

Abstract
CXCL9, an IFN‐γ inducible chemokine, has been reported to play versatile roles in tumor‐host interrelationships. However, little is known about its role in intrahepatic cholangiocarcinoma (iCCA). Here, we aimed to elucidate the prognostic and biological implications of CXCL9 in iCCA. Endogenous CXCL9 expression and the number of tumor‐infiltrating lymphocytes were immunohistochemically assessed in resection specimens. These data were validated in mice treated by silencing CXCL9 with short hairpin RNA. In addition, the induction of endogenous CXCL9 and the effects of CXCL9 on tumor biological behaviors were evaluated in human cholangiocarcinoma cell lines. Immunohistochemical analyses revealed that high CXCL9 expression was closely correlated with prolonged postoperative survival and a large number of tumor‐infiltrating natural killer (NK) cells. In fact, due to the trafficking of total and tumor necrosis factor‐related apoptosis‐inducing ligand‐expressing NK cells into tumors, CXCL9‐sufficient cells were less tumorigenic in the liver than CXCL9‐deficient cells in mice. Although CXCL9 involvement in tumor growth and invasion abilities differed across cell lines, it did not exacerbate these abilities in CXCL9‐expressing cell lines. We showed that CXCL9 was useful as a prognostic marker. Our findings also suggested that CXCL9 upregulation might offer a therapeutic strategy for treating CXCL9‐expressing iCCA by augmenting anti–tumor immune surveillance.
Keywords: CXCL9, intrahepatic cholangiocarcinoma, natural killer cell, tumor necrosis factor‐related apoptosis‐inducing ligand, tumor‐infiltrating lymphocyte
CXCL9, an IFN‐γ inducible chemokine, plays versatile roles in the tumor‐host interrelationship. In this study, we demonstrated that elevated intratumoral CXCL9 expression was associated with a large number of tumor‐infiltrating NK cells, leading to favorable postoperative survival in patients with intrahepatic cholangiocarcinoma. Upregulation of CXCL9 might be an immunotherapeutic approach for treating intrahepatic cholangiocarcinoma.

1. INTRODUCTION
Intrahepatic cholangiocarcinoma (iCCA) is the second most common primary hepatic malignancy that originates from intrahepatic bile epithelium. Although iCCA is a relatively infrequent entity, alarming rises in iCCA incidence and iCCA‐related mortality have been reported worldwide over the past few decades.1, 2 In addition, surgical resection and transplantation remain the only potentially curative approaches, and limited chemotherapeutic treatment options and frequent treatment resistance are unduly prominent in patients with iCCA.3, 4, 5, 6 Hence, the iCCA prognosis continues to be pessimistic, with a 5‐year survival rate of <10%,7 and rapid, fatal deterioration.
It has become increasingly important to understand the tumor‐host interrelationship to help overcome malignancies. Recent accumulated evidence has shown that the tumor immune microenvironment (TIME) intensely dictates tumor fate. Favorable cancer‐immune editing can provide new therapeutic directions that might potentially eradicate tumors, exemplified by adoptive immune cell therapy8 or immune check‐point blockage.9 However, recent studies have shown that only a small subset of patients with iCCA derive therapeutic benefits from immune‐modulating drugs.10, 11 Developing effective therapies that can alter TIME remains challenging.
Chemokines comprise a large family of small, functionally divergent molecules that play crucial roles in orchestrating TIME, tumor growth and metastases. Among the various types of chemokines, CXCL9 is a member of the ELR‐motif negative‐CXC chemokine family, which is induced by IFN‐γ. CXCL9 is produced by macrophages, endothelial cells, hepatocytes and tumors. As a CXCR3 ligand, CXCL9 mainly acts as a chemoattractant to activated immune cells, including T cells and natural killer (NK) cells.12 We previously reported that intra‐graft CXCL9 mRNA was an effective biomarker of acute cellular rejection (ACR) after liver transplantation.13 In this study, we showed that intra‐graft CXCL9 was most upregulated in patients with ACR, and its expression was sensitive to ACR treatments, indicating that CXCL9 was a key regulator of liver cellular immunity. Gorbachev et al14 showed that CXCL9‐deficient tumor cells were more tumorigenic than CXCL9‐sufficient counterparts because CXCL9‐deficient tumor cells disrupted activated T‐cell and NK‐cell recruitment into tumors in murine cutaneous fibrosarcoma. In addition, CXCL9 directly influenced tumor biological properties by binding to the CXCR3 receptor. Importantly, there are two CXCR3 splice variants, CXCR3A and CXCR3B, with pro–tumor and anti–tumor functions, respectively. Studies have shown that CXCR3A contributed to tumor invasion and metastases, and that CXCR3B suppressed these activities in cancer settings.15, 16, 17, 18 Furthermore, previous studies revealed that elevated CXCL9 expression was associated with favorable survival following curative surgery in ovarian and colorectal cancers,19, 20 whereas it was connected to unfavorable postoperative outcome in renal cell carcinoma.21 These findings indicated that CXCL9 plays various roles, some contradictory, depending on the cancer type. Few studies have explored the prognostic and biological significance of CXCL9 in iCCA.
We hypothesized that endogenous CXCL9 expression might regulate TIME and tumor behaviors, and, thus, it might be associated with patient prognosis in iCCA. Accordingly, the present study aimed to evaluate the prognostic significance of endogenous CXCL9 expression and its impact on TIME and tumor biological behaviors in iCCA.
2. MATERIALS AND METHODS
2.1. Cell lines and cell culture
For in vitro experiments, we used four human CCA cell lines: MzChA‐1, TFK‐1, HuCCT‐1 and CCLP‐1.22, 23, 24, 25 For in vivo experiments, we used a murine iCCA cell line, 1‐1a, derived from C57BL6/J mice. MzChA‐1, CCLP‐1 and murine 1‐1a cell lines were generously provided by Professor Gregory J. Gores (Mayo Clinic). TFK‐1 and HuCCT‐1 cell lines were obtained from the Riken BioResource Center. Cells were cultured in D‐MEM supplemented with 10% FBS and 100 units/mL penicillin. Cell cultures were maintained in 5% CO2 at 37°C.
2.2. Patients and tissue samples
We analyzed tissue samples from 70 consecutive patients who underwent potentially curative resections for iCCA, between September 1998 and January 2015 at Osaka University Hospital. In all patients, iCCA was histologically confirmed by expert pathologists at our institute. Clinicopathological patient characteristics are summarized in Table S1. In 41 patients, tumors were classified as the peripheral type, and approximately half were in stages ≤ II based on the 8th edition of the UICC TNM classification system.26 The use of clinical samples was approved by the Human Ethics Review Committee of Osaka University Graduate School of Medicine (no. 15149). Informed consent forms were signed by each patient before participation.
2.3. Histological analysis
Specimens were fixed with 10% formalin and embedded in paraffin. Next, 2.5‐µm sections were cut, mounted on slides, and stained for immunohistochemistry (IHC) analyses. Slides were deparaffinized, and antigens were retrieved with sodium citrate buffer (pH 6) and autoclave heating. Next, slides were incubated overnight at 4°C with primary Abs specific for CXCL9, CD8, NCR1, FOXP3 and cytokeratin (CK) 19, or incubated at room temperature for 30 minutes with a primary Ab specific for T‐bet. Primary Abs and dilutions were listed in Table S2. Slides were subsequently stained with Dako DAB+ substrate buffer (Agilent) and counterstained with hematoxylin. Microscopic evaluations were conducted by two independent observers (YF and MK), who had no knowledge of clinicopathological factors in each patient.
2.4. CXCL9 knockdown with shRNA‐mediated gene silencing
MISSION shRNA, frozen in bacterial glycerol stocks, were purchased from Sigma‐Aldrich. These sequence‐verified shRNA lentiviral TRC2‐pLKO‐puro plasmids were designed to silence murine CXCL9. In this study, we used two shRNA that targeted different sequences in the CXCL9 coding regions:
CCGGCATCATCTTCCTGGAGCAGTGCTCGAGCACTGCTCCAGGAAGATGATGTTTTTTG‐(Clone ID:NM_008599.4‐86s21c1, referred to as “sh‐CXCL9 1”) and
CCGGCTAGATCCGGACTCGGCAAATCTCGAGATTTGCCGAGTCCGGATCTAGTTTTTTG‐(Clone ID:NM_008599.4‐276s21c1, referred to as “sh‐CXCL9 2”).
An empty vector, SHC002 (a non–targeting shRNA that activates the RNA interference pathway without targeting any known murine gene), was also purchased from Sigma‐Aldrich (referred to as “empty shRNA”). Each bacterial stock sample was streaked onto a Luria‐Bertani agar plate (Thermo Fisher Scientific). After a 20‐hour incubation, a single colony was selected and propagated in Luria‐Bertani medium for 16 hours. Then, the plasmid DNA was purified with the QIAGEN Plasmid Midi Kit (Qiagen), according to the manufacturer’s recommendations. To produce a lentivirus that encoded an shRNA against CXCL9, we mixed the plasmid DNA and two packaging vectors (pCAG‐HIVgp and pCMV‐VSV‐G‐RSV‐Rev, provided by Dr M. Konno, Osaka University, Osaka, Japan) at a ratio of 2:1:1, respectively, with P3000 and Lipofectamine 3000 Transfection Reagents (Thermo Fisher Scientific). This mixture was transfected into HEK293Ta cells. After 48 hours of transfection, the released lentivirus particles were collected in the supernatant. The purified shRNA‐encoding lentivirus was then transduced into a 40% confluent murine iCCA cell line with polybrene (Nacalai Tesque). Transduced cells were selected with puromycin (Thermo Fisher Scientific). Subclones were obtained with the limiting dilution method.
2.5. Flow cytometry and other methods
Trimmed mouse liver tumors were dissociated to single cells with the Tumor Dissociation Kit (Miltenyl Biotec), according to the manufacturer’s recommendations. Dissociated cells were hemolyzed with BD Pharm Lyse (BD Biosciences). Leukocyte populations were isolated with 35% Percoll (GE Healthcare) and blocked with anti–CD16/32 Ab (clone 2.4G2, Bio X Cell). Cell viability was simultaneously assessed by staining with the Zombie Red Fixable Viability Kit (BioLegend) at room temperature for 15 minutes. Cells were then stained with Abs (Table S2) for 30 minutes at 4°C. After washing, cells were analyzed with an LSR Fortessa flow cytometer (BD Biosciences). Additional in vitro methods are provided in Appendix S1.
2.6. In vivo experiment
The animal experimental protocol was approved by the Animal Experiments Committee at Osaka University (no. 30‐081‐001). The animal experiments were conducted according to the National Institutes of Health guidelines for the use of experimental animals. Seven‐week‐old C57BL/6 mice were purchased from Charles River. We anesthetized the mice with intraperitoneal injections of midazolam, medetomidine and butorphanol, and then randomly inoculated 100‐μL suspensions of 1‐1a cells transduced with empty shRNA, sh‐CXCL9 1 or sh‐CXCL9 2 (1.5 × 106 cells per mouse) into mouse spleens, followed by splenectomy to induce liver tumor formation. To deplete NK cells, mice were injected intraperitoneally with 200 (day −3) and 300 μg (day −1) InVivoMab anti–mouse NK1.1 mAb (clone PK136, Bio X Cell). Depletion efficiency was evaluated by injecting treated sentinel mice with the anti–mouse NK1.1 mAb and counting labeled cells with flow cytometry. Successful NK removal was defined as >90% depletion (data not shown). To maintain depletion, mice were injected with 150 μg anti–mouse NK1.1 mAb every 7 days after 1‐1a cell inoculations.
2.7. Statistical analysis
Statistical analyses were performed with JMP software (SAS Institute). Continuous variables were expressed as the median (range) or mean ± SD. Variables were compared between groups with the Student’s t test, Welch’s t test or Wilcoxon signed‐rank test, as appropriate. Survival curves were estimated using the Kaplan‐Meier method, and compared using the log‐rank test. Univariate and multivariate analyses were carried out using a Cox proportional hazards model and any variable deemed significant (P < 0.05) in the univariate analyses could be a candidate for multivariate analyses. P‐values < 0.05 were considered significant.
3. RESULTS
3.1. High endogenous CXCL9 expression was correlated with favorable postoperative survival
We evaluated endogenous CXCL9 expression in 70 resected specimens with IHC. CXCL9 was predominantly stained in tumor cell cytoplasm. Weak background staining was observed in normal bile duct epithelial cells (Figure 1A, lower left insets). Patients were first grouped according to IHC intensity, as follows (Figure 1A): strongly (n = 6), moderately (n = 25) and weakly (n = 30) positive staining, or negative (n = 9) staining. In the first two groups, staining intensity was higher in tumor cells than in corresponding normal bile duct epithelial cells. Patients were further classified in terms of high CXCL9 expression (CXCL9high; strongly or moderately positive; n = 31) or low CXCL9 expression (CXCL9low; weakly positive or negative; n = 39). CXCL9high tumors were less aggressive than CXCL9low tumors (smaller tumor size, fewer metastatic nodes and less vascular invasion; Table S3).
Figure 1.
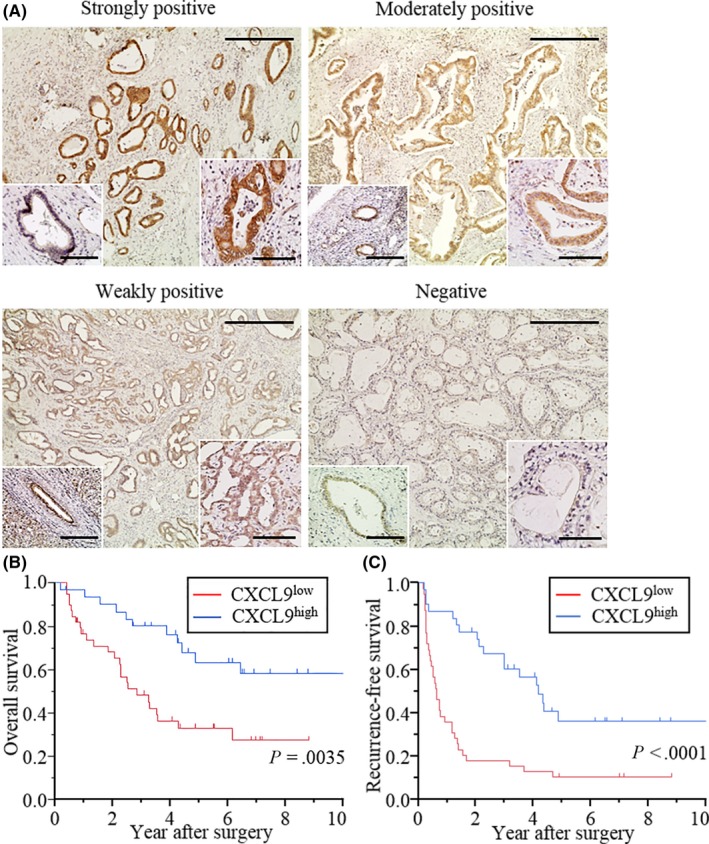
Endogenous CXCL9 expression in intrahepatic cholangiocarcinoma and postoperative survival. A, Representative images of CXCL9 expression in resected immunohistochemistry (IHC) stained specimens. CXCL9 expression was categorized as strongly (n = 6), moderately (n = 25) or weakly positive (n = 30), or negative (n = 9), according to IHC staining intensity. (Lower right inset) Examples of staining at high magnification. (Lower left inset) Examples of normal bile duct epithelial cells, at high magnification, weakly stained with anti–CXCL9 Ab. Scale bars = 400 μm for low magnification and 100 μm for high magnification. B, Kaplan‐Meier curves show postoperative overall survival (OS) in patients with intrahepatic cholangiocarcinoma (iCCA), based on CXCL9 expression. OS was significantly better in patients classified as CXCL9high (strongly or moderately positive; n = 31), compared to those classified as CXCL9low (weakly positive or negative; n = 39); P = 0.0035. C, Kaplan‐Meier curves show postoperative recurrence‐free survival (RFS) in patients with iCCA, based on CXCL9 expression. RFS was significantly better in patients classified as CXCL9high, compared to those classified as CXCL9low (P < 0.0001)
Patients with CXCL9high showed favorable postoperative overall survival (OS; P = 0.0035; Figure 1B) and recurrence‐free survival (RFS; P < 0.0001; Figure 1C) compared to those with CXCL9low. A multivariate analysis revealed that CXCL9high was an independent prognostic factor for prolonged RFS (hazard ratio [HR]: 0.51; 95% confidence interval [95% CI]: 0.26‐0.99, P = 0.048) (Table 1). In contrast, no significant factors were associated with overall survival (Table S4).
Table 1.
Clinicopathological factors that potentially affect recurrence‐free survival in patients with intrahepatic cholangiocarcinoma
| Factor | Univariate analysis | Multivariate analysis | ||||
|---|---|---|---|---|---|---|
| HR | 95% CI | P‐value | HR | 95% CI | P‐value | |
| Age | ||||||
| ≥65 vs <65 y | 0.97 | 0.51‐1.87 | 0.91 | |||
| Gender | ||||||
| Male vs female | 0.90 | 0.47‐1.75 | 0.75 | |||
| Hepatitis | ||||||
| Presence vs absence | 0.58 | 0.29‐1.06 | 0.078 | |||
| Tumor origin | ||||||
| Hilar vs peripheral | 2.17 | 1.13‐4.19 | 0.021 | 1.23 | 0.63‐2.40 | 0.53 |
| Tumor size | ||||||
| ≥50 vs <50 mm | 2.95 | 1.65‐5.16 | 0.000040 | N/A | ||
| Tumor number | ||||||
| Multiple vs single | 2.81 | 1.43‐5.26 | 0.0035 | N/A | ||
| CA19‐9 | ||||||
| ≥100 vs <100 U/mL | 1.75 | 0.98‐3.05 | 0.058 | |||
| CEA | ||||||
| ≥5 vs <5 ng/mL | 5.78 | 2.81‐11.58 | <0.0001 | 1.76 | 1.32‐6.74 | 0.0092 |
| Adjuvant chemotherapy | ||||||
| Presence vs absence | 0.86 | 0.50‐1.51 | 0.59 | |||
| Vascular invasion | ||||||
| Presence vs absence | 3.79 | 2.12‐6.81 | <0.0001 | N/A | ||
| Tumor differentiation | ||||||
| Por vs Tub, muc | 1.55 | 0.69‐3.16 | 0.27 | |||
| Pathological T factor | ||||||
| T3, 4 vs T1a, b, 2 | 2.93 | 1.67‐5.14 | 0.00020 | N/A | ||
| Pathological N factor | ||||||
| N1 vs N0 | 3.63 | 2.04‐6.44 | <0.0001 | N/A | ||
| Pathological stage | ||||||
| IIIA, B vs IA, B, II | 3.28 | 1.86‐5.97 | <0.0001 | 2.09 | 1.08‐4.13 | 0.029 |
| CXCL9 expression | ||||||
| High vs low | 0.33 | 0.18‐0.59 | 0.00010 | 0.51 | 0.26‐0.99 | 0.048 |
Abbreviations: CI, confidence interval; HR, hazard ratio; N/A, not applicable.
3.2. High endogenous CXCL9 expression was associated with high numbers of tumor‐infiltrating natural killer cells
Next, we analyzed the extent to which endogenous CXCL9 expression affected the abundance of tumor‐infiltrating T‐helper‐1 (Th1) cells, cytotoxic T cells, NK cells and regulatory T (Treg) cells. These cells typically express CXCR3 and were identified by staining with anti–T‐bet, anti–CD8, anti–NCR1 and anti–FOXP3 Abs, respectively (Figure 2A‐D). The average number of DAB‐stained cells was counted in four randomly selected fields with a light microscope at 200× magnification. Although the number of T‐bet+ Th1 cells was significantly higher in patients with CXCL9high and a trend toward significance was detected with the number of CD8+ cytotoxic T cells in all iCCA stages, the difference was not statistically significant when early (stages ≤II) and advanced (stages ≥III) cancer stages were considered separately (Figure 2E,F). NCR+ NK cells were significantly enriched in patients with CXCL9high compared to those with CXCL9low, in both early and advanced cancer stages (Figure 2G). FOXP3+ Treg cells were comparable between patients with CXCL9high and CXCL9low (Figure 2H).
Figure 2.
