

Abstract
Patients with lower‐risk myelodysplastic syndromes (LR‐MDS) as defined by the International Prognostic Scoring System (IPSS) have more favorable prognosis in general, but significant inter‐individual heterogeneity exists. In this study, we examined the molecular profile of 15 MDS‐relevant genes in 159 patients with LR‐MDS using next‐generation sequencing. In univariate COX regression, shorter overall survival (OS) was associated with mutation status of ASXL1 (P = .001), RUNX1 (P = .031), EZH2 (P = .049), TP53 (P = .016), SRSF2 (P = .046), JAK2 (P = .040), and IDH2 (P = .035). We also found significantly shorter OS in patients with an adjusted TET2 variant allele frequency (VAF) ≥18% versus those with either an adjusted TET2 VAF <18% or without TET2 mutations (median: 20.4 vs 47.8 months; P = .020; HR = 2.183, 95%CI: 1.129‐4.224). After adjustment for IPSS, shorter OS was associated with mutation status of ASXL1 (P < .001; HR = 4.306, 95% CI: 2.144‐8.650), TP53 (P = .004; HR = 4.863, 95% CI: 1.662‐14.230) and JAK2 (P = .002; HR = 5.466, 95%CI: 1.848‐16.169), as well as adjusted TET2 VAF ≥18% (P = .008; HR = 2.492, 95% CI: 1.273‐4.876). Also, OS was increasingly shorter as the number of mutational factors increased (P < .001). A novel prognostic scoring system incorporating the presence/absence of the four independent mutational factors into the IPSS further stratified LR‐MDS patients into three prognostically different groups (P < .001). The newly developed scoring system redefined 10.1% (16/159) of patients as a higher‐risk group, who could not be predicted by the currently prognostic models. In conclusion, integration of the IPSS with mutation status/burden of certain MDS‐relevant genes may improve the prognostication of patients with LR‐MDS and could help identify those with worse‐than‐expected prognosis for more aggressive treatment.
Keywords: lower‐risk myelodysplastic syndromes, mutation burden, mutation status, prognosis, variant allele frequency
Shorter OS was associated with mutation status of ASXL1, EZH2 and JAK2, as well as TET2 VAF ≥18%. A novel prognostic scoring system based on the IPSS and the presence/absence of the four independent mutational factors further stratified LR‐MDS patients into three prognostically different groups. Integration of IPSS with mutation status/burden of certain MDS‐relevant genes may improve the prognostication of patients with LR‐MDS and could help identify those with worse‐than‐expected prognosis for more aggressive treatment.

1. INTRODUCTION
Myelodysplastic syndromes (MDS) are a group of clonal hematological malignancies with significant clinical heterogeneity and genetic diversity.1, 2 The life expectancy of MDS patients ranges from several months to several years; thus, accurate survival prediction is critical for treatment decision‐making.3 The International Prognostic Scoring System (IPSS) is the most commonly used tool for prognostic assessment of patients with untreated primary MDS in clinical practice, trial eligibility and treatment recommendation.4, 5 Patients with low risk (IPSS score: 0) and intermediate 1 (Int‐1) risk (IPSS score: 0.5‐1.0) are grouped together as the lower‐risk (LR) group, and generally tend to have an indolent course of disease progression. However, a significant subset of LR‐MDS patients have a worse outcome and a much higher frequency of progression into secondary acute myeloid leukemia (sAML).6, 7, 8
Attempts have been made to identify patients with greater‐than‐expected risk in LR‐MDS patients. The MD Anderson Lower‐Risk Prognostic Scoring System (LR‐PSS) could stratify LR‐MDS into three prognostically distinct groups.9 The recently revised IPSS (IPSS‐R) reassigns MDS patients into a lower‐ versus a higher‐risk group (IPSS‐R score of ≤3.5 vs >3.5).10 Despite these refinements, a significant proportion of patients with poor outcomes could not be identified.8
Recurrent mutations in MDS are associated with overall survival (OS), leukemic transformation, and response to hypomethylating agents (HMA).11, 12, 13, 14, 15 In a pivotal study of LR‐MDS, ASXL1, EZH2, TP53, and RUNX1 mutations were shown to be independent prognostic predictors.16 Also, combination of EZH2 mutation status with LR‐PSS could predict a subset of LR‐MDS patients with poor prognosis not identified otherwise.16
Mutation burden has also been associated with prognosis in MDS patients.3, 5 In general, increasing number of mutations is associated with progressively poorer prognosis.13, 17, 18, 19 For mutations of a specific gene, higher TP53 mutation allelic burden was negatively associated with OS in both unselected MDS and LR‐MDS;20, 21, 22 higher TET2 clonal burden predicted an increased response to decitabine in MDS.15 In addition to MDS, variant allele frequency (VAF) has been associated with prognosis in patients with AML.23, 24
In the current study, we examined the mutation status and burden of 15 MDS‐relevant genes in a group of patients with LR‐MDS by next‐generation sequencing (NGS). The results showed association between poor prognosis with ASXL1 mutant type (MT), TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%. A scoring system that combines IPSS and mutation status/burden was further developed to identify LR‐MDS patients with greater‐than‐predicted risk.
2. SUBJECTS AND METHODS
2.1. Patients
This study included all patients with LR‐MDS (IPSS score ≤1.0) upon initial diagnosis who were treated at the First Affiliated Hospital, Zhejiang University School of Medicine during a period between February 2011 and January 2018. The diagnosis of MDS was based on the 2016 World Health Organization (WHO) classification.25 Patients with IPSS score >1.0 were excluded from data analysis. Metaphase cytogenetic and mutational analysis was conducted prior to treatment in all subjects. Single nucleotide polymorphism array (SNP‐A) analysis was further carried out on patients with TET2 mutations confirmed by NGS. This study was approved by the Ethics Committee of the First Affiliated Hospital, Zhejiang University School of Medicine. The study was conducted in compliance with the Helsinki Declaration. All patients provided informed consent for the use of samples for research purposes.
2.2. Metaphase cytogenetic analysis
Unstimulated bone marrow cells were obtained upon initial diagnosis (prior to treatment). Cytogenetic slides were prepared using a standard protocol, and then R‐banded. Twenty metaphases were analyzed and the karyotypes were described according to the current International System for Human Cytogenetic Nomenclature.26
2.3. Mutational analysis
Genomic DNA was extracted from bone marrow mononuclear cells. Sample integrity was verified using standard NGS criteria (≥50 ng/μL, and OD 260/280:1.8‐2.0). We used a custom targeted NGS approach that combined multiplex PCR‐based target enrichment and library generation with ultra‐deep high‐throughput parallel sequencing using the Ion Proton Platform or MiSeq.27, 28 A total of 15‐MDS‐relevant genes, including TET2, SF3B1, U2AF1, ASXL1, SRSF2, DNMT3A, RUNX1, EZH2, JAK2, NRAS, TP53, CBL, ETV6, IDH1, and IDH2, were covered, and exons with coding regions known to be hotspots or related with MDS (average depth >800 X) were targeted. The raw sequence data are available on the Sequence Read Archive (SRA) (PRJNA550098).
2.4. SNP‐A‐based karyotyping
DNA was extracted from bone marrow. Sample quantity and purity were assessed by spectrophotometry (Nanodrop 2000, Thermo Fisher Scientific). CytoScan 750K arrays and reagents (Thermo Fisher Scientific) were used for SNP‐A testing. The array analysis and interpretation were carried out using Chromosome Analysis Suite (ChAS; Thermo Fisher Scientific) software version 4.0. Copy number variations (CNV) called by the ChAS software algorithm are denoted as true aberrations, with the exception of those known to be normal genomic variants based on a publicly available database (http://dgv.tcag.ca/dgv/app/home). We used SNP‐A to screen for microdeletions and uniparental disomy (UPD) at chromosome 4q/24 involving TET2 locus.
2.5. Mutation VAF
Variant allele frequency (referred to as raw VAF) is defined as the number of variant reads divided by the number of total reads and reported as a percentage. Variants with a VAF of <1% were excluded from analysis. Mutations were annotated using multiple databases, including 1000 genomes, COSMIC, PolyPhen‐2, and dbSNP.
Adjusted VAF was acquired with the adjustment of raw VAF based on copy number and zygosity confirmed by SNP‐A. VAF of homozygous mutation was reduced to as half the value of raw VAF. Hemizygous mutation VAF was adjusted based on the formula “adjusted VAF = a/1 + a (a = raw VAF value).” There is no adjustment for heterozygous or compound heterozygous mutations.
The R language‐based web tool Cutoff Finder (http://molpath.charite.de/cutoff/) was used to determine the optimal VAF cutoff in a given gene for survival stratification.29 In cases with multiple mutations of a certain gene, we chose the higher/highest VAF for calculation. For each cutoff, survival was examined in the two separated groups using the function survfit from the R package survival. Finally, the optimal cutoff for differences in survival was selected (lowest P value under log‐rank test).29
2.6. Follow up and response assessment
Overall survival was defined as the period from the date of initial diagnosis to the date of death regardless of the cause. Data were censored at the last follow up. Response to decitabine was assessed using the modified International Working Group (IWG) criteria.30 Patients with complete remission (CR), partial remission (PR), marrow CR (mCR), or hematological improvement (HI) were regarded as responders. Patients with stable disease, failure, or disease progression were regarded as non‐responders.
2.7. Statistical analysis
Survival analysis was carried out using the Kaplan‐Meier method followed by the log‐rank test. Univariate COX regression was used to select mutational variables for entry into multivariate COX regression with stepwise backward selection (P < .05). For the genes of which P ≥ .05 in the univariate analysis, the R language‐based web tool Cutoff Finder was then used to find the optimal VAF cutoff for differences in survival. Independent variables in the multivariate regression also included: age (≥ vs <60 years), gender, and currently common prognostic scoring systems (IPSS, IPSS‐R or LR‐PSS excluding age). Associations between mutation status and leukemic conversion were evaluated by Chi‐squared test or Fisher’s exact test. Bonferroni correction was applied for multiple testing. All statistical analyses were carried out using SPSS 23.0 software. P < .05 (2‐sided) was considered statistically significant. Venn diagram was generated by BioVenn.31
3. RESULTS
3.1. Clinical characteristics of the study population
A total of 159 LR‐MDS patients (median age: 58 years, range: 14‐89 years; 95 men and 64 women) were included in data analysis (Table S1). Based on the IPSS score, 29 (18.2%) patients had low risk and the remaining 130 (81.8%) had Int‐1 risk (Table 1). The 2016 WHO MDS types included: MDS with single lineage dysplasia (MDS‐SLD; n = 25, 15.7%), MDS with ring sideroblasts (MDS‐RS; n = 16, 10.1%), MDS with multilineage dysplasia (MDS‐MLD; n = 76, 47.8%), MDS with excess blast‐1 (MDS‐EB‐1; n = 33, 20.8%), MDS‐EB‐2 (n = 3, 1.9%), MDS unclassifiable (MDS‐U; n = 5, 3.1%), and MDS with isolated del (5q) (n = 1, 0.6%). Forty‐five (28.3%) patients had abnormal karyotypes. The four most frequent abnormal karyotypes were trisomy 8 (13.2%), del (20q) (4.4%), del (5q) (3.7%), and del (7) (2.5%) (Figure 1A).
Table 1.
Demographic and baseline characteristics of the study population
| Variable | Baseline distribution in cohort |
|---|---|
| Demographics | |
| Age (y), median (range) | 58 (14‐89) |
| Male gender, N (%) | 95 (59.7) |
| 2016 WHO classification, N (%) | |
| MDS‐SLD | 25 (15.7) |
| MDS‐RS | 16 (10.1) |
| MDS‐MLD | 76 (47.8) |
| MDS‐EB‐1 | 33 (20.8) |
| MDS‐EB‐2 | 3 (1.9) |
| MDS‐U | 5 (3.1) |
| MDS with isolated del(5q) | 1 (0.6) |
| Blood counts at time of mutation analysis | |
| Hemoglobin level (g/dL), mean ± SD | 8.1 ± 2.9 |
| Neutrophil count (×109/L), mean ± SD | 1.9 ± 2.9 |
| Platelet count (×109/L), mean ± SD | 113.5 ± 155.2 |
| Bone marrow at time of mutation analysis | |
| Bone marrow blasts (%), mean ± SD | 3.1 ± 2.6 |
| Abnormal karyotype, N (%) | 45 (28.3) |
| IPSS, N (%) | |
| Low | 29 (18.2) |
| Int‐1 | 130 (81.8) |
| Treatment, N (%) | |
| Best supportive care | 99 (62.3) |
| HSCT | 14 (8.8) |
| Decitabine | 42 (26.4) |
| Chemotherapy | 4 (2.5) |
| Outcome, N (%) | |
| Leukemic transformation | 25 (15.7) |
| Death | 58 (36.5) |
Abbreviations: HSCT, hematopoietic stem cell transplantation; Int‐1, intermediate 1; IPSS, International Prognostic Scoring System; MDS‐EB‐1, myelodysplastic syndromes with excess blasts‐1; MDS‐EB‐2, MDS with excess blasts‐2; MDS‐MLD, MDS with multilineage dysplasia; MDS‐RS, MDS with ring sideroblasts; MDS‐SLD, MDS with single lineage dysplasia; MDS‐U, MDS unclassifiable; WHO, World Health Organization.
Figure 1.
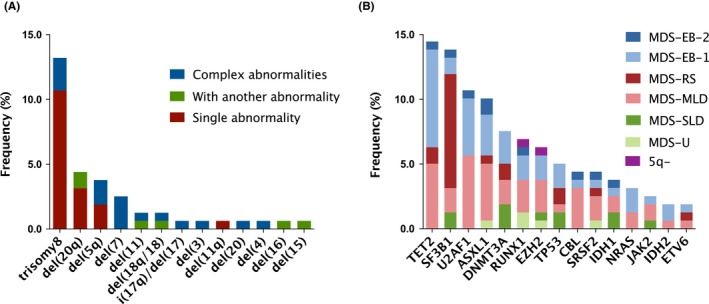
Cytogenetic and genomic spectrum in lower‐risk myelodysplastic syndromes (LR‐MDS). A, Frequency of abnormal karyotypes in 159 LR‐MDS patients. B, Frequency of 15 mutated genes in 159 LR‐MDS patients with different 2016 WHO subtypes. LR‐MDS, lower‐risk myelodysplastic syndromes; WHO, World Health Organization
Median follow up was 21.2 months (IQR: 13.6‐36.6 months). A total of 58 (36.5%) patients died during the follow up. Median OS was 47.8 months (95% CI: 38.0‐57.6 months). Leukemic conversion occurred in 25 (15.7%) patients during the follow up.
3.2. Gene mutational profile
Next‐generation sequencing revealed mutations in all 15‐target genes (Figure 1B; Table S2). The five most frequent mutated genes were TET2 (14.5%), SF3B1 (13.8%), U2AF1 (10.7%), ASXL1 (10.1%), and DNMT3A (7.5%). Genes less frequently mutated were RUNX1 (6.9%), EZH2 (6.3%), TP53 (5.0%), CBL (4.4%), SRSF2 (4.4%), IDH1 (3.8%), NRAS (3.1%), JAK2 (2.5%), IDH2 (1.9%), and ETV6 (1.9%). One hundred and one (63.5%) patients had at least one mutated gene. Fifty‐nine (37.1%) had one mutated gene only, 31 (19.5%) had two mutated genes, and 11 (6.9%) had at least three mutated genes.
3.3. Mutation status versus OS
Mutation status associated with OS was examined by univariate COX regression analysis: ASXL1 (P = .001), RUNX1 (P = .031), EZH2 (P = .049), TP53 (P = .016), SRSF2 (P = .046), JAK2 (P = .040), and IDH2 (P = .035) mutations were significantly associated with shorter OS (Table 2). Furthermore, mutation status of IDH2 was associated with conversion to sAML (P = .045 after Bonferroni correction; Table S3).
Table 2.
Univariate analyses of mutation status for overall survival
| Mutational variable | Mutation status | N | Median OS (months) | P value |
|---|---|---|---|---|
| TET2 | MT | 23 | 20.9 | .128 |
| WT | 136 | 47.8 | ||
| SF3B1 | MT | 22 | 39.6 | .322 |
| WT | 137 | 47.8 | ||
| U2AF1 | MT | 17 | 49.0 | .323 |
| WT | 142 | 47.8 | ||
| ASXL1 | MT | 16 | 20.4 | .001 |
| WT | 143 | 50.4 | ||
| DNMT3A | MT | 12 | NR | .841 |
| WT | 147 | 46.5 | ||
| RUNX1 | MT | 11 | 24.3 | .031 |
| WT | 148 | 49.0 | ||
| EZH2 | MT | 10 | 17.0 | .049 |
| WT | 149 | 49.0 | ||
| TP53 | MT | 8 | 7.8 | .016 |
| WT | 151 | 47.8 | ||
| CBL | MT | 7 | NR | .321 |
| WT | 152 | 47.8 | ||
| SRSF2 | MT | 7 | 25.6 | .046 |
| WT | 152 | 49.0 | ||
| IDH1 | MT | 6 | NR | .522 |
| WT | 153 | 46.5 | ||
| NRAS | MT | 5 | 56.8 | .824 |
| WT | 154 | 46.5 | ||
| JAK2 | MT | 4 | 16.8 | .040 |
| WT | 155 | 47.8 | ||
| IDH2 | MT | 3 | 24.9 | .035 |
| WT | 156 | 47.8 | ||
| ETV6 | MT | 3 | 16.6 | .388 |
| WT | 156 | 47.8 |
Abbreviations: MT, mutant type; NR, not reached; OS, overall survival; WT, wild type.
3.4. Mutation VAF versus OS
Using R language‐based web tool Cutoff Finder, a raw TET2 VAF threshold of 17.6% was the optimal cutoff for outcome prediction (P = .017; Figure S1A). By contrast, there was no significantly optimal raw VAF value for any other genes in the context of patients’ survival (Figure S1B‐H).
As raw VAF could not completely represent mutation burden which was also influenced by allelic status, we next carried out SNP‐A analysis on TET2‐mutated patients. NGS identified 23 patients with TET2 mutations, but DNA was available for SNP‐A analysis in 20 of these cases (Table 3; Table S4). Heterozygous TET2 mutations were found in 17 cases without 4q/24 aberration, hemizygous TET2 mutations were identified in two patients with 4q24 microdeletions, and compound heterozygous TET2 mutations were discovered in one case. VAF of TET2 mutations were then adjusted. For the three patients without SNP‐A analysis, whether the TET2 mutation was heterozygous, hemizygous, or homozygous, adjusted TET2 VAF was always <18% in one of the cases, whereas adjusted VAF was always ≥18% in the other two cases according to the adjustment method of VAF. After excluding three TET2‐mutated cases without SNP‐A analysis, we found an adjusted TET2 VAF threshold of 17.6% (rounded to 18%) remained the best cutoff for prognostic stratification by Cutoff Finder (P = .029; Figure S2).
Table 3.
Characteristics of TET2‐mutated patients
| Number | Cytogenetics | LOH4q24 | TET2 mutations | |||
|---|---|---|---|---|---|---|
| Consequence | State | Raw VAF | Adjusted VAF | |||
| 21 | Normal | NA | c.3393_3314insT | Unknown | 12.70 | <18a |
| 92 | Normal | Absence | c.3409 + 1G>A | Heterozygous | 44.50 | 44.50 |
| 109 | Normal | Absence | c.3315_3316insA | Heterozygous | 45.15 | 45.15 |
| 140 | Normal | Absence | c.2604T > G | Heterozygous | 54.20 | 54.20 |
| 156 | Normal | NA | c.2068C > T | Unknown | 52.20 | ≥18b |
| 157 | Normal | Absence | c.5543C > G | Heterozygous | 16.20 | 16.20 |
| 177 | Normal | Absence | c.2153delT | Heterozygous | 45.60 | 45.60 |
| 192 | 46,XX,‐11,+mar[10] | Absence | c.2440C > T | Heterozygous | 50.79 | 50.79 |
| 306 | Normal | NA | c.5476G > T | Unknown | 38.70 | ≥18b |
| 310 | Normal | del4q24 | c.3626T > C | Hemizygous | 19.00 | 15.97 |
| 346 | Normal | Absence | c.5618T > C | Heterozygous | 42.37 | 42.37 |
| 352 | Normal | Absence | c.4393C > T | Heterozygous | 51.83 | 51.83 |
| 355 | 46,XX,del(20)(q11)[3]/46,XX[7] | Absence | c.2604T > G | Heterozygous | 51.45 | 51.45 |
| 361 | Normal | Absence | c.3646C > T | Heterozygous | 20.22 | 20.22 |
| 366 | 47,XY,+mar[1]/45,XY,‐15[3]/46,XY[16] | Absence | c.4793delA | Compound | 32.20 | 32.20 |
| c.1664C > T | Heterozygous | 46.60 | 46.60 | |||
| 380 | 46,XY,der(20)(q11)[9]/46,XY[1] | Absence | c.2604T > G | Heterozygous | 48.10 | 48.10 |
| 382 | Normal | Absence | c.3955‐2A > G | Heterozygous | 4.70 | 4.70 |
| 384 | 47,XY,+8[9]/46,XY[1] | Absence | c.5298delC | Heterozygous | 40.40 | 40.40 |
| 386 | Normal | del4q24 | c.3626T > C | Hemizygous | 89.90 | 47.34 |
| 404 | 46,XX,der(5)(q32)[16]/46,XX[4] | Absence | c.2230C > T | Heterozygous | 40.80 | 40.80 |
| 412 | 47,XY,+8[10] | Absence | c.4546C > T | Heterozygous | 1.40 | 1.40 |
| 430 | Normal | Absence | c.5178delT | Heterozygous | 41.50 | 41.50 |
| 446 | Normal | Absence | c.5618T > C | Heterozygous | 42.40 | 42.40 |
Abbreviations: LOH: loss of heterozygosity; NA, not available; VAF, variant allele frequency.
Adjusted VAF was always <18% according to the adjustment method of VAF, whether the TET2 mutation was heterozygous, hemizygous, or homozygous.
Adjusted VAF was always ≥18% according to the adjustment method of VAF, whether the TET2 mutation was heterozygous, hemizygous, or homozygous.
3.5. TET2 VAF versus OS
Overall survival was not associated with TET2 mutation status per se (median: 20.9 vs 47.8 months; P = .128; Figure 2A), but with TET2 mutation burden (low, as defined by wild type plus adjusted TET2 VAF at <18% vs high, defined as adjusted TET2 VAF at ≥18%). Out of the 159 study subjects, TET2 mutation was identified in 23 patients, among which 19 (82.6%) had adjusted TET2 VAF at ≥18%. OS was significantly shorter in subjects with high TET2 mutation burden (adjusted VAF ≥18%) versus those with low TET2 mutation burden (adjusted VAF <18% plus TET2 wild type) (median: 20.4 vs 47.8 months; P = .020; HR = 2.183, 95% CI: 1.129‐4.224; Figure 2B). When compared with patients with no TET2 mutations alone, patients with an adjusted TET2 VAF ≥18% also had significantly shorter OS (median: 20.4 vs 47.8 months; P = .023; HR = 1.465, 95% CI: 1.053‐2.040; Figure S3).
Figure 2.
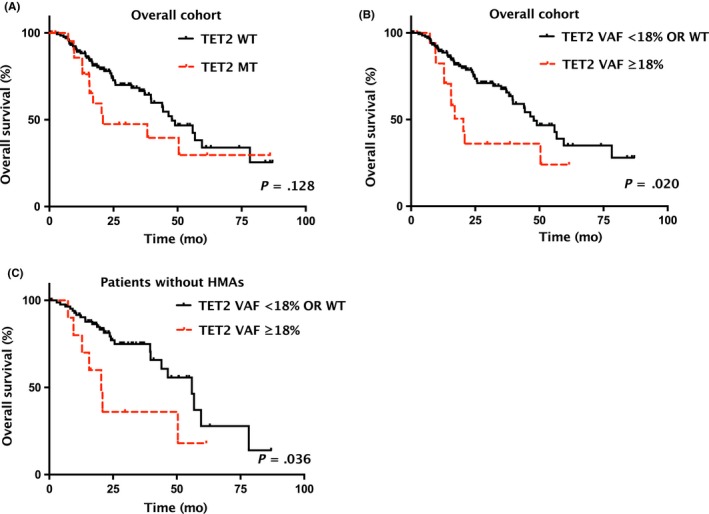
Overall survival (OS) by TET2 mutation status and TET2 variant allele frequency (VAF) in lower‐risk myelodysplastic syndromes (LR‐MDS). A, OS stratified by TET2 mutation status in the overall cohort. B, OS stratified by adjusted TET2 MT VAF <18% or WT vs adjusted TET2 MT VAF ≥18% in the overall cohort. C, OS stratified by adjusted TET2 MT VAF <18% or WT vs adjusted TET2 MT VAF ≥18% in 99 patients without hypomethylating agents (HMA). MT, mutant type; OS, overall survival; WT, wild type
In a subgroup analysis that included 99 subjects only receiving best supportive care without HMA, OS was also significantly shorter in patients with an adjusted TET2 VAF ≥18% (n = 11) (median: 20.4 vs 55.9 months; P = .036; HR = 2.484, 95% CI: 1.063‐5.805; Figure 2C). Response to decitabine also seemed to be associated with TET2 burden: the adjusted TET2 VAF value was 50.79%, 40.80%, and 41.50% in the three TET2‐mutated patients who responded to decitabine treatment, and <18%, 16.20%, and 20.22% in three TET2‐mutated patients who did not respond to decitabine (Table S5).
3.6. Predictors of OS
The following variables were entered as independent variables in the multivariate COX regression analysis: the IPSS risk group, age (≥ vs <60 years), gender, mutation status of ASXL1, RUNX1, EZH2, TP53, SRSF2, JAK2, and IDH2, and adjusted TET2 VAF (≥ vs <18%) (Table 4). The analysis showed that OS was independently associated with the following factors: IPSS Int‐1 risk (P = .014; HR = 3.626, 95% CI: 1.292‐10.174), ASXL1 mutations (P < .001; HR = 4.306, 95% CI: 2.144‐8.650), TP53 mutations (P = .004; HR = 4.863, 95% CI: 1.662‐14.230), JAK2 mutations (P = .002; HR = 5.466, 95% CI: 1.848‐16.169), and adjusted TET2 VAF ≥18% (P = .008; HR = 2.492, 95% CI: 1.273‐4.876).
Table 4.
Multivariate COX regression analysis for overall survival for IPSS
| Variable | P value | Hazard ratio (95% CI) |
|---|---|---|
| IPSS (Int‐1 vs low) | 0.014 | 3.626 (1.292‐10.174) |
| Age (≥ vs <60 y) | 0.161 | 1.579 (0.833‐2.991) |
| Gender (male vs female) | 0.240 | 1.443 (0.783‐2.662) |
| ASXL1 (MT vs WT) | < 0.001 | 4.306 (2.144‐8.650) |
| RUNX1 (MT vs WT) | 0.798 | 1.151 (0.391‐3.390) |
| EZH2 (MT vs WT) | 0.303 | 1.630 (0.643‐4.131) |
| TP53 (MT vs WT) | 0.004 | 4.863 (1.662‐14.230) |
| SRSF2 (MT vs WT) | 0.774 | 1.235 (0.293‐5.193) |
| JAK2 (MT vs WT) | 0.002 | 5.466 (1.848‐16.169) |
| IDH2 (MT vs WT) | 0.255 | 2.090 (0.587‐7.441) |
| Adjusted TET2 VAF (≥ vs <18%) | 0.008 | 2.492 (1.273‐4.876) |
Abbreviations: CI, confidence interval; Int‐1, intermediate 1; IPSS, International Prognostic Scoring System; MT, mutant type; VAF, variant allele frequency; WT, wild type.
In another similar model considering the LR‐PSS risk category (Table S6), mutational variables including ASXL1 mutations, TP53 mutations, JAK2 mutations, and adjusted TET2 VAF ≥18% remained as independent predictors of worse outcome. Similar results were also detected in the IPSS‐R risk group (Table S7).
Within the 43 subjects (27.0%) having at least one poor‐risk mutational factor (ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%), OS was increasingly shorter as the number of poor‐risk mutational factors increased (P < .001; Figure 3). OS was also significantly shorter in those with poor‐risk mutational factors in subgroup analysis that included patients with IPSS low risk only (P = .029; Figure 4A), or IPSS Int‐1 risk only (P < .001; Figure 4B). Similarly, in subgroup analysis that divided subjects using IPSS‐R (≤ vs >3.5), the presence of poor‐risk mutational factors was associated with a significantly shorter OS (P = .018 for IPSS‐R at ≤3.5; Figure 4C; P < .001 for IPSS‐R at >3.5; Figure 4D).
Figure 3.
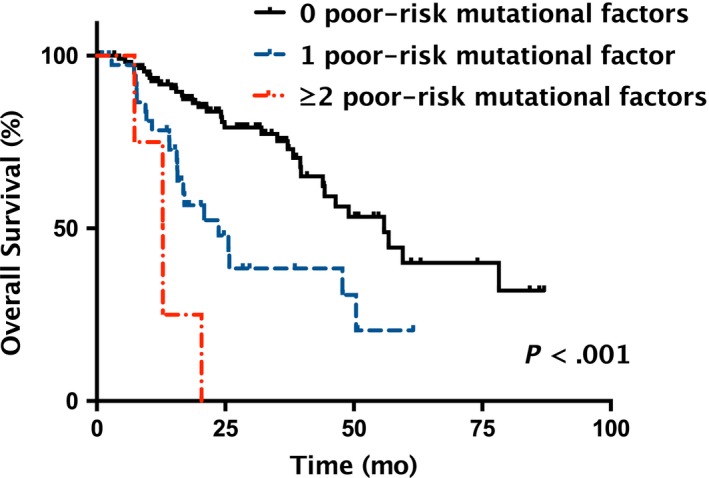
Overall survival (OS) by the number of poor‐risk mutational factors in lower‐risk myelodysplastic syndromes (LR‐MDS). OS stratified by the number (0, 1, and ≥2, respectively) of poor‐risk mutational factors of independent prognostic significance, including ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%. MT, mutant type
Figure 4.
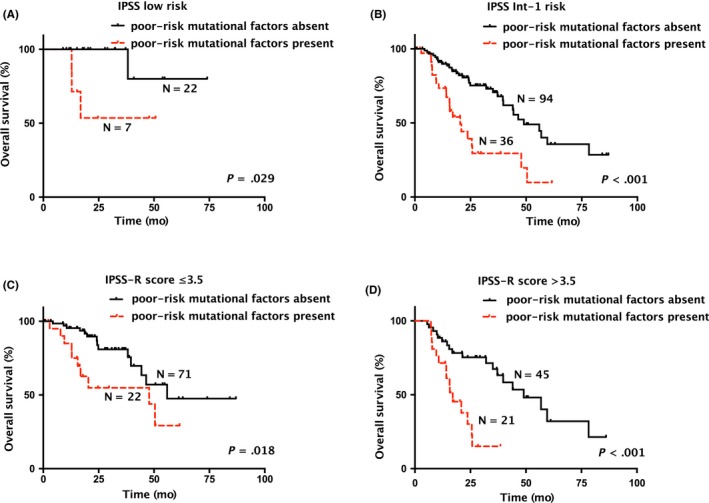
Overall survival (OS) by presence or absence of any poor‐risk mutational factors in lower‐risk myelodysplastic syndromes (LR‐MDS) with each risk category of the International Prognostic Scoring System (IPSS) or the revised IPSS (IPSS‐R). OS by presence or absence of any poor‐risk mutational factors of independent prognostic significance, including ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%, in LR‐MDS with (A) IPSS low risk, (B) IPSS Int‐1 risk, (C) an IPSS‐R score ≤3.5, or (D) an IPSS‐R score >3.5. Int‐1, intermediate 1; MT, mutant type
3.7. Development of a mutational factors‐based prognostic scoring system
Patients with IPSS Int‐1 risk had a significantly worse median OS versus patients with IPSS low risk (44.3 months vs median OS not reached, P = .029; Figure 5A). Furthermore, LR‐PSS category three patients had a significantly shorter median OS versus LR‐PSS category two patients (23.7 months vs 47.8 months; P = .001) or LR‐PSS category one patients (median OS not reached; P < .001) (Figure 5B). Moreover, patients with IPSS‐R >3.5 had a shorter median OS versus those with IPSS‐R ≤3.5 (37.1 months vs 55.9 months; P = .011; Figure 5C).
Figure 5.

Overall survival (OS) by prognostic models in lower‐risk myelodysplastic syndromes (LR‐MDS). (A) OS stratified by IPSS low risk vs Int‐1 risk, (B) LR‐PSS categories, (C) IPSS‐R score ≤3.5 vs >3.5, and (D) LR‐M‐PSS groups, respectively. Int‐1, intermediate 1; IPSS, International Prognostic Scoring System; IPSS‐R, revised IPSS; LR‐M‐PSS, Lower‐Risk Molecular Prognostic Scoring System; LR‐PSS, MD Anderson Lower‐Risk Prognostic Scoring System
We developed a prognostic scoring system, referred to as Lower‐Risk Molecular Prognostic Scoring System (LR‐M‐PSS below), using the five risk factors identified in the current study (IPSS Int‐1 risk, ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%). A score of 1 was assigned to each factor if present; otherwise, a score of 0 was assigned. The maximum possible overall score was 5.
The overall score was 0 in 21 subjects (13.2%), 1 in 102 subjects (64.2%), and ≥2 in the remaining 36 subjects (22.6%). The median OS was not reached in subjects with an overall score of 0, 49.0 months (95% CI: 36.8‐61.2 months) in those with an overall score of 1, and 19.8 months (95% CI: 13.0‐26.6 months) in those with an overall score of 2 or above (P < .001; Figure 5D).
Comparing the identification power of the higher‐risk patients, 10.3% (3/29) of patients were redefined as higher‐risk group by LR‐M‐PSS (overall score of ≥2) within the group of low IPSS scores, with a median OS of 12.9 months; whereas none could be upgraded to higher‐risk group by either the LR‐PSS or the IPSS‐R (LR‐PSS category 3 or IPSS‐R at >3.5). In the group of Int‐1 IPSS scores, 25.4% (33/130) of patients were identified as having poor survival by the LR‐M‐PSS, whereas 24.6% (32/130) were identified by LR‐PSS and 50.8% (66/130) by IPSS‐R.
Considering the overlap of the higher‐risk group recognized by these models (Figure 6), IPSS‐R obviously identified the largest number of the patients (41.5%, 66/159), covering most of the subjects identified by LR‐PSS (81.3%, 26/32) and half of the patients by LR‐M‐PSS (50%, 18/36). However, our new model could identify an additional 10.1% (16/159) of patients who neither identified by IPSS‐R nor by LR‐PSS, with a median OS of 20.4 months (95% CI: 11.2‐29.5 months). By contrast, the higher‐risk patients identified by IPSS‐R or LR‐PSS but not LR‐M‐PSS had a median OS of 49.0 months (95% CI: 31.7‐66.3 months) or 44.0 months (95% CI: 11.3‐76.7 months).
Figure 6.
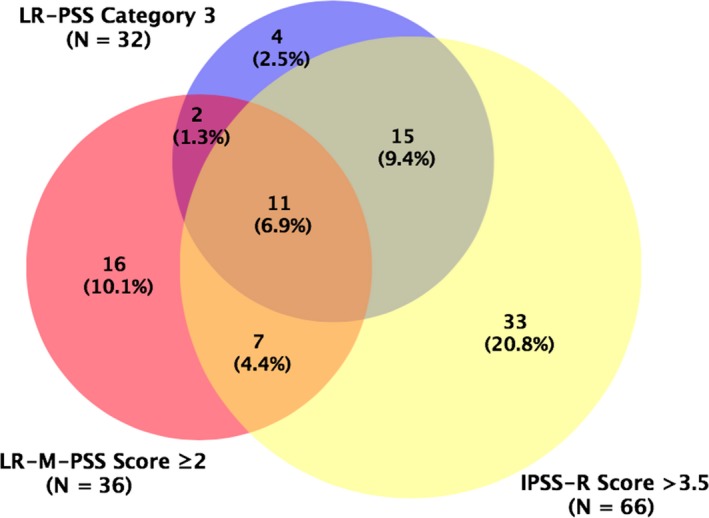
Venn diagram showing the overlap of lower‐risk myelodysplastic syndromes (LR‐MDS) patients with a poor prognosis identified by IPSS‐R (IPSS‐R at >3.5), LR‐PSS (category 3), and LR‐M‐PSS (overall score of ≥2), respectively. Created using BioVenn.31 IPSS‐R, revised International Prognostic Scoring System; LR‐M‐PSS, Lower‐Risk Molecular Prognostic Scoring System; LR‐PSS, MD Anderson Lower‐Risk Prognostic Scoring System
4. DISCUSSION
In the present study, ASXL1 MT, TP53 MT, JAK2 MT, and high TET2 mutation burden were found to be significantly associated with poorer prognosis independent of IPSS. A scoring system that combines these mutational factors with the IPSS could stratify LR‐MDS patients into three prognostically distinct groups and help identify patients with greater‐than‐predicted risk for early intervention.
A previous report showed that ASXL1 and EZH2 mutations were more common in LR‐MDS than higher‐risk MDS.11 A subsequent study from the same group of investigators showed that ASXL1, EZH2, and NRAS mutations were associated with poorer prognosis in LR‐MDS independent of IPSS; mutations of ASXL1, EZH2, TP53, and RUNX1 remained independently significant after adjusting for LR‐PSS.16 In the current study, we confirmed the association of ASXL1, EZH2, TP53, and RUNX1 with worse outcome in the univariate analysis and the independent prognostic significance of ASXL1 and TP53 mutations in the multivariate analysis. We failed to show an association of NRAS mutation with patient survival, probably due to the small number of patients with NRAS mutation in our cohort. JAK2 mutations have previously been reported to be associated with poor patient survival in MDS receiving hematopoietic stem cell transplantation (HSCT),32 but not in LR‐MDS.16 The current study extended such an association to LR‐MDS.
We also observed that LR‐MDS patients with IDH2 mutations were more likely to develop sAML, as previous studies of unselected MDS reported.14, 33 Mutations of genes involved in DNA methylation including IDH2 might represent early events in MDS and play an indirect role in disease progression through a multistep evolutionary process.34, 35 NARS mutations were reported to be associated with leukemic transformation of MDS in several studies of serial sequences.34, 35, 36 However, we failed to statistically find their relation in our study probably because the association was not obvious in the early stage of MDS.
A major finding in the current study is that the independent association of TET2 mutation burden with prognosis in LR‐MDS. TET2 is one of the most commonly mutated genes in MDS,11, 13, 16 and often found at the initiation and early progression of MDS.37, 38, 39 TET2 mutations had been reported to be associated with poorer survival32, 40, 41 and higher response to HMA15, 42 by some, but not all, studies11, 12, 16, 17, 43, 44 in either unselected MDS or LR‐MDS. In the current study, we found that TET2 mutation status had no association with OS per se, but with TET2 mutation burden. Specifically, the patients with an adjusted TET2 VAF ≥18% had a significantly worse survival, compared with patients with an adjusted TET2 VAF <18% or without TET2 mutations. Such an association remained statistically significant even after adjustment for HMA. Our finding is in line with a previous study that indicated higher TET2 VAF leading to worse survival in myeloid neoplasms.32 Another interesting finding is that all three TET2‐mutated subjects who responded to decitabine had >40% adjusted TET2 VAF, whereas all three TET2‐mutated subjects who did not respond to decitabine had adjusted TET2 VAF of approximately less than 20%. This preliminary finding is consistent with a previous study that suggested higher TET2 VAF in decitabine responders.15
Another major finding in the current study is that mutational factors (eg, ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%) could help to identify LR‐MDS patients with poor survival within each risk group defined by either IPSS or IPSS‐R. Patients with mutational factors had shorter OS in comparison with those without such mutational factors within the same IPSS or IPSS‐R risk group. Also, survival was increasingly worse as the number of these mutational factors increased.
LR‐PSS could stratify LR‐MDS into three categories with distinct outcomes, and thus represents an advance in more accurate risk prediction.9 Median OS in patients of LR‐PSS category 3 has been shown to be equivalent to that of patients of the IPSS intermediate 2 (Int‐2) risk group.16 IPSS‐R represents another attempt at more accurate risk prediction: it could reassign patients into a lower‐ or higher‐risk group based on a cutoff score of 3.5 points.10 The prognostic value of LR‐PSS and IPSS‐R had already been elaborately examined before,8, 16, 45 which was also confirmed by our lower‐risk MDS cohort. About 25% and 50% of patients were reassessed as higher‐risk group by LR‐PSS (category 3) or IPSS‐R (IPSS‐R at >3.5) respectively, within the IPSS Int‐1 risk group in the current study. However, neither the LR‐PSS nor the IPSS‐R could be used to recognize patients with poor OS within the IPSS low‐risk group, as reported previously.45 It is expected that additional molecular predictors will contribute to better risk stratification of LR‐MDS.
Combination of molecular data with the existing risk‐prediction systems has already been explored. For example, by combining EZH2 mutation status and LR‐PSS, 29% of LR‐MDS patients with either EZH2 mutation or LR‐PSS category 3 risk were identified as having a shorter‐than‐predicted OS.16 In the present study, we developed a novel prediction model (abbreviated as LR‐M‐PSS) by combing the four mutational factors, including ASXL1 MT, TP53 MT, JAK2 MT, and adjusted TET2 VAF ≥18%, with the IPSS risk group. LR‐M‐PSS stratified LR‐MDS patients into three groups with distinct prognosis (overall score of 0, 1, or ≥2). Approximately one‐fourth of the study subjects fell into the high‐risk group (overall LR‐M‐PSS score ≥2).
Comparing the identification power of the higher‐risk patients, IPSS‐R definitely had the advantage of identifying the largest number of patients as previously reported,8, 45 covering most of the patients identified by LR‐PSS and half of the patients by LR‐M‐PSS. However, our model made a more refined risk prediction by subdividing LR‐MDS patients into three prognostically distinct groups. LR‐M‐PSS could additionally identify more than 10% of patients who were failed to be recognized by both IPSS‐R and LR‐PSS. LR‐M‐PSS also could recognize these patients from the IPSS low‐risk group, whereas the other two models could not.45 Moreover, the higher‐risk patients identified by LR‐M‐PSS only had a definitely poor survival, whereas patients identified by IPSS‐R or LR‐PSS but not LR‐M‐PSS seemed to have a longer‐than‐predicted survival. These preliminary findings indicated that mutation status and burden of certain genes could improve prognostic stratification of LR‐MDS, and would be an important complement to the current prognostic models.
We acknowledged several limitations in this study. First, the optimal VAF cutoff was primarily determined based on the raw VAF data, which may lack biological significance to some extent. Thus, we further explored the prognostic significance of TET2 mutation burden with the adjustment of VAF for copy number and zygosity. Additional limitations included the relatively small sample size and the lack of an independent validation cohort. More studies are needed to confirm our findings, and to improve prognostic prediction of LR‐MDS. Notwithstanding the above limitations, our study allows for a new approach on the application of molecular data for prognostication of LR‐MDS.
In summary, this study shows that ASXL1 MT, TP53 MT, JAK2 MT, and high TET2 mutation burden are important predictors for poor survival in LR‐MDS. Our study highlights that integrating mutation status and burden of certain MDS‐relevant genes into IPSS may improve risk stratification of patients with LR‐MDS and help identify those with worse‐than‐expected prognosis for more aggressive treatment.
CONFLICTS OF INTEREST
Authors declare no conflicts of interest for this article.
AUTHOR CONTRIBUTIONS
HT and LJ conceived and designed the study. YL, SZ and LW analyzed and arranged the data. LM, HZ, CS, and WY carried out the mutation analysis. YR, XZ, CM, WX, LY, HY, and CL provided patient samples and data. JJ guided the research with valuable comments. HT provided critical revision and suggestions.
Supporting information
ACKNOWLEDGMENTS
This work was supported by grants from the National Natural Science Foundation of China Grants (81700121, 81800121, 81970117).
Jiang L, Luo Y, Zhu S, et al. Mutation status and burden can improve prognostic prediction of patients with lower‐risk myelodysplastic syndromes. Cancer Sci. 2020;111:580–591. 10.1111/cas.14270
Jiang and Luo contributed equally to this work.
REFERENCES
- 1. Raza A, Galili N. The genetic basis of phenotypic heterogeneity in myelodysplastic syndromes. Nat Rev Cancer. 2012;12:849‐859. [DOI] [PubMed] [Google Scholar]
- 2. Zeidan AM, Komrokji RS. There's risk, and then there's risk: the latest clinical prognostic risk stratification models in myelodysplastic syndromes. Curr Hematol Malig Rep. 2013;8:351‐360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Nazha A, Bejar R. Molecular data and the IPSS‐R: how mutational burden can affect prognostication in MDS. Curr Hematol Malig Rep. 2017;12:461‐467. [DOI] [PubMed] [Google Scholar]
- 4. Greenberg P, Cox C, LeBeau MM, et al. International scoring system for evaluating prognosis in myelodysplastic syndromes. Blood. 1997;89:2079‐2088. [PubMed] [Google Scholar]
- 5. Nazha A. The MDS genomics‐prognosis symbiosis. Hematol Am Soc Hemat. 2018;2018:270‐276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Greenberg PL, Tuechler H, Schanz J, et al. Revised international prognostic scoring system for myelodysplastic syndromes. Blood. 2012;120:2454‐2465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zeidan AM, Smith BD, Komrokji RS, Gore SD. Prognostication in myelodysplastic syndromes: beyond the International Prognostic Scoring System (IPSS). Am J Med. 2013;126:E25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zeidan AM, Sekeres MA, Wang XF, et al. Comparing the prognostic value of risk stratifying models for patients with lower‐risk myelodysplastic syndromes: Is one model better? Am J Hematol. 2015;90:1036‐1040. [DOI] [PubMed] [Google Scholar]
- 9. Garcia‐Manero G, Shan J, Faderl S, et al. A prognostic score for patients with lower risk myelodysplastic syndrome. Leukemia. 2008;22:538‐543. [DOI] [PubMed] [Google Scholar]
- 10. Pfeilstocker M, Tuechler H, Sanz G, et al. Time‐dependent changes in mortality and transformation risk in MDS. Blood. 2016;128:902‐910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bejar R, Stevenson K, Abdel‐Wahab O, et al. Clinical effect of point mutations in myelodysplastic syndromes. New Engl J Med. 2011;364:2496‐2506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes. Leukemia. 2014;28:241‐247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes. Blood. 2013;122:3616‐3627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lin PP, Luo YW, Zhu SH, et al. Isocitrate dehydrogenase 2 mutations correlate with leukemic transformation and are predicted by 2‐hydroxyglutarate in myelodysplastic syndromes. J Cancer Res Clin. 2018;144:1037‐1047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bejar R, Lord A, Stevenson K, et al. TET2 mutations predict response to hypomethylating agents in myelodysplastic syndrome patients. Blood. 2014;124:2705‐2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bejar R, Stevenson KE, Caughey BA, et al. Validation of a prognostic model and the impact of mutations in patients with lower‐risk myelodysplastic syndromes. J Clin Oncol. 2012;30:3376‐3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hou HA, Tsai CH, Lin CC, et al. Incorporation of mutations in five genes in the revised International prognostic scoring system can improve risk stratification in the patients with myelodysplastic syndrome. Blood Cancer J. 2018;8:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bejar R. Clinical and genetic predictors of prognosis in myelodysplastic syndromes. Haematologica. 2014;99:956‐964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xu YY, Li Y, Xu QY, et al. Implications of mutational spectrum in myelodysplastic syndromes based on targeted next‐generation sequencing. Oncotarget. 2017;8:82475‐82490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sallman DA, Komrokji R, Vaupel C, et al. Impact of TP53 mutation variant allele frequency on phenotype and outcomes in myelodysplastic syndromes. Leukemia. 2016;30:666‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Belickova M, Vesela J, Jonasova A, et al. TP53 mutation variant allele frequency is a potential predictor for clinical outcome of patients with lower‐risk myelodysplastic syndromes. Oncotarget. 2016;7:36266‐36279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wang W, Routbort MJ, Tang ZY, et al. Characterization of TP53 mutations in low‐grade myelodysplastic syndromes and myelodysplastic syndromes with a non‐complex karyotype. Eur J Haematol. 2017;99:536‐543. [DOI] [PubMed] [Google Scholar]
- 23. Hirsch CM, Nazha A, Kneen K, et al. Consequences of mutant TET2 on clonality and subclonal hierarchy. Leukemia. 2018;32:1751‐1761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Patel SS, Kuo FC, Gibson CJ, et al. High NPM1‐mutant allele burden at diagnosis predicts unfavorable outcomes in de novo AML. Blood. 2018;131:2816‐2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127:2391‐2405. [DOI] [PubMed] [Google Scholar]
- 26. Shaffer LG, Slovak ML, Campbell LJ. ISCN 2013: An International System for Human Cytogenetic Nomenclature. Basel: S Karger; 2013. [Google Scholar]
- 27. Bacher U, Kohlmann A, Haferlach T. Mutational profiling in patients with MDS: ready for every‐day use in the clinic? Best Pract Res Clin Haematol. 2015;28:32‐42. [DOI] [PubMed] [Google Scholar]
- 28. Kohlmann A, Grossmann V, Nadarajah N, Haferlach T. Next‐generation sequencing ‐ feasibility and practicality in haematology. Br J Haematol. 2013;160:736‐753. [DOI] [PubMed] [Google Scholar]
- 29. Budczies J, Klauschen F, Sinn BV, et al. Cutoff finder: a comprehensive and straightforward web application enabling rapid biomarker cutoff optimization. PLoS ONE. 2012;7:e51862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group (IWG) response criteria in myelodysplasia. Blood. 2006;108:419‐425. [DOI] [PubMed] [Google Scholar]
- 31. Hulsen T, de Vlieg J, Alkema W. BioVenn ‐ a web application for the comparison and visualization of biological lists using area‐proportional Venn diagrams. BMC Genom. 2008;9:488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem‐cell transplantation. New Engl J Med. 2017;376:536‐547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jin J, Hu C, Yu MX, et al. Prognostic value of isocitrate dehydrogenase mutations in myelodysplastic syndromes: a retrospective cohort study and meta‐analysis. PLoS ONE. 2014;9:e100206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Kim T, Tyndel MS, Kim HJ, et al. The clonal origins of leukemic progression of myelodysplasia. Leukemia. 2017;31:1928‐1935. [DOI] [PubMed] [Google Scholar]
- 35. Pellagatti A, Roy S, Di Genua C, et al. Targeted resequencing analysis of 31 genes commonly mutated in myeloid disorders in serial samples from myelodysplastic syndrome patients showing disease progression. Leukemia. 2016;30:247‐250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Makishima H, Yoshizato T, Yoshida K, et al. Dynamics of clonal evolution in myelodysplastic syndromes. Nat Genet. 2017;49:204‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nakajima H, Kunimoto H. TET2 as an epigenetic master regulator for normal and malignant hematopoiesis. Cancer Sci. 2014;105:1093‐1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kwok B, Hall JM, Witte JS, et al. MDS‐associated somatic mutations and clonal hematopoiesis are common in idiopathic cytopenias of undetermined significance. Blood. 2015;126:2355‐2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Lindsley RC. Uncoding the genetic heterogeneity of myelodysplastic syndrome. Hematology. 2017;2017:447‐452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bejar R, Stevenson KE, Caughey B, et al. Somatic mutations predict poor outcome in patients with myelodysplastic syndrome after hematopoietic stem‐cell transplantation. J Clin Oncol. 2014;32:2691‐2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu XL, Zhang GS, Yi Y, et al. Decreased 5‐hydroxymethylcytosine levels are associated with TET2 mutation and unfavorable overall survival in myelodysplastic syndromes. Leukemia Lymphoma. 2013;54:2466‐2473. [DOI] [PubMed] [Google Scholar]
- 42. Itzykson R, Kosmider O, Cluzeau T, et al. Impact of TET2 mutations on response rate to azacitidine in myelodysplastic syndromes and low blast count acute myeloid leukemias. Leukemia. 2011;25:1147‐1152. [DOI] [PubMed] [Google Scholar]
- 43. Bejar R, Papaemmanuil E, Haferlach T, et al. Somatic mutations in MDS patients are associated with clinical features and predict prognosis independent of the IPSS‐R: analysis of combined datasets from the International Working Group for Prognosis in MDS‐Molecular Committee. Blood. 2015;126:907. [Google Scholar]
- 44. Tefferi A, Lasho TL, Patnaik MM, et al. Targeted next‐generation sequencing in myelodysplastic syndromes and prognostic interaction between mutations and IPSS‐R. Am J Hematol. 2017;92:1311‐1317. [DOI] [PubMed] [Google Scholar]
- 45. Valcarcel D, Sanz G, Ortega M, et al. Use of newer prognostic indices for patients with myelodysplastic syndromes in the low and intermediate‐1 risk categories: a population‐based study. Lancet Haematol. 2015;2:E260‐E266. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials