

Abstract
Fibroblast growth factor receptors (FGFR) are a family of transmembrane receptor tyrosine kinases involved in regulating cellular processes. FGFR mutations are implicated in oncogenesis, representing therapeutic potential in the form of FGFR inhibitors. This phase I, first‐in‐human study in Japan evaluated safety and tolerability of E7090, a potent selective FGFR1‐3 inhibitor, in patients with advanced solid tumors. Dose escalation (daily oral dose of 1‐180 mg) was carried out to assess dose‐limiting toxicity (DLT), maximum tolerated dose, and pharmacokinetics. Pharmacodynamic markers (serum phosphate, fibroblast growth factor 23, and 1,25‐(OH)2‐vitamin D) were also evaluated. A total of 24 patients refractory to standard therapy or for whom no appropriate treatment was available were enrolled. No DLT were observed up to the 140‐mg dose; one patient in the 180‐mg cohort experienced a DLT (increased aspartate aminotransferase/alanine aminotransferase, grade 3). The maximum tolerated dose was not reached. Dose‐dependent increases in the maximum concentration and area under the curve from time 0 to the last measurable concentration were observed up to 180 mg. Dose‐dependent increases were observed in all pharmacodynamic markers and plateaued at 100‐140 mg, indicating sufficient FGFR pathway inhibition at doses ≥100 mg. In conclusion, E7090 showed a manageable safety profile with no DLT at doses ≤140 mg. Maximum tolerated dose was not determined. The recommended dose for the follow‐up expansion part, restricted to patients with tumors harboring FGFR alterations, was determined as 140 mg, once daily.
Keywords: fibroblast growth factor, maximum tolerated dose, pharmacokinetics, phase I clinical trial, safety
This phase I, first‐in‐human study in Japan evaluated the safety and tolerability of E7090, a selective fibroblast growth factor receptor (FGFR) 1‐3 inhibitor, for treatment of advanced solid tumors. A total of 24 patients refractory to standard therapy or for whom no appropriate treatment was available were enrolled; no dose‐limiting toxicities were observed up to 140 mg QD and the maximum tolerated dose was not reached. E7090 showed an acceptable safety profile with dose‐dependent pharmacokinetic/pharmacodynamic parameters; preliminary efficacy will be investigated in a follow‐up expansion part restricted to gastric cancer patients with FGFR2 amplification or cholangiocarcinoma with FGFR2 fusion.
![]()
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- DLT
dose‐limiting toxicity
- ECOG
Eastern Cooperative Oncology Group
- ELISA
enzyme‐linked immunosorbent assay
- FGF
fibroblast growth factor
- FGFR
fibroblast growth factor receptor
- FISH
fluorescence in situ hybridization
- IS
internal standard
- LC‐MS/MS
liquid chromatography‐tandem mass spectrometry
- MTD
maximum tolerated dose
- mTPI
modified toxicity probability interval
- PD
pharmacodynamics
- PK
pharmacokinetic
- PPE
palmar‐plantar erythrodysesthesia
- QD
once daily
- RIA
radioimmunoassay
- SAE
serious adverse event
- TEAE
treatment‐emergent adverse event
1. INTRODUCTION
Fibroblast growth factor receptors are a family of four highly conserved transmembrane receptor tyrosine kinases (FGFR1‐4) that can bind FGF ligands.1 FGF signaling is implicated in downstream transduction pathways that regulate several key cellular processes, including proliferation, differentiation, migration, and survival.2 The signaling pathway is frequently perturbed in cancer, often manifesting as FGFR amplification and mutation, oncogenic fusion, dysregulated FGF ligand signaling, and promotion of angiogenesis.3 Although anti‐FGFR therapy represents a promising targeted cancer treatment, early phase clinical trials have had mixed success, with response to therapy dependent on several factors, including cancer type, tumor histology, and presence or absence of certain biomarkers.4
Trials of non‐selective, multi‐target tyrosine kinase inhibitors have shown variable anti‐FGFR activity and broad‐spectrum off‐target inhibition of other tyrosine kinases, notably vascular endothelial growth factor, leading to toxicities.5 Off‐target inhibition has also been associated with several other AE, such as bone marrow suppression caused by platelet‐derived growth factor inhibition and skin rash caused by KIT inhibition.6 Thus, selective inhibitors may offer the benefit of reduced toxicity by eliminating concerns about such off‐target effects.
To date, several selective FGFR inhibitors have been assessed in early phase clinical testing. A phase I study of the selective FGFR1‐3 inhibitor AZD4547 in patients with squamous cell lung cancers confirmed target inhibition but failed to achieve its efficacy endpoint.7 In Japanese patients with advanced solid tumors, AZD4547 was well tolerated, with best response being stable disease (duration ≥4 weeks).8 BGJ398, another selective FGFR1‐3 inhibitor, showed antitumor activity in several tumor types and had a tolerable safety profile in a phase I study in patients with advanced solid tumors,9 whereas JNJ‐42756493, a pan‐FGFR inhibitor recently approved by the FDA for urothelial carcinoma,10 showed a clinical response with acceptable safety in a similar patient population.11 Similar findings have been reported for the pan‐FGFR inhibitors LY287445512 and ARQ 087.13 Clinical trials are ongoing for other highly selective FGFR inhibitors in development, such as TAS‐12014 and INCB054828,15 in which patients are screened for FGFR abnormalities using next‐generation sequencing or FISH techniques.
E7090 is an orally available and potent selective inhibitor of the tyrosine kinase activities of FGFR1, ‐2, and ‐3, developed at the Eisai Tsukuba Research Laboratories. Based on its unique binding kinetics with FGFR1, E7090 is classified as a type V kinase inhibitor; in contrast, the developmental agent AZD4547 is a common type I inhibitor.16 In a human gastric cancer cell line (SNU‐16) that expresses high levels of FGFR2 protein, E7090 inhibited phosphorylation of both FGFR (IC50 = 1.2 nmol/L) and downstream molecules including FRS2α, ERK1/2, and AKT in a dose‐dependent method.16 E7090 also showed antitumor activity in preclinical models (in vitro assays and mouse xenografts using gastric, lung, bladder, or breast cancer cells) harboring FGFR genetic alterations such as FGFR1 and/or FGFR2 amplification, FGFR1 fusion, FGFR2 mutation, FGFR3 fusion, and FGFR3 mutation.16
The aim of this first‐in‐human phase I study of E7090 was to evaluate the primary endpoints of safety and tolerability in patients with advanced solid tumors. Secondary endpoints included determination of the MTD of E7090 to identify the dose for future studies, and to establish its PK characteristics and preliminary antitumor activity. Exploratory objectives included identification of PD markers (including markers of FGFR pathway inhibition such as serum phosphate, FGF23, and 1,25‐(OH)2‐vitamin D)17 and pharmacogenomics of E7090; assessment of the relationships among PK variables, PD markers, and pharmacogenomics; and analysis of the plasma and urinary metabolites of E7090.
2. MATERIALS AND METHODS
2.1. Patients
Patients aged ≥20 years with histologically or cytologically confirmed advanced solid tumors refractory to standard therapy, or for whom no appropriate treatment was available, were eligible for the study. Other inclusion criteria included corrected serum calcium and phosphate ≤ upper limit of normal and ECOG performance status of 0 or 1. Patients with brain metastasis associated with clinical symptoms or requiring treatment, current evidence or history of ≥ grade 2 corneal disorder, or a history of clinically significant cardiovascular impairment were excluded. Patients who had previously been treated with FGFR inhibitors were also excluded.
2.2. Study design
This first‐in‐human phase I study conducted in Japan consisted of two parts: a single‐center dose‐escalation study to assess DLT and to determine MTD in patients with advanced solid tumors (Part 1), and a multicenter expansion part restricted to patients with tumors harboring FGFR alterations, including gastric cancer with FGFR2 amplification and cholangiocarcinoma with FGFR2 fusion (Part 2) (Figure S1). The current report describes results from Part 1; Part 2 of the study is ongoing and results will be described in a subsequent publication.
A mTPI design18 was used to determine the MTD of E7090, with each cohort assigned a dose of E7090 in accordance with the rules of the mTPI design based on a target DLT rate of 25% ± 5% (Table S1). E7090 was given orally on a QD continuous schedule in 28‐day cycles. In cycle 0, patients received a single oral dose of E7090 (1 mg; the starting dose was determined based on preclinical animal toxicity studies) on day 1, followed by a washout period (days 2‐7) for PK analysis. Cycle 1 started between day 8 and day 10. For cycle 1 and subsequent cycles, the cycle length was 28 days and patients received E7090 QD until any of the predefined discontinuation criteria were met. If no patients experienced DLT at the starting dose, dose escalation was permitted, as summarized in Figure S2.
This study was conducted in accordance with the Declaration of Helsinki and the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (Guideline for Good Clinical Practice), and was approved by the institutional review board at the study center. All patients provided written informed consent prior to participation. This study was registered in the Japan Pharmaceutical Information Center Clinical Trials Information registry (JapicCTI‐142740) and in ClinicalTrials.gov (NCT02275910).
2.3. Safety and tolerability
Dose‐limiting toxicities (monitored during cycle 0‐cycle 1) were defined as any of the following AE: grade 4 neutropenia persisting for more than 7 days or febrile neutropenia; grade 4 thrombocytopenia or grade 3 thrombocytopenia requiring blood transfusion; any ≥grade 3 non‐hematological toxicity with the exception of abnormal clinical laboratory values with no clinical significance and any events that could be managed and controlled to ≤grade 2 by maximal medical management; new ectopic de novo calcification with clinical significance confirmed by radiological images; hyperphosphatemia defined as serum phosphate >7 mg/dL persisting for >7 days despite giving phosphate‐lowering therapy or >9 mg/dL (single measurement) despite giving phosphate‐lowering therapy; and development of any toxicity considered to be related to E7090 and requiring treatment interruption for ≥8 days from cycle 0 to cycle 1. Phosphate binders were given when serum phosphate levels exceeded the institutional normal levels. During the DLT assessment period, concomitant therapies comprising any treatment, change in dosage, or changes in medication for the purpose of preventing the occurrence of a DLT were not permitted.
Adverse events were coded using the Medical Dictionary for Regulatory Activities v20.1, and severity grades were determined using the Common Terminology Criteria for Adverse Events v4.03.
2.4. Efficacy
Tumor assessment by computed tomography and/or magnetic resonance imaging was carried out according to RECIST v1.1 guidelines19 at screening, once every 8 weeks from day 1 of cycle 1, and at study discontinuation.
2.5. Pharmacokinetic analysis
Blood samples for PK analysis were obtained before dosing in all cycles and at specific time points after giving E7090 up to cycle 3. Plasma and urine concentrations of E7090 were measured by LC‐MS/MS. To isolate E7090 from plasma, 10 µL IS and 500 µL of 5 mmol/L ammonium hydrogen carbonate were added to 100 µL plasma and vortexed. Solid‐phase extraction was carried out with an Oasis HLB µElution plate (Waters Corporation). After conditioning with 700 µL methanol and equilibration with 700 µL of 5 mmol/L ammonium hydrogen carbonate, samples were loaded onto the plate and washed with distilled water/methanol (3/2, v/v, 100 µL) and eluted with methanol/formic acid (100/1, v/v, 50 µL). The elution process was repeated twice and the eluate was subjected to LC‐MS/MS analysis under the following conditions. Reversed‐phase chromatography was run on an L‐column2 ODS (2.1 mm internal diameter × 150 mm, 3 μm, Chemicals Evaluation and Research Institute) at 40°C using a mobile phase consisting of distilled water (A) and acetonitrile/distilled water (4/1, v/v) (B) containing 15 mmol/L ammonium hydrogen carbonate (A:B = 40:60) at a flow rate of 0.25 mL/minute. E7090 and IS were detected by MS/MS using multiple reaction monitoring under the positive ionization mode.
2.6. Pharmacodynamics, pharmacogenomics and biomarker assessments
Blood samples, stored tumor samples, and tumor biopsy samples were obtained before giving E7090 in cycle 0, cycle 1, subsequent odd‐numbered cycles, and after dosing in cycle 0 for days 1‐4. Markers of FGFR pathway inhibition included serum phosphate, FGF23, and 1,25‐(OH)2‐vitamin D. FGF23 was detected by solid‐phase sandwich ELISA (FGF‐23 ELISA Kit; Kainos) using mouse anti‐human FGF23 and peroxidase‐conjugated mouse anti‐human FGF23 monoclonal antibodies, and colorimetric detection. Measurement of 1,25‐(OH)2‐vitamin D was done using a double‐antibody RIA. Sheep anti‐1,25‐(OH)2‐vitamin D antibody was added to pretreated samples, incubated for 16‐18 hours, and an 125I‐labeled 1,25‐(OH)2‐vitamin D tracer was added, followed by a cellulose‐conjugated donkey anti‐sheep IgG antibody. After removal of unbound tracer, radioactivity was measured (ARC‐950/ARC‐8010 γ‐counter [Hitachi Ltd], 1460SRL [PerkinElmer Co., Ltd]).
2.7. Statistical analyses
Descriptive statistics were used to analyze demographic and other baseline characteristics and safety assessments. All patients who received at least one dose of E7090 were included in the safety analysis. These patients had one or more target lesions as defined by RECIST v1.1 and were included in the analysis of best overall response. Efficacy analysis set included patients in the safety analysis set who underwent tumor assessment at baseline and at least once post‐baseline. Plasma concentrations of E7090 were analyzed using a non‐compartmental analysis to determine PK parameters (maximum plasma concentration [C max], time to reach maximum concentration [T max], area under the concentration‐time curve [AUC], and clearance).
The number of patients required for inclusion in Part 1 of the study was estimated to be approximately 20, based on the recommended sample size to reach MTD using nine dose levels, each with a cohort size of two.
3. RESULTS
3.1. Patients
A total of 24 patients (11 males, 13 females) were enrolled in the study between 10 November 2014 and 21 February 2017. Baseline characteristics are summarized in Table 1. Eight patients (33%) had cholangiocarcinoma and there were three cases (13%) of pancreatic cancer, two cases (8%) of endometrial cancer, two cases (8%) of cancer of unknown primary, and nine (38%) patients with other solid tumors. All 24 patients had measurable disease by RECIST criteria at baseline and had received prior chemotherapy, with nine (38%) having received two or fewer lines and 15 (63%) three or more lines of chemotherapy. No assessment of FGFR status (mutation, amplification, or fusion) was carried out during Part 1 of the study, although if available, information on FGFR status was collected from patients who had obtained such data from other examinations outside the present clinical trial.
Table 1.
Patient demographic and baseline characteristics
| All patients N = 24 (%) | ||
|---|---|---|
| Age, years | Median (range) | 65 (42‐75) |
| Gender | Male | 11 (45.8) |
| Female | 13 (54.2) | |
| ECOG‐PS | 0 | 17 (70.8) |
| 1 | 7 (29.2) | |
| No. of prior lines of chemotherapy | ≤2 | 9 (37.5) |
| ≥3 | 15 (62.5) | |
| Cancer type | Cholangiocarcinoma | 8 (33.3) |
| Pancreatic cancer | 3 (12.5) | |
| Endometrial cancer | 2 (8.3) | |
| Cancer of unknown primary | 2 (8.3) | |
| Other | 9 (37.5) |
Abbreviation: PS, performance status.
3.2. Safety
No DLT was observed between 1 mg QD and 140 mg QD. Of the 24 patients treated, three experienced a SAE (dyspnea in one subject in the 8‐mg QD cohort, tumor pain in one subject in the 8‐mg QD cohort, and pyrexia in one subject in the 30‐mg cohort). None of the SAE was considered to be related to the study drug (Table 2).
Table 2.
Treatment‐related treatment‐emergent adverse events (occurring in ≥10% of the total study population)
| Preferred terma | 1‐30 mg (n = 12) | 60 mg (n = 3) | 100 mg (n = 3) | 140 mg (n = 3) | 180 mg (n = 3) | Total (N = 24) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Any grade | ≥Grade 3 | Any grade | ≥Grade 3 | Any grade | ≥Grade 3 | Any grade | ≥Grade 3 | Any grade | ≥Grade 3 | Any grade | ≥Grade 3 | |
| Blood creatinine increased | 2 (16.7) | – | 2 (66.7) | – | 1 (33.3) | – | 3 (100.0) | – | 1 (33.3) | – | 9 (37.5) | – |
| Hyperphosphatemia | – | – | – | – | 3 (100.0) | – | 3 (100.0) | – | 3 (100.0) | – | 9 (37.5) | – |
| ALT increased | – | – | 1 (33.3) | – | – | – | 3 (100.0) | – | 2 (66.7) | 2 (66.7) | 6 (25.0) | 2 (8.3) |
| Diarrhea | – | – | 2 (66.7) | – | – | – | 1 (33.3) | – | 3 (100.0) | – | 6 (25.0) | – |
| Lipase increased | 1 (8.3) | – | 1 (33.3) | – | 1 (33.3) | – | 1 (33.3) | – | 1 (33.3) | – | 5 (20.8) | – |
| Nausea | 1 (8.3) | – | 1 (33.3) | – | 1 (33.3) | – | 1 (33.3) | – | 1 (33.3) | – | 5 (20.8) | – |
| AST increased | – | – | – | – | – | – | 2 (66.7) | – | 2 (66.7) | 1 (33.3) | 4 (16.7) | 1 (4.2) |
| PPE syndrome | – | – | 1 (33.3) | – | – | – | 1 (33.3) | – | 2 (66.7) | – | 4 (16.7) | – |
| Anemia | – | – | 1 (33.3) | – | 1 (33.3) | – | 1 (33.3) | – | – | – | 3 (12.5) | – |
| Blood ALP increased | – | – | – | – | 1 (33.3) | – | 2 (66.7) | – | – | – | 3 (12.5) | – |
| Dysgeusia | – | – | – | – | – | – | 2 (66.7) | – | 1 (33.3) | – | 3 (12.5) | – |
| Retinal detachment | – | – | – | – | 2 (66.7) | – | 1 (33.3) | – | – | – | 3 (12.5) | – |
| Vomiting | – | – | 1 (33.3) | – | – | – | 1 (33.3) | – | 1 (33.3) | 1 (33.3) | 3 (12.5) | 1 (4.2) |
Values are shown as n (%); percentages are based on the number of treated patients in the relevant treatment cohort.
Abbreviations: ALP, alkaline phosphatase; ALT, alanine aminotransferase; AST, aspartate aminotransferase; PPE, palmar‐plantar erythrodysesthesia; –, no incidence.
Coded using the Medical Dictionary for Regulatory Activities, version 20.1.
No drug‐related deaths were reported, and no TEAE led to discontinuation of the study drug. TEAE leading to dose reductions were increased ALT (n = 2), increased AST (n = 1), and PPE syndrome (n = 2). Reasons for TEAE‐related dose interruptions included nausea (n = 3), vomiting (n = 2), anorexia (n = 2), nasopharyngitis (n = 1), and decreased neutrophil count (n = 1). Selective FGFR inhibition has been associated with an increased risk of hyperphosphatemia20 and eye toxicity, including retinal detachment.8, 13 In the present study, there were nine AE of hyperphosphatemia and three AE of retinal detachment, but all were <grade 3 in severity and did not lead to dose reduction or discontinuation.
The dose levels are shown in Figure S2. One patient experienced a DLT (grade 3 increased AST/ALT) in the 180‐mg cohort, whereas no DLT were reported at doses up to and including 140 mg. Because sufficient inhibition of the targeted signaling pathway was obtained at 100‐140 mg (as shown by PD markers described in the later section) and the safety profile at this dose level was acceptable, the recommended dose for Part 2 was determined as 140 mg QD.
3.3. Pharmacokinetics
Following both single and repeat oral administration, once‐daily E7090 was cleared from plasma with a mean terminal elimination phase half‐life (T 1/2) of 15‐27 hours. Dose‐dependent increases in C max and AUC(0‐ t ) were observed up to 180 mg E7090 (Figure 1). T max was similar among doses, with a median of 2‐5 hours. Excretion of unchanged form into urine was low (data not shown). The PK parameters are presented in Table 3 and include data only from patients who received ≥30 mg E7090, as plasma concentrations in the 1‐16‐mg cohorts at 72 hours after dosing were below the limit of quantification.
Figure 1.
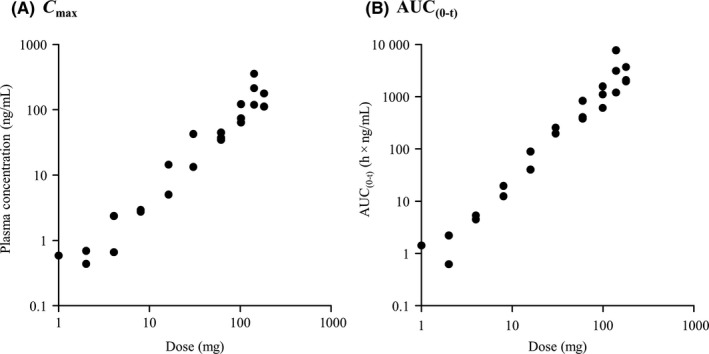
Relationship between E7090 dose and pharmacokinetic parameters. A, Maximum plasma concentration (C max). B, AUC(0‐t) (single‐dose study). AUC, area under the concentration‐time curve
Table 3.
Pharmacokinetic parameters of E7090
| 30 mg (n = 2) | 60 mg (n = 3) | 100 mg (n = 3) | 140 mg (n = 3) | 180 mg (n = 3) | |
|---|---|---|---|---|---|
| Single | |||||
| T 1/2 (h) | 23 | 23 ± 8 | 15 ± 2 | 27 ± 12 | 24 ± 10 |
| T max (h) | 3 (1‐5) | 3 (3‐5) | 5 (3‐5) | 5 (3‐5) | 2 (2‐5) |
| C max (ng/mL) | 28 | 39 ± 5 | 86 ± 31 | 227 ± 118 | 154 ± 38 |
| AUC(0‐t) (h × ng/mL) | 222 | 533 ± 257 | 1080 ± 474 | 3960 ± 3230 | 2570 ± 933 |
| AUC(0‐inf) (h × ng/mL) | 239 | 554 ± 247 | 1120 ± 479 | 4050 ± 3340 | 2610 ± 921 |
| Vz/F (L) | 4180 | 3790 ± 1240 | 2310 ± 1360 | 1670 ± 617 | 2550 ± 1550 |
| CL/F (L/h) | 127 | 121 ± 43 | 103 ± 48 | 59 ± 50 | 74 ± 22 |
| Repeated | |||||
| T max (h) | 4 (3‐5) | 3 (2‐3)a | 3 (2‐5) | 5 (3‐5) | 5 (3‐5) |
| C max (ng/mL) | 13 | 51a | 116 ± 4 | 372 ± 173 | 337 ± 60 |
| AUC(0‐t) (h × ng/mL) | 162 | 507a | 1340 ± 437 | 4700 ± 3380 | 3860 ± 764 |
| C min (ng/mL) | 5 | 11a | 22 ± 9 | 117 ± 116 | 65 ± 19 |
| C avg (ng/mL) | 7 | 21a | 56 ± 18 | 196 ± 141 | 161 ± 32 |
Data are shown as mean ± SD, except T max; for T max, median (range) is shown.
Abbreviations: AUC, area under the concentration‐time curve; C avg, average plasma concentration; CL/F, total clearance; C max, maximum plasma concentration; C min, minimum plasma concentration; T 1/2, terminal elimination phase half‐life; T max, time to reach maximum plasma concentration; Vz/F, volume of distribution.
n = 2; data are missing for one patient who discontinued treatment prior to cycle 1, day 22 because of progressive disease.
3.4. Pharmacodynamics
E7090 administration induced dose‐dependent increases in serum phosphate, FGF23, and 1,25‐(OH)2‐vitamin D, and these increases reached a maximum at approximately 100‐140 mg QD. Percent changes from baseline of PD markers of FGFR pathway inhibition (cycle 1, day 15) are shown in Figure 2.
Figure 2.
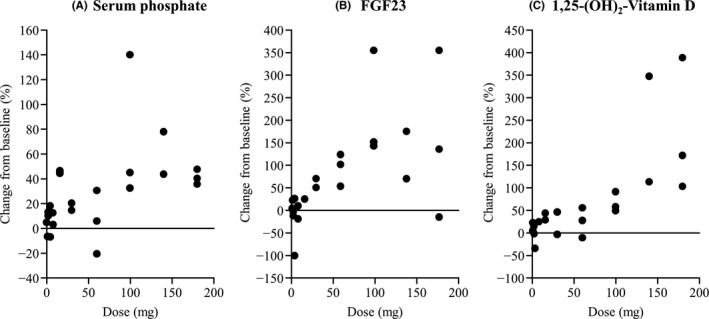
Percent change from baseline of pharmacodynamic markers of fibroblast growth factor receptor (FGFR) pathway inhibition. A, Serum phosphate. B, Fibroblast growth factor 23 (FGF23). C, 1,25‐(OH)2‐vitamin D (cycle 1 day 15)
3.5. Antitumor activity
One patient achieved a partial response and seven patients achieved stable disease (Table S2). Fourteen patients had a best overall response of progressive disease, and two patients were not evaluable. The patient (a 45‐year‐old woman) who showed a partial response was in the 180‐mg cohort, had diffuse‐type gastric cancer with para‐aortic lymph node and bone metastases (poorly differentiated adenocarcinoma) and FGFR2 amplification (copy number 51 by next‐generation sequencing), and had undergone three prior lines of chemotherapy. A 71% reduction in diameter of the target lesion (from 38 to 11 mm) was observed at cycle 3 day 1 (Figure 3). The patient discontinued treatment on cycle 4, day 8 because of progressive disease.
Figure 3.
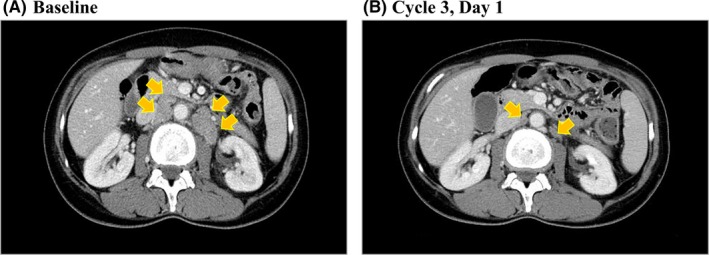
Computed tomography images showing antitumor activity of E7090. A, Baseline. B, Cycle 3, day 1 in the patient who achieved a partial response. The target lesion (para‐aortic lymphadenopathy) was reduced by 71% (from 38 to 11 mm). Arrows show the target lesion
4. DISCUSSION
In the present study, E7090 was evaluated as monotherapy at total daily doses ranging from 1 to 180 mg in 24 patients with advanced solid tumors. Based on the study findings, E7090 appears to have a tolerable and manageable safety profile, with tumor/FGFR gene alteration‐specific efficacy.
Pharmacokinetic data indicated rapid absorption of E7090; exposure as measured by C max and AUC(0‐t) increased with dose escalation. Dose‐dependent increases in PD markers of FGFR pathway inhibition17 were observed for serum phosphate, FGF23, and 1,25‐(OH)2‐vitamin D and reached a plateau at 100‐140 mg, indicating that doses ≥100 mg QD sufficiently inhibited the FGFR pathway. PD data from previous phase I studies in other specific FGFR inhibitors have been inconclusive. For example, treatment with JNJ‐42756493 was associated with increased phosphate and 1,25‐(OH)2‐vitamin D but not FGF23 in studies in solid tumors,11, 21 whereas AZD4547 has also been shown to increase serum phosphate but not FGF23.7
The toxicity profile of E7090 was generally consistent with previous findings in non‐clinical studies. In the present study, SAE occurred in three patients (although none was considered related to the study drug), and no drug‐related deaths or TEAE requiring discontinuation of the study drug were reported. For Part 2, 140 mg QD was selected as the recommended dose based on the dose‐dependent increases in PK parameters and in serum phosphate, FGF23, and 1,25‐(OH)2‐vitamin D observed following E7090 administration, which reached a maximum at approximately 100‐140 mg QD; and the absence of any AE ≥grade 3 observed at this dose. The dosage schedule (once‐daily continuous dosing) for Part 2 was also considered to be appropriate, based on the elimination half‐life of the drug; for safety reasons, intermittent dosing was not considered a suitable regimen for E7090. Although MTD was not reached, E7090 is a molecular targeted agent and sufficient inhibition of the targeted signaling pathway was achieved between 100‐140 mg QD. In the context of selecting an appropriate dose for Part 2, determination of MTD is therefore not essential. FGFR signaling is involved in the regulation of FGF23 expression,17 and increased FGF23 production has been implicated in the development of several hypophosphatemic diseases.22, 23, 24 Selective FGFR inhibitors are typically associated with on‐target toxicities such as hyperphosphatemia, which appears to play a role in FGF23 signaling20 and eye toxicity such as macular edema and retinal detachment, as reported previously.8, 13 No clear relationship between these toxicities and the inhibition of FGFR has been found. Skin and eye toxicities are a concern as a class effect of FGFR inhibition1 and although PPE syndrome and retinal detachment AE occurred in this study, they were <grade 3, manageable, and did not lead to dose discontinuation. Similarly, in a phase I study of JNJ‐42756493, detachment of retinal pigment epithelium <grade 3 was the only DLT observed, and occurred only at the highest dose investigated (12 mg).21 However, in a phase I study of AZD4547, grade 3 central serous retinopathy was reported in two patients in a dose‐escalation cohort.7
With respect to efficacy, promising antitumor activity was observed in one gastric cancer patient with an FGFR2 amplification. Although E7090 did not show potent antitumor activity in patients with cholangiocarcinoma with FGFR2 fusion in the 30‐mg cohort, this could be attributable to insufficient dosing. In preclinical models of tumors harboring FGFR abnormalities, E7090 elicited a significantly prolonged survival effect in mice with tumors metastasized to the lung.16 In a previous study of AZD4547, a selective FGFR1‐3 inhibitor, in patients with advanced gastric cancer showing FGFR2 polysomy or gene amplification, no significant effect on progression‐free survival was observed by AZD4547 compared with paclitaxel, although considerable intratumor heterogeneity was observed in the study population.25 However, phase II data on the efficacy of JNJ‐42756493 in patients with advanced urothelial cancer showing specific FGFR genetic alterations indicated promising outcomes, including a 32% objective response rate and median response duration of 5.4 months.10
A limitation of this first‐in‐human study was the small number of patients included, an inherent feature of early‐phase trials. A notable feature of the present study was the use of a modified toxicity probability interval design, which has been shown to be a safer and more robust alternative to the standard 3 + 3 design for phase I dose‐escalation oncology trials.18
In conclusion, this first‐in‐human phase I study indicates that E7090 has an acceptable safety profile; dose‐dependent PK parameters; and the potential for clinical efficacy in FGFR2‐amplified gastric cancer. Further evaluation (in Part 2) of patients with FGFR2‐amplified gastric cancer and FGFR2‐fusion‐positive cholangiocarcinoma is ongoing. It is becoming increasingly apparent that FGFR aberrations vary according to tumor histology, meaning that a more detailed understanding of the role of FGF/FGFR in cancer is required.
CONFLICTS OF INTEREST
Tatsuya Sasaki was an employee of the study sponsor during the conduct of Part 1 of the present study, and is currently an employee of Astellas Pharma Inc. Toshio Shimizu has received honoraria from Boehringer Ingelheim, Chugai Pharmaceutical, Taiho Pharma, Ono Pharmaceutical, and Ono Pharma Taiwan; and funds for research expenses from Bristol‐Myers Squibb, Daiichi Sankyo, Takeda‐Millennium, FivePrime, PharmaMar, and 3D Medicine. Satoru Iwasa has received grants from Daiichi Sankyo, Chugai Pharmaceutical, Bristol‐Myers Squibb, Eisai, Novartis, and Merck Serono. Yutaka Fujiwara has received consulting fees from Bristol‐Myers Squibb and Ono Pharmaceutical; grants from AbbVie, AstraZeneca, Bristol‐Myers Squibb, Chugai Pharmaceutical, Daiichi Sankyo, Eisai, Eli Lilly, Incyte, Merck Serono, MSD, and Novartis; and honoraria from AstraZeneca, Bristol‐Myers Squibb, MSD, Ono Pharmaceutical, and Taiho Pharma. Shigehisa Kitano has received grants from Astellas Pharma, Eisai, Gilead Sciences, and Regeneron; and honoraria from Ono Pharmaceutical, Bristol‐Myers Squibb, AstraZeneca, Chugai Pharmaceutical, Pfizer, Sanofi, Nippon Kayaku, Boehringer Ingelheim, Meiji Seika Pharma, Taiho Pharma, Novartis, Daiichi Sankyo, MSD, Kyowa Hakko Kirin, Celgene, and Sumitomo Dainippon Pharma. Noboru Yamamoto has received consulting fees from Eisai, Takeda Pharmaceutical, OncoTherapy Science, Otsuka, and Boehringer Ingelheim; and grants from Quintiles, Astellas Pharma, Chugai Pharmaceutical, Eisai, Taiho Pharma, Bristol‐Myers Squibb, Pfizer, Novartis, Daiichi Sankyo, Bayer, Boehringer Ingelheim, Kyowa‐Hakko Kirin, Takeda Pharmaceutical, and Ono Pharmaceutical; and honoraria from Bristol‐Myers Squibb, Pfizer, AstraZeneca, Eli Lilly, Ono Pharmaceutical, and Chugai Pharmaceutical. Junji Furuse has received consulting fees/honoraria from Eisai related to the present work; grants from J‐Pharma, Taiho Pharma, Sumitomo Dainippon Pharma, Janssen Pharmaceutical, Daiichi Sankyo, MSD, Yakult, Takeda Pharmaceutical, Chugai Pharmaceutical, Ono Pharmaceutical, Astellas Pharma, Zeria, Novartis, Nanocarrier, Shionogi, OncoTherapy Science, Eli Lilly Japan, Bayer, Bristol‐Myers Squibb, Merck Serono, Kyowa Hakko Kirin, Eisai, Mochida, Baxalta, and Sanofi; and honoraria from Taiho Pharma, Chugai Pharmaceutical, Yakult, Sumitomo Dainippon Pharma, Eli Lilly Japan, Astellas Pharma, Ono Pharmaceutical, Pfizer, Bayer, Novartis, Merck Serono, Takeda Pharmaceutical, Eisai, MSD, Shionogi, J‐Pharma, Daiichi Sankyo, Mochida, Nippon Kayaku, EA Pharma, Sawai, and Teijin Pharma. Takafumi Koyama, Shunsuke Kondo, Kan Yonemori, and Akihiko Shimomura have no conflicts of interest to declare. Sakura Iizumi is currently an employee of AstraZeneca KK.
AUTHOR CONTRIBUTIONS
Noboru Yamamoto, Junji Furuse, and Tatsuya Sasaki designed the study. All authors except Junji Furuse and Tatsuya Sasaki were involved in data collection. All authors contributed to the study conduct, data analysis, and manuscript writing.
Supporting information
ACKNOWLEDGMENTS
This study was funded by Eisai Co., Ltd, Tokyo, Japan. The authors would like to thank all of the patients as well as investigators and their teams who participated in this study. We thank Clare Cox, PhD, and Mary Richardson, MSc, of Edanz Medical Writing for providing medical writing support, which was funded by Eisai Co., Ltd through EMC KK in accordance with Good Publication Practice (GPP3) guidelines (http://www.ismpp.org./gpp3).
Koyama T, Shimizu T, Iwasa S, et al. First‐in‐human phase I study of E7090, a novel selective fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. Cancer Sci. 2020;111:571–579. 10.1111/cas.14265
Some results of this study were previously presented as an abstract at the AACR‐NCI‐EORTC International Conference: Molecular Targets and Cancer Therapeutics; October 26‐30, 2017; Philadelphia, PA, USA.
Clinical Trial Registration: JapicCTI‐142740; NCT02275910.
DATA AVAILABILITY STATEMENT
The data for this study will not be shared in a publicly available repository.
REFERENCES
- 1. Babina IS, Turner NC. Advances and challenges in targeting FGFR signalling in cancer. Nat Rev Cancer. 2017;17:318‐332. [DOI] [PubMed] [Google Scholar]
- 2. Porta R, Borea R, Coelho A, et al. FGR a promising druggable target in cancer: molecular biology and new drugs. Crit Rev Oncol Hematol. 2017;113:256‐267. [DOI] [PubMed] [Google Scholar]
- 3. Carter EP, Fearon AE, Grose RP. Careless talk costs lives: fibroblast growth factor receptor signalling and the consequences of pathway malfunction. Trends Cell Biol. 2015;25:221‐233. [DOI] [PubMed] [Google Scholar]
- 4. Chae YK, Ranganath K, Hammerman PS, et al. Inhibition of the fibroblast growth factor receptor (FGFR) pathway: the current landscape and barriers to clinical application. Oncotarget. 2017;8:16052‐16074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Brooks AN, Kilgour E, Smith PD. Molecular pathways: fibroblast growth factor signaling: a new therapeutic opportunity in cancer. Clin Cancer Res. 2012;18:1855‐1862. [DOI] [PubMed] [Google Scholar]
- 6. Dy GK, Adjei AA. Understanding, recognizing, and managing toxicities of targeted anticancer therapies. CA Cancer J Clin. 2013;63:249‐279. [DOI] [PubMed] [Google Scholar]
- 7. Paik PK, Shen R, Berger MF, et al. A phase 1b open label multicentre study of AZD4547 in patients with advanced squamous cell lung cancers. clinical cancer research. Clin Cancer Res. 2017;23:5366‐5373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Saka H, Kitagawa C, Kogure Y, et al. Safety, tolerability and pharmacokinetics of the fibroblast growth factor receptor inhibitor AZD4547 in Japanese patients with advanced solid tumours: a Phase I study. Invest New Drugs. 2017;35:451‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Nogova L, Sequist LV, Perez Garcia JM, et al. Evaluation of BGJ398, a fibroblast growth factor receptor 1–3 kinase inhibitor, in patients with advanced solid tumors harboring genetic alterations in fibroblast growth factor receptors: results of a global phase I, dose‐escalation and dose‐expansion study. J Clin Oncol. 2017;35:157‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. US Food & Drug Administration . FDA grants accelerated approval to erdafitinib for metastatic urothelial carcinoma. 2019. https://www.fda.gov/drugs/resources-information-approved-drugs/fda-grants-accelerated-approval-erdafitinib-metastatic-urothelial-carcinoma. Accessed June 21, 2019.
- 11. Tabernero J, Bahleda R, Dienstmann R, et al. Phase I dose‐escalation study of JNJ‐42756493, an oral pan‐fibroblast growth factor receptor inhibitor, in patients with advanced solid tumors. J Clin Oncol. 2015;33:3401‐3408. [DOI] [PubMed] [Google Scholar]
- 12. Michael M, Bang YJ, Park YS, et al. A phase 1 study of LY2874455, an oral selective pan‐FGFR inhibitor, in patients with advanced cancer. Target Oncol. 2017;12:463‐474. [DOI] [PubMed] [Google Scholar]
- 13. Papadopoulos KP, El‐Rayes BF, Tolcher AW, et al. A phase 1 study of ARQ 087, an oral pan‐FGFR inhibitor in patients with advanced solid tumours. Br J Cancer. 2017;117:1592‐1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Meric‐Bernstam F, Arkenau H, Tran B, et al. Efficacy of TAS‐120, an irreversible fibroblast growth factor receptor (FGFR) inhibitor, in cholangiocarcinoma patients with FGFR pathway alterations who were previously treated with chemotherapy and other FGFR inhibitors. Ann Oncol. 2018;29(Suppl 5):v100‐v110 (abstract). [Google Scholar]
- 15. Liu PCC, Lehmann BD, Ruggeri B, et al. Activity of the selective FGFR 1, 2 and 3 inhibitor INCB054828 in genetically‐defined models of triple‐negative breast cancer. Cancer Res. 2017;77(Suppl):531 (abstract 531). [Google Scholar]
- 16. Miyano SW, Yamamoto Y, Kodama K, et al. E7090, a novel selective inhibitor of fibroblast growth factor receptors, displays potent antitumor activity and prolongs survival in preclinical models. Mol Cancer Ther. 2016;15:2630‐2639. [DOI] [PubMed] [Google Scholar]
- 17. Wöhrle S, Bonny O, Beluch N, et al. FGF receptors control vitamin D and phosphate homeostasis by mediating renal FGF‐23 signaling and regulating FGF‐23 expression in bone. J Bone Miner Res. 2011;26:24862497. [DOI] [PubMed] [Google Scholar]
- 18. Ji Y, Wang SJ. Modified toxicity probability interval design: a safer and more reliable method than the 3+3 design for practical phase I trials. J Clin Oncol. 2013;31:1785‐1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eisenhauer EA, Therasse P, Bogaerts J, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228‐247. [DOI] [PubMed] [Google Scholar]
- 20. Hierro C, Rodon J, Tabernero J. Fibroblast growth factor (FGF) receptor/FGF inhibitors: novel targets and strategies for optimization of response of solid tumors. Semin Oncol. 2015;42:801‐819. [DOI] [PubMed] [Google Scholar]
- 21. Nishina T, Takahashi S, Iwasawa R, et al. Safety, pharmacokinetic, and pharmacodynamics of erdafitinib, a pan‐fibroblast growth factor receptor (FGFR) tyrosine kinase inhibitor, in patients with advanced or refractory solid tumors. Invest New Drugs. 2018;36:424‐434. [DOI] [PubMed] [Google Scholar]
- 22. The ADHR Consortium . Autosomal dominant hypophosphataemic rickets is associated with mutations in FGF23. Nat Genet. 2000;26:345‐348. [DOI] [PubMed] [Google Scholar]
- 23. Shimada T, Mizutani S, Muto T, et al. Cloning and characterization of FGF23 as a causative factor of tumor‐induced osteomalacia. Proc Natl Acad Sci USA. 2001;98:6500‐6505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. White KE, Carn G, Lorenz‐Depiereux B, et al. Autosomal‐dominant hypophosphatemic rickets (ADHR) mutations stabilize FGF‐23. Kidney Int. 2001;60:2079‐2086. [DOI] [PubMed] [Google Scholar]
- 25. Van Cutsem E, Bang YJ, Mansoor W, et al. A randomized, open‐label study of the efficacy and safety of AZD4547 monotherapy versus paclitaxel for the treatment of advanced gastric adenocarcinoma with FGFR2 polysomy or gene amplification. Ann Oncol. 2017;28:1316‐1324. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data for this study will not be shared in a publicly available repository.