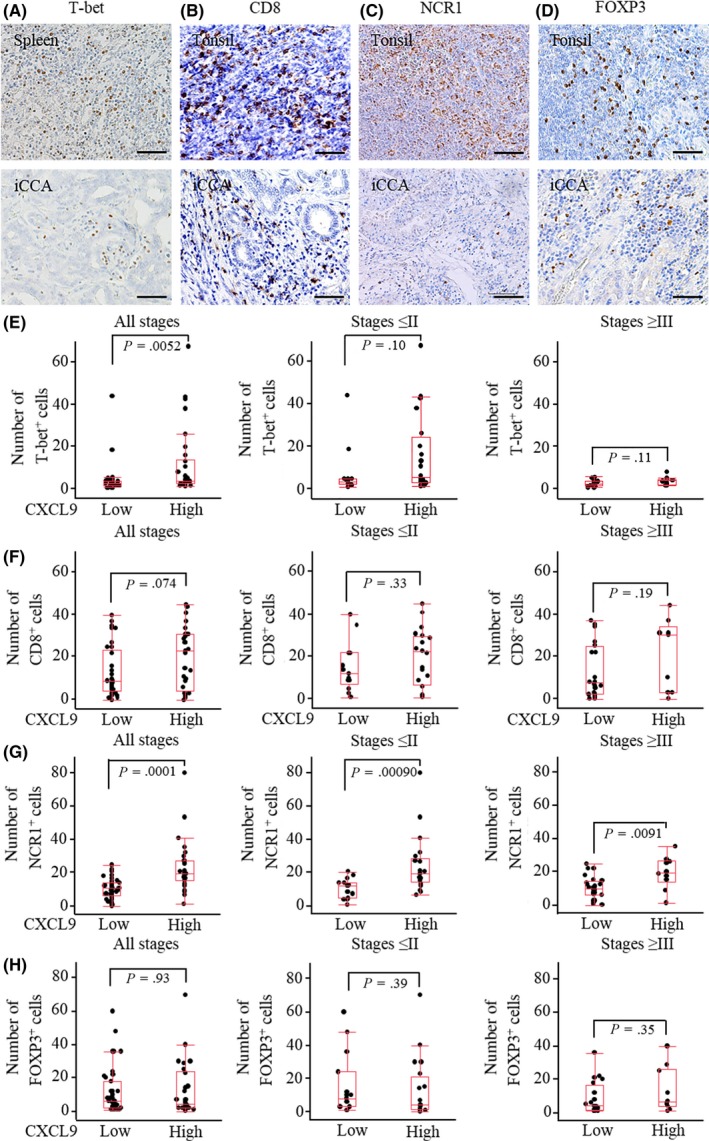
Correlation between CXCL9 expression and tumor‐infiltrating lymphocytes. A, (Lower panel) Representative image of tumor‐infiltrating Th1 cells stained with anti–T‐bet Ab; (upper panel) positive control: spleen; B, lower panel, representative image of tumor‐infiltrating cytotoxic T cells stained with anti–CD8 Ab; (upper panel) positive control: tonsil; C, (lower panel) representative image of tumor‐infiltrating natural killer (NK) cells stained with anti–NCR1 Ab; (upper panel) positive control: tonsil; D, (lower panel) representative image of tumor‐infiltrating Treg cells stained with anti–FOXP3 Ab; (upper panel) positive control: tonsil. Scale bars = 200 µm. E, The number of T‐bet+ Th1 cells was significantly larger in patients with CXCL9high compared to those with CXCL9low in all stages; however, the difference was not significant when cancer stages were analyzed separately (ie, stages ≤II or stages ≥III). F, The number of CD8+ cytotoxic T cells tended to be larger in patients with CXCL9high compared to those with CXCL9low in all stages. G, The number of NCR+ NK cells was significantly larger in patients with CXCL9high compared to those with CXCL9low in all stages, in stages ≤II, and in stages ≥III. H, The number of FOXP3+ Treg cells was similar between patients with CXCL9high and CXCL9low in all stages, in stages ≤II, and in stages ≥III
3.3. CXCL9 knockdown led to greater tumor burden by disrupting natural killer cell recruitment into tumors in mice
To validate the impact of endogenous CXCL9 expression on tumor burden, we compared tumor volumes between murine iCCA 1‐1a cells that expressed CXCL9 (empty shRNA) and 1‐1a cells with knocked‐down CXCL9 (sh‐CXCL9 1 or sh‐CXCL9‐2) (Figure S1A,B). Cell proliferation was similar across these three cell lines (Figure S2). We inoculated all three cell lines into the spleen of mice, and 35 days later, tumor‐bearing mice were killed. Tumors in the liver were manually, meticulously trimmed (Figure 3A). Tumor volumes (% of whole liver) were significantly higher in mice inoculated with the two CXCL9‐deficient cell lines compared to mice inoculated with control cells (Figure 3B). All tumors were strongly stained with the CK19 Ab, a marker of bile duct epithelial cells, but CXCL9 expression was higher in control tumors compared to CXCL9‐deficient tumors (Figure S3A,B). Next, we investigated the influence of endogenous CXCL9 expression on tumor‐infiltrating lymphocytes. After gating the CD45+, CD3− (Figure 3C) and CD11b− cell populations, we compared NK cell populations isolated from CXCL9‐sufficient tumors to those from CXCL9–deficient tumors. In addition, we investigated the NK cell subset that expressed tumor necrosis factor‐related apoptosis‐inducing ligand (TRAIL). This NK cell subset can kill target cells through extrinsic apoptotic machinery and plays a critical role in enhancing NK cell‐mediated anti–tumor immunity in the liver. We found that total NK cells and TRAIL+ NK cells were significantly more abundant in CXCL9‐sufficient tumors compared to CXCL9‐deficient tumors (Figure 3D‐F and Figure S4A,B). In contrast, endogenous CXCL9 was not involved in the recruitment of CD4+ or CD8+ T cells (Figure 3G,H and Figure S4C,D). These results were consistent with the IHC results in clinical samples. Furthermore, total CXCR3+ cells and CXCR3+ NK cells among total lymphocytes were significantly reduced in CXCL9‐deficient tumors compared to CXCL9‐sufficient tumors (Figure 3I,J and Figure S4E,F). To examine whether tumor‐infiltrating NK cells substantially contributed to tumor enlargement, we compared volumes between CXCL9‐sufficient and CXCL9‐deficient tumors under NK‐cell depleted conditions. We found that the NK depletion eliminated the volume differences between CXCL9‐sufficient and CXCL9‐deficient tumors (Figure 3K).
Figure 3.
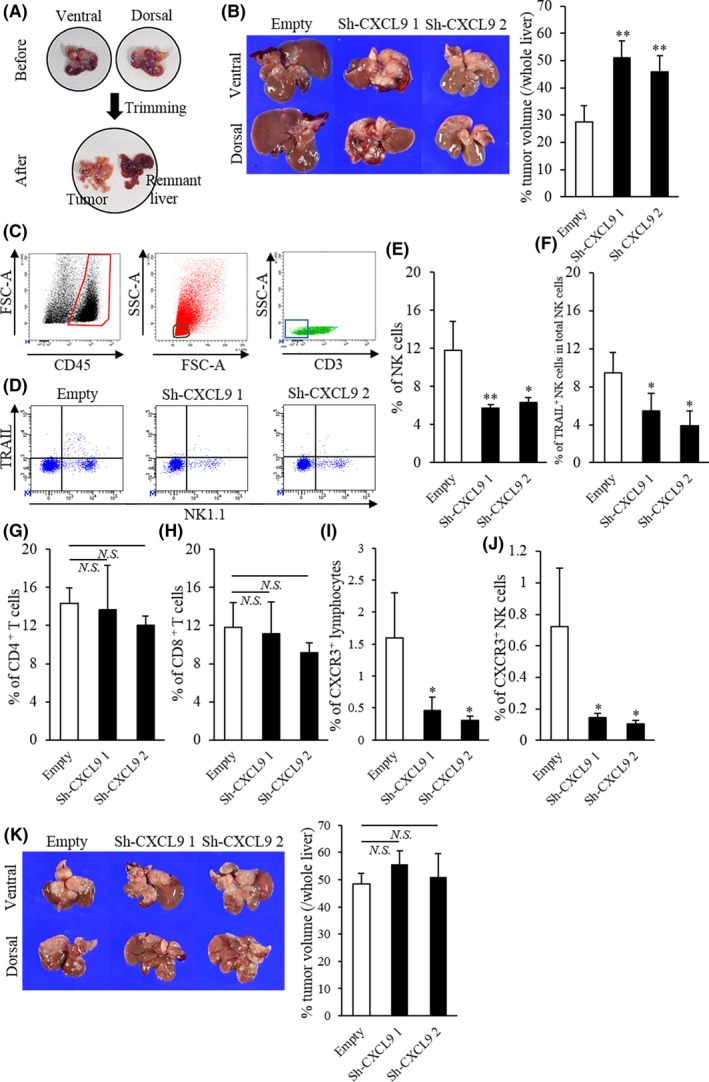
Comparison of liver tumors and tumor‐infiltrating lymphocytes with CXCL9‐sufficient and CXCL9‐deficient cells in mice. A, Tumor‐bearing livers were meticulously dissected to isolate tumors. B, (Left) Representative images of liver tumors, 35 d after 1‐1a cells carrying the empty shRNA, sh‐CXCL9 1 or sh‐CXCL9 2 were inoculated into the spleen; (upper panels) ventral view; (lower panels) dorsal view. (Right) Percentage of tumor/whole liver volumes were significantly higher in CXCL9‐deficient mice compared to CXCL9‐sufficient mice (empty: n = 6; sh‐CXCL9 1: n = 5, sh‐CXCL9 2: n = 6). C, Representative flow cytometry results illustrate gating. (Left) Leukocytes were gated as CD45+ cells (enclosed in red). (Middle) Lymphocytes were identified by low forward scatter (FSC) and low side scatter (SSC) gates (circled in black) and by CD11b expression (data not shown). (Right) The NK and T cell populations were purified from CD3− (boxed in blue) and CD3+ cells, respectively. D, Representative flow cytometry plots show NK1.1 (x‐axis) and TRAIL (y‐axis)‐stained tumor‐infiltrating lymphocytes in tumors from mice with CXCL9‐sufficiency or CXCL9‐deficiencies. E‐J, The percentages of tumor‐infiltrating (E) natural killer (NK) cells, (G) CD4+ T cells, (H) CD8+ T cells, (I) CXCR3+ lymphocytes and (J) CXCR3+ NK cells in total lymphocytes and (F) the percentage of TRAIL+ NK cells in total NK cells are shown for tumors from mice inoculated with 1‐1a cells that carried empty vs sh‐CXCL9 1 or sh‐CXCL9 2 shRNA. (K, Left) Representative images show (top row) ventral and (bottom row) dorsal views of liver tumors at 32 d after cells that carried empty, sh‐CXCL9 1 or sh‐CXCL9 2 shRNA were inoculated into the spleens of NK‐cell‐depleted mice. (Right) Percentages of tumor/whole liver volumes in tumors with CXCL9‐sufficiency or CXCL9‐deficiencies (all groups: n = 4). Isotype controls were used as negative controls to help differentiate non–specific background signal from anti–CD3, NK1.1, TRAIL and CXCR3 Abs. All data are the mean ± SD. *P < 0.05, **P < 0.01, N.S., not significant
3.4. CXCL9 was released in response to inflammatory stimuli in cholangiocarcinoma cell lines
Our IHC and in vivo analyses indicated that increased CXCL9 expression might improve patient survival by regulating TIME. However, we also aimed to elucidate the impact of CXCL9 on tumor cell biological properties because previous studies demonstrated that CXCL9 could promote tumor growth.16 First, we stimulated four CCA cell lines with different concentrations of IFN‐γ and TNF‐α. When cells were stimulated with 10 ng/mL IFN‐γ, the anti–CXCL9 Ab showed positive staining in the cytoplasm of MzChA‐1, TFK‐1 and HuCCT‐1 cells but not CCLP‐1 cells (Figure S5). IFN‐γ stimulation dose‐dependently increased CXCL9 release in MzChA‐1, TFK‐1 and HuCCT‐1 cells. In addition, IFN‐γ and TNF‐α showed synergistic CXCL9 induction in these three cell lines, despite the failure of TNF‐α alone to induce CXCL9 production. In contrast, stimulating with any concentration of IFN‐γ and/or TNF‐α could not induce CXCL9 release in CCLP‐1 cells (Figure 4A).
Figure 4.
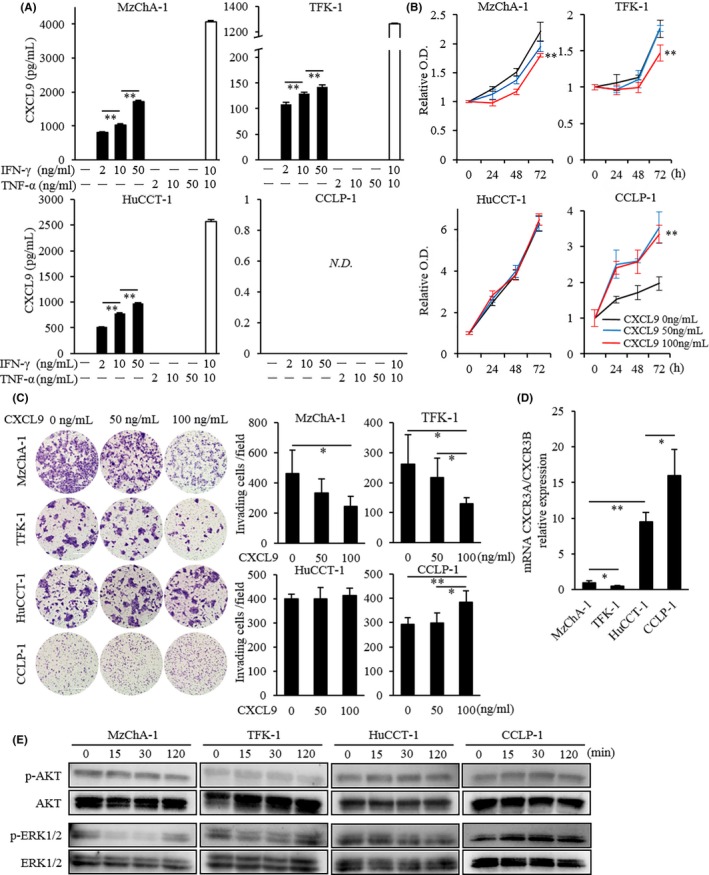
Induction of CXCL9 by inflammatory stimuli and the influence of CXCL9 on biological tumor behaviors in human cholangiocarcinoma cell lines. A, CXCL9 release after stimulation with IFN‐γ and/or TNF‐α in four holangiocarcinoma (CCA) cell lines. CXCL9 induction by IFN‐γ was dose‐dependent in MzChA‐1, TFK‐1 and HuCCT‐1 cells. In addition, 10 ng/mL TNF‐α and IFN‐γ synergistically induced CXCL9 expression in these three cell lines. In contrast, CXCL9 was undetectable (ND not detected) in CCLP‐1 cells after stimulation with IFN‐γ and/or TNF‐α at any concentration. B, Cell proliferation assay in four CCA cell lines. Cells were stimulated with different concentrations of CXCL9 (0, 50 and 100 ng/mL), then incubated with CCK‐8 at 0, 24, 48 and 72 h after CXCL9 stimulation. After 72 h of CXCL9 stimulation, 100 ng/mL CXCL9 significantly inhibited cell growth in MzChA‐1 and TFK‐1 cells, but it significantly promoted growth in CCLP‐1 cells, and it did not affect growth in HuCCT‐1 cells. C, Cell invasion assay in four CCA cell lines. (Left) Representative microscopic images show cells that migrated to the underside of the invasion chamber membrane. (Right) The means of six randomly‐selected microscopic fields show that 100 ng/mL CXCL9 significantly inhibited invasion in MzChA‐1 and TFK‐1 cells, significantly stimulated invasion in the CCLP‐1 line, and did not affect invasion in HuCCT‐1 cells. D, Ratios of CXCR3A‐to‐CXCR3B mRNA expression in four CCA cell lines. Results are the fold‐change relative to the ratio observed in MzChA‐1 cells. E, Western blot analysis shows the effects of 100 ng/mL of CXCL9 stimulation on cell signaling pathways. The AKT signaling pathway was unaltered in all four CCA cell lines. In contrast, ERK1/2 phosphorylation was downregulated in MzChA‐1 and TFK‐1 cells and upregulated in CCLP‐1 cells at 15 and 30 min. No alteration was observed in HuCCT‐1 cells. All data are the mean ± SD. *P < 0.05, **P < 0.01
3.5. CXCL9 did not promote cell growth or cell invasion in CXCL9‐expressing cholangiocarcinoma cell lines
To determine whether CXCL9 affected the biological properties of CCA, we treated four CCA cell lines with different concentrations of rhCXCL9 and investigated the proliferation and invasion abilities. At 72 hours after adding 100 ng/mL CXCL9, cell growth was significantly inhibited in MzChA‐1 and TFK‐1 cells but significantly promoted in CCLP‐1 cells. Similarly, adding 100 ng/mL of CXCL9 to the invasion chambers caused a significant reduction in MzChA‐1 and TFK‐1 cell invasion and a significant increase in CCLP‐1 cell invasion. No changes were observed in HuCCT‐1 cell growth or invasion capabilities (Figure 4B,C). We reasoned that the variability in cell growth and invasion abilities across these cell lines might be attributable to the different levels of CXCR3A and CXCR3B expression. We found that the expression of CXCR3A mRNA was lowest in TFK‐1 cells, and increased progressively in MzChA‐1, HuCCT1 and CCLP‐1 cells. On the other hand, CXCR3B expression was highest in TFK‐1 cells and decreased progressively in MzChA‐1, CCLP‐1 and HuCCT‐1 cells (Figure S6A,B). The CXCR3A/CXCR3B gene expression ratio was lowest in TFK‐1 cells and increased progressively in MzChA‐1, HuCCT‐1 and CCLP‐1 cells (Figure 4D). Finally, we screened two signaling pathways, the PI3K/AKT pathway and the ERK1/2 pathway, which were reported to be activated via the CXCL9‐CXCR3 axis in different cancer settings.16, 27 Administration of 100 ng/mL CXCL9 did not alter the AKT signaling pathway in any of our four cell lines. In contrast, after 15 and 30 minute exposures to 100 ng/mL CXCL9, ERK1/2 phosphorylation was downregulated in MzChA‐1 and TFK‐1 cells and upregulated in CCLP‐1 cells. No alteration was observed in the ERK1/2 signaling pathway in HuCCT‐1 cells (Figure 4E).
4. DISCUSSION
Chemokines are inextricably linked with cancers. Chemokines produced by cancer cells can dictate their fate through autocrine and paracrine signaling. The distinct chemokines produced in different tumors lead to substantial differences in prognosis, due to differences in their control of the tumor microenvironment and tumor behaviors. The present study is the first to imply that endogenous CXCL9 modulated tumor‐infiltrating NK cells, which influenced tumor progression and postoperative survival in patients with iCCA.
It has been demonstrated that tumor‐derived CXCL9 is a tumor suppressor12; hence, CXCL9 was implicated in a favorable prognosis19, 20, 28 and good responsiveness to chemotherapy.29 CXCL9 stimulated lymphocyte trafficking into the tumor and enhanced anti–cancer immune surveillance, even though the targeted lymphocyte phenotypes differed, depending on the cancer type. Mlecnik et al28 demonstrated that CXCL9 was a chemoattractant to cytotoxic T cells, memory cells and TCRαβ T cells in surgically resected colorectal cancer tumors. In addition, Walser et al30 showed that NK cells and CD4+ T cells were critical to the mechanism by which CXCL9 limited metastasis and local growth in a murine breast cancer model. In our study, tumor‐derived CXCL9 was involved in the accumulation of tumor‐infiltrating NK cells, based on IHC staining of resected specimens and flow cytometry of cells from a murine iCCA tumor model. In this model, we showed that tumors with CXCL9‐sufficient cells were associated with high frequencies of all tumor‐infiltrating NK cells and of CXCR3+ NK cells. This result might be explained by the immune system, with the recruitment of a CXCR3‐dependent cell population driving further entry of CXCR3‐negative subsets, as described by Groom et al.31 NK cells are abundant in the liver, where they serve as immune correlates to protect against cancer cells invading the liver. NK cells induce cancer cell death by apoptosis via several cytotoxic pathways, including granule‐mediated, FasL‐mediated and TNF‐mediated pathways. Of these apoptotic pathways, previous studies have shown that TRAIL+ NK cells expressed CXCR3. They also showed that the frequency of TRAIL+ NK cells among liver lymphocytes markedly decreased after a hepatectomy through the downregulation of the CXCL9‐CXCR3 axis.32, 33 Consistent with those findings, in the present study, we found that TRAIL+ NK cell trafficking into tumors was regulated by CXCL9 release from tumor cells and the limited recruitment of TRAIL+ NK cells most likely contributed to tumor enlargement in CXCL9‐deficient tumors. In contrast, FOXP3+ Treg cells that expressed CXCR3 could migrate into tumor sites, which then limited the host immune response.34 Indeed, CXCL9 expression was positively correlated with FOXP3+ infiltration in ovarian cancer.19 However, we found no distinct relationship between CXCL9 and FOXP3+ Treg cells in our series.
Prior reports have demonstrated that CXCL9 plays pivotal roles in tumor behaviors, via a specific CXCR3 isoform.12 Some studies show that dominant isoform switching from CXCR3B to CXCR3A is characteristic of cancer cells (compared to corresponding normal cells) and of elevated cancer grades.16, 17 In addition, CXCR3A is a dominant participant in the exacerbation of tumor migration and invasion abilities, via ERK1/2 activation and its cognate downstream pathways, in gastric and liver cancer cells.15, 16 CXCR3A is involved in motility and invasion abilities; CXCR3B overexpression reduces these abilities in prostate cancer.17 In the current study, we used cell lines that represented four human CCA subtypes. CCLP‐1 is the most malignant cell line with epithelial‐mesenchymal transformation traits. HuCCT‐1 is a metastatic, moderately differentiated cell line. MzChA‐1 is a metastatic, highly differentiated cell line. TFK‐1 is a primary, papillary cell line.22, 23, 24, 25 Our data indicated that upregulation of the CXCR3A‐to‐CXCR3B ratio was accompanied by elevated CCA tumor grading and enhanced tumor growth and invasion abilities via ERK1/2 signaling.
Manipulating TIME through positive modulation of CXCL9 release is a promising immunotherapeutic approach.12 Cytotoxic chemotherapies, molecular targeted therapies and cytokine therapies have shown potential in upregulating CXCL9 release.35, 36 Hannesdóttir et al35 demonstrated that lapatinib and doxorubicin, two key drugs for breast cancer, upregulated the production of endogenous CXCL9, thereby attracting CD8+ T cells in signal transducer and activator of transcription 1‐dependent HER2‐positive breast cancer. In lung cancer, Andersson et al36 demonstrated that IL‐7/IL‐7Rα‐Fc therapy induced M1 macrophage phenotype, inhibited tumor growth and prolonged survival via upregulation of endogenous CXCL9. Moreover, another group revealed that COX inhibitors could effectively upregulate the production of CXCL9 in ovarian and breast cancers since PGE2, a downstream product of COX, negatively regulates CXCL9 expression.19, 37 Focusing on COX inhibitors should be appealing because it has been well recognized as a useful candidate for repositioning drugs in treating and preventing cancers.38, 39 Furthermore, overexpression of COX, especially COX‐2 isoform, is deemed oncogenic; it has contributed to immune evasion in many types of cancer.40 Indeed, previous reports demonstrated that COX‐2 inhibitors induced cell growth inhibition through cell cycle arrest and apoptosis in CCA.41, 42 Notably, our preliminary data showed that low‐dose celecoxib, a COX‐2 selective inhibitor, significantly enhanced CXCL9 release in all CXCL9‐expressing CCA cell lines (data not shown); however, in vivo testing is needed to demonstrate the effectiveness of low‐dose celecoxib in iCCA. Moreover, further investigation could provide effective drugs that have the potential to alter TIME via upregulation of endogenous CXCL9 in iCCA.
The current study has several limitations. First, we used single‐labelling IHC staining for the detection of a certain subset of tumor‐infiltrating immune cells in iCCA tissues. However, each primary Ab used for analysis could stain multiple subsets of immune cells. Second, we did not investigate which types of immune cells were predominantly involved in IFN‐γ production. As endogenous CXCL9 can be released in the presence of immune cell‐derived IFN‐γ, clarifying this point is of great importance to better understand the impact of CXCL9 on TIME in iCCA. Third, in this study, we did not clarify the extent to which the different CXCR3 isoforms contributed to cell growth and invasion abilities induced by CXCL9, nor did we examine the downstream of the ERK1/2 signaling pathway, which might alter tumor behaviors. However, importantly, we showed that CXCL9 was unlikely to exacerbate tumor aggressiveness in CXCL9‐expressing CCA.
In conclusion, we showed that CXCL9 had prognostic significance in iCCA, and that it was correlated with tumor‐infiltrating NK cells. Our results also suggested that upregulation of CXCL9 is a promising therapeutic approach by inducing immunopotentiation without promoting tumor aggressiveness in patients with CXCL9‐expressing iCCA.
DISCLOSURE
The authors have no conflict of interest.
Supporting information
ACKNOWLEDGMENTS
The authors wish to thank Kumiko Goto (SHIONOGI) for technical support in performing flow cytometry in this study.
Fukuda Y, Asaoka T, Eguchi H, et al. Endogenous CXCL9 affects prognosis by regulating tumor‐infiltrating natural killer cells in intrahepatic cholangiocarcinoma. Cancer Sci. 2020;111:323–333. 10.1111/cas.14267
REFERENCES
- 1. Bridgewater J, Galle PR, Khan SA, et al. Guidelines for the diagnosis and management of intrahepatic cholangiocarcinoma. J Hepatol. 2014;60:1268‐1289. [DOI] [PubMed] [Google Scholar]
- 2. Shaib YH, Davila JA, McGlynn K, El‐Serag HB. Rising incidence of intrahepatic cholangiocarcinoma in the United States: a true increase? J Hepatol. 2004;40:472‐477. [DOI] [PubMed] [Google Scholar]
- 3. Colvin H, Mizushima T, Eguchi H, Takiguchi S, Doki Y, Mori M. Gastroenterological surgery in Japan: the past, the present and the future. Ann Gastroenterol Surg. 2017;1:5‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yamashita Y‐I, Shirabe K, Beppu T, et al. Surgical management of recurrent intrahepatic cholangiocarcinoma: predictors, adjuvant chemotherapy, and surgical therapy for recurrence: a multi‐institutional study by the Kyushu Study Group of Liver Surgery. Ann Gastroenterol Surg. 2017;1:136‐142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Valle J, Wasan H, Palmer DH, et al. Cisplatin plus gemcitabine versus gemcitabine for biliary tract cancer. N Engl J Med. 2010;362:1273‐1281. [DOI] [PubMed] [Google Scholar]
- 6. Ebata T, Hirano S, Konishi M, et al. Randomized clinical trial of adjuvant gemcitabine chemotherapy versus observation in resected bile duct cancer. Br J Cancer. 2018;105:192‐202. [DOI] [PubMed] [Google Scholar]
- 7. Dhanasekaran R, Hemming AW, Zendejas I, et al. Treatment outcomes and prognostic factors of intrahepatic cholangiocarcinoma. Oncol Rep. 2013;29:1259‐1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dai H, Wang Y, Lu X, Han W. Chimeric antigen receptors modified T‐cells for cancer therapy. J Natl Cancer Inst. 2016;108:pii: djv439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer. 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Fontugne J, Augustin J, Pujals A, et al. PD‐L1 expression in perihilar and intrahepatic cholangiocarcinoma. Oncotarget. 2017;8:24644‐24651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kriegsmann M, Roessler S, Kriegsmann K, et al. Programmed cell death ligand 1 (PD‐L1, CD274) in cholangiocarcinoma – correlation with clinicopathological data and comparison of antibodies. BMC Cancer. 2019;19:72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ding Q, Lu P, Xia Y, et al. CXCL9: evidence and contradictions for its role in tumor progression. Cancer Med. 2016;5:3246‐3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Asaoka T, Marubashi S, Kobayashi S, et al. Intragraft transcriptome level of CXCL9 as biomarker of acute cellular rejection after liver transplantation. J Surg Res. 2012;178:1003‐1014. [DOI] [PubMed] [Google Scholar]
- 14. Gorbachev AV, Kobayashi H, Kudo D, et al. CXC chemokine ligand 9/monokine induced by IFN‐gamma production by tumor cells is critical for T cell‐mediated suppression of cutaneous tumors. J Immunol. 2007;178:2278‐2286. [DOI] [PubMed] [Google Scholar]
- 15. Ding Q, Xia Y, Ding S, Lu P, Sun L, Liu M. An alternatively spliced variant of CXCR3 mediates the metastasis of CD133+ liver cancer cells induced by CXCL9. Oncotarget. 2016;7:14405‐14414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yang C, Zheng W, Du W. CXCR3A contributes to the invasion and metastasis of gastric cancer cells. Oncol Rep. 2016;36:1686‐1692. [DOI] [PubMed] [Google Scholar]
- 17. Wu Q, Dhir R, Wells A. Altered CXCR3 isoform expression regulates prostate cancer cell migration and invasion. Mol Cancer. 2012;11:3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Datta D, Banerjee P, Gasser M, Waaga‐Gasser AM, Pal S. CXCR3‐B can mediate growth‐inhibitory signals in human renal cancer cells by down‐regulating the expression of heme oxygenase‐1. J Biol Chem. 2010;285:36842‐36848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bronger H, Singer J, Windmüller C, et al. CXCL9 and CXCL10 predict survival and are regulated by cyclooxygenase inhibition in advanced serous ovarian cancer. Br J Cancer. 2016;115:553‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Wu Z, Huang X, Han X, et al. The chemokine CXCL9 expression is associated with better prognosis for colorectal carcinoma patients. Biomed Pharmacother. 2016;78:8‐13. [DOI] [PubMed] [Google Scholar]
- 21. Liu W, Liu Y, Fu Q, et al. Elevated expression of IFN‐inducible CXCR3 ligands predicts poor prognosis in patients with non–metastatic clear‐cell renal cell carcinoma. Oncotaget. 2016;7:13976‐13983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Cardinale V, Renzi A, Carpino G, et al. Profiles of cancer stem cell subpopulations in cholangiocarcinomas. Am J pathol. 2015;185:1724‐1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Miyagiwa M, Ichida T, Tokiwa T, Sato J, Sasaki H. A new human cholangiocellular carcinoma cell line (HuCC‐T1) producing carbohydrate antigen 19/9 in serum‐free medium. Vitro Cell Dev Biol. 1989;25:503‐510. [DOI] [PubMed] [Google Scholar]
- 24. Knuth A, Gabbert H, Dippold W, et al. Biliary adenocarcinoma. Characterisation of three new human tumor cell lines. J Hepatol. 1985;1:579‐596. [DOI] [PubMed] [Google Scholar]
- 25. Saijyo S, Kudo T, Suzuki M, et al. Establishment of a new extrahepatic bile duct carcinoma cell line, TFK‐1. Tohoku J Exp Med. 1995;177:61‐71. [DOI] [PubMed] [Google Scholar]
- 26. Lee AJ, Chun YS. Intrahepatic cholangiocarcinoma: the AJCC/UICC 8th edition updates. Chin Clin Oncol. 2018;7(5):52. [DOI] [PubMed] [Google Scholar]
- 27. Li Z, Liu J, Li L, et al. Epithelial mesenchymal transition induced by the CXCL9/CXCR3 axis through AKT activation promotes invasion and metastasis in tongue squamous cell carcinoma. Oncol Rep. 2018;39:1356‐1368. [DOI] [PubMed] [Google Scholar]
- 28. Mlecnik B, Tosolini M, Charoentong P, et al. Biomolecular network reconstruction identifies T‐cell homing factors associated with survival in colorectal cancer. Gastroenterology. 2010;138:1429‐1440. [DOI] [PubMed] [Google Scholar]
- 29. Denkert C, Loibl S, Noske A, et al. Tumor‐associated lymphocytes as an independent predictor of response to neoadjuvant chemotherapy in breast cancer. J Clin Onocl. 2010;28:105‐113. [DOI] [PubMed] [Google Scholar]
- 30. Walser TC, Ma X, Kundu N, Dorsey R, Goloubeva O, Fulton AM. Immune‐mediated modulation of breast cancer growth and metastasis by the chemokine Mig (CXCL9) in a murine model. J Immunother. 2007;30:490‐498. [DOI] [PubMed] [Google Scholar]
- 31. Groom JR, Luster AD. CXCR3 in T cell function. Exp Cell Res. 2011;317:620‐631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yano T, Ohira M, Nakano R, Tanaka Y, Ohdan H. Hepatectomy leads to loss of TRAIL‐expressing liver NK cells via downregulation of the CXCL9‐CXCR3 axis in mice. PLoS ONE. 2017;12:e0186997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Ohira M, Ohdan H, Mitsuta H, et al. Adoptive transfer of TRAIL‐expressing natural killer cells prevents recurrence of hepatocellular carcinoma after partial hepatectomy. Transplantation. 2006;82:1712‐1719. [DOI] [PubMed] [Google Scholar]
- 34. Redjimi N, Raffin C, Raimbaud I, et al. CXCR3+ T regulatory cells selectively accumulate in human ovarian carcinomas to limit type I immunity. Cancer Res. 2012;72:4251‐4260. [DOI] [PubMed] [Google Scholar]
- 35. Hannesdóttir L, Tymoszuk P, Parajuli N, et al. Lapatinib and doxorubicin enhance the Stat1‐dependent antitumor immune response. Eur J Immunol. 2013;43:2718‐2729. [DOI] [PubMed] [Google Scholar]
- 36. Andersson A, Srivastava MK, Harris‐White M, et al. Role of CXCR3 ligands in IL‐7/IL‐7R alpha‐Fc‐mediated antitumor activity in lung cancer. Clin Cancer Res. 2011;17:3660‐3672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Bronger H, Kraeft S, Schwarz‐Boeger U, et al. Modulation of CXCR3 ligand secretion by prostaglandin E2 and cyclooxygenase inhibitors in human breast cancer. Breast Cancer Res. 2012;14:R30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tołoczko‐Iwaniuk N, Dziemiańczyk‐Pakieła D, Nowaszewska BK, Celińska‐Janowicz K, Miltyk W. Celecoxib in cancer therapy and prevention – review. Curr Drug Targets. 2019;20:302‐315. [DOI] [PubMed] [Google Scholar]
- 39. Arber N, Eagle CJ, Spicak J, et al. Celecoxib for the prevention of colorectal adenomatous polyps. N Engl J Med. 2006;355:885‐895. [DOI] [PubMed] [Google Scholar]
- 40. Markosyan N, Chen EP, Smyth EM. Targeting COX‐2 abrogates mammary tumorigenesis: breaking cancer‐associated suppression of immunosurveillance. Oncoimmunology. 2014;30:e29287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Han C, Leng J, Demetris AJ, Wu T. Cyclooxygenase‐2 promotes human cholangiocarcinoma growth: evidence for cyclooxygenase‐2‐independent mechanism in celecoxib‐mediated induction of p21waf1/cip1 and p27kip1 and cell cycle arrest. Cancer Res. 2004;64:1369‐1376. [DOI] [PubMed] [Google Scholar]
- 42. Wu T, Leng J, Han C, Demetris AJ. The cyclooxygenase‐2 inhibitor celecoxib blocks phosphorylation of Akt and induces apoptosis in human cholangiocarcinoma cells. Mol Cancer Ther. 2004;3:299‐307. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials