

Abstract
The incidence of epidermal growth factor receptor uncommon mutation (EGFRum) is relatively low and patients harboring EGFRum are resistant to the first‐generation tyrosine kinase inhibitors (TKI). However, the mechanism of primary resistance remains unclear. Medical records of 98 patients who had never been treated by TKI and who accepted icotinib treatment were collected and followed. The circulating tumor DNA (ctDNA) were detected and analyzed using the next‐generation sequencing (NGS) platform after progression on icotinib. The potential primary resistance mechanism of icotinib was explored. A total of 21 (21.4%) and 48 (49%) patients developed primary and acquired resistance to icotinib, respectively. The median progression‐free survival (PFS) of primary resistance patients was 1.8 months (0.5‐2.3, 95% CI = 1.50‐2.10). Before treatment, 52.4% (11/21) of patients carried S768I, 23.8% (5/21) L861Q, 14.3% (3/21) G719X and 14.3% (3/21) exon 20‐ins mutations. Approximately 23.8% (5/21) of patients harbored the combined pattern mutations and 76.2% (16/21) of patients harbored the single pattern mutations. The combined pattern with EGFR classical mutation (EGFRcm) had worse PFS than the combined with EGFRum and single pattern (P < .05). There were 6 (28.57%) patients with acquired EGFR extracellular domain mutation, 5 (23.81%) with BCL2L11 loss (BIM deletion polymorphism), 3 (14.29%) with MET amplification, 1 (4.76%) with ERBB2 amplification, 1 (4.76%) with MYC amplification, 1 (4.76%) with PTEN mutation, 1 (4.76%) with PIK3CA mutation and 3 (14.29%) with unknown status. EGFR extracellular domain mutation, BCL2L11 loss, PI3K‐AKT‐mTOR signaling pathway (PTEN and PIK3CA mutations), MET amplification, ERBB2 amplification or MYC amplification might contribute to molecular mechanisms of primary resistance to icotinib in patients with advanced non‐small cell lung cancer harboring uncommon mutant epidermal growth factor receptor. Combined targeted therapy or chemotherapy should be considered in this population.
Keywords: ctDNA, epidermal growth factor receptor, icotinib, next‐generation sequencing, non–small cell lung cancer
In a large‐scale multi‐center real‐world study in China, we detected and analyzed potential primary resistance to icotinib in EGFRum patients using the next‐generation sequencing (NGS) platform. EGFR extracellular domain mutation, BCL2L11 loss, PI3K‐AKT‐mTOR signaling pathway (PTEN, PIK3CA mutations), MET amplification, ERBB2 amplification or MYC amplification might contribute to primary resistance of icotinib in EGFRum NSCLC patients.

1. INTRODUCTION
Activating epidermal growth factor receptor (EGFR) mutant lung cancer has a remarkable response to tyrosine kinase inhibitors (TKI), which have replaced chemotherapy as the first‐line therapy.1 Approximately one‐tenth of all EGFR mutations are EGFR uncommon mutation (EGFRum) carriers in advanced non–small cell lung cancer (NSCLC) and their response and primary resistance to TKI were understudied from July 2013 to November 2016.2, 3, 4 Icotinib is a quinazoline derivative that reversibly binds to the ATP binding site of EGFR protein and stops tumor cells from overgrowing.5 It biologically belongs to the first‐generation TKI and is mainly prescribed in China.6
There is no strict definition of primary resistance to TKI, but disease progression within 3 months from initial treatment could be considered primary resistance in a clinical trial.7 It is currently believed that the primary resistance mechanism to TKI might be the activation of other gene mutations or bypass pathway signals that coexist with EGFR‐sensitive mutations.8 The resistance mechanisms in EGFR classical mutation (EGFRcm) include de novo T790M mutations, exon 20 insertion (20‐ins) mutation, PI3K/AKT, IGF1R, NF‐κB‐dependent pathway and loss of the proapoptotic protein BIM gene polymorphism.9, 10, 11 Based on studies of the primary resistance mechanism of TKI in EGFRcm, whether a similar situation exists in EGFRum remains to be shown.
Finding new effective targets is the key strategy to overcome drug resistance in advanced NSCLC patients. Traditional genomic mutation tests do not meet the current clinical needs. Next‐generation sequencing (NGS) is a relatively new genomic testing platform that brings added high throughput, sensitivity and efficiency, and is widely used in clinical practice and scientific research.12 Circulating tumor DNA (ctDNA) in peripheral blood is becoming increasingly popular in comparison with tumor biopsy. There are several reasons why ctDNA is superior to tumor tissue biopsy: it is relatively non–invasive, efficient and economical, and a promising tool to monitor dynamically and could possibly replace tissue biopsy in future.13, 14 Other specimens including tumor cells in malignant pleural effusion could also be used to analyze genetic profiling if ctDNA is unavailable.
Therefore, we conducted an observational study of the clinical response and putative primary resistance mechanism of icotinib in advanced NSCLC patients with EGFRum. Tumor biopsy and ctDNA either from plasma or pleural effusion were collected and profiled by 170 cancer‐relevant genes panel using next‐generation sequencing. We further compared the difference between primary and acquired resistance groups in clinical‐pathological characteristics and accompanied mutations after disease progression.
2. METHODS
2.1. Patients and follow up
We retrospectively enrolled and collected medical data of 3117 patients who were diagnosed with lung adenocarcinoma from multi‐cancer centers in China during the period from July 2013 to November 2016. The EGFRum status was screened and those who had been treated by TKI were excluded. After the median follow up of 6.2 months, 21 EGFRum patients treated by icotinib (125 mg, tid) whose disease developed quickly and progressed within 3 months during the follow‐up time were analyzed in this study. Patients’ demographic data are summarized in Table 1. Samples from tumor tissue and ctDNA in plasma or pleural effusion were collected for genetic profiling by NGS before and after disease progression following resistance to icotinib therapy in both primary and acquired resistance groups. A combined pattern of mutation was defined as the coexistence of two different EGFR‐mutant types. Clinical responses were evaluated using the standard version of response evaluation criteria in solid tumors (RECIST, v1.1)15 based on regular imagine detection. PFS was referred to as the time from the beginning of taking icotinib to disease progression confirmed by RECIST criteria or death (whichever comes first). Patients whose disease did not progress were censored at the last follow up. Formal consent was obtained, and the project was approved by the hospitals’ ethics committees.
Table 1.
Baseline characteristics in icotinib primary and acquired resistance EGFR uncommon mutation NSCLC patients
| Characteristic | Primary resistance | Acquired resistance |
|---|---|---|
| N = 21 (%) | N = 48 (%) | |
| Median age (y) | ||
| <65 | 13 (61.90) | 30 (62.5) |
| ≥65 | 8 (38.1) | 18 (37.5) |
| Sex | ||
| Male | 10 (47.62) | 21 (43.8) |
| Female | 11 (52.38) | 27 (56.2) |
| Smoking status | ||
| Present or former smoker | 5 (23.81) | 14 (29.2) |
| Non–smoker | 16 (76.19) | 34 (70.8) |
| ECOG PS | ||
| 0‐1 | 18 (85.71) | 40 (83.3) |
| 2‐3 | 3 (14.29) | 8 (16.7) |
| Histology | ||
| Adenocarcinoma | 17 (80.95) | 45 (93.8) |
| Non–adenocarcinoma | 4 (19.05) | 3 (6.2) |
| Treatment lines | ||
| First | 0 (0) | 1 (2.1) |
| Second | 1 (4.76) | 4 (8.3) |
| Third and more | 20 (95.24) | 43 (89.6) |
Abbreviations: ECOG PS, Eastern Cooperative Oncology Group performance status; EGFR, epidermal growth factor receptor; NSCLC, non–small cell lung cancer.
2.2. Targeted next‐generation sequencing
Genomic DNA sequencing libraries were prepared using the protocols recommended for the Illumina TruSeq DNA Library Preparation Kit. For samples close to the minimum input requirement, additional pre–capture PCR cycles were performed to generate sufficient PCR product for hybridization. The libraries were hybridized to custom‐designed probes (Integrated DNA Technology), including all exons of 170 genes and selected introns of ALK, RET and ROS1 for the detection of Genomic rearrangements. DNA sequencing was performed on a HiSeq3000 sequencing system (Illumina) with 2 × 75 bp paired‐end reads. The reads were aligned to the human genome build GRCh37 using a Burrows‐Wheeler Aligner (BWA). Somatic single nucleotide variant and indel calls were generated using MuTect and GATK, respectively. Somatic copy number alterations were identified with CONTRA. Genomic rearrangements were identified using the software developed in‐house for analyzing chimeric read pairs.
2.3. Statistical analysis
Clinical and mutational characteristic data were analyzed using SPSS software (Version 22.0, SPSS). Categorical variables were compared between the EGFR‐mutant subgroups using χ2 and Fisher’s exact tests. PFS rates were estimated using the Kaplan‐Meier method and examined using the log‐rank test. Differences were confirmed by two‐sided P < .05.
3. RESULTS
3.1. Patients characteristics
The clinical characteristics of 21 primary resistance and 48 acquired resistance EGFRum advanced NSCLC are summarized in Table 1. More than half of the primary resistance patients (61.9%) were younger than 65 years, female (52.4%) and non–smokers (76.2%). Adenocarcinoma was the most common histology (81%) and most patients received icotinib as the second line therapy or later (95.2%). The clinicopathological characteristics are not significantly different from those of the acquired resistance group (Table 1). In the primary resistance group, 52.4% (11/21) of patients had S768I, 23.8% (5/21) had L861Q, 14.3% (3/21) had G719X and 14.3% (3/21) had exon 20‐ins mutations. Approximately 23.8% (5/21) of the patients with combined pattern and 76.2% (16/21) with single pattern mutations. Two cases have a combined pattern with EGFRcm L858R and one case with exon 19del. The median PFS time of 21 patients was 1.8 months (0.5‐2.3, 95% CI = 1.50‐2.10) (Figure 1). The combined pattern with EGFRcm had worse median PFS than combined with EGFRum and single pattern (P < .05, Figure 2).
Figure 1.
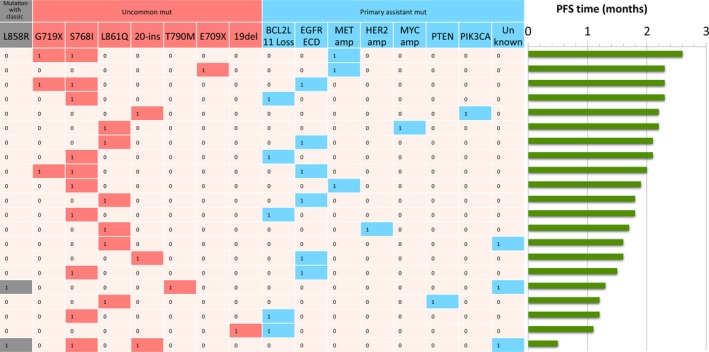
Plasma circulating tumor DNA sequencing results for 21 EGFRum NSCLC patients with primary drug resistance. The heat map shows the baseline EGFRum patterns (grey and red), genetic profiling of progression from disease (blue) and the PFS time (green). EGFRum, epidermal growth factor receptor uncommon mutation; NSCLC, non–small cell lung cancer; PFS, progression‐free survival
Figure 2.
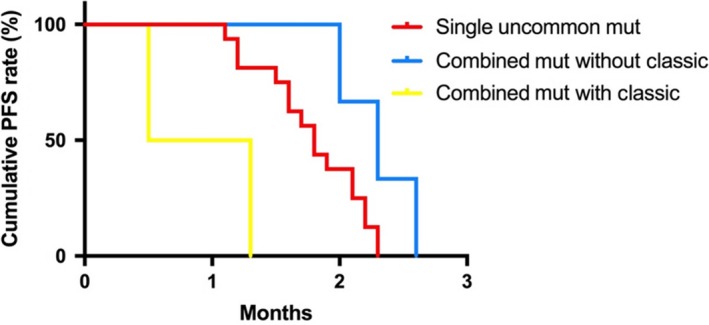
Comparisons of PFS rate in EGFRum patients by mutation patterns. Combined mutation without EGFR classic mutant (blue) carriers has better PFS than single‐pattern (red) and combined mutation with EGFR classic mutant (yellow) (P < .05). EGFRum, epidermal growth factor receptor uncommon mutation; PFS, progression‐free survival
3.2. Potential mechanisms that confer primary resistance to icotinib
To identify potential mechanisms that confer primary resistance to icotinib treatment, we further compared mutation profiles of patients. The most commonly acquired alteration was 28.6% (6/21) EGFR ECD, followed by 21.8% (5/21) BCL2L11 deletion, 14.3% (3/21) MET amplification, and 33.3% (7/21) others, including ERBB2 and MYC amplification, PTEN deletion, PIK3CA mutation and unknown mutation. No significant correlation was found between the mutation carried before icotinib treatment and the mutant genetic alterations after disease progression following resistance to icotinib (Table 2, Figure 1). The potential mechanisms that confer primary resistance to icotinib are listed in Table 2.
Table 2.
Genetic profiling and potential activated pathway of 21 EGFR uncommon mutant NSCLC patients with primary resistance to icotinib
| EGFR mutation | N | Genetic alteration | Potential pathway |
|---|---|---|---|
| S768I | 4 | BCL2L11 loss1 | The Bcl‐2‐regulated apoptotic pathway2 |
| S768I | 2 | EGFR ECD3 | Canonical ligand‐dependent EGFR signaling pathway4 |
| S768I | 1 | MET amp5 | MET pathway6 |
| S768I + G719X | 2 | EGFR ECD3 | Canonical ligand‐dependent EGFR signaling pathway4 |
| S768I + G719X | 1 | MET amp5 | MET pathway6 |
| S768I + L858R+20‐ins | 1 | Unknown | — |
| L861Q | 1 | EGFR ECD3 | Canonical ligand‐dependent EGFR signaling pathway4 |
| L861Q | 1 | ERBB2 amp7 | ERBB2 pathway8 |
| L861Q | 1 | MYC amp9 | MYC‐associated pathway10 |
| L861Q | 1 | PTEN11 | Phosphatidylinositide 3‐kinase pathway12 |
| L861Q | 1 | Unknown | — |
| E709X | 1 | MET amp5 | MET pathway6 |
| 20‐ins | 1 | PIK3CA13 | PI3K‐Akt‐mTOR signaling pathway14 |
| 20‐ins | 1 | EGFR ECD3 | Canonical ligand‐dependent EGFR signaling pathway4 |
| T790M + L858R | 1 | Unknown | — |
| A750P + L747_E749del | 1 | BCL2L11 Loss1 | The Bcl‐2‐regulated apoptotic pathway2 |
Abbreviations: ECD, extracellular domain; EGFR, epidermal growth factor receptor; ERBB2, erb‐b2 receptor tyrosine kinase 2; NSCLC, non–small cell lung cancer.
3.3. Comparison of accompanied mutant genes and patterns between primary and acquired resistance groups
Apart from the potential actionable acquired genetic alterations, the primary resistance group was also found to harbor significantly more accompanied mutations than the acquired resistance group (159 vs 114 in all and 6 vs 2 per patient, respectively, P < .0001, Figure 3). In the primary resistance group, the most common accompanied mutant genes and patterns included TP53 (61.9%, 13/21), NF1 (23.8%, 5/21), DNMT3A (19.04%, 4/21), deletion mutations (81.8%, 130/159) and nonsense mutations (9.4%, 15/159), respectively. In the acquired resistance group, the most common accompanied mutant genes and patterns included TP53 (45.8%, 22/48), SMARTA4 (8.3%, 4/48), CDKN2A (8.3%, 4/48) and deletion mutations (75.4%, 86/114) and nonsense mutations (12.3%, 14/114) (Figure 4).
Figure 3.
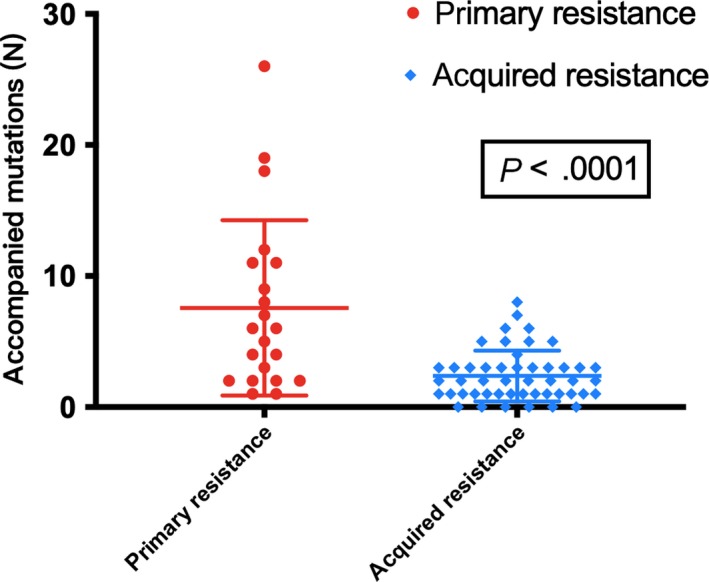
Comparison of the accompanied mutations between 21 primary and 48 acquired resistance to icotinib in EGFRum advanced non–small cell lung cancer (NSCLC) patients. The primary resistance group presented significantly more accompanied mutations after progression than the acquired resistance group, which means the potential resistance mechanism may be complicated
Figure 4.
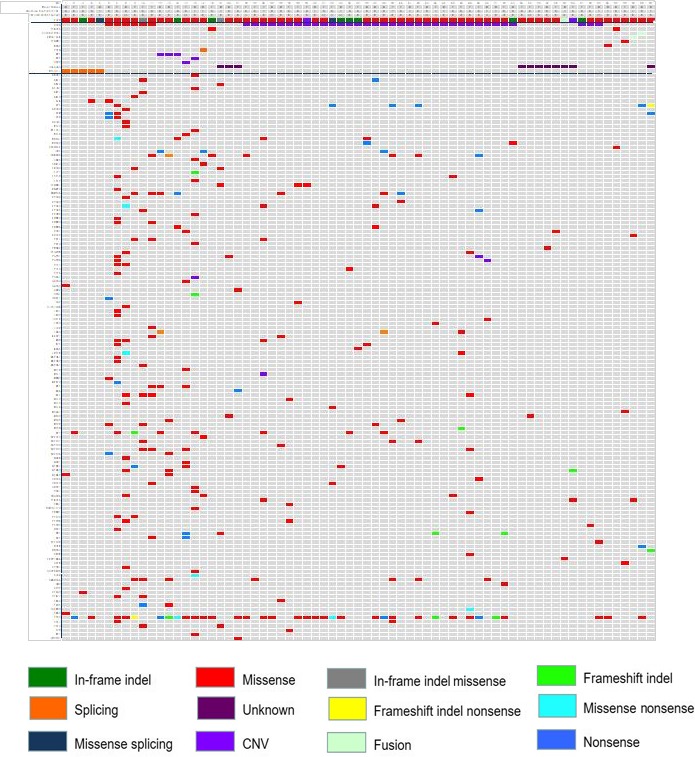
The canonical characteristics and mutation profiles of 21 primary and 48 acquired resistance to icotinib in EGFRum advanced NSCLC patients. B, blood; CNV, Copy number variation; EGFRum, epidermal growth factor receptor uncommon mutation; F, female; M, male; NSCLC, non–small cell lung cancer. Smoking history (N, no; Y, yes). Type of sample (H, histology; Ht, hydrothorax)
4. DISCUSSION
Heterogeneity in tumors is an important cause of primary resistance to EGFR‐targeted therapy in advanced NSCLC patients.16 Molecular heterogeneity and complicated mutation patterns are well known in EGFRum NSCLC patients;17, 18 however, few studies have addressed the problem of primary resistance to icotinib in this rare population. This study presents a comprehensive mutation profiling of 69 EGFRum patients who developed resistance to icotinib using ctDNA samples for genetic analysis and focusing on primary resistance. G719X, S768I and L861Q were the most common de novo mutations in the primary resistance group. The bypass pathway activation was the predominant alteration in this group, including EGFR ECD mutations, BCL2L11 deletion and MET amplification. The primary resistance group harbored more accompanied mutations than the acquired resistance group after disease progress following resistance to icotinib, which was consistent with the much more complicated resistance mechanism in the former group.
In our study, approximately 21.4% (21/98) EGFRum patients developed resistance to icotinib within 3 months of initial therapy. One study reported that approximately 21.7% (15/69) of patients presented primary resistance to icotinib in NSCLC, including some EGFRum carriers.19 The G719X, S768I and L861Q carriers contributed most primary resistance patients in our study. Robust evidence has shown the poor response to the first‐generation TKI in G719X/S768I/L861Q carriers.20, 21 Therefore, second generation TKI, such as afatinib, have been recommended to treat these patients.22 We also found three exon 20ins carriers, with one of them harboring S768I + L858R + D770delinsGY who developed disease progression within 0.5 months. One study showed that an EGFR V769_D770insASV carrier treated by TKI had TTP and OS of 19.8 months and 24 months, respectively,23 and another study showed that the OS was 16 months and worse than of EGFRcm carriers.24 Although the poor response to icotinib in our case would be possibly impacted by the concurrent S768I mutation, insensitivity of this point mutation could be assumed regarding the shortest PFS.
EGFRum NSCLC patients usually harbor combined mutations and concurrent with EGFRcm would be expected to harvest better response to TKI than single EGFRum.4 However, the EGFRum combined with EGFRcm group was shown to have the worst median PFS compared to other mutant patterns in our study. One of them carried de novo T790M + L858R. The incident of de novo T790M mutation varies from 1% to 65% in different studies 25 and often appears with combined with EGFRcm.26, 27 Tu et al28 studied the efficacy of TKI and chemotherapy in 218 patients with EGFRum and found that T790M carriers had the shortest median PFS compared with other rare mutations and composite mutations, of only 1 month (95% CI 0.0‐2.2) even combined with L858R. Osimertinib, a third generation TKI has been recommended as effective TKI in this situation.29
The EGFR extracellular domain (ECD) mutation and BCL2L11 deletion appeared in almost half of the primary resistance patients in our study, with median PFS of only 1.8 months. Recently, a novel EGFR ECD somatic mutation M277E in lung adenocarcinoma was found and proved to be a carcinogenic‐driven mutation in vitro.30 We have found one ECD M277E mutation but others coexisting with S768I, if there is a synergetic role in promoting primary resistance in icotinib, need to be studied in the future. In vitro studies have also found that downregulation of BIM expression is associated with primary resistance to gefitinib in EGFR‐mutant lung cancer cells.31 BCL2L11 deletion has been (P = .027). Personalizing therapy with BH3 analogs could possibly overcome BIM‐polymorphism–associated TKI resistance.32 Of note, 4 in 5 patients who acquired BCL2L11 deletion in our study were S768I carriers before treatment. Whether there is a rational connection between S768I and acquired BCL2L11 deletion, which contributes to the primary resistance to icotinib, remains to be further studied.
The major limitations of our study are the retrospective design and the small sample size. Despite the low incidence of EGFRum NSCLC patients, we undertook comprehensive genetic profiling and compared results between patients with primary and acquired resistance to icotinib. We listed all the de novo and acquired genetic alterations in detail and the presumed activated pathways according to previous literature.
The mechanisms of primary resistance to icotinib in EGFRum NSCLC may be highly heterogeneous. De novo T790M mutations in EGFR and its activated bypass pathways are significantly related to primary resistance of icotinib. Combined targeted therapy is one actionable option other than chemotherapy in this population.
DISCLOSURE
The authors have no conflict of interest to declare.
ACKNOWLEDGMENTS
This study was supported in part by grants from the Medical Scientific Research Foundation of Zhejiang Province of China (2019RC027) and Xisike‐Hanson Cancer Research Foundation (Y‐HS2019‐20).
Lei L, Wang W‐X, Zhu Y‐C, et al. Potential mechanism of primary resistance to icotinib in patients with advanced non–small cell lung cancer harboring uncommon mutant epidermal growth factor receptor: A multi‐center study. Cancer Sci. 2020;111:679–686. 10.1111/cas.14277
Lei and Wang contributed equally to this work.
Contributor Information
Chun‐wei Xu, Email: xuchunweibbb@163.com.
Xiao‐jia Wang, Email: wxiaojia0803@163.com.
REFERENCES
- 1. Russo A, Franchina T, Ricciardi GRR, et al. A decade of EGFR inhibition in EGFR‐mutated non small cell lung cancer (NSCLC): Old successes and future perspectives. Oncotarget. 2015;6:26814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wu JY, Yu CJ, Chang YC, et al. Effectiveness of tyrosine kinase inhibitors on “uncommon” epidermal growth factor receptor mutations of unknown clinical significance in non–small cell lung cancer. Clin Cancer Res. 2011;17:3812‐3821. [DOI] [PubMed] [Google Scholar]
- 3. Massarelli E, Johnson FM, Erickson HS, et al. Uncommon epidermal growth factor receptor mutations in non–small cell lung cancer and their mechanisms of EGFR tyrosine kinase inhibitors sensitivity and resistance. Lung Cancer. 2013;80:235‐241. [DOI] [PubMed] [Google Scholar]
- 4. Keam B, Kim DW, Park JH, et al. EGFR Kinase Rare and complex mutations of epidermal growth factor receptor, and efficacy of tyrosine kinase inhibitor in patients with non–small cell lung cancer. Int J Clin Oncol. 2014;19:594‐600. [DOI] [PubMed] [Google Scholar]
- 5. Liu D, Zhang L, Wu Y, et al. Clinical pharmacokinetics, safety, and preliminary efficacy evaluation of icotinib in patients with advanced non–small cell lung cancer. Lung Cancer. 2015;89:262‐267. [DOI] [PubMed] [Google Scholar]
- 6. Shi Y, Zhang L, Liu X, et al. Icotinib versus gefitinib in previously treated advanced non–small‐cell lung cancer (ICOGEN): a randomised, double‐blind phase 3 non–inferiority trial. Lancet Oncol. 2013;14:953‐961. [DOI] [PubMed] [Google Scholar]
- 7. Jackman D, Pao W, Riely GJ, et al. Clinical definition of acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitors in non–small‐cell lung cancer. J Clin Oncol. 2010;28:357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Wang J, Wang B, Chu H, et al. Intrinsic resistance to EGFR tyrosine kinase inhibitors in advanced non–small‐cell lung cancer with activating EGFR mutations. Onco Targets Ther. 2016;9:3711‐3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Inukai M, Toyooka S, Ito S, et al. Presence of epidermal growth factor receptor gene T790M mutation as a minor clone in non–small cell lung cancer. Cancer Res. 2006;66:7854‐7858. [DOI] [PubMed] [Google Scholar]
- 10. Wu D, Chen C, Chu C, et al. Paxillin confers resistance to tyrosine kinase inhibitors in EGFR‐mutant lung cancers via modulating BIM and Mcl‐1 protein stability. Oncogene. 2016;35:621. [DOI] [PubMed] [Google Scholar]
- 11. Yasuda H, Park E, Yun CH, et al. Structural, biochemical, and clinical characterization of epidermal growth factor receptor (EGFR) exon 20 insertion mutations in lung cancer. Sci Transl Med. 2013;5:216ra177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Watanabe M, Kawaguchi T, Isa SI, et al. Ultra‐sensitive detection of the pretreatment EGFR T790M Mutation in non–small cell lung cancer patients with an EGFR‐activating mutation using droplet digital PCR. Clin Cancer Res. 2015;21:3552‐3560. [DOI] [PubMed] [Google Scholar]
- 13. Lohinai Z, Hoda MA, Fabian K, et al. Distinct epidemiology and clinical consequence of classic versus rare EGFR mutations in lung adenocarcinoma. J Thorac Oncol. 2015;10:738‐746. [DOI] [PubMed] [Google Scholar]
- 14. Li K, Yang M, Liang N, et al. Determining EGFR‐TKI sensitivity of G719X and other uncommon EGFR mutations in non–small cell lung cancer: Perplexity and solution. Oncol Rep. 2017;37:1347‐1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Schwartz LH, Litiere S, de Vries E, et al. RECIST 1.1‐Update and clarification: from the RECIST committee. Eur J Cancer. 2016;62:132‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Morgillo F, Della Corte CM, Fasano M, et al. Mechanisms of resistance to EGFR‐targeted drugs: lung cancer. ESMO Open. 2016;1:e000060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Arcila ME, Nafa K, Chaft JE, et al. EGFR Exon 20 insertion mutations in lung adenocarcinomas: prevalence, molecular heterogeneity, and clinicopathologic characteristics. Mol Cancer Ther. 2013;12:220‐229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kobayashi Y, Mitsudomi T. Not all epidermal growth factor receptor mutations in lung cancer are created equal: perspectives for individualized treatment strategy. Cancer Sci. 2016;107:1179‐1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Jin Y, Shi X, Zhao J, et al. Mechanisms of primary resistance to EGFR targeted therapy in advanced lung adenocarcinomas. Lung Cancer. 2018;124:110‐116. [DOI] [PubMed] [Google Scholar]
- 20. Yang JC, Sequist LV, Geater SL, et al. Clinical activity of afatinib in patients with advanced non–small‐cell lung cancer harbouring uncommon EGFR mutations: a combined post–hoc analysis of LUX‐Lung 2, LUX‐Lung 3, and LUX‐Lung 6. Lancet Oncol. 2015;16:830‐838. [DOI] [PubMed] [Google Scholar]
- 21. Chen D, Song Z, Cheng G. S Clinical efficacy of first‐generation EGFR‐TKIs in patients with advanced non–small‐cell lung cancer harboring EGFR exon 20 mutations. Onco Targets Ther. 2016;9:4181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wu YL, Hirsh V, Sequist LV, et al. Does EGFR mutation type influence patient‐reported outcomes in patients with advanced EGFR mutation‐positive non–small‐cell lung cancer? Analysis of two large, phase III studies comparing Afatinib with chemotherapy (LUX‐Lung 3 and LUX‐Lung 6). Patient. 2018;11:131‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Naidoo J, Sima CS, Rodriguez K, et al. Epidermal growth factor receptor exon 20 insertions in advanced lung adenocarcinomas: Clinical outcomes and response to erlotinib. Cancer. 2015;121:3212‐3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Oxnard GR, Lo PC, Nishino M, et al. Natural history and molecular characteristics of lung cancers harboring EGFR exon 20 insertions. J Thorac Oncol. 2013;8:179‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y, Sun L, Xiong ZC, et al. Meta‐analysis of the impact of de novo and acquired EGFR T790M mutations on the prognosis of patients with non–small cell lung cancer receiving EGFR‐TKIs. Onco Targets Ther. 2017;10:2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Thomas A, Xi L, Carter CA, et al. Concurrent molecular alterations in tumors with germ line epidermal growth factor receptor T790M mutations. Clin Lung Cancer. 2013;14:452‐456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Yamane H, Ochi N, Yasugi M, et al. Docetaxel for non–small‐cell lung cancer harboring the activated EGFR mutation with T790M at initial presentation. Onco Targets Ther. 2013;6:155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tu HY, Ke E, Yang JJ, et al. A comprehensive review of uncommon EGFR mutations in patients with non–small cell lung cancer. Lung Cancer. 2017;114:96‐102. [DOI] [PubMed] [Google Scholar]
- 29. Lamb YN, Scott LJ. Osimertinib: a review in T790M‐positive advanced non–small cell lung cancer. Targeted Oncol. 2017;12:555‐562. [DOI] [PubMed] [Google Scholar]
- 30. Yu S, Zhang Y, Pan Y, et al. The non–small cell lung cancer EGFR extracellular domain mutation, M277E, is oncogenic and drug‐sensitive. Onco Targets Ther. 2017;10:4507‐4515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Li Z, Zhou S, Zhang L, et al. BIM induction of apoptosis triggered by EGFR‐sensitive and resistance cell lines of non–small‐cell lung cancer. Med Oncol. 2011;28:572‐577. [DOI] [PubMed] [Google Scholar]
- 32. Ng KP, Hillmer AM, Chuah CT, et al. A common BIM deletion polymorphism mediates intrinsic resistance and inferior responses to tyrosine kinase inhibitors in cancer. Nat Med. 2012;18:521. [DOI] [PubMed] [Google Scholar]