

Abstract
To date, there is no clear agreement regarding which is the best method to detect a connective tissue disease (CTD) during the initial diagnosis of interstitial lung diseases (ILD). The aim of our study was to explore the impact of a systematic diagnostic strategy to detect CTD-associated ILD (CTD-ILD) in clinical practice, and to clarify the significance of interstitial pneumonia with autoimmune features (IPAF) diagnosis in ILD patients.
Consecutive patients evaluated in an ILD Diagnostic Program were divided in 3 groups: IPAF, CTD-ILD, and other ILD forms. Clinical characteristics, exhaustive serologic testing, high resolution computed tomography (HRCT) images, lung biopsy specimens, and follow-up were prospectively collected and analyzed.
Among 139 patients with ILD, CTD was present in 21 (15.1%), 24 (17.3%) fulfilled IPAF criteria, and 94 (67.6%) were classified as other ILD forms. Specific systemic autoimmune symptoms such as Raynaud phenomenon (19%), inflammatory arthropathy (66.7%), and skin manifestations (38.1%) were more frequent in CTD-ILD patients than in the other groups (all P < .001). Among autoantibodies, antinuclear antibody was the most frequently found in IPAF (42%), and CTD-ILD (40%) (P = .04). Nonspecific interstitial pneumonia, detected by HRCT scan, was the most frequently seen pattern in patients with IPAF (63.5%), or CTD-ILD (57.1%) (P < .001). In multivariate analysis, a suggestive radiological pattern by HRCT scan (odds ratio [OR] 15.1, 95% confidence interval [CI] 4.7–48.3, P < .001) was the strongest independent predictor of CTD-ILD or IPAF, followed by the presence of clinical features (OR 14.6, 95% CI 4.3–49.5, P < .001), and serological features (OR 12.4, 95% CI 3.5–44.0, P < .001).
This systematic diagnostic strategy was useful in discriminating an underlying CTD in patients with ILD. The defined criteria for IPAF are fulfilled by a considerable proportion of patients referred for ILD.
Keywords: autoimmune disease, idiopathic interstitial pneumonias, interstitial pneumonia, nonspecific interstitial pneumonia, undifferentiated connective tissue disease
1. Introduction
Connective tissue diseases (CTD) encompass a heterogeneous group of autoimmune disorders including systemic lupus erythematosus, systemic sclerosis, idiopathic inflammatory myopathies, Sjögren syndrome, and rheumatoid arthritis. Interstitial lung disease (ILD) is a well-recognized and severe manifestation of many CTD that impacts on the prognosis and management in many of these patients.[1–4] However, there is no clear agreement regarding which is the best method to detect a CTD during the initial diagnosis of ILDs. Besides, despite recent technological advances, the precise prevalence rates of ILD among the various CTDs are not known and are influenced by the methods of detection.[5–9] Although serological testing is recommended by international guidelines, [10,11] it is not clear which antibodies must be included in the initial evaluation of an ILD patient in order to detect an underlying CTD accurately. A rheumatoid factor (RF), anti-cyclic citrullinated peptide (CCP), and antinuclear antibody (ANA) titer and pattern seemed to be generally accepted until now.[1] However, it has not been established whether adding other autoantibodies to the initial serologic tests could improve the diagnosis of CTD in patients with ILD. This could be relevant as a large number of patients who would have previously been categorized as having idiopathic interstitial pneumonia (IIP) by the recently revised IIP criteria are now likely to have an underlying CTD. Therefore, identifying occult or incomplete forms of CTD in patients with presumed IIP can be challenging.[1,12]
Moreover, it is not uncommon for ILDs to be the initial manifestation of an underlying CTD, even preceding extrapulmonary symptoms by several years.[13] This “lung-dominant” presentation could complicate the diagnosis, as many patients present a constellation of clinical, serologic, or morphological findings that belong to the spectrum of CTDs, but do not fulfill the classification criteria for CTD. [14–20] Such patients are now considered to have a syndrome recently named as “interstitial pneumonia with autoimmune features” (IPAF).[21] Although IPAF has been recently defined by a recent International Consensus Task Force made up of experts from the European Respiratory Society (ERS) and the American Thoracic Society (ATS), the proposed criteria for this entity has not been tested or validated so far.
Thus, the present study was conducted to explore the impact of a systematic diagnostic strategy to detect CTD-ILD during an ILD diagnostic program that included an extensive assessment of clinical signs, symptoms, and autoantibodies. Furthermore, we attempted to clarify the incidence and the clinical characteristics of IPAF at the initial assessment of ILD diagnosis.
2. Methods
2.1. Study design and population
This is a clinical descriptive study of a total of 146 patients evaluated at an ILD Diagnostic Program of a tertiary referral center. Data from all patients prospectively and consecutively included in the ILD Diagnostic Program from January 2013 to January 2014 was assessed. As part of their initial assessment, a complete and systematic anamnesis and physical examination, including a broad range of autoimmune disease symptoms and signs, and a wide serologic testing for CTD, was obtained from all subjects at the time of initial diagnosis and periodically during 1-year follow-up. The study was approved by the Ethics Committee of the Hospital Clinic of Barcelona, and informed consent from all participants was obtained.
2.2. Autoimmune clinical features
All patients were assessed for extrathoracic specific features suggestive of an underlying CTD, including Raynaud phenomenon, arthralgia/arthritis, morning stiffness, skin manifestations (cutaneous sclerosis, distal digital fissuring or tip ulceration, telangiectasia, Gottron sign, heliotrope rash), oral ulceration, and digital edema. Other nonspecific features, but commonly related to CTD, such as nonandrogenic alopecia, dry eyes or dry mouth, photosensitivity, unintentional weight loss, dysphagia, recurrent unexplained fever, gastroesophageal reflux, or proximal muscle weakness, were also explored.
2.3. Autoantibody tests
All patients underwent serologic tests for autoantibodies at the time of diagnosis. ANA and antineutrophil cytoplasmic antibodies (ANCA) were tested using indirect immunofluorescence (IIF). Anti-Ro (SS-A), anti-La (SS-B), anti-ribonucleoprotein, anti-Smith, anti-double-stranded DNA (dsDNA), anti-cyclic CCP, anti-Scl-70, anti-centromere, anti-Ro52, and anti-Jo1 antibodies, were tested using enzyme-linked immunosorbent assay (ELISA). RF was measured using latex agglutination. Specific ANCA (MPO and PR3) was also determined by ELISA. Moreover, anti-dsDNA (Crithidia luciliae) antibodies were also tested by IIF, anti-Mi2, anti-Ku, anti-PM-Scl (75 and 100 subunits), anti-PL7, anti-PL12, anti-OJ, anti-EJ, and anti-SRP antibodies were measured using Dot-Blot method. The serologic tests were considered positive if the circulating autoantibody levels were above the manufacturer reference value, with the exception of ANA and RF. ANA was considered positive if the titer were 1:320 or higher with diffuse, speckled, homogeneous patterns, or any titer with nucleolar or centromere pattern. RF was considered positive if the titer was ≥2x upper limit of normal.
2.4. High-resolution CT scan evaluation
A high-resolution CT (HRCT) scan was performed at the time of initial evaluation in all patients. HRCT examination of the lungs was performed on 1.0-or-1.5-mm-thick sections to evaluate radiographic abnormalities. Chest radiologists experienced in the interpretation of diffuse lung disease reviewed all HRCT scan images. HRCT diagnosis was made by the definitions accepted by international guidelines.[10,11,22] If there was disagreement in the assessment of the findings, consensus was reached via discussion between the investigators.
2.5. Histopathology
Lung biopsy was performed in selected patients after a multidisciplinary discussion. All lung biopsy specimens were evaluated by an expert pulmonary pathologist with experience and advanced training in the evaluation of ILDs, and were classified using the histopathologic patterns described in the international guidelines.[10,11,22] All specimens were evaluated prospectively as an integral part of clinical care.
2.6. Diagnostic criteria and clinical follow-up
Rheumatic and autoimmune diseases were defined according to the updated classification criteria.[23–28] Patients with complete or incomplete forms of antisynthetase syndrome (ASSD), were assessed using the American and European NEtwork of Antisynthetase Syndrome international collaborative group definition.[29]
Proposed classification criteria for IPAF were taken into consideration after the “ERS/ATS research statement of IPAF” was published.[21] Patients were considered to have a diagnosis of IPAF if they met all of the a priori requirements, and had at least 1 feature from at least 2 of the proposed clinical, serologic or morphologic domain. The proposed clinical and serological features for the diagnosis criteria of IPAF were considered to be specific features suggestive of CTD.[21] The radiologic patterns considered to be suggestive of CTD were nonspecific interstitial pneumonia (NSIP), organizing pneumonia (OP), NSIP with OP, and lymphoid interstitial pneumonia (LIP), as defined by ERS/ATS research statement of IPAF.[21] Diagnosis of other forms of ILD was based on history, clinical assessment, HRCT scan, and histologic examination when performed, in accordance with the ATS/ERS Consensus Classification.[10,11,22] Diagnosis was confirmed by consensus of a multidisciplinary panel of ILD experts.
Improvement/deterioration of ILD was defined as a change of ≥10% in the forced vital capacity (FVC) and/or ≥15% in the diffusing capacity of carbon monoxide.[10,30] Survival status was evaluated at 1-year follow-up.
2.7. Multidisciplinary diagnosis
Diagnosis of ILDs was done through multidisciplinary discussion based on clinical characteristics along with HRCT and lung biopsy patterns if appropriate. Multidisciplinary discussions were conducted between pulmonologists, radiologists, and pathologists experienced in the diagnosis of ILD. Bronchoscopists, thoracic surgeons, and autoimmune specialists were also included.
2.8. Statistical analysis
Analyses were performed using the Predictive Analytics Software Statistics statistical software (version 18.0, SPSS Japan Inc, Tokyo, Japan). Continuous variables were presented as mean ± standard deviation if normally distributed, while non-normally distributed continuous variables were summarized as median and interquartile range. Kolmogorov–Smirnov testing identified states of non-normality. The Chi-squared test (or Fisher exact test) was used to compare categorical data. Continuous variables were compared using the analysis of variance with post hoc Bonferroni correction. The survival probability of etiological groups was estimated using the Kaplan–Meyer method and compared with Log-rank test. Survival was evaluated from the date of inclusion, which corresponds to the date of the initial diagnosis, until the end of the follow-up period.
Multivariate logistic regression was performed to assess the predictive value of different variables for a CTD-ILD or IPAF diagnosis. Univariate analysis was performed first, and all the variables that exhibited a P < .05 or those clinically relevant were entered in the multivariate model. Model fit was assessed using receiver operating characteristic curves analyses. Cox and Snell R2 and NagelkerkeR2 were used to identify the amount of variation in the dependent variable explained by the model. Also, we set out to construct a decision tree analysis, a nonparametric statistical method used to assess the incidence of CTD-ILD or IPAF, and to screen for the most important relevant predictive factors. The decision tree model was developed using those significant or clinically relevant variables previously reported. A P-value < .05 was established as the level of statistical significance for all tests.
3. Results
3.1. Multidisciplinary evaluation in the ILD program and prevalence of CTD or IPAF
Among 146 consecutive patients evaluated in the ILD Program (Fig. 1), 7 patients were excluded because another lung disease, different from ILD, was finally diagnosed. Among the 139 patients assessed, 10 (7.2%) initially fulfilled the criteria of a known CTD.[23–28] Autoimmune disease specialists evaluated 72 (51.8%) patients because they presented any symptoms or any antibody suggestive of CTD. Of these, 11 (7.9%) patients fulfilled the CTD criteria, and 32 (23%) fulfilled IPAF criteria, and were presented again at the multidisciplinary committee. A total of 24 (17.3%) patients were finally classified as IPAF. From the group of IPAF patients, 16 fulfilled at least 1 serologic and 1 morphologic feature, 3 presented at least 1 serologic and 1 clinical feature, 2 presented at least 1 clinical and 1 morphologic feature, and 3 patients presented at least 1 feature from each domain. A total of 94 (67.6%) patients were finally diagnosed with other forms of ILD. Among the group of patients presenting other forms of ILD, 15 (10.8%) were diagnosed with idiopathic pulmonary fibrosis (IPF), 6 (4.3%) with possible IPF, 13 (9.4%) with idiopathic NSIP, 13 (9.4%) with hypersensitivity pneumonitis, 9 (6.5%) with sarcoidosis, 8 (5.8%) with OP, 3 (2.2%) with Langerhans cell's granulomatosis, and 27 (19.4%) with other forms of ILD.
Figure 1.
