

Abstract
MicroRNAs (miRNAs) fine‐tune cellular signaling by regulating expression of signaling proteins, and aberrant expression of miRNAs is observed in many cancers. The tyrosine kinase c‐Src is upregulated in various human cancers, but the molecular mechanisms underlying c‐Src‐mediated tumor progression remain unclear. In previous investigations of miRNA‐mediated control of c‐Src‐related oncogenic pathways, we identified miRNAs that were downregulated in association with c‐Src transformation and uncovered the signaling networks by predicting their target genes, which might act cooperatively to control tumor progression. Here, to further elucidate the process of cell transformation driven by c‐Src, we analyzed the expression profiles of miRNAs in a doxycycline‐inducible Src expression system. We found that miRNA (miR)‐129‐1‐3p was downregulated in the early phase of c‐Src‐induced cell transformation, and that reexpression of miR‐129‐1‐3p disrupted c‐Src‐induced cell transformation. In addition, miR‐129‐1‐3p downregulation was tightly associated with tumor progression in human colon cancer cells/tissues. Expression of miR‐129‐1‐3p in human colon cancer cells caused morphological changes and suppressed tumor growth, cell adhesion, and invasion. We also identified c‐Src and its critical substrate Fer, and c‐Yes, a member of the Src family of kinases, as novel targets of miR‐129‐1‐3p. Furthermore, we found that miR‐129‐1‐3p‐mediated regulation of c‐Src/Fer and c‐Yes is important for controlling cell adhesion and invasion. Downregulation of miR‐129‐1‐3p by early activation of c‐Src increases expression of these target genes and synergistically promotes c‐Src‐related oncogenic signaling. Thus, c‐Src‐miR‐129‐1‐3p circuits serve as critical triggers for tumor progression in many human cancers that harbor upregulation of c‐Src.
Keywords: colon cancer, malignancy, microRNA, signaling, Src
We found that microRNA (miR)‐129‐1‐3p was repressed in the early phase of c‐Src‐induced cell transformation, and that reexpression of miR‐129‐1‐3p disrupted c‐Src‐induced cell transformation. MicroRNA‐129‐1‐3p was repressed in various colon cancer tissues. In addition, we showed that miR‐129‐1‐3p directly targets c‐Src, Fer, and c‐Yes, which are required for cancer cell adhesion and invasion. These findings indicate that downregulation of miR‐129‐1‐3p by early activation of c‐Src synergistically promotes c‐Src‐related oncogenic signaling.
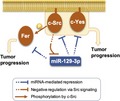
1. INTRODUCTION
c‐src, the first protooncogene to be identified, encodes a nonreceptor type tyrosine kinase.1 c‐Src is a pivotal component of multiple signaling pathways, including cell proliferation, survival, cytoskeletal organization, intercellular contact, adhesion, and migration, which are tightly associated with tumor progression.2, 3 c‐Src is frequently overexpressed and activated in a wide range of human cancers, suggesting a crucial role in tumor progression.4, 5, 6, 7 In normal cells, the kinase activity of c‐Src is strictly controlled by the C‐terminal Src kinase (Csk); therefore, even when c‐Src is abundantly expressed, its oncogenic potential is suppressed.8 In some cancer cells, the c‐src gene is not mutated, but c‐Src function is nonetheless upregulated.9 It is thought that disruption of the strict regulation of c‐Src signaling could trigger cancer progression; however, the underlying mechanisms remain unclear. Once activated, c‐Src acts as a common relay point for several downstream cascades from extracellular signals, such as growth factors and integrins, to intracellular signaling pathways.5, 10
c‐Src is a member of the Src family of kinases (SFKs), which comprises 8 members in mammals: c‐Src, Fyn, c‐Yes, Lyn, Lck, Hck, c‐Fgr, and Blk.11 Among those, c‐Src and c‐Yes are frequently upregulated in a variety of human cancers.5, 7 The distinctive expression patterns and functional redundancy of SFK members have hampered concurrent analyses of their contributions to cancer progression. Previously, we showed that the oncogenic function of c‐Src is spatially regulated, and that c‐Src‐mediated cell transformation is initiated from nonraft compartments.12, 13 Based on these findings, we recently identified a critical substrate for c‐Src in nonraft compartments and showed that Fer tyrosine kinase is a key mediator of c‐Src‐induced cell transformation.14 In addition, we found that Fer is involved in tumorigenesis and invasiveness in some cancers in which c‐Src is upregulated. Indeed, upregulation of Fer has been implicated in tumor progression in various human cancers; however, the mechanism underlying upregulation remains unknown.15, 16, 17, 18, 19
MicroRNAs (miRNAs) are a family of small, endogenous, evolutionarily conserved noncoding RNAs involved in the regulation of expression of target mRNAs.20, 21 MicroRNAs control diverse cellular functions and fine‐tune various signaling pathways.22 MicroRNAs are extensively dysregulated in several human cancers and act as key regulators of complex signaling networks by altering expression of oncogenes or tumor suppressor genes.23, 24, 25
To verify the molecular mechanisms underlying c‐Src‐mediated cell transformation, we previously developed a model system using Csk‐deficient mouse embryonic fibroblasts (Csk−/− cells), which can be transformed by c‐Src.26 A series of studies showed that this system is useful for the identification of critical pathways leading to c‐Src‐induced cell transformation. Using this system, we focused on the contribution of miRNAs and uncovered miRNA‐mediated c‐Src oncogenic signaling and cross‐talk between c‐Src and other oncogenic signaling networks, including the focal adhesion‐mediated pathways, microRNA (miR)‐542‐3p‐ILK, miR‐27b‐paxillin, and the mTOR pathways, and miR‐99a‐mTOR and miR‐424/503‐Rictor.27, 28, 29, 30, 31 In contrast, we also found that expression of c‐Src is regulated by miR‐137, which is substantially downregulated in many cancers.32 Downregulation of miR‐137 is induced in the early phase of tumor progression, which results in the upregulation of c‐Src signaling. MicroRNA‐137‐mediated upregulation of c‐Src signaling induces expression of c‐Src‐regulated miRNAs such as miR‐542‐3p, ‐27b, ‐99a, and ‐424/503. These studies showed that oncogenic c‐Src signaling is regulated by multiple miRNA‐mediated mechanisms in tumors in which oncogenic signaling was maintained at steady state; however, the trigger for miRNA‐mediated signaling in c‐Src‐induced transformation remains unknown.
In this study, to verify the mechanisms underlying the processes of c‐Src‐induced transformation, we analyzed the expression of miRNAs in the early phases of cell transformation using an inducible c‐Src expression system in Csk‐deficient fibroblasts. Expression profiles of miRNAs in this system revealed that previously identified miRNAs such as miR‐99a, ‐322 (‐424 in human), ‐503, and ‐542‐3p were definitely downregulated in the later phases of cell transformation. In particular, based on its significant downregulation in earlier phases of c‐Src transformation, we focused our analysis on miR‐129‐1‐3p. We found that miR‐129‐1‐3p is downregulated in human colon cancers harboring c‐Src upregulation and directly targets c‐Src, c‐Yes, and Fer. Functional analysis of miR‐129‐1‐3p suggests that miR‐129‐1‐3p functions as a suppressor of tumor progression by controlling cell adhesion and invasion mediated by Src‐related oncogenic pathways.
2. MATERIALS AND METHODS
2.1. MicroRNA microarray analysis
MicroRNA microarray experiments were carried out using Agilent Mouse miRNA microarrays, catalogued in the Sanger database version 16.0 (design ID 031184). Briefly, 100 ng total RNA was labeled with pCp‐Cy3 using the miRNA Labeling Reagent and Hybridization Kit (Agilent Technologies). Images were extracted using Agilent Feature Extraction software (version 10.7.3.1). Agilent GeneSpring GX software (version 14.9.1) was then used to calculate the between‐sample fold change by one‐way ANOVA (P < .1) with Benjamini‐Hochberg correction for multiple testing.
2.2. Gene expression microarray analysis
Briefly, 500 ng total RNA was reverse transcribed into double‐stranded cDNA using AffinityScript multiple temperature reverse transcriptase (Agilent Technologies). The resulting complementary RNAs were labeled with cyanine‐3 (Perkin Elmer) using the Low Input Quick‐Amp Labeling kit (Agilent Technologies). Hybridizations were undertaken on Agilent SurePrint G3 Mouse GE 8 × 60K Microarrays (design ID 028005). Agilent GeneSpring GX software (version 14.9.1) was used to calculate the between‐sample fold change (P < .1; two‐tailed Student’s t test).
Other methods are described in Document S1.
3. RESULTS
3.1. MicroRNA‐129‐1‐3p is downregulated by Src activation
To analyze the processes of cell transformation induced by c‐Src upregulation in the early phase of tumor progression, we used a doxycycline (Dox)‐inducible expression system in Csk‐deficient fibroblasts (Csk −/− MEF/pBKT2‐c‐Src).33, 34 Inducible expression of c‐Src and its concomitant activation by Dox treatment peaked at 6 hours (Figure 1A). Using these cells, we undertook microarray analysis to compare the expression profiles of miRNAs as c‐Src‐induced transformation progressed. Following induction of Src activation, miRNAs including previously identified miR‐99a, ‐322, ‐503, and ‐542‐3p were definitely downregulated at 72 hours, while 5 miRNAs showed a twofold or greater downregulation at 24 hours after Dox induction (Figure S1) were newly identified. Among them, 3 miRNAs including mmu‐miR‐129‐1‐3p, ‐200b, and ‐149‐3p are conserved between humans and mice. MicroRNA‐200b has been well studied as an epithelial‐mesenchymal transition‐related miRNA35 and miR‐149‐3p has been reported to be involved in some types of cancers,36, 37 whereas the role of miR‐129‐1‐3p has not been clearly elucidated. Thus, we focused on miR‐129‐1‐3p. Quantitative real‐time PCR (qRT‐PCR) analyses confirmed downregulation of miR‐129‐1‐3p in the earlier phase of c‐Src‐induced transformation (at 24 hours) by Dox treatment (Figure 1B). To verify c‐Src‐induced repression of miR‐129‐1‐3p, we used dasatinib to inhibit Src family kinases. Quantitative RT‐PCR analysis revealed that dasatinib increased miR‐129‐1‐3p expression (Figure 1C). To assess the correlation of miR‐129‐1‐3p expression with c‐Src‐induced transformation, we removed Dox from the culture medium in c‐Src‐induced Csk −/− cells, causing c‐Src expression to decrease (Figure 1D). Under conditions in which morphological transformation reverted to normal 4 days after removal of Dox, miR‐129‐1‐3p expression was mostly restored (Figure 1D,E), suggesting that c‐Src activation accompanied by transformation plays a role in the repression of miR‐129‐1‐3p.
Figure 1.
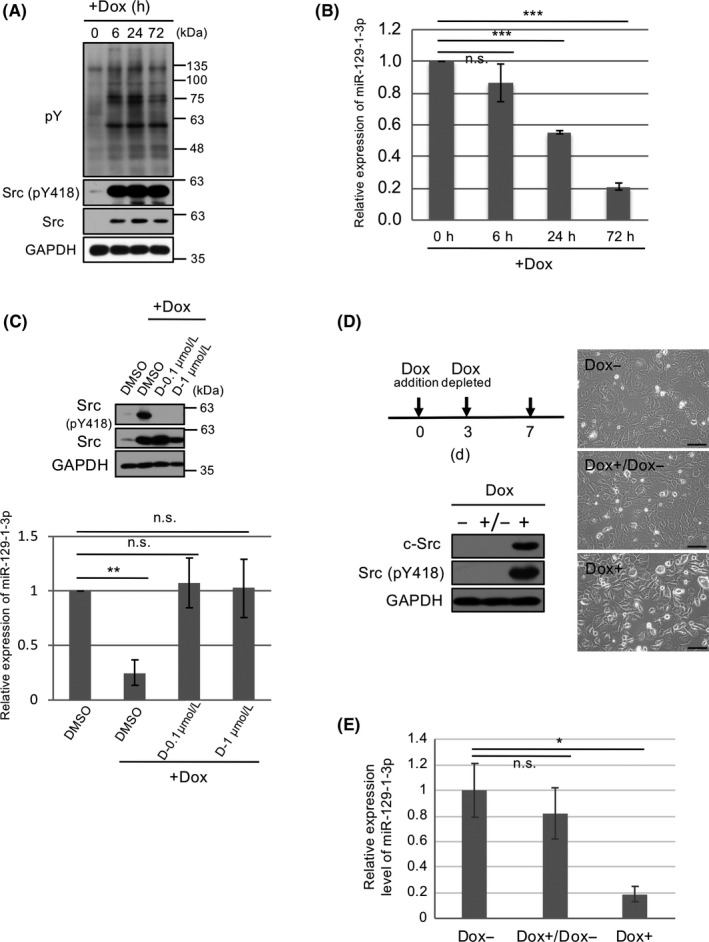
MicroRNA (miR)‐129‐1‐3p is downregulated in c‐Src‐induced cell transformation in a doxycycline (Dox)‐inducible expression system. A, Csk −/− murine embryonic fibroblasts harboring pBKT2‐c‐Src (CskKO/pBKT2‐c‐Src) were incubated with or without 1 μg/mL Dox for the indicated time periods. Total cell lysates were immunoblotted with the indicated Abs. pY, phosphorylated tyrosine residue. B, Expression of miR‐129‐1‐3p in (A) was analyzed by quantitative real‐time (qRT)‐PCR. C, CskKO/pBKT2‐c‐Src cells were treated with or without dasatinib (D) at the indicated concentrations in the presence of Dox for 48 h, and total cell lysates were immunoblotted with the indicated Abs. Levels of miR‐129‐1‐3p were analyzed by qRT‐PCR. D, CskKO/pBKT2‐c‐Src cells were cultured without (−) or with (+) Dox for 3 days. The c‐Src‐induced transformed cells were further cultured in the absence (−) or presence (+) of Dox for 4 days, and total cell lysates were immunoblotted with the indicated Abs. Morphology of the indicated cells was observed by phase‐contrast microscopy. Scale bar = 100 µm. E, CskKO/pBKT2‐c‐Src cells were cultured with or without Dox as indicated in (D), and the expression levels of miR‐129‐1‐3p were assessed by qRT‐PCR. Relative values ± SD were obtained from 3 independent assays (B, C, E). *P < .05, **P < .01, and ***P < .001 by Student’s t test. n.s., not significant
3.2. MicroRNA‐129‐1‐3p suppresses c‐Src‐mediated cell transformation
To assess the role of miR‐129‐1‐3p downregulation in the early Src activation, we investigated whether miR‐129‐1‐3p could suppress c‐Src‐induced transformation (Figure 2A). Soft‐agar colony formation assays revealed that reexpression of miR‐129‐1‐3p markedly decreased the colony numbers in c‐Src‐inducible Csk −/− cells in the presence of Dox (Figure 2B). Reexpression of miR‐129‐1‐3p also suppressed c‐Src‐induced morphological transformation after Dox treatment (Figure 2C). To determine the physiological relevance of miR‐129‐1‐3p expression, we examined the contribution of epidermal growth factor (EGF) receptor signaling involving c‐Src activation. When murine embryonic fibroblasts (MEFs) were stimulated with EGF, c‐Src activity was increased (Figure 2D), and the cells underwent morphological transformation (Figure 2E, upper panels). In these EGF‐transformed cells, miR‐129‐1‐3p expression was appreciably downregulated by EGF stimulation (Figure 2F). To evaluate the effect of miR‐129‐1‐3p on the EGF‐induced cell transformation, we transfected MEFs with miR‐129‐1‐3p and stimulated with EGF. Reexpression of miR‐129‐1‐3p completely blocked EGF‐induced morphological transformation. Thus, miR‐129‐1‐3p is downregulated by the Src‐induced pathway, and expression of miR‐129‐1‐3p per se suppresses Src‐mediated cell transformation.
Figure 2.
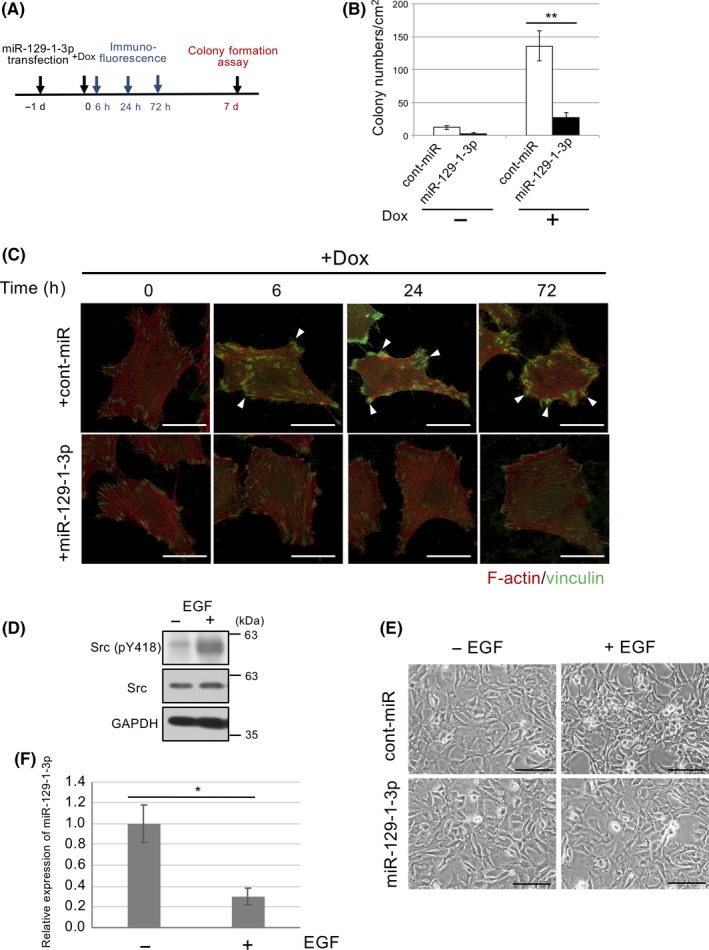
MicroRNA (miR)‐129‐1‐3p suppresses c‐Src‐induced cell transformation. A, CskKO/pBKT2‐c‐Src cells transfected with miR‐129‐1‐3p or control (cont‐miR) were incubated with doxycycline (Dox) for the indicated time periods. B, Soft‐agar colony formation assays of CskKO/pBKT2‐c‐Src cells treated with miR‐129‐1‐3p or cont‐miR in the presence of Dox. C, Actin filaments (red) and vinculin (green) as markers of focal contact at the indicated times were analyzed by immunostaining. Scale bar = 10 µm. D, Murine embryonic fibroblasts (MEFs) were treated with or without 10 ng/mL epidermal growth factor (EGF) for 4 days, and total cell lysates were immunoblotted with the indicated Abs. E, MEFs transfected with miR‐129‐1‐3p or control were treated with or without 10 ng/mL EGF for 4 days, and the morphology of indicated cells was observed by phase‐contrast microscopy. F, Expression of miR‐129‐1‐3p in (D) was analyzed by quantitative real‐time PCR. Scale bar = 100 µm. Relative values ± SD were obtained from 3 independent assays (B, E). *P < .05 and **P < .01 by Student’s t test
3.3. MicroRNA‐129‐1‐3p in human cancer cells
To elucidate the role of miR‐129‐1‐3p in the growth of colon cancer cells in which Src is upregulated, we examined the effects of miR‐129‐1‐3p expression on the tumor growth of HCT116 and HT29 cells (Figure S2A). The qRT‐PCR analysis indicated that miR‐129‐1‐3p was greatly reduced in both cell lines compared with those in normal cells (Figure S2B). Expression of miR‐129‐1‐3p suppressed colony‐forming activity in these cells (Figure 3A). Tumorigenesis of HT29 cells in nude mice was potently suppressed by miR‐129‐1‐3p expression (Figure 3B), suggesting that miR‐129‐1‐3p plays a crucial role in regulating tumor growth when Src is upregulated. We next examined the impact of miR‐129‐1‐3p expression on the responsiveness of HT29 cells to dasatinib. Colony formation assays revealed that dasatinib suppressed Src activity and colony formation in a dose‐dependent manner, and that introduction of miR‐129‐1‐3p significantly sensitized these cells to the suppressive effect of dasatinib (Figure 3C).
Figure 3.
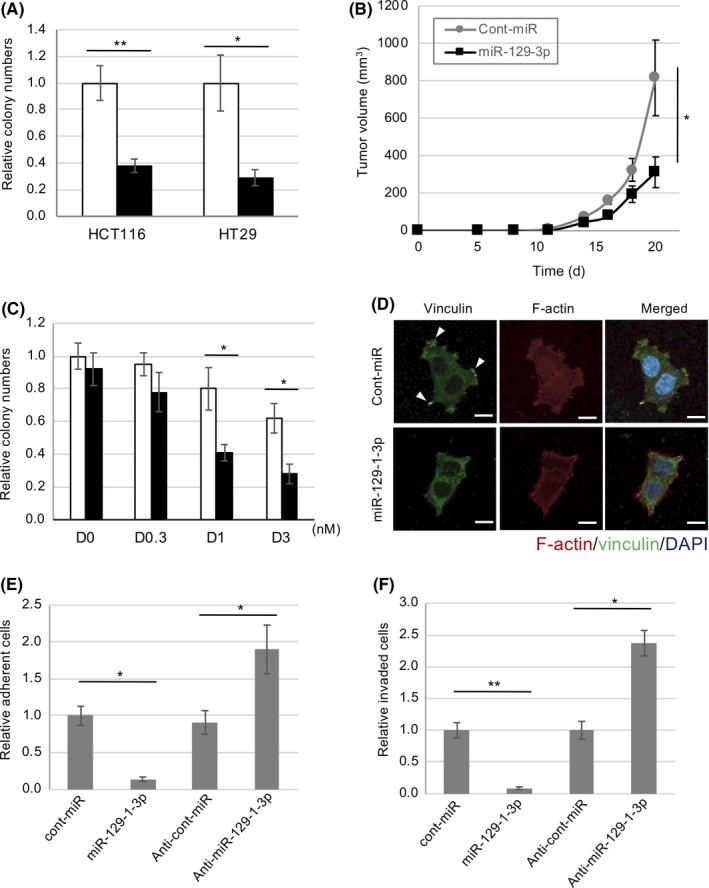
MicroRNA (miR)‐129‐1‐3p as a suppressor of tumor growth and malignancy in human colon cancer cells. A, HT29 and HCT116 cells were transfected with miR‐129‐1‐3p (black bar) or control‐miR (white bar) and subjected to soft‐agar colony formation assays. Relative colony numbers ± SD were obtained from 3 independent experiments. B, HT29 cells transfected with or without miR‐129‐1‐3p were inoculated s.c. into nude mice. Means ± SD of tumor volumes (mm3) obtained from 5 mice are plotted vs. time after inoculation (d). C, miR‐129‐1‐3p increased the sensitivity of HT29 cell growth to dasatinib (D). HT29 cells transfected with control (white bar) or miR‐129‐1‐3p (black bar) were treated with dasatinib at different concentrations, and cell growth was analyzed 3 days after dasatinib treatment. D, HCT116 cells were transfected with miR‐129‐1‐3p or control‐miR, and then subjected to immunocytochemistry. Actin filaments (red), vinculin, a marker of focal contact (green), and DAPI (blue) were analyzed by immunostaining HCT116 cells grown on fibronectin‐coated dishes. Scale bar = 20 µm. E, Cell adhesion assay on fibronectin of HCT116 cells treated with control, miR‐129‐1‐3p, or anti‐miR‐129‐1‐3p. F, In vitro invasiveness of the HCT116 cells used in (E). After 48 h, membranes were detached and cells were stained and counted. Relative number of cells per mm2 ± SD was obtained from 3 independent experiments (A–C, E, F). *P < .05, **P < .01 by Student’s t test
Because Src activity is involved in cancer malignancies, we examined the effects of miR‐129‐1‐3p on the morphology, cell adhesion, and invasiveness of HCT116 cells. Introduction of miR‐129‐1‐3p into HCT116 cells induced disruption of stress fibers (F‐actin) and suppressed formation of focal adhesions (vinculin; Figure 3D). Furthermore, because focal adhesions are crucial for adhesion, motility, and invasion by cancer cells, we examined the effect of miR‐129‐1‐3p expression on these behaviors in HCT116 cells. Consistent with the changes in cell morphology, adhesion of HCT116 cells to fibronectin‐coated dishes was remarkably suppressed by miR‐129‐1‐3p expression (Figure 3E). Conversely, introduction of miR‐129‐1‐3p enhanced adhesion in HCT116 cells, which express a low level of miR‐129‐1‐3p (Figures 3E and S2B). Likewise, introduction of miR‐129‐1‐3p potently suppressed the invasiveness of these cells (Figure 3F). These findings suggest that miR‐129‐1‐3p suppresses tumor growth and malignancies of cancer cells harboring Src upregulation.
3.4. MicroRNA‐129‐1‐3p targets c‐Src/c‐Yes/Fer
To verify the function of miR‐129‐1‐3p, we examined the effect of miR‐129‐1‐3p on Src‐related signaling. In HCT116 cells, as well as Src‐transformed cells (Csk −/−+c‐Src), miR‐129‐1‐3p expression suppressed tyrosine phosphorylation of cellular proteins (Figure 4A). Therefore, we searched for potential targets of miR‐129‐1‐3p in Src‐related molecules reportedly involved in tumor growth and malignancies with help of TargetScan and miRDB (Figure S3). Consequently, c‐Src and c‐Yes, which are members of the SFKs, were found to have miR‐129‐1‐3p target sites in their 3′‐UTR. Their protein levels or specific activities of c‐Src and c‐Yes are frequently upregulated in multiple human cancers.5, 7 Fer, a nonreceptor kinase and key substrate of c‐Src that transduces oncogenic Src signals through autophosphorylation,14 is also identified to have the target site in its ORFs.
Figure 4.
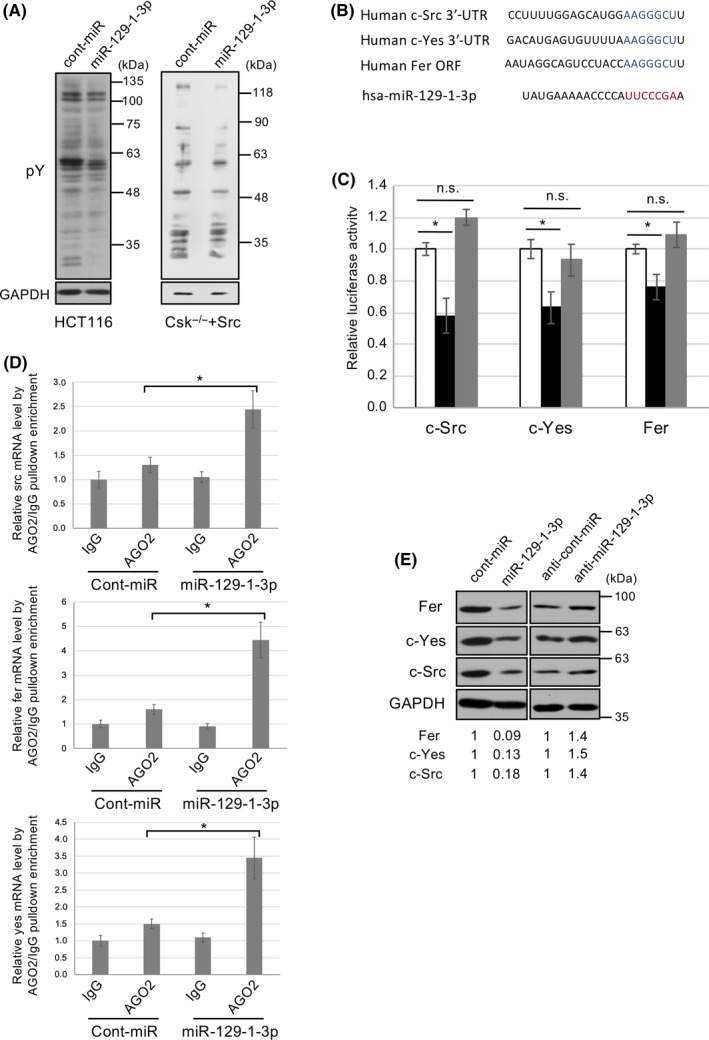
MicroRNA (miR)‐129‐1‐3p targets c‐Src, c‐Yes, and Fer. A, HCT116 cells or c‐Src‐transformed cells were transfected with control or miR‐129‐1‐3p and subjected to immunoblotting with the indicated Abs. pY, phosphorylated tyrosine residue. B, Alignment of RNA sequences of human miR‐129‐1‐3p and potential miR‐129‐1‐3p binding sequences in the c‐Src and c‐Yes 3′‐UTRs, or Fer ORF. Seed sequences of miR‐129‐1‐3p are shown in red, and binding sequences are shown in blue. C, Luciferase reporter constructs containing WT (white and black) or mutated (mt, gray) c‐Src 3′‐UTRs, c‐Yes 3′‐UTR, or Fer ORF were transfected into HCT116 cells. Cells were treated with control (white) or miR‐129‐1‐3p (black or gray), and relative luciferase activity was determined. Relative Renilla luciferase expression was standardized to a transfection control. D, Ribonucleoprotein immunoprecipitation (RIP) assay of miR‐129‐1‐3p interaction with c‐src, c‐yes, or fer mRNA. Each mRNA coimmunoprecipitated by normal IgG or anti‐AGO2 RIP are shown. E, HCT116 cells were transfected with cont‐miR, miR‐129‐1‐3p, anti‐cont‐miR, or anti‐miR‐129‐1‐3p. Total cell lysates were immunoblotted with the indicated Abs. The relative expression level of each miR‐129‐1‐3p target is shown at the bottom of the panels. Relative values ± SD were obtained from 3 independent assays (C, D). *P < .01 by Student’s t test. n.s., not significant
To verify the relationship between each candidate and miR‐129‐1‐3p, we validated our findings using luciferase reporter assays in HCT116 cells. The luciferase activities of constructs containing the predicted target sites c‐Src, c‐Yes, and Fer were significantly reduced in miR‐129‐1‐3p‐transfected cells (Figure 4B,C). The miR‐129‐1‐3p‐mediated reduction of luciferase activities was abolished by mutation of the recognition sites, confirming that miR‐129‐1‐3p interacts specifically with these target sequences. We further undertook ribonucleoprotein immunoprecipitation (RIP) assay and proved that c‐src, c‐yes, and fer mRNA was enriched in RNA‐induced silencing complex compared with the IgG control (Figure 4D). Consistent with the results of the luciferase assays and RIP assay, western blot analysis revealed that c‐Src, c‐Yes, and Fer protein levels were decreased following miR‐129‐3p treatment of HCT116 cells (Figure 4E, left panels). Conversely, anti‐miR‐129‐1‐3p increased the levels of these proteins (Figure 4E, right panels). These findings suggest that miR‐129‐1‐3p suppresses tumor growth and progression by targeting multiple genes related to SFK‐related signaling. Consistent with this, in the Dox‐inducible Src activation system, the set of 51 genes upregulated by Src activation (P < .01 and fold change greater than 1.4) included c‐Src, c‐Yes, and Fer (Figure S4). Interestingly, when we compared the results of the miRNA and mRNA expression profiles, we found that the expression change in each gene was observed simultaneously with, or within 24 hours after, reduced expression of miR‐129‐1‐3p, suggesting that the expression of miR‐129‐1‐3p is linked to expression of c‐Src/c‐Yes/Fer (Figures S1 and S4).
3.5. MicroRNA‐129‐1‐3p‐mediated regulation of c‐Src/c‐Yes/Fer is important for cell adhesion and invasion
We next examined whether the tumor‐suppressive effect of miR‐129‐1‐3p is mediated by the c‐Src/c‐Yes/Fer pathway. When ORF cDNAs of c‐Src, c‐Yes, and Fer mutant, which are resistant to miR‐129‐1‐3p, were introduced into miR‐129‐1‐3p‐treated HCT116 cells, colony‐forming activity was only moderately rescued (Figure 5A,B). These results suggest that c‐Src/c‐Yes/Fer promote tumor growth of colon cancer cells, but the growth suppressive effect of miR‐129‐1‐3p cannot be solely attributed to downregulation of c‐Src/c‐Yes/Fer. Because SFK‐related signaling is involved in cancer malignancies,6 we examined the effects of the miR‐129‐1‐3p‐c‐Src/c‐Yes/Fer pathway on the morphology, adhesion, and invasiveness of colon cancer cells. As mentioned above, miR‐129‐1‐3p caused morphological changes and reduced the number of focal adhesions, concomitant with suppression of cell adhesion and invasive activity of HCT116 cells (Figure 3). Expression of c‐Src/c‐Yes/Fer in miR‐129‐1‐3p‐treated cells almost completely reverted cytoskeletal organization and formation of focal contacts (Figure 5C) and successfully restored the activity of cell adhesion and invasion of HCT116 cells (Figure 5D,E). Taken together, these findings suggest that the miR‐129‐1‐3p‐c‐Src/c‐Yes/Fer circuit plays crucial roles in controlling adhesion and invasive potential in cancer cells.
Figure 5.
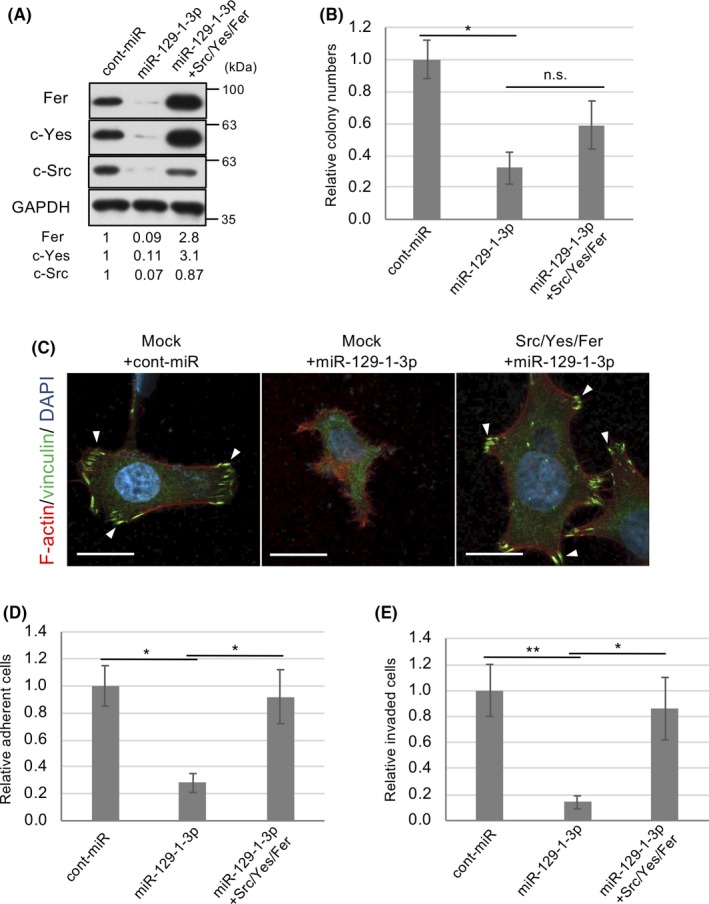
c‐Src/c‐Yes/Fer are crucial for microRNA (miR)‐129‐1‐3p‐mediated regulation of tumor progression. A, HCT116 cells transfected with the c‐Src and c‐Yes ORFs and a Fer mutant resistant to miR‐129‐1‐3p cDNA were treated with control (cont)‐miR or miR‐129‐1‐3p, and total cell lysates were immunoblotted with the indicated abs. The relative expression levels of each protein are shown below the panels. B, Soft‐agar colony formation assays of the cells in (A). Relative colony numbers ± SD were obtained from 3 independent experiments. C, HCT116 cells in (A) were subjected to immunocytochemistry. F‐actin (red), vinculin, a marker of focal contact (green), and DAPI (blue) were analyzed by immunostaining HCT116 cells grown on fibronectin‐coated dishes. Scale bar = 20 µm. D, HCT116 cells used in (A) were subjected to adhesion assay on fibronectin‐coated dishes. E, In vitro invasiveness of the HCT116 cells in (A). Cells were seeded in Matrigel invasion chambers. After 48 h, the membranes were detached, and the cells were stained and counted. Relative numbers of invading cells per mm2 ± SD were obtained from 3 independent experiments (B, D, E). *P < .05, **P < .01 by Student’s t test. n.s., not significant
3.6. MicroRNA‐129‐1‐3p is downregulated in human cancer tissues
Finally, we examined the role of miR‐129‐1‐3p in human cancers by assessing miR‐129‐1‐3p expression in 10 pairs of primary colon tumors and adjacent noncancerous tissues using qRT‐PCR. MicroRNA‐129‐1‐3p was markedly downregulated, relative to the level in adjacent noncancerous tissues, in tumors from 10 of 10 patients examined (Figure 6A). Moreover, western blot analysis of tissue samples showed that activity of SFK (Src pY418) was greatly elevated in 9 of 10 tumor tissues compared to noncancerous tissues (Figure 6B). These observations suggest an inverse correlation between the activity of c‐Src and the expression of miR‐129‐1‐3p in human cancer tissues. To further confirm the importance of miR‐129‐1‐3p, we examined Gene Expression Omnibus data. Because the expression level is very low, only a few datasets include expression measurements for miR‐129‐1‐3p. We chose microarray datasets (GSE33125) from among the available miRNA profiles for malignant cancer tissues and reanalyzed them (see Materials and Methods). In 9 paired noncancerous/cancer colon tissues, miR‐129‐1‐3p was significantly downregulated relative to paired noncancerous tissues (P = .02373) (Figure 6C). These observations suggest that downregulation of miR‐129‐3p is associated with a wide array of human cancers in which SFK signaling is upregulated.
Figure 6.
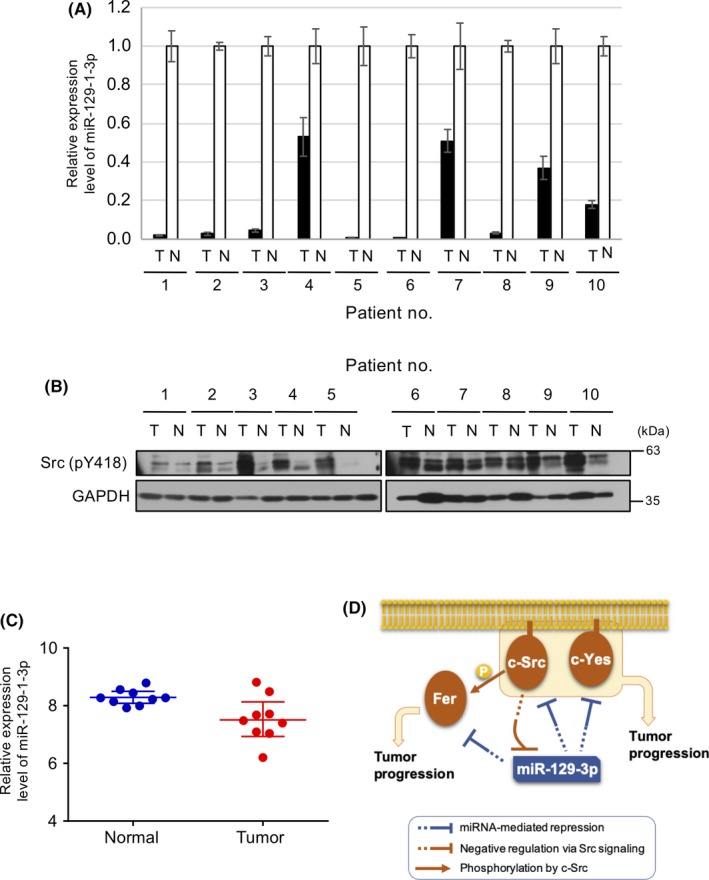
MicroRNA (miR)‐129‐1‐3p is downregulated in human colon cancer cells harboring Src upregulation. A, Relative expression level of miR‐129‐1‐3p in human colon tumors (T) and corresponding noncancerous tissue (N) from individual patients (#1‐10) was analyzed by quantitative real‐time PCR. Relative values ± SD were obtained from 3 independent assays. B, Whole‐tissue lysates in (A) were analyzed by immunoblotting with the indicated Abs. C, miRNA expression profiling in colon cancer tissue vs. paired noncancerous colon tissue, based on a reanalysis of dataset http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE33125. Vertical axis represents the log2 transformed expression level of miR‐129‐1‐3p. Wilcoxon test, P = .0273; fold change, −1.68. D, Schematic model of miR‐129‐1‐3p‐mediated regulation of tumor progression. In normal cells expressing miR‐129‐1‐3p, expression of c‐Src/c‐Yes/Fer is limited to avoid unregulated cell growth and/or motility. Once miR‐129‐1‐3p expression is downregulated by c‐Src activation, expression of c‐Src/c‐Yes/Fer is elevated, resulting in upregulation of Src family of kinase‐mediated oncogenic signaling and stimulation of cell adhesion and invasion
4. DISCUSSION
Previously, to elucidate the molecular mechanisms underlying the early phases of c‐Src‐induced transformation, we established an experimental system using an inducible c‐Src expression system in Csk‐deficient cells.33 Here, we used this system to study the potential roles of miRNAs in the early phases of c‐Src‐induced tumor progression. MicroRNA profiling revealed that the expression of various miRNAs decreased at different times after Src activation. Our previous studies indicated that 7 miRNAs were downregulated in stably c‐Src‐transformed cells, among which 5 miRNAs are implicated in cancers in which c‐Src is upregulated.27, 28, 29, 30 Because the miRNA species identified in our previous studies, including miR‐99a, ‐322 (‐424 in human), ‐503, and ‐542‐3p, decreased at relatively later phases of c‐Src transformation (72 hours after Src activation), we tried to identify miRNAs downregulated at earlier phases of transformation. Of those miRNAs, we focused on miR‐129‐1‐3p, because it was conserved among several species. We showed that reexpression of miR‐129‐3p suppressed c‐Src‐induced transformation, including acquisition of anchorage‐independent growth and morphological changes. Our findings in this study revealed that miR‐129‐1‐3p is a critical mediator of oncogenic potential of c‐Src. A schematic model for the miR‐129‐1‐3p‐mediated regulation of tumor progression is depicted in Figure 6D. When c‐Src is activated by external stimuli such as EGF and ECM, miR‐129‐1‐3p is downregulated. The downregulation of miR‐129‐1‐3p results in upregulation of SFKs such as c‐Src and c‐Yes and the c‐Src substrate Fer, which are required for growth, adhesion, and invasion by cancer cells. These findings suggest that c‐Src‐induced miR‐129‐1‐3p downregulation initiates a positive feedback loop that activates SFK‐mediated oncogenic signaling by inducing expression of SFK and its substrates; this loop might contribute broadly to promotion of cancer malignancy.
The substantial downregulation of miR‐129‐1‐3p in human colon tumors in which c‐Src is activated suggests that miR‐129‐1‐3p is a critical regulator of these cancers. Downregulation of miR‐129‐1‐3p has been observed in some types of cancers, including gastric, hepatocellular, and pancreatic cancer.38, 39, 40, 41 Previous studies showed that miR‐129‐1‐3p was downregulated by DNA methylation,41 but the mechanisms downstream of c‐Src that lead to changes in miR‐129‐1‐3p gene expression remain unclear. Methylation‐specific PCR showed that the methylation status of CpG islands in the promoter region of the miR‐129‐1 gene in human colon cancer cells (Figure S5). Further analysis will be necessary to elucidate the precise mechanisms underlying miR‐129‐1‐3p downregulation in cancers with c‐Src upregulation.
Multiple studies reported c‐Src upregulation in a broad range of cancer types, including colon, lung, breast, prostate, pancreas, head and neck carcinoma, glioma, and melanoma. Overall, the Src‐mediated pathway is activated in 80% of human colon tumors, suggesting that Src activation plays a central role in the initiation and promotion of tumors.42, 43 We confirmed that the expression of miR‐129‐1‐3p was greatly reduced in colon, lung, and pancreas cancer cells, where c‐Src is activated (Figures S2B and S6). Among SFKs, protein levels or specific activities of c‐Src and c‐Yes are frequently upregulated in a variety of human cancers.5, 6 Reintroduction of miR‐129‐1‐3p in colon cancer cells downregulated c‐Src/c‐Yes/Fer expression and suppressed tumorigenesis, cell adhesion, and in vitro invasive activity. In addition, the miR‐129‐1‐3p‐mediated downregulation of c‐Src/c‐Yes/Fer suppressed tyrosine phosphorylation of cellular proteins in human cancer cells, suggesting that miR‐129‐1‐3p repression is required for activation of Src‐related oncogenic signals. These lines of evidence reveal strong correlations of miR‐129‐1‐3p and c‐Src/c‐Yes/Fer with human cancers, and suggest that miR‐129‐1‐3p downregulation contributes to cancer progression not only through upregulation of c‐Src/c‐Yes/Fer themselves, but also through activation of the c‐Src‐mediated oncogenic pathway. Fer expression is also elevated in some cancers, but the underlying mechanisms remained unclear.15, 16, 17, 18, 19 In this study, we provided clear evidence that elevated expression of Fer in various cancers can be explained in some, if not all, cases by frequent downregulation of miR‐129‐1‐3p through Src activation.
In addition to the suppressive effect on tumor growth, expression of miR‐129‐1‐3p induced robust inhibition of integrin‐mediated cell adhesion. The expression of c‐Src/c‐Yes/Fer in miR‐129‐1‐3p‐treated cancer cells significantly rescued cell adhesion, but had more moderate effects on tumor growth. Therefore, it is likely that c‐Src/c‐Yes/Fer are targets of miR‐129‐1‐3p that are preferentially involved in cell adhesion signals. The insufficiency of c‐Src/c‐Yes/Fer for tumor growth suggests that additional miR‐129‐1‐3p targets exist that are required for complete control of tumor growth. Previous studies showed that miR‐129‐1‐3p targets cyclin‐dependent kinase 6 and Glypican‐3 and inhibits cell proliferation by inducing cell arrest, suggesting a potential role for miR‐129‐1‐3p in control of human cancer growth.38, 44 Further analysis of the contribution of such additional targets will be necessary to elucidate the whole picture of the c‐Src‐miR‐129‐1‐3p axis.
In this study, we proposed a new mechanism for the upregulation of Src‐related oncogenic signals: downregulation of c‐Src activation and upregulation of SFKs (c‐Src/c‐Yes) and its downstream target (Fer) through downregulation of miR‐129‐1‐3p. If this is the case for human cancers, upregulation of the c‐Src‐mediated pathway observed in various human cancers could be induced through a positive feedback loop involving miR‐129‐1‐3p and c‐Src that can be initiated by growth factor and integrin stimuli. Furthermore, recent work showed that c‐Src is also activated by Fer, suggesting that c‐Src activation is maintained by its substrate.45 Thus, upregulation of c‐Src might further amplify the positive feedback loop mediated by direct regulation of c‐Src‐related protein levels by miR‐129‐1‐3p and regulation of c‐Src kinase activity by Fer, thereby promoting tumor malignancy mediated by c‐Src activation. These signaling circuits could account for the frequent upregulation of c‐Src in various human cancers. Because the Src‐related oncogenic pathway is frequently activated in human cancers, the functional analysis of the c‐Src‐miR‐129‐1‐3p circuits presents a leap forward in our understanding of cancer etiology. Our study provides insights into the functions of new signaling circuits, and offers new opportunities for therapeutic intervention in a wide array of human cancers.
CONFLICT OF INTEREST
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
We are grateful to Dr F. Imamoto and Dr T. Akagi for their generous gifts of reagents. This work was supported by a Grant‐in‐Aid for Scientific Research (B) from the Ministry of Education, Culture, Sports, Science and Technology of Japan.
Okuzaki D, Yamauchi T, Mitani F, et al. c‐Src promotes tumor progression through downregulation of microRNA‐129‐1‐3p. Cancer Sci. 2020;111:418–428. 10.1111/cas.14269
REFERENCES
- 1. Jove R, Hanafusa H. Cell transformation by the viral src oncogene. Annu Rev Cell Biol. 1987;3:31‐56. [DOI] [PubMed] [Google Scholar]
- 2. Brown M, Cooper J. Regulation, substrates and functions of src. Biochim Biophys Acta. 1996;1287:121‐149. [DOI] [PubMed] [Google Scholar]
- 3. Playford MP, Schaller MD. The interplay between Src and integrins in normal and tumor biology. Oncogene. 2004;23(48):7928‐7946. [DOI] [PubMed] [Google Scholar]
- 4. Frame M. Src in cancer: deregulation and consequences for cell behaviour. Biochi Biophy Acta. 2002;1602:114‐130. [DOI] [PubMed] [Google Scholar]
- 5. Ishizawar R, Parsons S. c‐Src and cooperating partners in human cancer. Cancer Cell. 2004;6:209‐214. [DOI] [PubMed] [Google Scholar]
- 6. Summy JM, Gallick GE. Src family kinases in tumor progression and metastasis. Cancer Metastasis Rev. 2003;22(4):337‐358. [DOI] [PubMed] [Google Scholar]
- 7. Yeatman TJ. A renaissance for SRC. Nat Rev Cancer. 2004;4(6):470‐480. [DOI] [PubMed] [Google Scholar]
- 8. Okada M. Regulation of the SRC family kinases by Csk. Int J Biol Sci. 2012;8(10):1385‐1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Irby R, Yeatman T. Role of Src expression and activation in human cancer. Oncogene. 2000;19:5636‐5642. [DOI] [PubMed] [Google Scholar]
- 10. Ingley E. Src family kinases: regulation of their activities, levels and identification of new pathways. Biochim Biophys Acta. 2008;1784(1):56‐65. [DOI] [PubMed] [Google Scholar]
- 11. Parsons SJ, Parsons JT. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23(48):7906‐7909. [DOI] [PubMed] [Google Scholar]
- 12. Oneyama C, Hikita T, Enya K, et al. The lipid raft‐anchored adaptor protein Cbp controls the oncogenic potential of c‐Src. Mol Cell. 2008;30(4):426‐436. [DOI] [PubMed] [Google Scholar]
- 13. Oneyama C, Iino T, Saito K, Suzuki K, Ogawa A, Okada M. Transforming potential of Src family kinases is limited by the cholesterol‐enriched membrane microdomain. Mol Cell Biol. 2009;29(24):6462‐6472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Oneyama C, Yoshikawa Y, Ninomiya Y, Iino T, Tsukita S, Okada M. Fer tyrosine kinase oligomer mediates and amplifies Src‐induced tumor progression. Oncogene. 2016;35(4):501‐512. [DOI] [PubMed] [Google Scholar]
- 15. Allard P, Zoubeidi A, Nguyen LT, et al. Links between Fer tyrosine kinase expression levels and prostate cell proliferation. Mol Cell Endocrinol. 2000;159(1–2):63‐77. [DOI] [PubMed] [Google Scholar]
- 16. Kawakami M, Morita S, Sunohara M, et al. FER overexpression is associated with poor postoperative prognosis and cancer‐cell survival in non‐small cell lung cancer. Int J Clin Exp Pathol. 2013;6(4):598‐612. [PMC free article] [PubMed] [Google Scholar]
- 17. Li H, Ren Z, Kang X, et al. Identification of tyrosine‐phosphorylated proteins associated with metastasis and functional analysis of FER in human hepatocellular carcinoma cells. BMC Cancer. 2009;9:366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Menges CW, Chen Y, Mossman BT, Chernoff J, Yeung AT, Testa JR. A phosphotyrosine proteomic screen identifies multiple tyrosine kinase signaling pathways aberrantly activated in malignant mesothelioma. Genes Cancer. 2010;1(5):493‐505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Miyata Y, Kanda S, Sakai H, Greer PA. Feline sarcoma‐related protein expression correlates with malignant aggressiveness and poor prognosis in renal cell carcinoma. Cancer Sci. 2013;104(6):681‐686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ha M, Kim VN. Regulation of microRNA biogenesis. Nat Rev Mol Cell Biol. 2014;15(8):509‐524. [DOI] [PubMed] [Google Scholar]
- 21. Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835‐840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Mukherji S, Ebert MS, Zheng GX, Tsang JS, Sharp PA, van Oudenaarden A. MicroRNAs can generate thresholds in target gene expression. Nat Genet. 2011;43(9):854‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Di Leva G, Croce CM. miRNA profiling of cancer. Curr Opin Genet Dev. 2013;23(1):3‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kunej T, Godnic I, Horvat S, Zorc M, Calin GA. Cross talk between microRNA and coding cancer genes. Cancer J. 2012;18(3):223‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zhang B, Pan X, Cobb GP, Anderson TA. microRNAs as oncogenes and tumor suppressors. Dev Biol. 2007;302(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 26. Oneyama C, Hikita T, Nada S, Okada M. Functional dissection of transformation by c‐Src and v‐Src. Genes Cells. 2008;13(1):1‐12. [DOI] [PubMed] [Google Scholar]
- 27. Matsuyama R, Okuzaki D, Okada M, Oneyama C. MicroRNA‐27b suppresses tumor progression by regulating ARFGEF1 and focal adhesion signaling. Cancer Sci. 2016;107(1):28‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oneyama C, Ikeda J, Okuzaki D, et al. MicroRNA‐mediated downregulation of mTOR/FGFR3 controls tumor growth induced by Src‐related oncogenic pathways. Oncogene. 2011. [DOI] [PubMed] [Google Scholar]
- 29. Oneyama C, Kito Y, Asai R, et al. MiR‐424/503‐mediated Rictor upregulation promotes tumor progression. PLoS ONE. 2013;8(11):e80300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Oneyama C, Morii E, Okuzaki D, et al. MicroRNA‐mediated upregulation of integrin‐linked kinase promotes Src‐induced tumor progression. Oncogene. 2012;31(13):1623‐1635. [DOI] [PubMed] [Google Scholar]
- 31. Oneyama C, Okada M. MicroRNAs as the fine‐tuners of Src oncogenic signalling. J Biochem. 2015;157(6):431‐438. [DOI] [PubMed] [Google Scholar]
- 32. Kokuda R, Watanabe R, Okuzaki D, Akamatsu H, Oneyama C. MicroRNA‐137‐mediated Src oncogenic signaling promotes cancer progression. Genes Cells. 2018;23(8):688‐701. [DOI] [PubMed] [Google Scholar]
- 33. Inoue K, Sone T, Oneyama C, et al. A versatile nonviral vector system for tetracycline‐dependent one‐step conditional induction of transgene expression. Gene Ther. 2009;16(12):1383‐1394. [DOI] [PubMed] [Google Scholar]
- 34. Kajiwara K, Yamada T, Bamba T, et al. c‐Src‐induced activation of ceramide metabolism impairs membrane microdomains and promotes malignant progression by facilitating the translocation of c‐Src to focal adhesions. Biochem J. 2014;458(1):81‐93. [DOI] [PubMed] [Google Scholar]
- 35. Koutsaki M, Spandidos DA, Zaravinos A. Epithelial‐mesenchymal transition‐associated miRNAs in ovarian carcinoma, with highlight on the miR‐200 family: prognostic value and prospective role in ovarian cancer therapeutics. Cancer Lett. 2014;351(2):173‐181. [DOI] [PubMed] [Google Scholar]
- 36. Yang D, Du G, Xu A, Xi X, Li D. Expression of miR‐149‐3p inhibits proliferation, migration, and invasion of bladder cancer by targeting S100A4. Am J Cancer Res. 2017;7(11):2209‐2219. [PMC free article] [PubMed] [Google Scholar]
- 37. Liang Y, Hou L, Li L, et al. Dichloroacetate restores colorectal cancer chemosensitivity through the p53/miR‐149‐3p/PDK2‐mediated glucose metabolic pathway. Oncogene. 2020;39(2):469‐485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Yu X, Song H, Xia T, et al. Growth inhibitory effects of three miR‐129 family members on gastric cancer. Gene. 2013;532(1):87‐93. [DOI] [PubMed] [Google Scholar]
- 39. Wang D, Luo L, Guo J. miR‐129‐1‐3p inhibits cell migration by targeting BDKRB2 in gastric cancer. Med Oncol. 2014;31(8):98. [DOI] [PubMed] [Google Scholar]
- 40. Zhang Z, Pan B, Lv S, et al. Integrating microRNA expression profiling studies to systematically evaluate the diagnostic value of microRNAs in pancreatic cancer and validate their prognostic significance with the cancer genome atlas data. Cell Physiol Biochem. 2018;49(2):678‐695. [DOI] [PubMed] [Google Scholar]
- 41. Cui S, Zhang K, Li C, et al. Methylation‐associated silencing of microRNA‐129‐3p promotes epithelial‐mesenchymal transition, invasion and metastasis of hepatocelluar cancer by targeting Aurora‐A. Oncotarget. 2016;7(47):78009‐78028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Chen J, Elfiky A, Han M, Chen C, Saif MW. The role of Src in colon cancer and its therapeutic implications. Clin Colorectal Cancer. 2014;13(1):5‐13. [DOI] [PubMed] [Google Scholar]
- 43. Cartwright CA, Meisler AI, Eckhart W. Activation of the pp60c‐src protein kinase is an early event in colonic carcinogenesis. Proc Natl Acad Sci USA. 1990;87(2):558‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Maurel M, Jalvy S, Ladeiro Y, et al. A functional screening identifies five microRNAs controlling glypican‐3: role of miR‐1271 down‐regulation in hepatocellular carcinoma. Hepatology. 2013;57(1):195‐204. [DOI] [PubMed] [Google Scholar]
- 45. Stanicka J, Rieger L, O'Shea S, et al. FES‐related tyrosine kinase activates the insulin‐like growth factor‐1 receptor at sites of cell adhesion. Oncogene. 2018;37(23):3131‐3150. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials