathway mutations and expanded as a resistant clone. In the other 36 patients, resistant clones persisted in the original FLT3 mutations. In 5 of them, FLT3‐F691L mutation was additionally acquired in resistant cells harboring the original FLT3 mutation. Although FLT3‐M837K and FLT3‐C35S mutations were identified in one resistant patient each, both mutations were confirmed to be silent because mutant FLT3 cannot induce autonomous proliferation to Ba/F3 cells. In contrast to type II inhibitors such as quizartinib and sorafenib, the low frequency of additional FLT3 mutation might reflect the potency of gilteritinib against FLT3‐TKD. However, even if the additional FLT3 mutations identified after treatment did not have transforming activity, we should consider that resistance mutation in the FLT3 gene would be acquired during the gilteritinib treatment. Of note is the fact that RAS/MAPK pathway mutations including NRAS, KRAS, PTPN11, CBL and BRAF mutations were acquired in 10 of the 36 patients whose leukemia cells showed persistent FLT3 mutations (Table 2). Importantly, the FLT3‐F691L mutation and RAS/MAPK pathway mutations were mutually exclusive. These results collectively indicate that the off‐target resistance mechanism is more common with gilteritinib than type II inhibitors.
Table 2.
Off–target gene mutations identified in patients during treatments with gilteritinib and crenolanib
| Gilteritinib | Crenolanib | |
|---|---|---|
| RAS/MAPK pathway genes |
NRAS, KRAS, PTPN11 CBL, BRAF |
NRAS, KRAS, PTPN11 |
| Epigenetic‐modifying genes | IDH2 |
IDH1, IDH2 TET2, DNMT3A ASXL1, BCOR |
| Myeloid transcription factor genes |
CEBPA RUNX1 |
CEBPA RUNX1 |
| Spliceosome‐complex genes |
U2AF1 SF3B1 |
|
| Cohesion complex genes | STAG2 | |
| Others |
WT1 TBL1XR1 BCR‐ABL |
Characteristics of resistant clones after treatment with another type I inhibitor crenolanib were also different from those after type II inhibitors.79 In crenolanib‐treated patients, acquired mutations on the FLT3 gene were infrequent in residual AML cells after treatment. In 50 resistant patients treated with crenolanib, 5 FLT3 mutations (D200N, K429F, Y572C, L601F and F691L) were identified after treatment in 6 individuals, while D200N and L601F mutations did not lead to transforming activity. In contrast, several gene mutations, which are classified into epigenetic regulators, myeloid transcription factors and the cohesin complex, were identified in crenolanib poor responders. Notably, NRAS, STAG2, CEBPA, ASXL1 and IDH2 mutations arose mostly in FLT3 mutation‐independent clones, indicating that these clones escaped and expanded during crenolanib treatment. However, TET2, IDH1 and TP53 mutations predominantly co–occur in FLT3‐mutated clones during crenolanib treatment (Table 2).
8. COVALENT‐BINDING TYPE FLT3 INHIBITOR
Previous studies collectively suggested that on–target resistance frequently occurs in patients after treatment with type II inhibitors, but off–target resistance is frequent in those after treatment with type I inhibitors. Because these studies were mainly conducted using the samples from patients who were treated in clinical trials, the accumulation of resistance features derived from patients treated in daily clinical practice is necessary to fully understand the resistance mechanism of each FLT3 inhibitor. However, the further development of novel FLT3 inhibitors is required to overcome the mutation of the gatekeeper residue (F691) in the FLT3 gene. Many type II inhibitors are designed to favorably bind to the back pocket of the inactive conformation of FLT3 to increase affinity to the ATP‐binding site. In contrast, because many type I inhibitors are designed to bind to only the ATP‐binding site, they can bind to the active conformation of FLT3; however, they have more broad‐kinase inhibitory activities than type II inhibitors, due to the similarity of binding sites among kinases. To resolve these problems, we developed a novel FLT3 inhibitor, FF‐10101, in collaboration with FUJIFILM (Kanagawa, Japan), which was designed to form a covalent binding between the C695 residue of FLT3 (Figure 4).yyyy80 This covalent bond formation of FF‐10101 induces irreversible inhibition of FLT3 and potent and selective inhibitory activity, and maintains the binding ability both to the active and inactive conformations of FLT3. The unique binding of FF‐10101 also provides broad and potent inhibitory effects on various FLT3 mutations, including the gatekeeper mutation F691L. We are now conducting a phase 1/2a dose escalation and dose ranging study of FF‐10101 in subjects with relapsed or refractory acute myeloid leukemia to determine the safety, tolerability, PK and preliminary efficacy (NCT03194685).
9. CONCLUSION
It takes more than 20 years after the first identification of FLT3 mutations until FLT3 inhibitors become clinically available for AML patients with FLT3 mutations. Although many issues regarding the clinical use of FLT3 inhibitors have been resolved during the long development period, further efforts are required to establish the most suitable therapeutic strategy for each inhibitor and for each patient. Because AML is a genetically heterogenous disease and FLT3 mutation is a late event during leukemogenesis, mono‐therapy of FLT3 inhibitor has limitations for curing AML patients with FLT3 mutations. Although the RATIFY study demonstrated that the combination of chemotherapy and midostaurin improved the prognosis of AML patients with FLT3 mutations, many patients underwent allo‐HSCT during the treatment. In addition, the extent to which combination therapies of other FLT3 inhibitors with chemotherapy or other targeting agents improves the prognosis remains unclear. Further basic and clinical studies are required to establish biomarkers for selecting the best therapeutic strategy according to the characteristics of AML cells and patients’ conditions.
CONFLICT OF INTEREST
Hitoshi Kiyoi received research funding from Chugai Pharmaceutical, Kyowa Hakko Kirin, Zenyaku Kogyo, FUJIFILM, Daiichi Sankyo, Astellas Pharma, Otsuka Pharmaceutical, Nippon Shinyaku, Eisai, Pfizer Japan, Takeda Pharmaceutical, Novartis Pharma KK, Sumitomo Dainippon Pharma, Sanofi KK and Celgene, consulting fees from Astellas Pharma, Amgen Astellas BioPharma KK and Daiichi Sankyo, and honoraria from Bristol‐Myers Squibb, Astellas Pharma and Novartis Pharma KK. The other authors have no conflict of interest.
ACKNOWLEDGMENTS
We would like to thank Dr Tomoki Naoe of Nagoya Medical Center for continuous advice on and support of our studies, all colleagues in our laboratory for their extensive studies, and Ms Manami Kira and Ms Satomi Yamaji for their secretarial and technical assistance. This work was supported in part by Grants‐in‐Aid from the Newly Extended Technology Transfer Program (NexTEP) of the Japan Science and Technology Agency, the Practical Research for Innovative Cancer Control from the Japan Agency for Medical Research and Development, AMED (19ck0106251h0003), the Project for Development of Innovative Research on Cancer Therapeutics (P‐DIRECT) from the AMED (19cm0106562h0001), the Scientific Research Program of the Ministry of Education, Culture, Sports, Science and Technology of Japan (17K09921), and Nagoya University Hospital Funding for Clinical Research.
Kiyoi H, Kawashima N, Ishikawa Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020;111:312–322. 10.1111/cas.14274
REFERENCES
- 1. Rosnet O, Mattei MG, Marchetto S, Birnbaum D. Isolation and chromosomal localization of a novel FMS‐like tyrosine kinase gene. Genomics. 1991;9:380‐385. [DOI] [PubMed] [Google Scholar]
- 2. Rosnet O, Schiff C, Pebusque MJ, et al. Human FLT3/FLK2 gene: cDNA cloning and expression in hematopoietic cells. Blood. 1993;82:1110‐1119. [PubMed] [Google Scholar]
- 3. Lemmon MA, Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell. 2010;141:1117‐1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Small D, Levenstein M, Kim E, et al. STK‐1, the human homolog of Flk‐2/Flt‐3, is selectively expressed in CD34+ human bone marrow cells and is involved in the proliferation of early progenitor/stem cells. Proc Natl Acad Sci USA. 1994;91:459‐463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Hannum C, Culpepper J, Campbell D, et al. Ligand for FLT3/FLK2 receptor tyrosine kinase regulates growth of haematopoietic stem cells and is encoded by variant RNAs. Nature. 1994;368:643‐648. [DOI] [PubMed] [Google Scholar]
- 6. Lyman SD, James L, Vanden Bos T, et al. Molecular cloning of a ligand for the flt3/flk‐2 tyrosine kinase receptor: a proliferative factor for primitive hematopoietic cells. Cell. 1993;75:1157‐1167. [DOI] [PubMed] [Google Scholar]