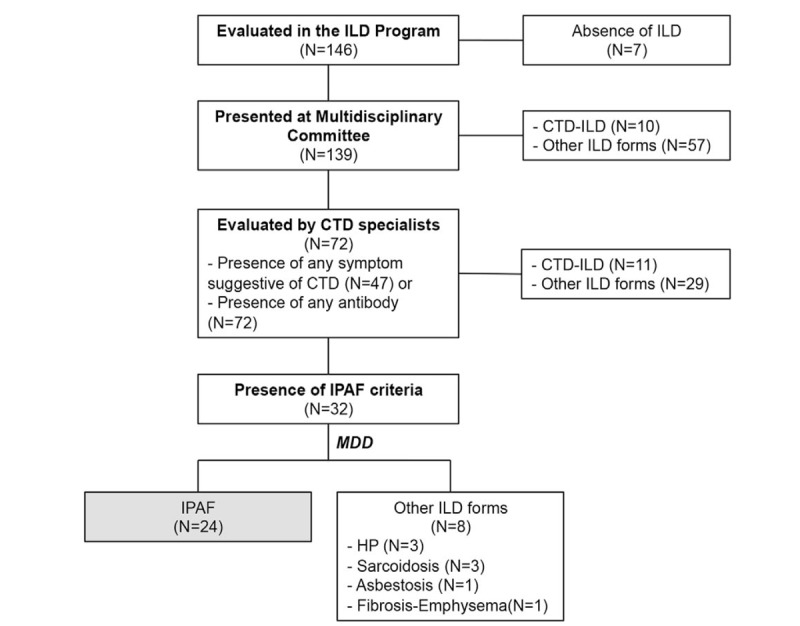
Flow diagram for diagnostic protocol used for the screening of connective tissue disease related interstitial lung disease. CTD = connective tissue disease, CTD-ILD = connective tissue disease-associated interstitial lung disease; HP = hypersensitivity pneumonia, ILD = interstitial lung disease, IPAF = interstitial pneumonia with autoimmune features, MDD = multidisciplinary committee.
3.2. Clinical characteristics, pulmonary function tests, and bronchoalveolar lavage
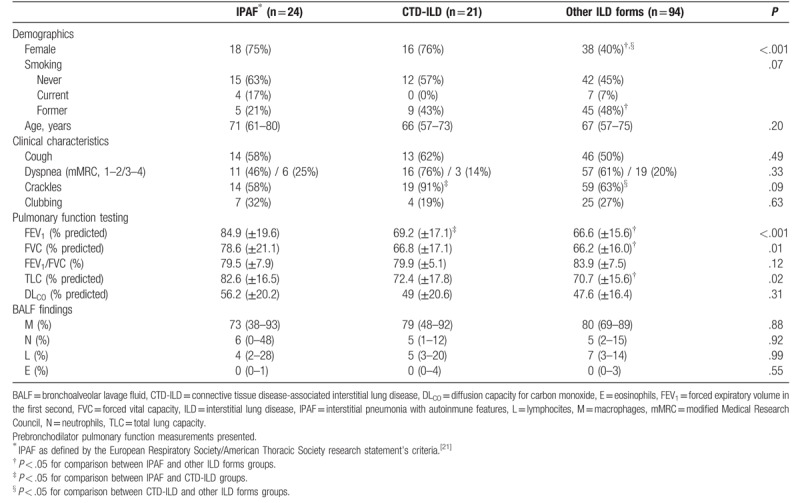
Baseline characteristics are summarised in Table 1. Patients were distributed in 3 groups according to the etiology of ILD: IPAF (n = 24), CTD-ILD (n = 21), and other ILD forms (n = 94). CTD-ILD patients were divided as follows: Sjögren syndrome (n = 6), systemic sclerosis (n = 5), rheumatoid arthritis (n = 6), dermatomyositis (n = 3, including 2 patients with ASSD), and systemic lupus erythematosus (n = 1). Patients with IPAF and CTD-ILD were more likely to be females than those with other forms of ILD (75% and 76% versus 40%, respectively; P < .001). Interestingly, there were significant differences in the results of pulmonary function tests, with a higher FVC (FVC % predicted) (P = .001), a higher forced expiratory volume in the first second (FEV1% predicted) (P = .008), and a higher total lung capacity (% predicted) (P = .02) in patients with IPAF.
Table 1.
Baseline characteristics according to etiological groups.
3.3. Systemic autoimmune symptoms and laboratory findings
Results are shown in Tables 2 and 3. Specific systemic autoimmune symptoms such as Raynaud phenomenon (19%), inflammatory arthropathy (66.7%), and skin manifestations (38.1%) were more frequent in CTD-ILD patients than in the other groups (all P < .001). The most frequent non-specific systemic autoimmune symptoms in CTD-ILD patients were proximal muscle weakness (19%), and recurrent unexplained fever (14.3%). By contrast, IPAF patients presented more frequently with sicca symptoms (dry eyes, 37.5%; dry mouth, 50%) and gastroesophageal reflux (71%).
Table 2.
Distribution of autoimmune symptoms according to etiological groups.
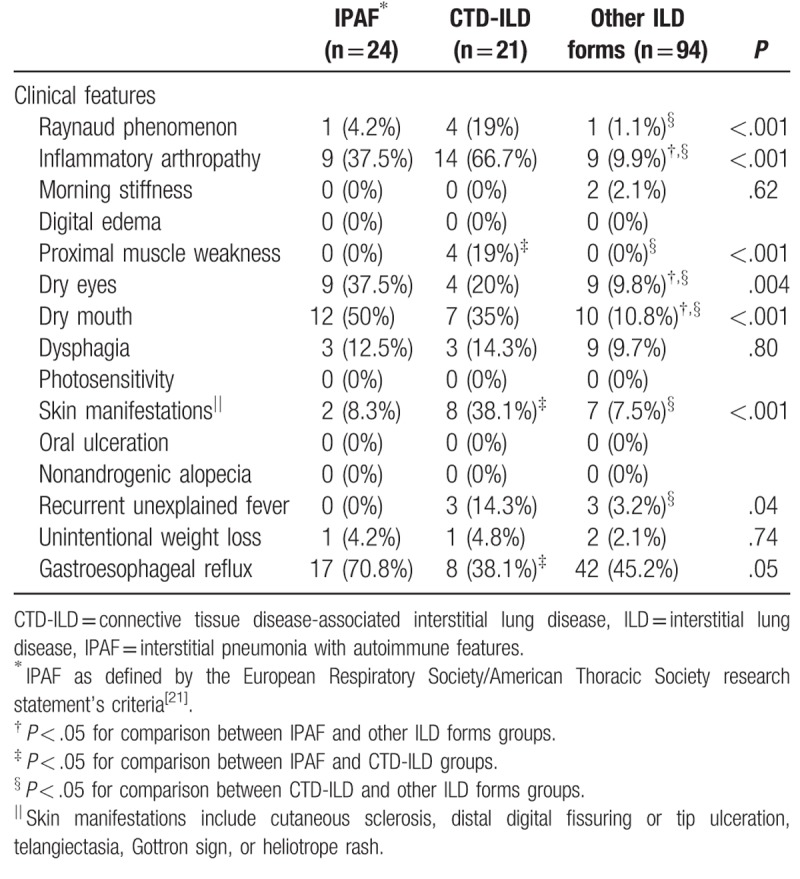
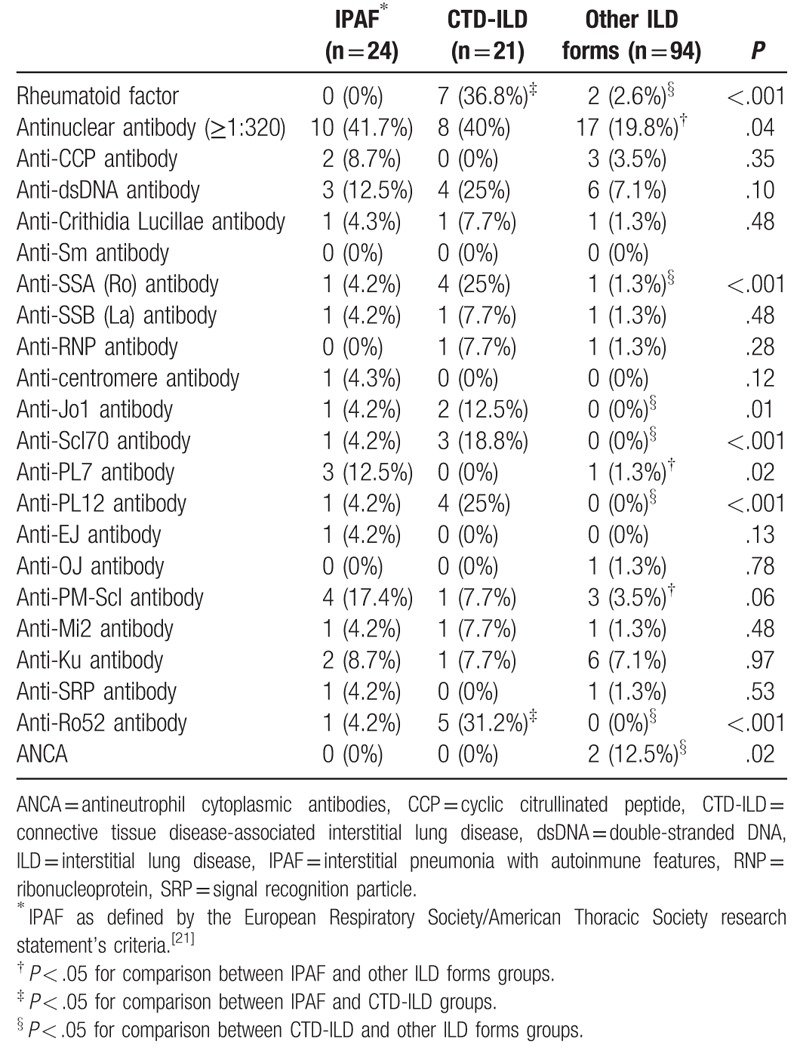
Table 3.
Distribution of autoimmune laboratory findings according to etiological groups.
The proportion of patients with at least 1 clinical autoimmune feature was higher in IPAF and CTD-ILD than that observed in other ILD forms (100% vs 15%, P = .001). Moreover, 52% of patients with CTD-ILD presented at least 2 clinical autoimmune features, a higher rate than that observed in patients with IPAF and other forms of ILD (44% and 15%, respectively, P = .001).
All patients with IPAF and CTD-ILD had at least 1 autoimmune antibody. Among autoantibodies, a positive ANA was the most frequently found in IPAF (42%), and CTD-ILD (40%). The incidence of positive ANA was significantly higher in IPAF and CTD-ILD patients than in other ILD forms (P = .04). In addition, the group of IPAF and CTD-ILD patients were more likely to have a relative frequency of higher ANA titres (1:320) (P < .05). The positive rates of RF (36.8%, P = .001), anti-Ro (25%, P = .001), anti-Jo1 antibody (12.5%, P = .01), anti-Scl70 (18.8%, P < .001), and anti-Ro52 (31.2%, P < .001) were significantly higher in CTD-ILD patients than in the other groups, as expected. Anti-PL7 antibody was positive in 12.5% of IPAF patients, with no ex novo manifestations of ASSD during the follow up.
3.4. Morphologic findings
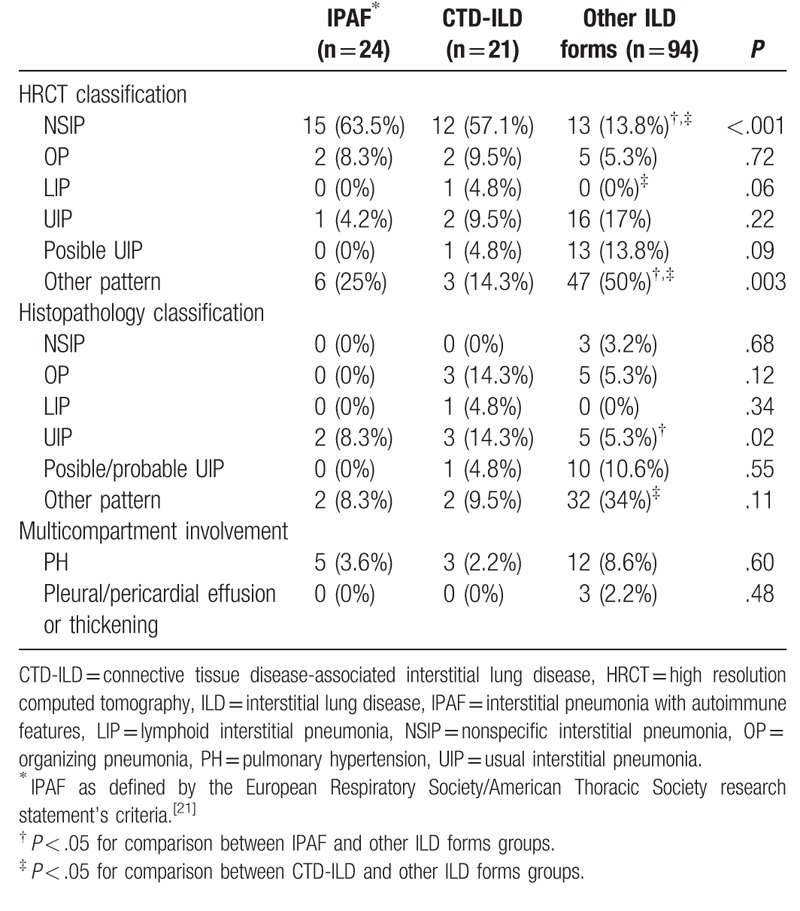
There were significant differences between the 3 groups regarding HRCT scan patterns (P = .001) (Table 4). A HRCT scan pattern suggestive of or consistent with NSIP was the most frequently seen in patients with IPAF (63.5%), or CTD-ILD (57.1%). Nevertheless, it is noteworthy that other patterns different from NSIP were present in patients with IPAF and CTD-ILD: usual interstitial pneumonia (UIP) (4.2% and 9.5%, respectively); and other patterns (25% and 14.3%, respectively). Significant differences were also seen between patients that required a pulmonary biopsy with a histopathologic pattern of UIP. This pattern was present in 2 (8.3%) IPAF patients, and 3 (14.3%) CTD-ILD patients (P = .02).
Table 4.
Morphological features according to etiological groups.
3.5. Predictive factors of CTD-ILD or IPAF diagnosis and decision tree analysis
Results of the multivariate analyses independent predictors of CTD-ILD or IPAF diagnosis showed that the strongest predictor was a suggestive radiological pattern by HRCT scan (odds ratio [OR] 3.32, 95% confidence interval [CI] 1.24–9.00; P < .001), followed by the presence of specific clinical features (OR 3.32, 95% CI 1.24–9.00; P < .001), and the presence of specific serological features (OR 12.37, 95% CI 3.46–44; P < .001) (Fig. 2). The regression model also had a very high Nagelkerke R2 of 0.66, and correctly classified 87.1% of patients. The area under the curve was 0.65 (95% CI 0.55–0.76; P = .004). This model had a high sensitivity (81.4%, 95% CI 67.4–90.3), high specificity (89.6%, 95% CI 81.9–94.2), with a positive predictive value (PPV) of 77.8% (95% CI 63.7–87.5), and a negative predictive value of 91.5% (95% CI 84.1–95.6), confirming the usefulness of excluding individuals with ILD and features suggestive of a CTD.
Figure 2.
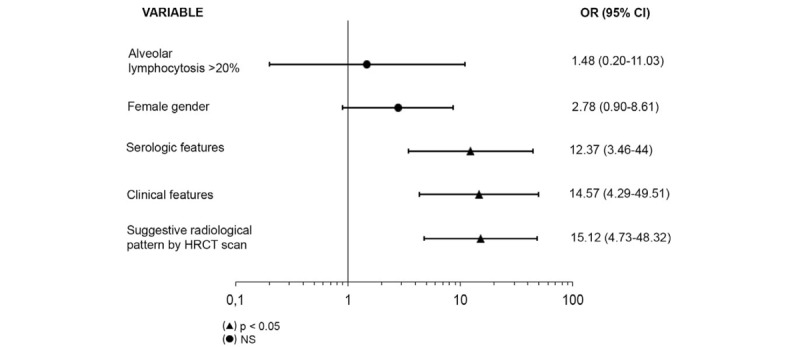
Multivariate analysis for factors associated with the diagnosis of connective tissue disease related interstitial pneumonia or interstitial pneumonia with autoimmune features. CI = confidence interval, HCRT = high resolution computed tomography, NS = not significant, OR = odds ratio.
The decision tree analysis showed that the most important predictive factor for CTD-ILD or IPAF diagnosis was a suggestive radiological pattern by HRCT scan, followed by the presence of specific clinical features and specific serologic features (Fig. 3). However, to be considered as having a CTD-ILD or IPAF, a patient with any other radiological pattern than NSIP, OP, NSIP with OP, or LIP, would need to have at least 1 clinical feature and 1 serologic feature.
Figure 3.
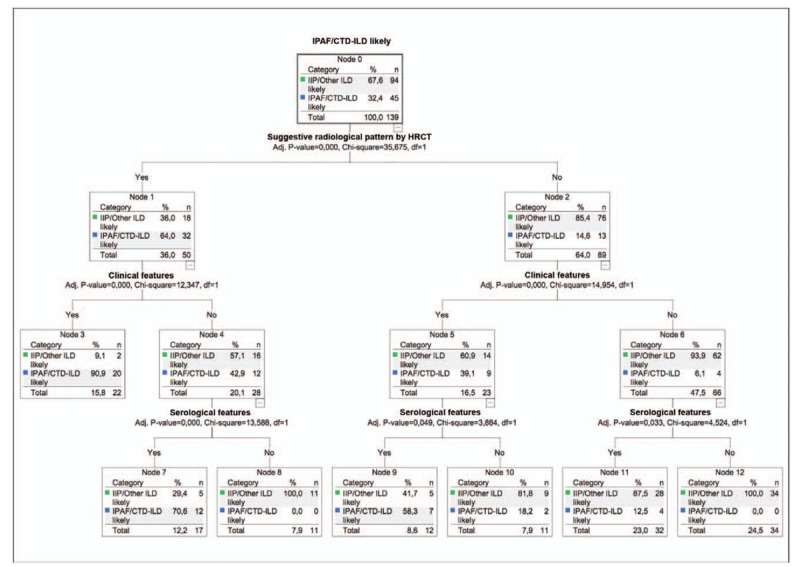
The assessment of factors associated with connective tissue disease related interstitial pneumonia or interstitial pneumonia with autoimmune features using decision tree analysis. CTD = connective tissue disease, CTD-ILD = connective tissue disease-associated interstitial lung disease, ILD = interstitial lung disease, IPAF = interstitial pneumonia with autoimmune features.
3.6. Clinical follow-up
As expected, the etiological groups differed in terms of the treatments received (Table 5). The CTD-ILD group had a significantly higher percentage of patients treated with mycophenolate mofetil (23.8%, P = .002) and hydroxychloroquine (9.5%, P = .02), compared with other patients. Conversely, no significant difference was observed between groups in terms of treatment with prednisone.
Table 5.
Treatment and outcomes according to etiological groups.
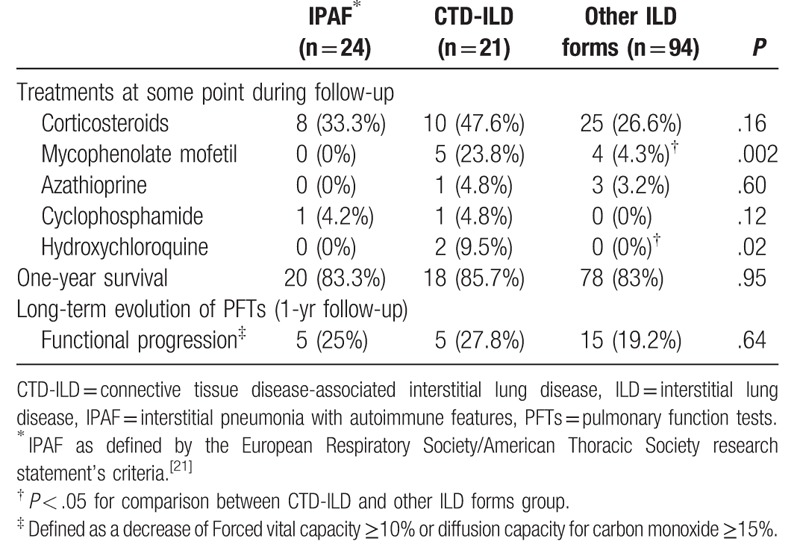
At the end of the study, side-by-side comparison showed that survival was similar across the 3 groups. Long-term functional follow-up (1 year) showed that, according to the etiological groups, functional progression was similar across the groups.
4. Discussion
To the best of our knowledge, this is the first prospective study to assess the impact of a systematic diagnostic strategy to detect CTD in patients presenting ILD. We have demonstrated that this systematic approach is useful, and also supports the notion that consideration for a classifiable CTD should be warranted in every patient with ILD. Indeed, 15.1% of patients were confirmed to have a new CTD-ILD diagnosis, which is similar to the percentage reported by Mittoo and colleagues.[13]
From the beginning of the study, as there was a lack of consensus concerning nomenclature and classification, we prospectively collected both, specific and nonspecific features. However, we agreed with other researchers that the presence of some non-specific serologic or clinical findings alone might be insufficient for a defined CTD diagnosis.[1,3,12,31] Thus, in order to properly classify patients with IPAF, we used only the specific criteria proposed in the ERS/ATS statement. We found that we were able to detect a lower percentage of patients with autoimmune features compared with other series using broader criteria for undifferentiated CTD (UCTD).[18,31,32]
In our study, the presence of specific clinical and serologic features in patients with ILD was considered significant independent prognostic factors of CTD-ILD or IPAF on multivariate analysis. More than 60% of UCTD patients in Kinder's study presented Raynaud phenomenon and arthralgia criteria.[18] Kondoh et al showed 13% of Raynaud phenomenon and 21% of arthralgia in UCTD patients.[31] Oldham and colleagues described that the most common clinical findings were Raynaud's phenomenon (27.8%) and inflammatory arthritis (17.4%) in their IPAF cohort.[33] Our results showed that the proportion of Raynaud phenomenon in IPAF is even lower (4.2%), and inflammatory arthropathy was present in 37.5% of patients with IPAF. In our cohort, we also found significant differences between some other specific and nonspecific features. Overall, we deduce that variability in population among studies may be a possible explanation for these differences. However, as no gold standard exists against which to validate IPAF criteria, we questioned whether using classification criteria for IPAF in patients with ILD leads to the exclusion of some patients who might be diagnosed as having a CTD.[34] Therefore, we suggest that non-specific features should not be excluded from the routine clinical practice.
On the other hand, we also showed that the detection of some ASSD antibodies, such as anti-PL7 or anti-PL12, in IPAF patients during the follow up may lead to patients’ misclassification. Although these antibodies are markers of ASSD, complete forms of the entity are characterized by the occurrence of myositis, ILD and arthritis, and there is a lack of well-established clinicoserological classification criteria.[29,35,36] In our study, at the end of the 1-year follow-up these patients were still classified as IPAF, but it has been suggested that this is generally a transient condition that may progress into a well-defined ASSD during a longer period. These considerations, together with the possible presence of minor features of ASSD, suggest that ASSD antibodies and an exhaustive multidisciplinary evaluation and follow-up should be carefully considered in all patients presenting with ILD. In accordance to this, our results support the need of newly established classification criteria for ASSD based on differential weights for various clinical, pathological, and serological features, as well as validated criteria for IPAF.
According to the criteria proposed by Fischer et al, we have also found that a patient without morphologic autoimmune features would need to have at least 1 feature from the other 2 domains (clinical or serologic features) to be considered more likely to have an IPAF or CTD-ILD.[21] Our study showed that NSIP pattern in HRCT scan was the most frequently found in CTD-ILD or IPAF, as suggested by other authors.[37] However, there were a considerable percentage of patients with other patterns, including UIP.[38,39] Consequently, these results reinforce the importance of screening IPAF or CTD-ILD in all patients with suspected IIPs, especially when IPF is suspected.
Previous studies suggested that UCTD or IPAF was not associated with improved survival.[3,40,41] However, a further study with a larger cohort demonstrated that the prognosis of UCTD in patients with UIP was decreased.[42] Similarly, Churg et al and Kim et al noted that the presence of UIP pattern is associated with worse survival when concomitant honeycombing is present in IPAF patients.[43,44] The present study showed that a diagnosis of IPAF had no influence on 1-year survival compared to other ILD patients. Besides, there were no significant differences in functional progression between the groups, perhaps due to the predominance of NSIP pattern and the effectiveness of immunosuppressive therapy in this group. Nonetheless, mainly regarding the relatively short period of follow-up, we consider that multicenter registers over a longer period of time are needed to study a possible evolution that may complete the clinical picture and demonstrate differences between IPAF and other ILD patients.
Our study has several strengths. In the first place, all the variables included in the analysis were collected prospectively. Second, all patients were assessed by a multidisciplinary panel of ILD experts and autoimmune specialists. Nevertheless, our study contains some limitations. Firstly, the fairly short duration of follow-up might increase the risk of missing patients with late development of CTD. Second, this study included a small population of a single center. Further studies are therefore needed to ascertain whether this result applies equally to other ethnic cohorts. Third, lung biopsy is rarely performed in the context of CTD, which might make it difficult to confirm suggestive patterns detected by the HRCT scan, especially the diagnosis of NSIP. This issue may detract from the predictive value of surgical lung biopsy patterns in the proposed algorithm, so we believe that its value needs to be further explored in future studies.
In conclusion, this study in a prospective cohort shows that the present systematic strategy to identify patients with a clear autoimmune flavor might well be useful when assessing ILD patients, and contributes to the evidence-based data. Moreover, this diagnostic approach seems to be both sensitive and specific for CTD-ILD or IPAF diagnosis. Because there are several implications of an early diagnosis of CTD-ILD, this study reminds us that a comprehensive and multidisciplinary approach, including specialists in autoimmune diseases, is essential to explore all potential aetiologies in an ILD of recent diagnosis.
Acknowledgments
The authors wish to dedicate this manuscript to the memory and the contribution of their co-author and friend Antoni Xaubet, MD, PhD, for his outstanding contribution to this study. They also thank the respiratory therapy and nursing staff, and the physicians attending the respiratory department, for their cooperation in this study.
Author contributions
Conceptualization: Fernanda Hernandez-Gonzalez, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Data curation: Fernanda Hernandez-Gonzalez, Sandra Cuerpo.
Formal analysis: Fernanda Hernandez-Gonzalez, Sergio Prieto-Gonzalez, Jacobo Sellares.
Funding acquisition: Jacobo Sellares.
Investigation: Fernanda Hernandez-Gonzalez, Pilar Brito-Zeron, Marcelo Sanchez, Jose Ramirez, Carmen M. Lucena, Marina Paradela, Ignacio Grafia, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Methodology: Fernanda Hernandez-Gonzalez, Sandra Cuerpo, Marcelo Sanchez, Jose Ramirez, Carlos Agustí, Carmen M. Lucena, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Project administration: Fernanda Hernandez-Gonzalez, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Resources: Fernanda Hernandez-Gonzalez, Carlos Agustí, Carmen M. Lucena, Marina Paradela, Ignacio Grafia, Gerard Espinosa, Sergio Prieto-Gonzalez.
Supervision: Pilar Brito-Zeron, Marcelo Sanchez, Jose Ramirez, Carlos Agustí, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Validation: Fernanda Hernandez-Gonzalez, Pilar Brito-Zeron.
Visualization: Fernanda Hernandez-Gonzalez, Sandra Cuerpo, Marcelo Sanchez, Jose Ramirez, Carlos Agustí, Carmen M. Lucena, Marina Paradela, Ignacio Grafia, Gerard Espinosa, Sergio Prieto-Gonzalez.
Writing – original draft: Fernanda Hernandez-Gonzalez.
Writing – review and editing: Pilar Brito-Zeron, Gerard Espinosa, Sergio Prieto-Gonzalez, Jacobo Sellares.
Footnotes
Abbreviations: ANA = antinuclear antibody, ANCA = antineutrophil cytoplasmic antibodies, ASSD = antisynthetase syndrome, CCP = anti-cyclic citrullinated peptide, CTD = connective tissue disease, FVC = forced vital capacity, HRCT = high resolution computed tomography, IIP = idiopathic interstitial pneumonia, ILD = interstitial lung disease, IPAF = interstitial pneumonia with autoimmune features, IPF = idiopathic pulmonary fibrosis, LIP = lymphoid interstitial pneumonia, NSIP = nonspecific interstitial pneumonia, OP = organizing pneumonia, PPV = positive predictive value, RF = rheumatoid factor, SLB = surgical lung biopsy, UCTD = undifferentiated connective tissue disease, UIP = usual interstitial pneumonia.
How to cite this article: Hernandez-Gonzalez F, Prieto-González S, Brito-Zeron P, Cuerpo S, Sanchez M, Ramirez J, Agustí C, Lucena CM, Paradela M, Grafia I, Espinosa G, Sellares J. Impact of a systematic evaluation of connective tissue disease on diagnosis approach in patients with interstitial lung diseases. Medicine. 2020;99:4(e18589).
This work has been financed with the grant SLT008/18/00176 and the support of the Department of Health of Generalitat de Catalunya, in the call for grants 2019–2021, under a competitive regime, for the financing of different programs and instrumental actions included in the Strategic Research and Innovation Plan in Health 2016–2020. It has also been financed by SEPAR, SOCAP, FUCAP and with the PhD4MD Programme of the Institute for Research in Biomedicine (IRB) Barcelona, Hospital Clinic of Barcelona and the Institut d’Investigacions Biomèdiques August Pi i Sunyer (IDIBAPS).
The authors have no conflicts of interest to disclose.
References
- [1].Fischer A, Brown KK. Interstitial lung disease in undifferentiated forms of connective tissue disease. Arthritis Care Res (Hoboken) 2015;67:4–11. [DOI] [PubMed] [Google Scholar]
- [2].Danieli MG, Fraticelli P, Salvi A, et al. Undifferentiated connective tissue disease: natural history and evolution into definite CTD assessed in 84 patients initially diagnosed as early UCTD. Clin Rheumatol 1998;17:195–201. [DOI] [PubMed] [Google Scholar]
- [3].Corte TJ, Copley SJ, Desai SR, et al. Significance of connective tissue disease features in idiopathic interstitial pneumonia. Eur Respir J 2012;39:661–8. [DOI] [PubMed] [Google Scholar]
- [4].Flaherty KR, Mumford JA, Murray S, et al. Prognostic implications of physiologic and radiographic changes in idiopathic interstitial pneumonia. Am J Respir Crit Care Med 2003;168:543–8. [DOI] [PubMed] [Google Scholar]
- [5].Fischer A, Lee JS, Cottin V. Interstitial lung disease evaluation: detecting connective tissue disease. Respiration 2015;90:177–84. [DOI] [PubMed] [Google Scholar]
- [6].Mosca M, Tavoni A, Neri R, et al. Undifferentiated connective tissue diseases: the clinical and serological profiles of 91 patients followed for at least 1 year. Lupus 1998;7:95–100. [DOI] [PubMed] [Google Scholar]
- [7].Luppi F, Wells AU. Interstitial pneumonitis with autoimmune features (IPAF): a work in progress. Eur Respir J 2016;47:1622–4. [DOI] [PubMed] [Google Scholar]
- [8].Jee AS, Adelstein S, Bleasel J, et al. Role of autoantibodies in the diagnosis of connective-tissue disease ILD (CTD-ILD) and interstitial pneumonia with autoimmune features (IPAF). J Clin Med 2017;6:E51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].de Lauretis A, Veeraraghavan S, Renzoni E. Review series: aspects of interstitial lung disease: connective tissue disease-associated interstitial lung disease: how does it differ from IPF? How should the clinical approach differ? Chron Respir Dis 2011;8:53–82. [DOI] [PubMed] [Google Scholar]
- [10].Travis WD, Costabel U, Hansell DM, et al. An official American Thoracic Society/European Respiratory Society statement: update of the international multidisciplinary classification of the idiopathic interstitial pneumonias. Am J Respir Crit Care Med 2013;188:733–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Raghu G, Collard HR, Egan JJ, et al. An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med 2011;183:788–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Fischer A, Chartrand S. Assessment and management of connective tissue disease-associated interstitial lung disease. Sarcoidosis Vasc Diffuse Lung Dis 2015;32:2–1. [PubMed] [Google Scholar]
- [13].Mittoo S, Gelber AC, Christopher-Stine L, et al. Ascertainment of collagen vascular disease in patients presenting with interstitial lung disease. Respir Med 2009;103:1152–8. [DOI] [PubMed] [Google Scholar]
- [14].Fischer A, du Bois R. Interstitial lung disease in connective tissue disorders. Lancet 2012;380:689–98. [DOI] [PubMed] [Google Scholar]
- [15].Castelino FV, Varga J. Interstitial lung disease in connective tissue diseases: evolving concepts of pathogenesis and management. Arthritis Res Ther 2010;12:213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Castelino FV, Goldberg H, Dellaripa PF. The impact of rheumatological evaluation in the management of patients with interstitial lung disease. Rheumatology (Oxford) 2011;50:489–93. [DOI] [PubMed] [Google Scholar]
- [17].Cottin V. Interstitial lung disease: are we missing formes frustes of connective tissue disease? Eur Respir J 2006;28:893–6. [DOI] [PubMed] [Google Scholar]
- [18].Kinder BW, Collard HR, Koth L, et al. Idiopathic nonspecific interstitial pneumonia: lung manifestation of undifferentiated connective tissue disease? Am J Respir Crit Care Med 2007;176:691–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Clegg DO, Williams HJ, Singer JZ, et al. Early undifferentiated connective tissue disease. II. The frequency of circulating antinuclear antibodies in patients with early rheumatic diseases. J Rheumatol 1991;18:1340–3. [PubMed] [Google Scholar]
- [20].Doria A, Mosca M, Gambari PF, et al. Defining unclassifiable connective tissue diseases: incomplete, undifferentiated, or both? J Rheumatol 2005;32:213–5. [PubMed] [Google Scholar]
- [21].Fischer A, Antoniou KM, Brown KK, et al. An official European Respiratory Society/American Thoracic Society research statement: interstitial pneumonia with autoimmune features. Eur Respir J 2015;46:976–87. [DOI] [PubMed] [Google Scholar]
- [22].American Thoracic Society, European Respiratory Society. American Thoracic Society/European Respiratory Society International Multidisciplinary Consensus Classification of the Idiopathic Interstitial Pneumonias. This joint statement of the American Thoracic Society (ATS), and the European Respiratory Society (ERS) was adopted by the ATS board of directors, June 2001 and by the ERS Executive Committee, June 2001. Am J Respir Crit Care Med 2002;165:277–304. [DOI] [PubMed] [Google Scholar]
- [23].Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American college of rheumatology/European league against rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–55. [DOI] [PubMed] [Google Scholar]
- [25].Alarcon-Segovia D, Cardiel MH. Comparison between 3 diagnostic criteria for mixed connective tissue disease. Study of 593 patients. J Rheumatol 1989;16:328–34. [PubMed] [Google Scholar]
- [26].Dalakas MC. Inflammatory muscle diseases. N Engl J Med 2015;372:1734–47. [DOI] [PubMed] [Google Scholar]
- [27].Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for Sjogren's syndrome: a revised version of the European criteria proposed by the American-European Consensus Group. Ann Rheum Dis 2002;61:554–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010;69:1580–8. [DOI] [PubMed] [Google Scholar]
- [29].Cavagna L, Nuno L, Scire CA, et al. Clinical spectrum time course in anti Jo-1 positive antisynthetase syndrome: results from an international retrospective multicenter study. Medicine (Baltimore) 2015;94:e1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Marie I, Josse S, Hatron PY, et al. Interstitial lung disease in anti-Jo-1 patients with antisynthetase syndrome. Arthritis Care Res (Hoboken) 2013;65:800–8. [DOI] [PubMed] [Google Scholar]
- [31].Kondoh Y, Johkoh T, Fukuoka J, et al. Broader criteria of undifferentiated connective tissue disease in idiopathic interstitial pneumonias. Respir Med 2015;109:389–96. [DOI] [PubMed] [Google Scholar]
- [32].Suda T, Kono M, Nakamura Y, et al. Distinct prognosis of idiopathic nonspecific interstitial pneumonia (NSIP) fulfilling criteria for undifferentiated connective tissue disease (UCTD). Respir Med 2010;104:1527–34. [DOI] [PubMed] [Google Scholar]
- [33].Oldham JM, Adegunsoye A, Valenzi E, et al. Characterisation of patients with interstitial pneumonia with autoimmune features. Eur Respir J 2016;47:1767–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Scire CA, Gonzalez-Gay MA, Selva-O’Callaghan A, et al. Clinical spectrum time course of interstitial pneumonia with autoimmune features in patients positive for antisynthetase antibodies. Respir Med 2017;132:265–6. [DOI] [PubMed] [Google Scholar]
- [35].Cavagna L, Castaneda S, Scire C, et al. Antisynthetase syndrome or what else? Different perspectives indicate the need for new classification criteria. Ann Rheum Dis 2018;77:e50. [DOI] [PubMed] [Google Scholar]
- [36].Cavagna L, Nuno L, Scire CA, et al. Serum Jo-1 autoantibody and isolated arthritis in the antisynthetase syndrome: review of the literature and report of the experience of AENEAS collaborative group. Clin Rev Allergy Immunol 2017;52:71–80. [DOI] [PubMed] [Google Scholar]
- [37].Ahmad K, Barba T, Gamondes D, et al. Interstitial pneumonia with autoimmune features: clinical, radiologic, and histological characteristics and outcome in a series of 57 patients. Respir Med 2017;123:56–62. [DOI] [PubMed] [Google Scholar]
- [38].Collins B, Raghu G. Interstitial pneumonia with autoimmune features: the new consensus-based definition for this cohort of patients should be broadened. Eur Respir J 2016;47:1293–5. [DOI] [PubMed] [Google Scholar]
- [39].Collins BF, Spiekerman CF, Shaw MA, et al. Idiopathic interstitial pneumonia associated with autoantibodies: a large case series followed over 1 year. Chest 2017;152:103–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Strand MJ, Sprunger D, Cosgrove GP, et al. Pulmonary function and survival in idiopathic vs secondary usual interstitial pneumonia. Chest 2014;146:775–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chartrand S, Swigris JJ, Stanchev L, et al. Clinical features and natural history of interstitial pneumonia with autoimmune features: a single center experience. Respir Med 2016;119:150–4. [DOI] [PubMed] [Google Scholar]
- [42].Kim HC, Ji W, Kim MY, et al. Interstitial pneumonia related to undifferentiated connective tissue disease: pathologic pattern and prognosis. Chest 2015;147:165–72. [DOI] [PubMed] [Google Scholar]
- [43].Chung JH, Montner SM, Adegunsoye A, et al. CT findings, radiologic-pathologic correlation, and imaging predictors of survival for patients with interstitial pneumonia with autoimmune features. AJR Am J Roentgenol 2017;208:1229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Kim HC, Lee JH, Chae EJ, et al. Long-term clinical course and outcome of interstitial pneumonia with autoimmune features. Respirology 2019;[Epub ahead of print]. [DOI] [PubMed] [Google Scholar]