

Abstract
There are increased opportunities in oncology clinics to identify multiple pancreatic ductal adenocarcinomas (PDAC) that co‐occur simultaneously or arise metachronously in the pancreatic parenchyma, yet their pathogenesis remains elusive. We hypothesized that two potential pathways, multicentric carcinogenesis and intrapancreatic metastasis, might contribute to forming multiple PDAC. Among 241 resected cases, we identified 20 cancer nodules from nine patients with multiple PDAC (six with synchronous PDAC, one with metachronous PDAC, and two with both synchronous and metachronous PDAC). Integrated clinical, pathological, and mutational analyses, using TP53 and SMAD4 immunostaining and targeted next‐generation sequencing of 50 cancer‐related genes, were conducted to examine the intertumor relationships. Four of the nine patients were assessed as having undergone multicentric carcinogenesis because of heterogeneity of immunohistochemical and/or mutation characteristics. In contrast, tumors in the other five patients showed intertumor molecular relatedness. Two of these five patients, available for matched sequencing data, showed two or more shared mutations. Moreover, all the smaller nodules in these five patients showed identical TP53 and SMAD4 expression patterns to the corresponding main tumors. Consequently, these five patients were considered to have undergone intrapancreatic metastasis. None of the five smaller nodules arising from intrapancreatic metastasis was accompanied by pancreatic intraepithelial neoplasia, and three of them were tiny (≤1mm). Patients whose tumors resulted from intrapancreatic metastasis appeared to have higher disease stages and worse outcome than those with tumors from multicentric carcinogenesis. Our results provide insight into pancreatic carcinogenesis, showing that the development of multiple PDAC involves distinct evolutionary paths that potentially affect patient prognosis.
Keywords: multicentric occurrence, multiple pancreatic cancers, next‐generation sequence, pancreatic carcinogenesis, pancreatic intraepithelial neoplasia
Integrated clinical, pathological, and mutational analyses on multiple pancreatic cancers identify distinct evolutionary paths: multicentric carcinogenesis and intrapancreatic metastasis.
Abbreviations
- FFPE
formalin‐fixed paraffin embedded
- IHC
immunohistochemistry
- NGS
next‐generation sequencing
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
1. INTRODUCTION
Pancreatic ductal adenocarcinoma develops through sequential genetic and epigenetic alterations in a number of driver genes, including KRAS, TP53, SMAD4, and CDKN2A.1, 2, 3, 4, 5, 6, 7 Accumulating evidence from engineered mouse models attests to the importance of PanIN as a precursor of PDAC.8, 9, 10, 11, 12, 13 Previous studies have shown that incidental multifocal PanIN are frequently observed in the adult pancreas within surgical specimens and autopsy cases.14, 15, 16 Strict surveillance of high‐risk groups, such as familial pancreatic cancer kindreds, those with hereditary neoplastic syndromes, and PDAC patients following pancreatectomy for PDAC, suggests that spatial and temporal multicentric carcinogenesis involves pancreatic cancer development 17, 18, 19, 20 in a manner similar to that of hepatocellular carcinoma and colorectal cancer.21, 22 Because of the aging population and recent advances in technology and treatments for PDAC, the number of newly diagnosed patients and long‐term survivors after curative surgery for PDAC is growing. Consequently, in oncology clinics, there are increased opportunities to identity synchronous and/or metachronous multiple PDAC.
The propensity of orthotopic colonization in several types of cancers, including malignant melanoma, lung cancer, and liver cancer, is exemplified by tissue tropisms of metastasizing tumor cells in a process known as the “seed and soil hypothesis”.23, 24 Previous reports have suggested that intrapancreatic metastasis may serve as another evolutionary scenario for multiple PDAC.25, 26 A recent study has suggested that precancerous intraepithelial neoplasms can spread contiguously or disseminate discontiguously along the pancreatic ductal system to form multifocal clonal lesions.27 Therefore, we hypothesized that two distinct pathways, multicentric carcinogenesis and intrapancreatic metastasis, might contribute to the formation of multiple PDAC. A previous study by Oguro et al reported that 21 of 393 patients had synchronous multiple PDAC as a result of intrapancreatic metastasis and that the presence of this histological finding was a worse prognostic factor,26 although this previous study was conducted based on histomorphological features without any molecular assessment. Two recent studies using genetic testing on pancreatic cancer arising in the remnant pancreas after pancreatectomy identified recurrent cases as a result of multicentric occurrence.17, 28 In order to expand these prior studies, we analyzed not only metachronous tumors but also synchronous cases and integrated clinicopathological characteristics of multiple PDAC with molecular information, using NGS and IHC. In addition, we further explored the prognostic associations of the results with patient outcome to verify the clinical usefulness of distinguishing intrapancreatic metastasis from multicentric occurrence.
2. MATERIALS AND METHODS
2.1. Study population and tissue samples
We reviewed 241 PDAC patients who underwent pancreatectomy at Keio University Hospital between 2000 and 2016. During routine pathological assessment, formalin‐fixed resected specimens were cut stepwise at 4‐ to 6‐mm intervals, and all specimens containing pancreatic tissue were embedded in paraffin. Specimens were then evaluated microscopically according to the World Health Organization classification and the Classification of Pancreatic Carcinoma of the Japan Pancreas Society.29, 30 Among the 241 patients, we identified 22 tumor nodules in 10 patients who had metachronous and/or synchronous multiple invasive adenocarcinomas in the pancreatic parenchyma. One of these 10 patients had co‐occurring PDAC and intraductal papillary mucinous neoplasm (IPMN) with an associated invasive carcinoma and was excluded from the current study. Consequently, we analyzed 20 cancer nodules from nine patients, comprising six patients (12 nodules) with synchronous PDAC, one patient (two nodules) with metachronous PDAC, and two patients (six nodules) with both synchronous and metachronous PDAC (Figure 1). We conducted a histological review and confirmed that all the synchronous lesions were more than or equal to 10 mm apart with no connection between them (Table 1). For the three patients with metachronous tumors, pancreatic cut margin statuses at the initial surgery were negative. NGS analysis of tumors from the nine patients found no GNAS mutation, a major driver of IPMN tumorigenesis.31, 32, 33 None of the nine patients had a family history of pancreatic cancer in first‐degree relatives. One patient (MPKO09) had a pancreaticobiliary maljunction.
Figure 1.