- 7. Stirewalt DL, Radich JP. The role of FLT3 in haematopoietic malignancies. Nat Rev Cancer. 2003;3:650‐665. [DOI] [PubMed] [Google Scholar]
- 8. Drexler HG, Meyer C, Quentmeier H. Effects of FLT3 ligand on proliferation and survival of myeloid leukemia cells. Leuk Lymphoma. 1999;33:83‐91. [DOI] [PubMed] [Google Scholar]
- 9. Nakao M, Yokota S, Iwai T, et al. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia. 1996;10:1911‐1918. [PubMed] [Google Scholar]
- 10. Yamamoto Y, Kiyoi H, Nakano Y, et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood. 2001;97:2434‐2439. [DOI] [PubMed] [Google Scholar]
- 11. Kiyoi H, Naoe T. Biology, clinical relevance, and molecularly targeted therapy in acute leukemia with FLT3 mutation. Int J Hematol. 2006;83:301‐308. [DOI] [PubMed] [Google Scholar]
- 12. Kihara R, Nagata Y, Kiyoi H, et al. Comprehensive analysis of genetic alterations and their prognostic impacts in adult acute myeloid leukemia patients. Leukemia. 2014;28:1586‐1595. [DOI] [PubMed] [Google Scholar]
- 13. Cancer Genome Atlas Research N , Ley TJ, Miller C, et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368:2059‐2074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kiyoi H. Flt3 inhibitors: recent advances and problems for clinical application. Nagoya J Med Sci. 2015;77:7‐17. [PMC free article] [PubMed] [Google Scholar]
- 15. Dohner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129:424‐447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Patel JP, Gonen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366:1079‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374:2209‐2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yokota S, Kiyoi H, Nakao M, et al. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia. 1997;11:1605‐1609. [DOI] [PubMed] [Google Scholar]
- 19. Kiyoi H, Naoe T, Yokota S, et al. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia. 1997;11:1447‐1452. [DOI] [PubMed] [Google Scholar]
- 20. Horiike S, Yokota S, Nakao M, et al. Tandem duplications of the FLT3 receptor gene are associated with leukemic transformation of myelodysplasia. Leukemia. 1997;11:1442‐1446. [DOI] [PubMed] [Google Scholar]
- 21. Steudel C, Wermke M, Schaich M, et al. Comparative analysis of MLL partial tandem duplication and FLT3 internal tandem duplication mutations in 956 adult patients with acute myeloid leukemia. Genes Chromosomes Cancer. 2003;37:237‐251. [DOI] [PubMed] [Google Scholar]
- 22. Taketani T, Taki T, Sugita K, et al. FLT3 mutations in the activation loop of tyrosine kinase domain are frequently found in infant ALL with MLL rearrangements and pediatric ALL with hyperdiploidy. Blood. 2004;103:1085‐1088. [DOI] [PubMed] [Google Scholar]
- 23. Kiyoi H, Naoe T. FLT3 in human hematologic malignancies. Leuk Lymphoma. 2002;43:1541‐1547. [DOI] [PubMed] [Google Scholar]
- 24. Stirewalt DL, Kopecky KJ, Meshinchi S, et al. FLT3, RAS, and TP53 mutations in elderly patients with acute myeloid leukemia. Blood. 2001;97:3589‐3595. [DOI] [PubMed] [Google Scholar]
- 25. Xu F, Taki T, Eguchi M, et al. Tandem duplication of the FLT3 gene is infrequent in infant acute leukemia Japan Infant Leukemia Study Group. Leukemia. 2000;14:945‐947. [DOI] [PubMed] [Google Scholar]
- 26. Shih LY, Kuo MC, Liang DC, et al. Internal tandem duplication and Asp835 mutations of the FMS‐like tyrosine kinase 3 (FLT3) gene in acute promyelocytic leukemia. Cancer. 2003;98:1206‐1216. [DOI] [PubMed] [Google Scholar]
- 27. Noguera NI, Breccia M, Divona M, et al. Alterations of the FLT3 gene in acute promyelocytic leukemia: association with diagnostic characteristics and analysis of clinical outcome in patients treated with the Italian AIDA protocol. Leukemia. 2002;16:2185‐2189. [DOI] [PubMed] [Google Scholar]
- 28. Duployez N, Marceau‐Renaut A, Boissel N, et al. Comprehensive mutational profiling of core binding factor acute myeloid leukemia. Blood. 2016;127:2451‐2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kawashima N, Akashi A, Nagata Y, et al. Clinical significance of ASXL2 and ZBTB7A mutations and C‐terminally truncated RUNX1‐RUNX1T1 expression in AML patients with t(8;21) enrolled in the JALSG AML201 study. Ann Hematol. 2019;98:83‐91. [DOI] [PubMed] [Google Scholar]
- 30. Speck NA, Gilliland DG. Core‐binding factors in haematopoiesis and leukaemia. Nat Rev Cancer. 2002;2:502‐513. [DOI] [PubMed] [Google Scholar]
- 31. Kelly LM, Kutok JL, Williams IR, et al. PML/RARalpha and FLT3‐ITD induce an APL‐like disease in a mouse model. Proc Natl Acad Sci USA. 2002;99:8283‐8288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Meyer SE, Qin T, Muench DE, et al. DNMT3A Haploinsufficiency transforms FLT3ITD myeloproliferative disease into a rapid, spontaneous, and fully penetrant acute myeloid leukemia. Cancer Discov. 2016;6:501‐515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Kiyoi H, Naoe T, Nakano Y, et al. Prognostic implication of FLT3 and N‐RAS gene mutations in acute myeloid leukemia. Blood. 1999;93:3074‐3080. [PubMed] [Google Scholar]
- 34. Yanada M, Matsuo K, Suzuki T, Kiyoi H, Naoe T. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: a meta‐analysis. Leukemia. 2005;19:1345‐1349. [DOI] [PubMed] [Google Scholar]
- 35. Dohner H, Estey EH, Amadori S, et al. Diagnosis and management of acute myeloid leukemia in adults: recommendations from an international expert panel, on behalf of the European LeukemiaNet. Blood. 2010;115:453‐474. [DOI] [PubMed] [Google Scholar]
- 36. Harada Y, Nagata Y, Kihara R, et al. Prognostic analysis according to the 2017 ELN risk stratification by genetics in adult acute myeloid leukemia patients treated in the Japan Adult Leukemia Study Group (JALSG) AML201 study. Leuk Res. 2018;66:20‐27. [DOI] [PubMed] [Google Scholar]
- 37. Sakaguchi M, Yamaguchi H, Najima Y, et al. Prognostic impact of low allelic ratio FLT3‐ITD and NPM1 mutation in acute myeloid leukemia. Blood Adv. 2018;2:2744‐2754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Luskin MR, Lee JW, Fernandez HF, et al. Benefit of high‐dose daunorubicin in AML induction extends across cytogenetic and molecular groups. Blood. 2016;127:1551‐1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Schlenk RF, Dohner K, Krauter J, et al. Mutations and treatment outcome in cytogenetically normal acute myeloid leukemia. N Engl J Med. 2008;358:1909‐1918. [DOI] [PubMed] [Google Scholar]
- 40. Kiyoi H, Ohno R, Ueda R, Saito H, Naoe T. Mechanism of constitutive activation of FLT3 with internal tandem duplication in the juxtamembrane domain. Oncogene. 2002;21:2555‐2563. [DOI] [PubMed] [Google Scholar]
- 41. Kiyoi H, Towatari M, Yokota S, et al. Internal tandem duplication of the FLT3 gene is a novel modality of elongation mutation which causes constitutive activation of the product. Leukemia. 1998;12:1333‐1337. [DOI] [PubMed] [Google Scholar]
- 42. Hayakawa F, Towatari M, Kiyoi H, et al. Tandem‐duplicated Flt3 constitutively activates STAT5 and MAP kinase and introduces autonomous cell growth in IL‐3‐dependent cell lines. Oncogene. 2000;19:624‐631. [DOI] [PubMed] [Google Scholar]
- 43. Kelly LM, Liu Q, Kutok JL, Williams IR, Boulton CL, Gilliland DG. FLT3 internal tandem duplication mutations associated with human acute myeloid leukemias induce myeloproliferative disease in a murine bone marrow transplant model. Blood. 2002;99:310‐318. [DOI] [PubMed] [Google Scholar]
- 44. Kelly LM, Yu JC, Boulton CL, et al. CT53518, a novel selective FLT3 antagonist for the treatment of acute myelogenous leukemia (AML). Cancer Cell. 2002;1:421‐432. [DOI] [PubMed] [Google Scholar]
- 45. Stone RM, DeAngelo DJ, Klimek V, et al. Patients with acute myeloid leukemia and an activating mutation in FLT3 respond to a small‐molecule FLT3 tyrosine kinase inhibitor, PKC412. Blood. 2005;105:54‐60. [DOI] [PubMed] [Google Scholar]
- 46. Smith BD, Levis M, Beran M, et al. Single‐agent CEP‐701, a novel FLT3 inhibitor, shows biologic and clinical activity in patients with relapsed or refractory acute myeloid leukemia. Blood. 2004;103:3669‐3676. [DOI] [PubMed] [Google Scholar]
- 47. Fiedler W, Serve H, Dohner H, et al. A phase 1 study of SU11248 in the treatment of patients with refractory or resistant acute myeloid leukemia (AML) or not amenable to conventional therapy for the disease. Blood. 2005;105:986‐993. [DOI] [PubMed] [Google Scholar]
- 48. Ravandi F, Cortes JE, Jones D, et al. Phase I/II study of combination therapy with sorafenib, idarubicin, and cytarabine in younger patients with acute myeloid leukemia. J Clin Oncol. 2010;28:1856‐1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Levis M, Brown P, Smith BD, et al. Plasma inhibitory activity (PIA): a pharmacodynamic assay reveals insights into the basis for cytotoxic response to FLT3 inhibitors. Blood. 2006;108:3477‐3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Levis M, Ravandi F, Wang ES, et al. Results from a randomized trial of salvage chemotherapy followed by lestaurtinib for patients with FLT3 mutant AML in first relapse. Blood. 2011;117:3294‐3301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rollig C, Serve H, Huttmann A, et al. Addition of sorafenib versus placebo to standard therapy in patients aged 60 years or younger with newly diagnosed acute myeloid leukaemia (SORAML): a multicentre, phase 2, randomised controlled trial. Lancet Oncol. 2015;16:1691‐1699. [DOI] [PubMed] [Google Scholar]
- 52. Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377:454‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Burchert A, Bug G, Finke J, et al. Sorafenib as maintenance therapy post allogeneic stem cell transplantation for FLT3‐ITD positive AML: results from the randomized, double‐blind, placebo‐controlled multicentre sormain trial. Blood. 2018;132.29866817 [Google Scholar]
- 54. Mathew NR, Baumgartner F, Braun L, et al. Sorafenib promotes graft‐versus‐leukemia activity in mice and humans through IL‐15 production in FLT3‐ITD‐mutant leukemia cells. Nat Med. 2018;24:282‐291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mori M, Kaneko N, Ueno Y, et al. Gilteritinib, a FLT3/AXL inhibitor, shows antileukemic activity in mouse models of FLT3 mutated acute myeloid leukemia. Invest New Drugs. 2017;35:556‐565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lee LY, Hernandez D, Rajkhowa T, et al. Preclinical studies of gilteritinib, a next‐generation FLT3 inhibitor. Blood. 2017;129:257‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Park IK, Mishra A, Chandler J, Whitman SP, Marcucci G, Caligiuri MA. Inhibition of the receptor tyrosine kinase Axl impedes activation of the FLT3 internal tandem duplication in human acute myeloid leukemia: implications for Axl as a potential therapeutic target. Blood. 2013;121:2064‐2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Park IK, Mundy‐Bosse B, Whitman SP, et al. Receptor tyrosine kinase Axl is required for resistance of leukemic cells to FLT3‐targeted therapy in acute myeloid leukemia. Leukemia. 2015;29:2382‐2389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first‐in‐human, open‐label, phase 1–2 study. Lancet Oncol. 2017;18:1061‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Perl AE, Martinelli G, Cortes JE, et al. Gilteritinib or chemotherapy for relapsed or refractory FLT3‐mutated AML. N Engl J Med. 2019;381:1728‐1740. [DOI] [PubMed] [Google Scholar]
- 61. Karaman MW, Herrgard S, Treiber DK, et al. A quantitative analysis of kinase inhibitor selectivity. Nat Biotechnol. 2008;26:127‐132. [DOI] [PubMed] [Google Scholar]
- 62. Zarrinkar PP, Gunawardane RN, Cramer MD, et al. AC220 is a uniquely potent and selective inhibitor of FLT3 for the treatment of acute myeloid leukemia (AML). Blood. 2009;114:2984‐2992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Cortes JE, Tallman MS, Schiller GJ, et al. Phase 2b study of 2 dosing regimens of quizartinib monotherapy in FLT3‐ITD‐mutated, relapsed or refractory AML. Blood. 2018;132:598‐607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cortes J, Perl AE, Dohner H, et al. Quizartinib, an FLT3 inhibitor, as monotherapy in patients with relapsed or refractory acute myeloid leukaemia: an open‐label, multicentre, single‐arm, phase 2 trial. Lancet Oncol. 2018;19:889‐903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Galanis A, Levis M. Inhibition of c‐Kit by tyrosine kinase inhibitors. Haematologica. 2015;100:e77‐79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Cortes JE, Khaled S, Martinelli G, et al. Quizartinib versus salvage chemotherapy in relapsed or refractory FLT3‐ITD acute myeloid leukaemia (QuANTUM‐R): a multicentre, randomised, controlled, open‐label, phase 3 trial. Lancet Oncol. 2019;20:984‐997. [DOI] [PubMed] [Google Scholar]
- 67. Galanis A, Ma H, Rajkhowa T, et al. Crenolanib is a potent inhibitor of FLT3 with activity against resistance‐conferring point mutants. Blood. 2014;123:94‐100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Smith CC, Lasater EA, Lin KC, et al. Crenolanib is a selective type I pan‐FLT3 inhibitor. Proc Natl Acad Sci U S A. 2014;111:5319‐5324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Cortes JE, Kantarjian HM, Kadia TM, et al. Crenolanib besylate, a type I pan‐FLT3 inhibitor, to demonstrate clinical activity in multiply relapsed FLT3‐ITD and D835 AML. J Clin Oncol. 2016;34(Supplement):7008. [Google Scholar]
- 70. Weisberg E, Sattler M, Ray A, Griffin JD. Drug resistance in mutant FLT3‐positive AML. Oncogene. 2010;29:5120‐5134. [DOI] [PubMed] [Google Scholar]
- 71. Sato T, Yang X, Knapper S, et al. FLT3 ligand impedes the efficacy of FLT3 inhibitors in vitro and in vivo. Blood. 2011;117:3286‐3293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Traer E, Martinez J, Javidi‐Sharifi N, et al. FGF2 from marrow microenvironment promotes resistance to FLT3 inhibitors in acute myeloid leukemia. Cancer Res. 2016;76:6471‐6482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Chang YT, Hernandez D, Alonso S, et al. Role of CYP3A4 in bone marrow microenvironment‐mediated protection of FLT3/ITD AML from tyrosine kinase inhibitors. Blood Adv. 2019;3:908‐916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Smith CC, Wang Q, Chin CS, et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature. 2012;485:260‐263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Daver N, Schlenk RF, Russell NH, Levis MJ. Targeting FLT3 mutations in AML: review of current knowledge and evidence. Leukemia. 2019;33:299‐312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Chen F, Ishikawa Y, Akashi A, Naoe T, Kiyoi H. Co–expression of wild‐type FLT3 attenuates the inhibitory effect of FLT3 inhibitor on FLT3 mutated leukemia cells. Oncotarget. 2016;7:47018‐47032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Smith CC, Paguirigan A, Jeschke GR, et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single‐cell analysis. Blood. 2017;130:48‐58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. McMahon CM, Ferng T, Canaani J, et al. Clonal selection with RAS pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9:1050‐1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Zhang H, Savage S, Schultz AR, et al. Clinical resistance to crenolanib in acute myeloid leukemia due to diverse molecular mechanisms. Nat Commun. 2019;10:244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Yamaura T, Nakatani T, Uda K, et al. A novel irreversible FLT3 inhibitor, FF‐10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood. 2018;131:426‐438. [DOI] [PubMed] [Google Scholar]