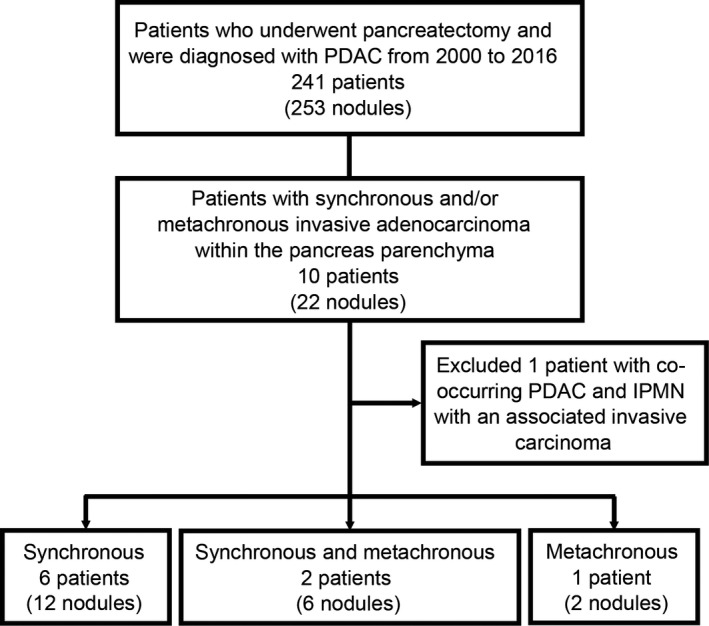
Flow diagram of the study population. IPMN, intraductal papillary mucinous neoplasm; PDAC, pancreatic ductal adenocarcinoma
Table 1.
Clinicopathological features and shared mutations according to 20 tumors from nine patients with synchronous and/or metachronous pancreatic ductal adenocarcinomas
| Size (mm) | Tumor location | Distance from main tumor (mm) | Months after initial surgery | Tumor grade | Tumor‐associated PanINa | Immunohistochemistry | Shared mutations by targeted NGS | Intertumor relationship | ||
|---|---|---|---|---|---|---|---|---|---|---|
| TP53 | SMAD4 | |||||||||
| MPKO01 (70s, Male) | ||||||||||
| T1: main | 29 | Tail | G3 | Present | Intact | Lost | None | Independent | ||
| T2: synchronous | 16 | Body | 35 | G2 | Present | Altered | Intact | |||
| MPKO02 (70s, Female) | ||||||||||
| T1: main | 30 | Tail | G1 | Present | Intact | Lost | None | Independent | ||
| T2: synchronous | 20 | Body | 45 | G2 | Present | Intact | Lost | |||
| MPKO03 (60s, Female) | ||||||||||
| T1: main | 21 | Head | G1 | Present | Intact | Intact | NAb | Independent | ||
| T2: metachronous | 50 | Body | 132 | G2 | Present | Altered | Intact | |||
| MPKO04 (60s, Male) | ||||||||||
| T1: main | 24 | Tail | G3 | Absent | Intact | Lost | None between T1/T2; | Independent | ||
| T2: synchronous | 8 | Body | 50 | G3 | Present | Intact | Intact | None between T2/T3; | ||
| T3: metachronous | 35 | Head | 43 | G3 | Present | Intact | Lost | KRAS p.Q61H between T1/T3 | ||
| MPKO05 (40s, Male) | ||||||||||
| T1: main | 28 | Head | NAc | Absent | Altered | Lost | NAb | T2 and T3 are likely related; indeterminate for T1 | ||
| T2: metachronous | 22 | Body | 17 | G2 | Absent | Altered | Lost | KRAS p.G12V and | ||
| T3: metachronous | 17 | Tail | 17 | G2 | Absent | Altered | Lost | TP53 3' splice site (exon 5) between T2/T3 | ||
| MPKO06 (60s, Male) | ||||||||||
| T1: main | 12 | Body | G3 | Absent | Intact | Intact | NAb | Likely related | ||
| T2: synchronous | <1 | Body | 10 | G3 | Absent | Intact | Intact | |||
| MPKO07 (30s, Male) | ||||||||||
| T1: main | 35 | Body | G3 | Absent | Altered | Lost | NAb | Likely related | ||
| T2: synchronous | <1 | Tail | 25 | G3 | Absent | Altered | Lost | |||
| MPKO08 (70s, Female) | ||||||||||
| T1: main | 32 | Body | G3 | Present | Altered | Lost | NAb | Likely related | ||
| T2: synchronous | 1 | Tail | 10 | G3 | Absent | Altered | Lost | |||
| MPKO09 (80s, Male) | ||||||||||
| T1: main | 30 | Tail | G2 | Absent | Altered | Lost | TP53 p.Y205C, CDKN2A p.E120ter, PTEN p.Q245ter, and KDR p.Q119K | Likely related | ||
| T2: synchronous | 7 | Body | 20 | G2 | Absent | Altered | Lost | |||
Abbreviation: NA, not available; NGS, next‐generation sequencing; PanIN, pancreatic intraepithelial neoplasia; PDAC, pancreatic ductal adenocarcinoma.
Tumor‐associated PanIN was determined by the presence of PanIN2 or PanIN3 lesion in or around each PDAC nodule.
Mutation profiles of tumor T1 or T2 were unable to be obtained because of low DNA quality and/or considerably limited tumor size/purity.
Tumor grade was not assigned because tumor cell morphology was severely modified by preoperative chemoradiotherapy.
Histological examinations were reviewed by two study pathologists (YM and MS). Tumor‐associated PanIN was defined by the presence of PanIN2 or PanIN3 lesion in or around each PDAC nodule. Intraductal carcinoma spread (as known as “cancerization”) was carefully examined and was not considered as the presence of tumor‐associated PanIN.16, 34 The current study included two patients (MPKO07 and MPKO08) who were described in a previous case report25 in which these two patients were diagnosed with intrapancreatic metastases on the basis of the following histopathological criteria: lesions were at least 10 mm apart with no connection between them; each lesion showed similar histological findings; there was no PanIN lesion in or around the smaller, recessive lesions; and the histological type of the smaller lesions was uniform. The TNM staging system used in this study was that defined by the American Joint Committee on Cancer, 8th edition.35 The study was approved by the institutional review board of Keio University, and was conducted in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
2.2. Immunohistochemistry of TP53 and SMAD4
We conducted immunohistochemical analysis using an automated staining system (Bond Max; Leica Biosystems) with the use of the Bond Polymer Refine Detection kit (Leica Biosystems). An anti‐TP53 mouse monoclonal antibody (clone DO7, dilution, 1:2000; Agilent) and an anti‐SMAD4 mouse monoclonal antibody (clone B‐7, dilution, 1:100; Santa Cruz Biotechnologies) were used as the primary antibodies. On the basis of previously established criteria,36 tumors that showed weak, heterogeneous nuclear TP53 staining were defined as “intact”, whereas those showing strong nuclear staining were classified as “altered”. In the current dataset, no tumor showed complete absence of nuclear TP53 expression. For SMAD4, tumors were classified as “intact” if there was nuclear/cytoplasmic staining within the tumor, or “lost” if there was complete absence of staining, as previously described.37, 38 No tumor had missing information on IHC data. Table S1 shows correlations between immunohistochemical expression patterns and mutation statuses for TP53 and SMAD4. All the eight tumors with altered TP53 expression had mutations in the TP53 allele, whereas six of 11 tumors with loss of SMAD4 protein expression were called “wild type” by NGS analysis.
2.3. Next‐generation sequencing
After macroscopic dissection, genomic DNA was extracted from tumor and non‐tumor areas of FFPE tissue sections using a QIAamp DNA FFPE Tissue Kit (Qiagen).39, 40 Integrity of purified DNA from FFPE samples was assessed using the TaqMan RNase P Detection Reagents kit and the FFPE DNA QC Assay (Thermo Fisher Scientific). Barcoded libraries were generated from 10 ng DNA per sample using the Ion AmpliSeq Cancer Hotspot Panel v2 and the Ion AmpliSeq Library Kit 2.0 (Thermo Fisher Scientific) following the manufacturer’s protocol. This panel contains 207 primer pairs in a single tube and targets hotspot regions in the following 50 oncogenes and tumor suppressor genes: ABL1, AKT1, ALK, APC, ATM, BRAF, CDH1, CDKN2A, CSF1R, CTNNB1, EGFR, ERBB2, ERBB4, EZH2, FBXW7, FGFR1, FGFR2, FGFR3, FLT3, GNA11, GNAS, GNAQ, HNF1A, HRAS, IDH1, IDH2, JAK2, JAK3, KDR, KIT, KRAS, MET, MLH1, MPL, NOTCH1, NPM1, NRAS, PDGFRA, PIK3CA, PTEN, PTPN11, RB1, RET, SMAD4, SMARCB1, SMO, SRC, STK11, TP53, and VHL. Sequencing of templates after emulsion PCR was carried out as previously described.41, 42
2.4. Identification of somatic mutations
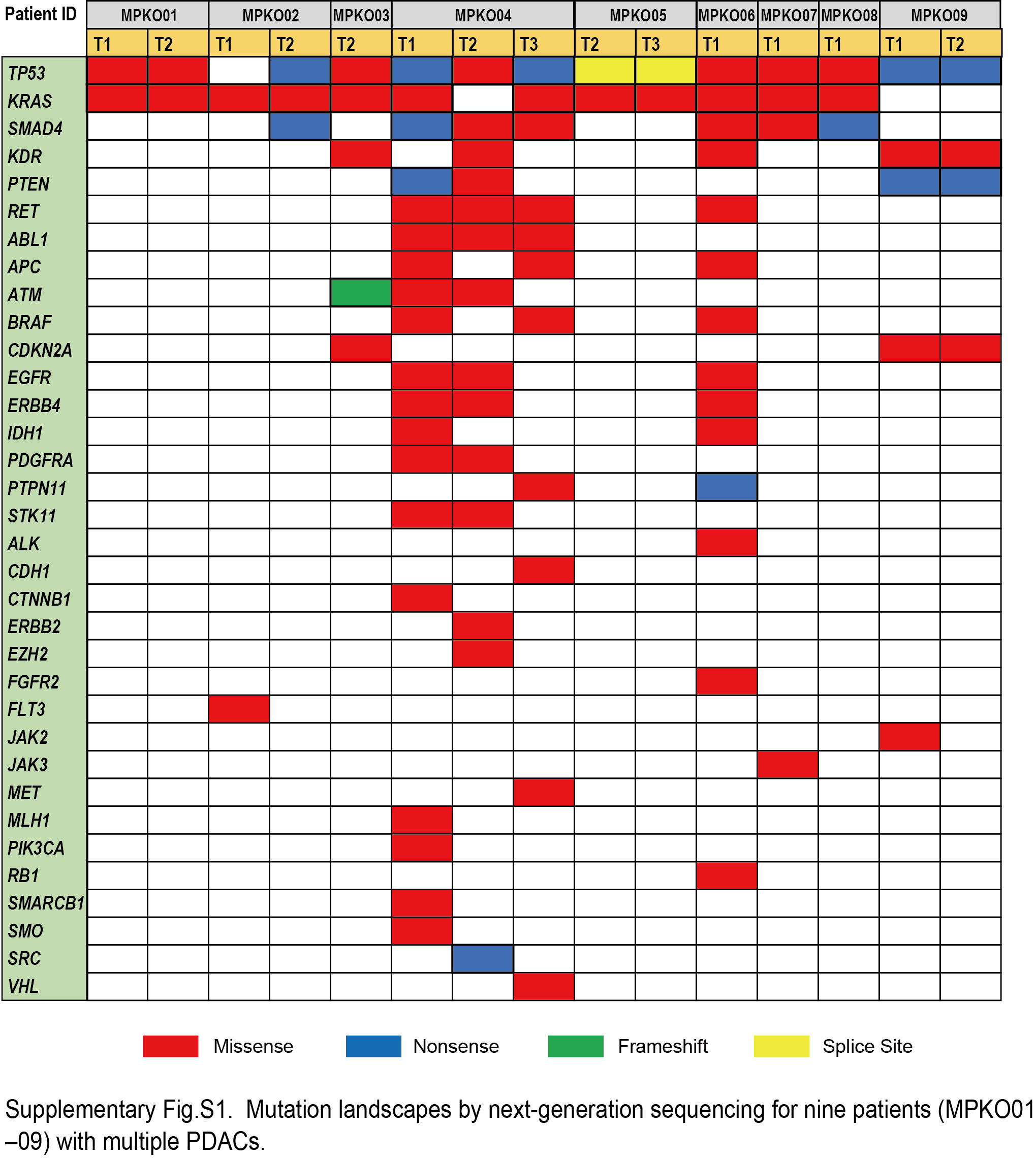
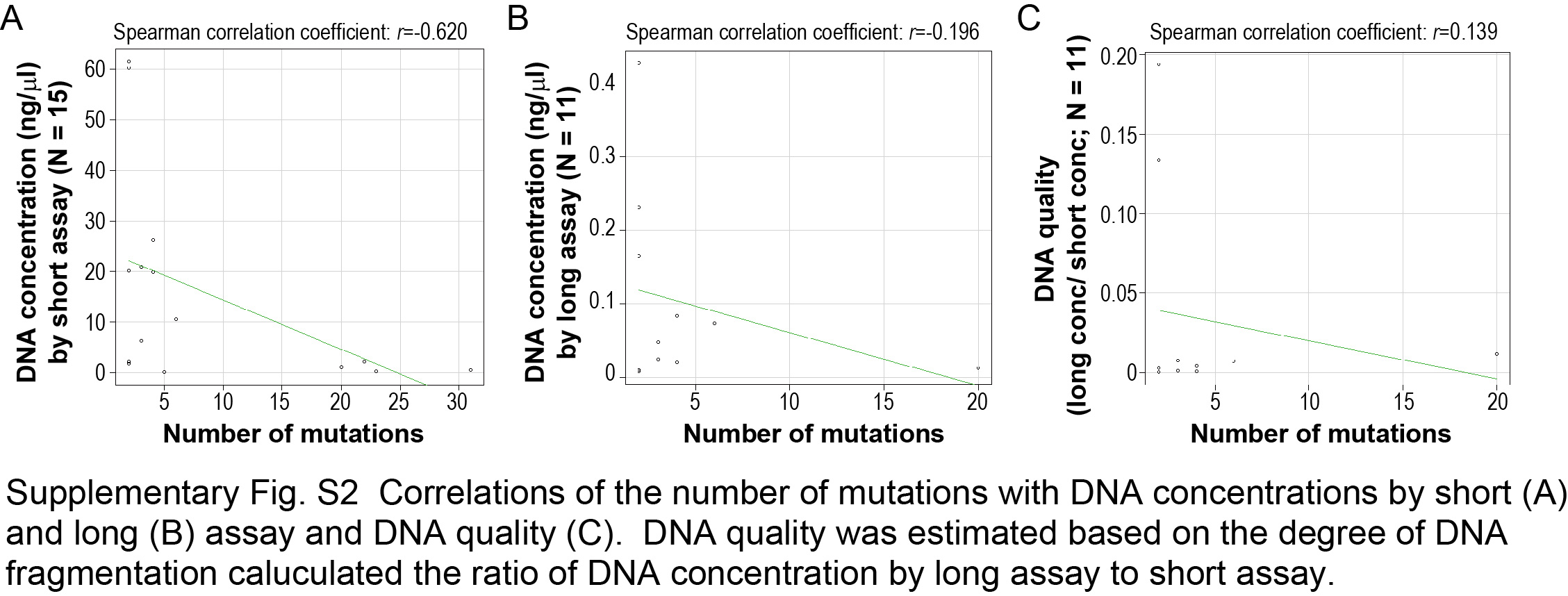
Signal processing, mapping to the reference genome (hg19), and quality control were carried out in Torrent Suite version 5.0 (Thermo Fisher Scientific). Sequence variants (point mutations, insertions, and deletions) were called using the Ion Reporter software 5.0 AmpliSeq Cancer Hotspot Panel v2 tumor‐normal pair workflow (Thermo Fisher Scientific) with default settings. A sequencing coverage of 100× and a minimum variant frequency of 15% of the total number of distinct tags were used as cutoffs. Mutations were called if they occurred in <0.1% of reads in the normal control and were absent from dbSNP 138 and the 1000 Genomes Project database. Alignment was visually inspected with the Integrative Genomics Viewer software (http//http://www.broadinstitute.org/igv). We were not able to obtain mutation profiles from five nodules as a result of low DNA quality (one specimen was surgically resected more than 10 years ago), limited tumor size (three nodules were less than or equal to 1 mm in diameter), or tumor purity (one nodule had less than 5% tumor purity because of neoadjuvant therapy). Of the 15 tumor nodules available for NGS data, 12 (80%) had somatic mutations in KRAS gene. Detailed information on detected mutations are summarized in Table S2 and Figure S1. To evaluate DNA quality, we conducted TaqMan FFPE DNA QC Assay (Thermo Fisher Scientific), following the manufacturer’s protocol. In this assay, we examined the degree of DNA fragmentation by comparing DNA concentrations measured by quantitative PCR with the use of two different primer sets for short (87 bp) and long (256 bp) amplicons in the RNase P gene. There was no positive correlation between the number of mutations and DNA concentration or quality (Spearman correlation coefficient r < .14; Figure S2).
2.5. Statistical analysis
Survival curves were estimated using the Kaplan‐Meier method. The survival curves for patients with multicentric carcinogenesis and intrapancreatic metastasis were compared using the log‐rank test. Outcome endpoint for distant metastasis‐free survival was the diagnosis of metastasis to distant organs (other than the pancreas or peripancreatic lesions). Death as a result of pancreatic cancer was the endpoint for cancer‐specific survival and death as a result of other causes was censored. Survival time was defined as the period from the date of surgery that showed multiple PDAC to event (death or recurrence) or to the end of follow up. Fisher’s exact test was used to compare the differences in mutation rates between patients with multicentric carcinogenesis and those with intrapancreatic metastasis. For all statistical analyses, P values were two‐sided, and P < .05 was defined as significant. All statistical analyses were carried out using EZR version 1.37 (Saitama Medical Center, Jichi Medical University),43 which is a graphic user interface for R (The R Foundation for Statistical Computing).
3. RESULTS
3.1. Clinicopathological and genetic relationships between tumors in patients with multiple PDAC
We analyzed 20 tumors from nine patients who had synchronous and/or metachronous PDAC. Table 1 summarizes the results of integrated clinical, pathological, and mutational analyses. For patient MPKO01, NGS analysis showed that the two synchronous tumors had different mutational patterns in KRAS and TP53 (Table S2). Moreover, immunohistochemical examination showed distinct patterns between these two synchronous tumors in terms of TP53 and SMAD4 statuses. Therefore, we considered that the two tumors from MPKO01 were independent and likely arose from multifocal origins. Similarly, NGS data indicated no shared mutations between two synchronous tumors in patient MPKO02, which led us to postulate their origins to be independent. Tumor T2 in patient MPKO03 was clinically regarded as a second primary tumor, because it developed in the remnant pancreas 11 years after R0 surgery without any history of recurrence. Indeed, the two tumors from MPKO03 showed different TP53 immunostaining patterns, and both tumors had tumor‐associated PanIN lesions.
For patient MPKO04, no shared mutations were detected between T2 and T1 or T3, indicating that tumor T2 was genetically independent of the other two tumors. Among the 50 genes in the NGS panel, we found one mutation in KRAS shared between T1 and T3 in MPKO04; however, the mutation patterns were different in 22 other genes (31 and 20 mutations were detected in tumors T1 and T3, respectively). In particular, the mutation patterns were different in TP53 and SMAD4, which are major driver genes essential for the progression to carcinoma.44 In addition, the pancreatic resection margin for the initial surgery for MPKO04 was negative, and the anatomical locations were some distance apart (T1, tail; T3, head). Consequently, we considered the three tumors from MPKO04 as likely being independent.
Mutation analysis uncovered strong intertumor relationships between the two tumors in patient MPKO09, whose tumors shared four mutations in TP53, CDKN2A, PTEN, and KDR. In MPKO05, we also found tight molecular associations between T2 and T3, which shared mutations in KRAS and TP53 and aberrant immunostaining patterns for TP53 and SMAD4. The IHC findings of tumors T2 and T3 in MPKO05 were analogous with those of T1; however, T1 was resected after chemoradiotherapy, which made it challenging to determine its relatedness to T2 or T3 because of the absence of NGS data and the severely restricted histomorphological analysis.
The smaller tumors (T2) from patients MPKO06, MPKO07, and MPKO08 were all tiny (≤1 mm). These smaller tumors showed identical TP53 or SMAD4 expression pattern, and had similar histological findings, to those of the corresponding main tumors. Consequently, we considered these tiny nodules from patients MPKO06, MPKO07, and MPKO08 to be orthotopic metastatic daughters.
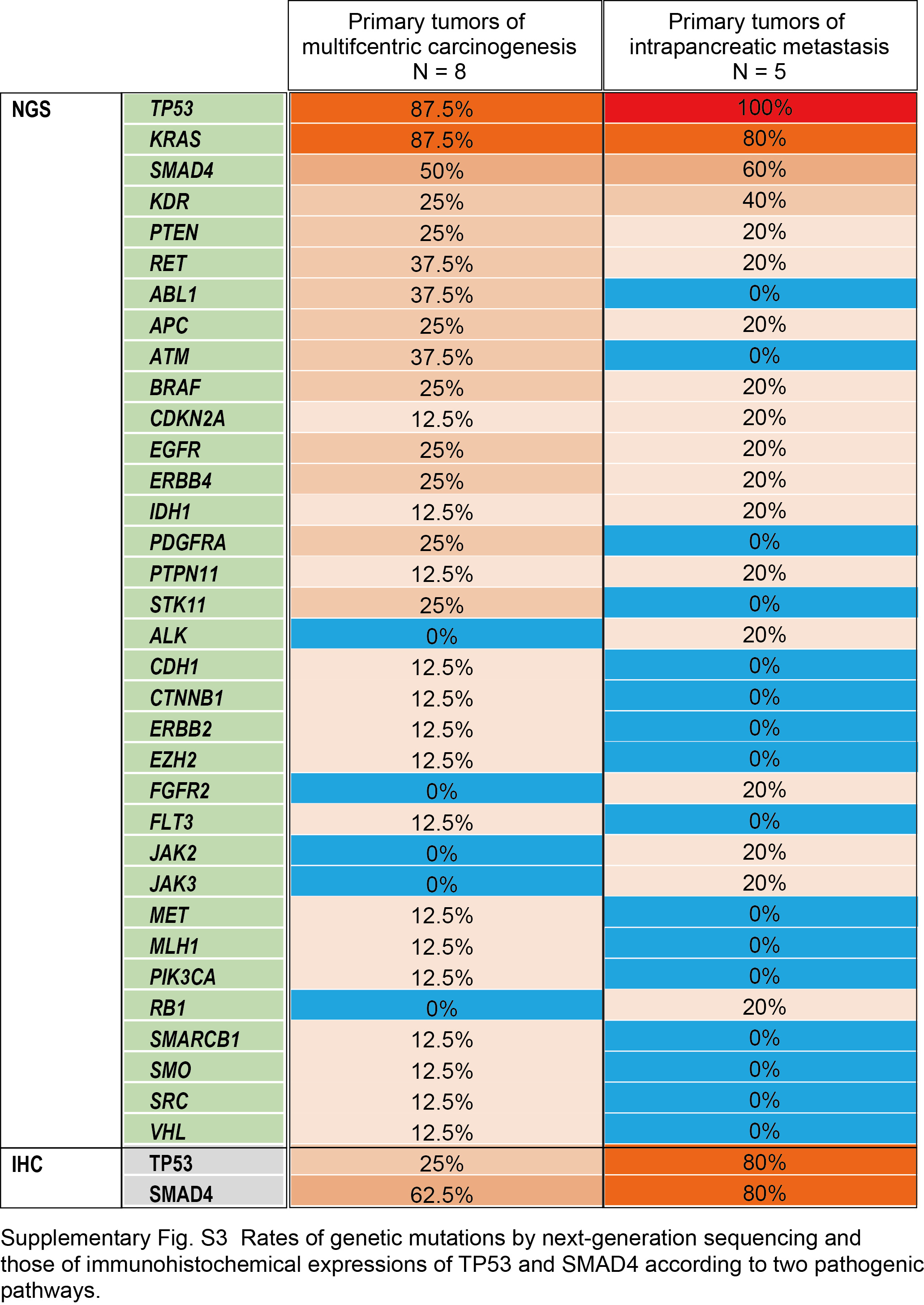
In summary, we identified four patients (MPKO01‐04) who had independent tumors (regarded as “multicentric carcinogenesis”) and five patients (MPKO05‐09) with multiple tumors likely associated with “intrapancreatic metastasis”. In all the four patients with tumors resulting from multicentric carcinogenesis, eight (89%) of the nine tumor nodules were accompanied by tumor‐associated PanIN. In contrast, none of the five smaller nodules in the patients with postulated intrapancreatic metastasis had any PanIN lesion in or around the nodules. The immunohistochemical results for TP53 and SMAD4 were totally consistent between the main and non‐main tumors for the five intrapancreatic metastasis cases. Morphologically, the intrapancreatic metastatic nodules showed similar histology to partial components of their corresponding main tumors and, therefore, all these five postulated metastatic nodules met histological criteria proposed by previous studies.25, 26 We did not observe significant differences in mutation rates for any genes examined between the two possible evolutionary paths (Figure S3). Examples of the characteristics of tumors resulting from multicentric carcinogenesis (MPKO02) and intrapancreatic metastasis (MPKO09) are shown in Figure 2.
Figure 2.
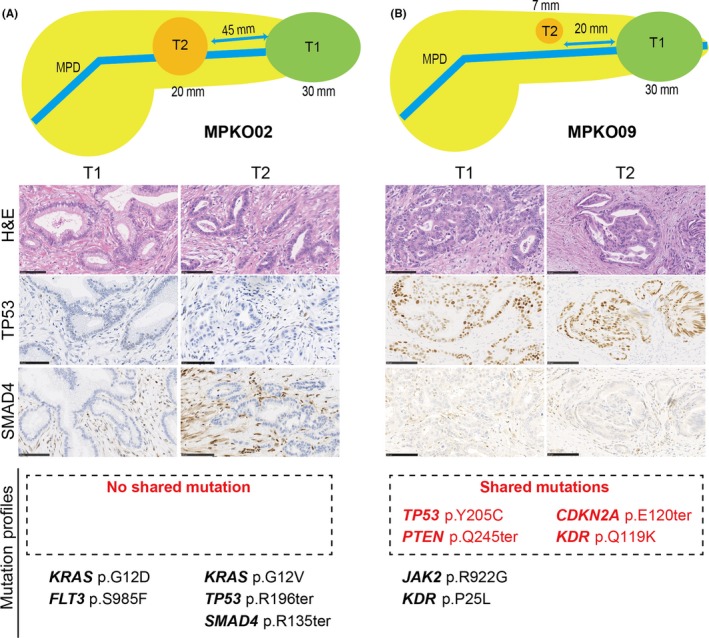
Histological findings and mutation profiles in a case of multicentric carcinogenesis (A, MPKO02) and in a case of intrapancreatic metastasis (B, MPKO09) (scale bar = 100 µm). MPD, main pancreatic duct
3.2. Differences in clinical behavior by etiologies of multiple PDAC
We then compared outcomes and found that disease stages appeared to be higher for intrapancreatic metastasis‐associated tumors than tumors resulting from multicentric carcinogenesis (Table 2). All the five patients with intrapancreatic metastasis died of pancreatic cancer, whereas one of the four patients whose tumors resulted from multicentric carcinogenesis died of this disease. Patients with intrapancreatic metastasis had statistically shorter distant metastasis‐free survival and cancer‐specific survival than those harboring tumors resulting from multicentric carcinogenesis (P = .047 and P = .049, respectively; by log‐rank test; Figure 3).
Table 2.
Outcomes of nine patients with multiple pancreatic ductal adenocarcinomas
| Patient ID | Postulated pathogenesis | UICC 8th | First distant metastatic organ | Distant recurrence timed (months) | Status | Survival timed (months) | ||
|---|---|---|---|---|---|---|---|---|
| T | N | Stage | ||||||
| MPKO01 | Multicentric carcinogenesis | 2/1 | 1 | IIBa | NAc | NAc | NED | 53 |
| MPKO02 | Multicentric carcinogenesis | 2/1 | 1 | IIBa | Lung | 18 | AWD | 48 |
| MPKO03 | Multicentric carcinogenesis |
2 3 |
0 1 |
IBb IIBb |
Lung, bone, peritonea | 9 | DOD | 20 |
| MPKO04 | Multicentric carcinogenesis |
2/1 2 |
1 1 |
IIBb |
NAc | NAc | DOO | 50 |
| MPKO05 | Intrapancreatic metastasis |
2 2/1 |
1 0 |
IIBb IBb |
Lymph node | 11 | DOD | 18 |
| MPKO06 | Intrapancreatic metastasis | 1c | 1 | IIB | Lung | 11 | DOD | 44 |
| MPKO07 | Intrapancreatic metastasis | 2 | 2 | III | Liver | 7 | DOD | 27 |
| MPKO08 | Intrapancreatic metastasis | 2 | 2 | III | Liver | 16 | DOD | 35 |
| MPKO09 | Intrapancreatic metastasis | 2 | 2 | III | Liver | 6 | DOD | 15 |
Abbreviation: AWD, alive with disease; DOD, died of disease; DOO, died of other causes; NA, not applicable; NED, no evidence of disease.
Stages for synchronous multicentric tumors were defined by the worst T factors and the N factors.
Stages for metachronous tumors were assigned to each timing.
Not applicable because there was no recurrence (MPKO01) or distant metastasis (MPKO04).
Survival time was defined as the period from the date of surgery that showed multiple pancreatic cancers to event (death or recurrence) or to the end of follow up.
Figure 3.
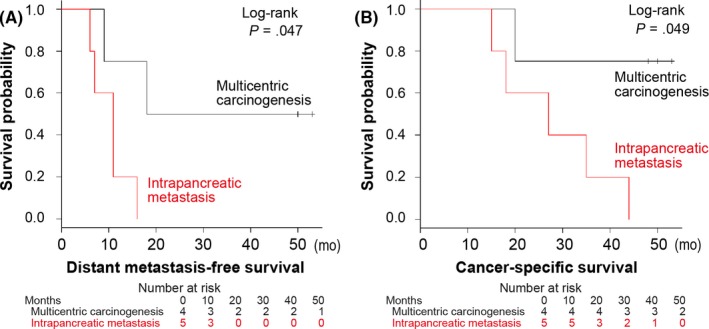
Kaplan‐Meier curves for distant metastasis‐free survival (A) and cancer‐specific survival (B) according to two pathogenic pathways. Tables show the number of patients who remained alive and at risk of the event at each time point
4. DISCUSSION
Integrated clinical, pathological, and mutational analyses on synchronous and/or metachronous pancreatic cancers suggested two pathogenic pathways for the formation of multiple PDAC: multicentric carcinogenesis and intrapancreatic metastasis. Among the nine patients with multiple PDAC in the current study, we found four cases (44%) of multicentric tumors and five cases (56%) of intrapancreatic metastasis. A recent study analyzed six PDAC arising in the remnant pancreas after surgical resection of primary pancreatic cancers using mutation testing for 11 pancreatic cancer‐related genes, and showed that three (50%) of six patients had “true” recurrence whereas two (33%) had independent tumors.17 These findings are generally consistent with our results. Another recent study analyzed RAS mutations in 14 patients with metachronous PDAC and identified two patients (14%) as having shared mutations;45 however, the mutation rate for KRAS was considerably low (21%) in that study cohort compared to previous large studies and our current study.46, 47, 48 These lines of evidence together with our findings indicate that the two distinct evolutionary paths may contribute to the formation of synchronous and/or metachronous PDAC in clinical practice.
Evidence from engineered mouse models and human tissue‐based studies has indicated that precursor PanIN initiate multifocally, and several of them can progress to carcinomas that eventually become invasive and metastatic.8, 9, 10, 11, 12, 13 We confirmed the presence of tumor‐associated PanIN in eight of the nine tumors resulting from multicentric carcinogenesis‐associated PDAC, but did not observe any PanIN lesion in or around the postulated metastatic nodules. In a number of cancer types, such as colorectal and hepatocellular carcinoma, the primary invasive cancers at early carcinogenic stages are frequently accompanied by preinvasive lesions from which the primary tumors may have originally arisen.49, 50 Hence, histological surveillance for the presence of tumor‐associated PanIN is likely useful to differentiate whether a pancreatic cancer nodule in multiple PDAC is primary or metastatic.
We determined the pathogenesis of tumors T1 and T3 in patient MPKO04 as multicentric carcinogenesis because they showed differing mutation statuses in 22 of the 23 mutated genes, including major drivers. However, the finding that these two tumors had a common mutation, KRAS p.Q61H, one of common KRAS variants in PDAC populations (prevalence rate, 5.8%),36 is intriguing. Given the markedly high mutation rates in tumors of patient MPKO04, a by‐chance scenario of this matched mutation is most likely. A recent study has suggested that the precancerous cells in PanIN move through the pancreatic ductal system and initiate multicentric nodules in the pancreas.27 Therefore, we presume an alternative scenario where a precancerous ancestor harboring KRAS p.Q61H disseminated via the pancreatic duct to form precursors of tumors T1 and T3, followed by sequential accumulation of mutations in different sets of driver genes, including TP53 and SMAD4, resulting in the formation of genetically independent invasive cancers.
The current study showed that patients with intrapancreatic metastasis had statistically shorter distant metastasis‐free survival and shorter pancreatic cancer‐specific survival than did patients with multiple PDAC of multicentric carcinogenesis. Based on pathological findings, Oguro et al identified 21 patients with intrapancreatic metastasis and showed that PDAC patients with intrapancreatic metastasis are associated with worse outcomes than those without intrapancreatic metastasis.26 Because of limited statistical power of these studies, further investigations with larger sample sizes are clearly needed to validate the prognostic utility of intrapancreatic metastasis. Such knowledge would inform the development of clinical strategies for assessing multiple PDAC.
In the present study, six of 11 tumors with loss of SMAD4 protein expression were called “wild type” by NGS analysis. The SMAD4 gene has been inactivated in 35% of PDAC populations by homozygous deletion and in 20% by loss of one allele coupled with a mutation in the second allele.29 Although SMAD4 immunohistochemistry has been a powerful tool to detect both types of genetic alteration,51 it is possible that targeted sequencing is not suitable for identification of homozygous deletion of SMAD4, which might cause this discrepancy. In addition, uncommon SMAD4 mutations are not covered by the Ion AmpliSeq Cancer Hotspot Panel v2 (data not shown), which would have also influenced the results.
We recognize certain limitations in this study. First, we were not able to obtain NGS data from some tissue samples despite repeated DNA extractions using archival FFPE and frozen tissues. However, in such cases, clinicopathological findings from IHC and morphological investigation helped us to clarify the intertumor relationships. Second, the number of cases was limited in this retrospective study. Nonetheless, our primary hypothesis testing was whether two potential pathways (multicentric carcinogenesis and intrapancreatic metastasis) might contribute to the formation of multiple PDAC. In our hypothesis testing, we were able to validate the distinct carcinogenic paths in the nine patients with multiple PDAC. Future prospective studies are warranted to determine which etiology is dominant in the pathogenesis of multiple PDAC.
Determining whether multiple PDAC are multicentric or metastatic is becoming increasingly important for clinical practice. From a practical viewpoint, histomorphological judgement with concurrent IHC examination on TP53 and SMAD4 are suggested to distinguish multicentric occurrence from intrapancreatic metastasis. However, in cases with uncertain intertumor relationships by these pathological methods, implementation of NGS analyses should be considered to determine the evolutionary paths for precise staging of multiple PDAC.
The genomic characterization of PDAC is beginning to be implemented in oncology clinics in an effort to harness potential therapeutic opportunities against this deadly cancer.52 Our data suggest that the clinicopathological and genetic assessments of multiple PDAC are helpful to distinguish tumors resulting from multifocal carcinogenesis from those resulting from intrapancreatic metastasis; the latter type likely have a dismal prognosis. This study provides insights into the origins of multiple PDAC and, once validated, could inform clinical practice by guiding treatment strategies against pancreatic cancer in the era of precision medicine.
CONFLICTS OF INTEREST
YK has received scholarship endowments from Taiho Pharmaceutical Co., Ltd, Chugai Pharmaceutical Co., Ltd, Tsumura & Co., Otsuka Pharmaceutical Co., Ltd, AsahiKASEI Co., Ltd, Eisai Co., Ltd, Otsuka Pharmaceutical Factory Inc., Takeda Pharmaceutical Co., Ltd, Medicon Inc., Pfizer Japan Inc., Ono Pharmaceutical Co., Ltd, Shionogi & Co., Ltd, and Daiichi Sankyo Co., Ltd, received lecture fees/honoraria from AsahiKASEI Co., Ltd, Ono Pharmaceutical Co., Ltd, Ethicon Inc., Taiho Pharmaceutical Co., Ltd, and Chugai Pharmaceutical Co., Ltd, and is an officer/advisor for medical corporation Keiyoukai Keiai Clinic, Hirata Clinic, and Ageo Central General Hospital. Ma.S. has received research funds from Taiho Pharmaceutical Co., Ltd, Shionogi & Co., Ltd, and Eisai Co., Ltd. The other authors declare that they have no conflicts of interest.
Supporting information
{kind=link}
{kind=link}
{kind=link}
ACKNOWLEDGMENTS
This work was supported by an endowed chair from Chugai Pharmaceutical Co., Ltd to the Department of Surgery, Keio University School of Medicine, and by KAKENHI from the Japan Society for the Promotion of Science (Grant number 18K15094 to YM and 16H06279 to TT). We are grateful to the Fourth Laboratory of the Department of Pathology in Keio University School of Medicine for assistance with tissue processing and staining.
Fujita Y, Matsuda S, Sasaki Y, et al. Pathogenesis of multiple pancreatic cancers involves multicentric carcinogenesis and intrapancreatic metastasis. Cancer Sci. 2020;111:739–748. 10.1111/cas.14268
Fujita and Matsuda contributed equally to this study.
Use of standardized official symbols: We use HUGO (Human Genome Organization)‐approved official symbols for genes and gene products, which are described at www.genenames.org. Gene names are italicized, and gene product names are non‐italicized.
Contributor Information
Yohei Masugi, Email: masugi@z6.keio.jp.
Minoru Kitago, Email: dragonpegasus427@gmail.co.jp.
REFERENCES
- 1. Notta F, Hahn SA, Real FX. A genetic roadmap of pancreatic cancer: still evolving. Gut. 2017;66:2170‐2178. [DOI] [PubMed] [Google Scholar]
- 2. Kleeff J, Korc M, Apte M, et al. Pancreatic cancer. Nat Rev Dis Primers. 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 3. Jones S, Zhang X, Parsons DW, et al. Core signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science. 2008;321:1801‐1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Campbell PJ, Yachida S, Mudie LJ, et al. The patterns and dynamics of genomic instability in metastatic pancreatic cancer. Nature. 2010;467:1109‐1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cicenas J, Kvederaviciute K, Meskinyte I, Meskinyte‐Kausiliene E, Skeberdyte A, Cicenas J. KRAS, TP53, CDKN2A, SMAD4, BRCA1, and BRCA2 mutations in pancreatic cancer. Cancers. 2017;9(12):42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Yachida S, Jones S, Bozic I, et al. Distant metastasis occurs late during the genetic evolution of pancreatic cancer. Nature. 2010;467:1114‐1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hruban RH, Gaida MM, Thompson E, et al. Why is pancreatic cancer so deadly? The pathologist’s view. J Pathol. 2019;248:131‐141. [DOI] [PubMed] [Google Scholar]
- 8. Hingorani SR, Petricoin EF, Maitra A, et al. Preinvasive and invasive ductal pancreatic cancer and its early detection in the mouse. Cancer Cell. 2003;4:437‐450. [DOI] [PubMed] [Google Scholar]
- 9. Hingorani SR, Wang L, Multani AS, et al. Trp53R172H and KrasG12D cooperate to promote chromosomal instability and widely metastatic pancreatic ductal adenocarcinoma in mice. Cancer Cell. 2005;7:469‐483. [DOI] [PubMed] [Google Scholar]
- 10. Lesina M, Kurkowski MU, Ludes K, et al. Stat3/Socs3 activation by IL‐6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell. 2011;19:456‐469. [DOI] [PubMed] [Google Scholar]
- 11. Collins MA, Bednar F, Zhang Y, et al. Oncogenic Kras is required for both the initiation and maintenance of pancreatic cancer in mice. J Clin Invest. 2012;122:639‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Criscimanna A, Duan LJ, Rhodes JA, et al. PanIN‐specific regulation of Wnt signaling by HIF2alpha during early pancreatic tumorigenesis. Cancer Res. 2013;73:4781‐4790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tsutsumi M, Konishi Y. Precancerous conditions for pancreatic cancer. J Hepatobiliary Pancreat Surg. 2000;7:575‐579. [DOI] [PubMed] [Google Scholar]
- 14. Brune K, Abe T, Canto M, et al. Multifocal neoplastic precursor lesions associated with lobular atrophy of the pancreas in patients having a strong family history of pancreatic cancer. Am J Surg Pathol. 2006;30:1067‐1076. [PMC free article] [PubMed] [Google Scholar]
- 15. Matsuda Y, Furukawa T, Yachida S, et al. The prevalence and clinicopathological characteristics of high‐grade pancreatic intraepithelial neoplasia: autopsy study evaluating the entire pancreatic parenchyma. Pancreas. 2017;46:658‐664. [DOI] [PubMed] [Google Scholar]
- 16. Basturk O, Hong SM, Wood LD, et al. A revised classification system and recommendations from the Baltimore consensus meeting for neoplastic precursor lesions in the pancreas. Am J Surg Pathol. 2015;39:1730‐1741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Luchini C, Pea A, Yu J, et al. Pancreatic cancer arising in the remnant pancreas is not always a relapse of the preceding primary. Mod Pathol. 2019;32:659‐665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Ibrahim I, Sibinga Mulder BG, Bonsing B, et al. Risk of multiple pancreatic cancers in CDKN2A‐p16‐Leiden mutation carriers. Eur J Hum Genet. 2018;26:1227‐1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Canto MI, Almario JA, Schulick RD, et al. Risk of neoplastic progression in individuals at high risk for pancreatic cancer undergoing long‐term surveillance. Gastroenterology. 2018;155(740–51):e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hata T, Suenaga M, Marchionni L, et al. Genome‐wide somatic copy number alterations and mutations in high‐grade pancreatic intraepithelial neoplasia. Am J Pathol. 2018;188:1723‐1733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61:759‐767. [DOI] [PubMed] [Google Scholar]
- 22. Sakamoto M, Effendi K, Masugi Y. Molecular diagnosis of multistage hepatocarcinogenesis. Jpn J Clin Oncol. 2010;40:891‐896. [DOI] [PubMed] [Google Scholar]
- 23. Fidler IJ. The pathogenesis of cancer metastasis: the 'seed and soil' hypothesis revisited. Nat Rev Cancer. 2003;3:453‐458. [DOI] [PubMed] [Google Scholar]
- 24. Vakoc CR, Tuveson DA. Soils and seeds that initiate pancreatic cancer metastasis. Cancer Discov. 2017;7:1067‐1068. [DOI] [PubMed] [Google Scholar]
- 25. Fujita Y, Kitago M, Masugi Y, et al. Two cases of pancreatic ductal adenocarcinoma with intrapancreatic metastasis. World J Gastroenterol. 2016;22:9222‐9228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Oguro S, Shimada K, Ino Y, et al. Pancreatic intraglandular metastasis predicts poorer outcome in postoperative patients with pancreatic ductal carcinoma. Am J Surg Pathol. 2013;37:1030‐1038. [DOI] [PubMed] [Google Scholar]
- 27. Makohon‐Moore AP, Matsukuma K, Zhang M, et al. Precancerous neoplastic cells can move through the pancreatic ductal system. Nature. 2018;561:201‐205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hashimoto D, Arima K, Nakagawa S, et al. Pancreatic cancer arising from the remnant pancreas after pancreatectomy: a multicenter retrospective study by the Kyushu Study Group of Clinical Cancer. J Gastroenterol. 2018;18(4):S61‐S62. [DOI] [PubMed] [Google Scholar]
- 29. Bosman F, Carneiro F, Hruban R, Theise N. WHO Classification of Tumours of the Digestive System. Lyon, France: IARC Press; 2010. [Google Scholar]
- 30. Japan Pancreas Society . Classification of Pancreatic Carcinoma, 4th English edn. Tokyo: Kanehara; 2017:70–79. [Google Scholar]
- 31. Omori Y, Ono Y, Tanino M, et al. Pathways of progression from intraductal papillary mucinous neoplasm to pancreatic ductal adenocarcinoma based on molecular features. Gastroenterology. 2019;156(647–61):e2. [DOI] [PubMed] [Google Scholar]
- 32. Furukawa T, Kuboki Y, Tanji E, et al. Whole‐exome sequencing uncovers frequent GNAS mutations in intraductal papillary mucinous neoplasms of the pancreas. Sci Rep. 2011;1:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wu J, Matthaei H, Maitra A, et al. Recurrent GNAS mutations define an unexpected pathway for pancreatic cyst development. Sci Transl Med. 2011;3:92ra66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hruban RH, Adsay NV, Albores‐Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579‐586. [DOI] [PubMed] [Google Scholar]
- 35. Chun YS, Pawlik TM, Vauthey JN. 8th edition of the AJCC cancer staging manual: pancreas and hepatobiliary cancers. Ann Surg Oncol. 2018;25:845‐847. [DOI] [PubMed] [Google Scholar]
- 36. Qian ZR, Rubinson DA, Nowak JA, et al. Association of alterations in main driver genes with outcomes of patients with resected pancreatic ductal adenocarcinoma. JAMA Oncol. 2018;4:e173420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Yamazaki K, Masugi Y, Effendi K, et al. Upregulated SMAD3 promotes epithelial‐mesenchymal transition and predicts poor prognosis in pancreatic ductal adenocarcinoma. Lab Invest. 2014;94:683‐691. [DOI] [PubMed] [Google Scholar]
- 38. Masugi Y, Abe T, Ueno A, et al. Characterization of spatial distribution of tumor‐infiltrating CD8(+) T cells refines their prognostic utility for pancreatic cancer survival. Mod Pathol. 2019;32:1495‐1507. [DOI] [PubMed] [Google Scholar]
- 39. Kitago M, Ueda M, Aiura K, et al. Comparison of K‐ras point mutation distributions in intraductal papillary‐mucinous tumors and ductal adenocarcinoma of the pancreas. Int J Cancer. 2004;110:177‐182. [DOI] [PubMed] [Google Scholar]
- 40. Nakano Y, Kitago M, Matsuda S, et al. KRAS mutations in cell‐free DNA from preoperative and postoperative sera as a pancreatic cancer marker: a retrospective study. Br J Cancer. 2018;118:662‐669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Nakagaki T, Tamura M, Kobashi K, et al. Profiling cancer‐related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget. 2017;8:59113‐59122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Nakagaki T, Tamura M, Kobashi K, et al. Targeted next‐generation sequencing of 50 cancer‐related genes in Japanese patients with oral squamous cell carcinoma. Tumour Biol. 2018;40:1010428318800180. [DOI] [PubMed] [Google Scholar]
- 43. Kanda Y. Investigation of the freely available easy‐to‐use software 'EZR' for medical statistics. Bone Marrow Transplant. 2013;48:452‐458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hosoda W, Chianchiano P, Griffin JF, et al. Genetic analyses of isolated high‐grade pancreatic intraepithelial neoplasia (HG‐PanIN) reveal paucity of alterations in TP53 and SMAD4. J Pathol. 2017;242:16‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hashimoto D, Arima K, Nakagawa S, et al. Pancreatic cancer arising from the remnant pancreas after pancreatectomy: a multicenter retrospective study by the Kyushu Study Group of Clinical Cancer. J Gastroenterol. 2019;54:437‐448. [DOI] [PubMed] [Google Scholar]
- 46. Bailey P, Chang DK, Nones K, et al. Genomic analyses identify molecular subtypes of pancreatic cancer. Nature. 2016;531:47. [DOI] [PubMed] [Google Scholar]
- 47. Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor‐ and stroma‐specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet. 2015;47:1168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med. 2011;17:500‐503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Je IJ, Vermeulen L, Meijer GA, Dekker E. Serrated neoplasia‐role in colorectal carcinogenesis and clinical implications. Nat Rev Gastroenterol Hepatol. 2015;12:401‐409. [DOI] [PubMed] [Google Scholar]
- 50. Chuma M, Sakamoto M, Yamazaki K, et al. Expression profiling in multistage hepatocarcinogenesis: identification of HSP70 as a molecular marker of early hepatocellular carcinoma. Hepatology. 2003;37:198‐207. [DOI] [PubMed] [Google Scholar]
- 51. Wilentz RE, Su GH, Dai JL, et al. Immunohistochemical labeling for dpc4 mirrors genetic status in pancreatic adenocarcinomas: a new marker of DPC4 inactivation. Am J Pathol. 2000;156:37‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Aguirre AJ, Nowak JA, Camarda ND, et al. Real‐time genomic characterization of advanced pancreatic cancer to enable precision medicine. Cancer Discov. 2018;8:1096‐1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials