

Abstract
Leigh syndrome (also called Leigh disease or subacute necrotizing encephalomyelopathy) is a rare inherited neurometabolic disorder, which affects the central nervous system. This meta-study systematically analyzed clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations.
Literature was searched for publications in MEDLINE, EMBASE, and the China National Knowledge Infrastructure database for meta-analyses of the incidence of clinical symptoms, laboratory assessments, imaging data, muscle biopsy histochemical staining, activity of the mitochondrial respiratory chain enzyme complex, gene mutations, and the association between age at disease onset and type of gene mutations.
This study included 5 studies with 385 Leigh syndrome patients. The most common clinical features of Leigh syndrome included elevated blood and/or cerebrospinal fluid (CSF) levels of lactate (72%), developmental retardation (57%), hypotonia (42%), followed by respiratory dysfunction (34%), epileptic seizures (33%), poor feeding (29%), and weakness (27%). Approximately 80% of the patients had deficiencies of the respiratory chain enzyme complex or isolated complex I deficiency (35%), 32% had mitochondrial DNA (mtDNA) mutations, and 38% had nuclear DNA (nDNA) mutations. Patients with nDNA mutations were younger than those with mtDNA mutations (8.82 ± 13.88 vs 26.20 ± 41.11 years, P = .007).
The data from the current meta-analysis demonstrated a variety of clinical and molecular manifestations of Leigh syndrome, with upregulated lactate levels in the blood or CSF being the most common feature. Diagnosis of Leigh syndrome could be confirmed using combined enzymatic and genetic analyses.
Keywords: clinical features, gene mutations, Leigh syndrome, mitochondrial respiratory chain enzyme complex
1. Introduction
Leigh syndrome (also called Leigh disease or subacute necrotizing encephalomyelopathy) is a rare inherited neurometabolic disorder and affects the central nervous system. Leigh syndrome was first described by Denis Leigh in 1951 and characterized by focal, bilaterally symmetrical, and subacute necrotic lesions in the thalamus, the brainstem, and the posterior columns of the spinal cord.[1] Genetically, alterations or mutations of the mitochondrial respiratory enzyme complex or pyruvate dehydrogenase complex are believed to be responsible for the development of Leigh syndrome.[2,3] However, to date, there is no single specific criterion to diagnose Leigh syndrome. Diagnosis is based on findings of clinical manifestations, family history, laboratory assessments, imaging, muscle biopsy with histochemical staining, activity of the mitochondrial respiratory chain enzyme, and identification of mitochondrial DNA (mtDNA) or nuclear DNA (nDNA) mutations.[3] Given the limitations and nonspecificity of diagnostic criteria, Leigh syndrome should be suspected and further investigation is warranted when symmetrical lesions are evident on neuroimaging in one or more areas of the central nervous system.[4,5] Computed tomography (CT) or magnetic resonance imaging (MRI) typically shows lesions in the basal ganglia, diencephalon, brainstem, cerebellum, and spinal cord with hypodensity on CT scan, a hyperintense signal on T2-weighted and hypointense signal on T1-weighted MRI.[6,7] Furthermore, clinical manifestations may include motor delay, mental retardation, and/or progressive cognitive decline, hypotonia, dyskinesia, akinesia, ataxia, dystonia, and brainstem dysfunction. Usually, brainstem dysfunction manifests as respiratory symptoms and abnormalities in swallowing, ophthalmology, and thermoregulation.[4] The neurologic manifestations may begin in infancy or early childhood, progressively worsen, and eventually lead to death in early childhood[2,3]; however, Leigh syndrome can also occur at any age, including adolescence or adulthood[3] and with appropriate treatment options, patients can survive for many years after diagnosis.[4,5,8–10]
Leigh syndrome is rare and heterogeneous, so it is difficult to obtain a large cohort of patients. To date, although there are numerous published studies on the biochemical and molecular features of the syndrome, very few studies address clinical diagnosis in the setting of negative biochemical or molecular findings.[11] As a result, clinical diagnostic criteria have not been established or validated. Therefore, in this meta-analysis, we systematically reviewed and analyzed clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations for better diagnosis based on published studies. We sought to provide a better assessment of Leigh syndrome for future diagnosis and control of the disease.
2. Materials and methods
All procedures were approved by the Ethics committee of First Hospital of Shanxi Medical University (Taiyuan, China).
2.1. Literature search
Two reviewers (XC and YW) independently searched MEDLINE (1966 to January 2017), EMBASE (1980 to January 2017), Wanfang date (2000 to January 2017), and China National Knowledge Infrastructure (1999 to January 2017) using various search terms (ie, Leigh syndrome, mtDNA, nDNA, and mitochondrial respiratory chain enzyme complex) to identify eligible studies. Then we reviewed relevant publications cited by the original studies and included the described patients, if any, in the systematic review and meta-analysis.
2.2. Study selection
Our inclusion criteria for patients and reports were:
-
(1)
A diagnosis of Leigh syndrome based on their clinical symptoms, laboratory assessment, imaging, muscle biopsy, and/or genetic analysis;
-
(2)
Genetic analysis showing mtDNA and/or nDNA mutations;
-
(3)
Assessment of alterations in the mitochondrial respiratory enzyme complex;
-
(4)
Publication in English or Chinese; and
-
(5)
Availability of full publications, including published and in-press journal articles, conference articles, and academic articles.
Our exclusion criteria were:
-
(1)
Patients who did not have genetic analysis;
-
(2)
Incomplete clinical data, such as that found in a statistical analysis from which it was impossible to extract detailed data for our analysis; and
-
(3)
Publications that were duplicated or partial data that appeared in another study.
We resolved any disagreements through discussion and then reached consensus of our investigators according to the above inclusion and exclusion criteria.
2.3. Data extraction and study assessment
Two investigators (XC and YW) independently selected studies and extracted data, including each study title, first author's name, journal, year of publication, study populations, sample size, and patient characteristics, such as age, sex, clinical symptoms, laboratory assessments, imaging data, muscle biopsy results, assessment of the mitochondrial respiratory chain enzyme complex, gene mutations, association of phenotypes or age with genotypes, and so on. Any discrepancies were resolved by consensus of our investigators through discussion. Any missing data in the publications were requested from the authors and analyzed, whenever possible.
The quality of each was assessed by using the published grading system described in previous studies.[12,13] Thus, a “point” system was used to grade the quality of each study in terms of:
-
(1)
Appropriate incidence of each clinical feature, the respiratory chain enzyme complex deficiency, and gene mutation;
-
(2)
Representation of patients for the general Leigh syndrome features;
-
(3)
Consecutive or random sampling in subjects for the respiratory chain enzyme complex deficiency and gene mutation;
-
(4)
Description of relevant clinical features; and
-
(5)
Less than 20% of ineligible subjects in the study.
We used this quality grading system to assess each study and explore the effect of study quality on prevalence values, as described previously.[14] Two investigators (XC and YW) independently performed quality assessment, and we resolved any disagreements through discussion or with a third review author (JG) if necessary. We maximally avoided potential publication bias thereafter.
2.4. Statistical analysis
In this study, we used R2.15.3 software (https://www.r-project.org/) to perform the meta-analyses. For example, the incidence of each clinical symptom was calculated as the legit transform, and the merging rate was assessed using the weight of sample size. The “incidence” and 95% confidence interval were ultimately obtained. A homogeneity test was used to assess the heterogeneity, with α = 0.1 used as the standard and I2 used to judge the degree of heterogeneity. A P-value <.10 and I2 >50% suggested heterogeneity between the results of studies and we then performed the random effects model for meta-analysis of the data; otherwise, we used the fixed effect model for meta-analysis of the data. The publication bias was also assessed by using Eggers’ test. In addition, a sensitivity analysis was performed by calculating the merging rate after excluding the lowest quality studies. The level of meta-analysis was α = 0.05. The age at disease onset and clinical phenotypes were compared between 2 groups defined by nDNA mutations versus mtDNA mutations. The differences between the groups were assessed with the Student t test using the Statistical Package for the Social Sciences version 23.0 (SPSS, version 23.0) for Windows (SPSS, Chicago, IL). A 2-tailed P ≤ .05 was considered statistically significant.
3. Results
3.1. Description of studies
In this meta-analysis, we initially identified 2040 potential publications after the search of different databases and the manual search of cited references. We excluded 1897 studies after reviewing the title and abstract of each publication. We then extracted and reviewed the full text of the remaining 143 studies and excluded 138 of them due to incomplete clinical data, genetic analysis, and/or ineligible publications, such as reviews or commentaries. In the end, for this systematic review and meta-analysis, we obtained 5 studies,[11,15–18] which included 385 patients with Leigh syndrome. Among these 385 patients, 162 were male, 117 were female and the gender information of 106 patients was not clearly indicated; 204 of them had data on the age of disease onset (77.5% patients were younger than 2-years-old when the disease occurred); and 98 patients had mtDNA or nDNA mutations at diagnosis. Patients with nDNA mutations were younger than those with mtDNA mutations (8.82 ± 13.88 vs 26.20 ± 41.11 years-old, P = .007).
3.2. Quality of studies
Study quality of each publication was then assessed independently by 2 investigators (XC and YW) using a 5-point scale (Table 1). Only 1 study[11] received a quality score of 5, and the 4 other studies[15–18] received a lower quality score of 4. The main reason for the low scores was that the studies did not analyze the respiratory chain enzyme complex and gene mutations in consecutive or random sampling of study subjects.
Table 1.
Characteristics of the 5 included studies.

3.3. Clinical features
3.3.1. Clinical characteristics at disease onset
Four studies[11,15,16,18] assessed clinical symptoms at disease onset (Table 2), but our meta-analysis was unable to conclusively determine the initial clinic symptoms at onset due to inconsistency in data, although developmental delay/retardation was the most common feature. Other common features included epileptic seizures, feeding/sucking difficulties, weakness, fatigue, abnormal ocular findings, and ataxia.
Table 2.
Initial clinical manifestations in 4 studies.
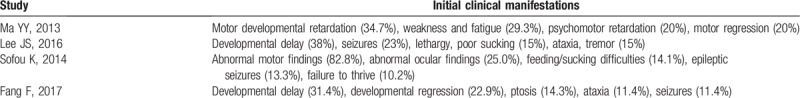
3.3.2. Clinical characteristics throughout the disease course
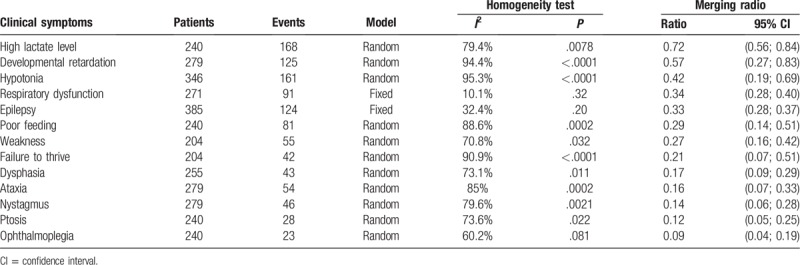
During progression of the disease, the developmental delay was the most common clinical sign (57%), while hypotonia was the most common motor finding (42%), followed by respiratory dysfunction (34%), epileptic seizures (33%), poor feeding (29%), and weakness (27%) (Table 3). Moreover, there were 4 studies[11,16–18] showing lactate in the blood and/or cerebrospinal fluid (CSF). Ogawa et al[17] reported that the mean serum and CSF L/P ratio was much higher than that of normal individuals. Lee et al[15] revealed that the most common nonneurological manifestations were ophthalmological and cardiac. The frequently abnormal ocular findings (Table 3) were nystagmus (14%), followed by ptosis (12%), and ophthalmoplegia (9%). Sofou et al[11] reported that the most common cardiac dysfunction was hypertrophic cardiomyopathy, followed by arrhythmia/conduction defects and dilated cardiomyopathy.
Table 3.
Meta-analysis of clinical symptoms from all 5 studies.
3.3.3. Alterations in the mitochondrial respiratory chain enzyme complex
Abnormality of the mitochondrial respiratory enzyme complex was assessed in 364 of 385 patients. The data showed that 74 of them had normal enzyme activities of the mitochondrial respiratory enzyme complex, whereas 79.7% patients had deficiencies in the respiratory chain enzyme complex, that is, an isolated complex I deficiency (35%), complex IV deficiency (16%), and complex V deficiency (11%) or a combined complex deficiency (22%) (Table 4). The most frequently occurring combined deficiency was of complexes I and IV (10%).
Table 4.
Meta-analysis of the respiratory chain enzyme complex deficiency.

3.4. Gene mutation
3.4.1. mtDNA mutations
The mtDNA mutations were analyzed in 330 patients and the data showed that 96 patients (32%) possessed mtDNA mutations, among which, mutations of the MT-ND and MT-ATP6 gene were the most frequent (20% and 11%, respectively) Table 5. The m.8993T>G/C in the MT-ATP6 gene, m.10191T>C and m.10197 G>A in the ND3 gene, m. 13513G>A in the ND5 gene and 14487T>C in the ND6 gene were the most common genotypes of Leigh syndrome. One study[15] demonstrated that mutations of ND genes were associated with the isolated complex I deficiency, while another study[18] showed that mutations of the MT-ATP6 genes were associated with the isolated complex V deficiency.
Table 5.
Meta-analysis of gene mutations.

3.4.2. nDNA mutations
A total of 241 patients were subjected to nDNA mutation analysis, resulting in 89 (38%) patients with nDNA mutations. We did not find any hot spot mutations in nDNA. Mutations of the SURF1 were the most frequent (23 patients, 9.5%) and were associated with the complex IV deficiency.[18] Thus, mutations of nDNA were more common than those mtDNA (38% vs 32%, respectively).
3.5. Radiological abnormalities
Lee et al[15] analyzed the initial findings of brain MRIs and showed that structural involvement occurred in the midbrain (38%), thalamus (21%), white matter (13%), brainstem (8%), and cerebellum (8%), in addition to the basal ganglia. A total of 19 patients showed a lactate peak in the brain magnetic resonance spectroscopy (MRS) but only 4 patients had elevated serum lactate levels. Moreover, Fang et al[18] assessed patients’ neuroimaging and found involvement in the brainstem (77%) and/or basal ganglia (69%), as well as other areas, such as the thalamus (45.7%), medulla oblongata (40%), cerebellum (22.9%), and white matter (20%). Most patients with involvement of the medulla oblongata (P = .001) and cerebellum (P = .041) also had nDNA variations.
3.6. Muscle biopsy abnormalities
Sofou et al[11] performed muscle biopsies in 104 patients and found that 57 had abnormal histological findings. Lee et al[15] assessed muscle biopsies from 39 patients. Light-microscopy did not reveal any specific abnormalities, but transmission electron microscope showed sarcoplasmic accumulation in both normal and abnormal mitochondrial morphology (46%) or large/swollen mitochondria with or without abnormal cristae (22%); however, approximately 20% of the biopsies had nonspecific degenerative changes.
3.7. Acute exacerbations and survival
Sofou et al[11] followed patients for a median of 9.6 years and found that 56.9% of them experienced at least 1 acute exacerbation-related hospitalization, the leading causes of which were infection (60.8%), respiratory complications (13.5%), stroke-like episodes (4.0%), and/or poor nutrition or dehydration (4.0%). This study also showed that the presence of pathological signs at birth and a history of epileptic seizures were significantly associated with acute exacerbations and/or relapses. At the end of follow-up, 40.8% of patients were alive, 39.2% were deceased, and the remaining 20.0% were lost to follow-up. The median age at death was 2.4 years (range 1 month–21 years), while the median elapsed median duration from disease onset to death was 1.8 years. The main causes of death were respiratory complications (51.0%), disease progression (17.6%), or infection (17.6%). The multivariate analysis demonstrated that factors associated with poor survival were: disease onset before 6 months of age, failure to thrive, brainstem lesions visible on neuroimaging, and admission to an intensive care.
4. Discussion
Leigh syndrome is a rare and under-documented neurodegenerative disorder that generally affects infants and young children (younger than 2-years-old). It progresses rapidly and often causes death.[2,19,20] However, Leigh syndrome also shows significant clinical heterogeneity.[21,22] Thus, our current systematic review and meta-analysis assessed clinical features and showed that the most common of these were: elevated lactate levels in the blood and/or CSF (72%), developmental delay (57%), hypotonia (42%), respiratory dysfunction (34%), epileptic seizures (33%), poor feeding (29%), and weakness (27%).
Indeed, Leigh disease is characterized by focal and bilateral lesions in the brainstem, basal ganglia, cerebellum, and other regions of the brain. Pathologically, the lesions show demyelination, spongiosis, gliosis, necrosis, and capillary proliferation.[3] Since the brain stem controls and maintains basic life functions—like breathing, swallowing, and blood circulation—and the basal ganglia and cerebellum control body movement and balance, damage to these tissues could result in the major clinical manifestations (see above). Moreover, Leigh syndrome is induced genetically by aberrant mitochondrial activity, due to mutations of cytochrome c oxidase and other respiratory chain enzyme complex proteins.[3] Such alterations cause oxidative phosphorylation deficiencies that lead to buildup of pyruvate in the body and lactate accumulation in the blood stream and CSF.[3] Thus, 1 study showed that increased CSF lactate levels were associated with disease severity and short survival,[11] which could be a prognostic marker, although up to 25% of patients had normal lactate levels in the blood or CSF throughout the disease course.[11] Another study showed an elevated CSF lactate peak in all patients assessed by MRS, but 15 of these 19 patients had normal serum lactate levels.[15] However, this study did not stratify patients by disease severity. However, another study also suggested that the presence of the MRS lactate peak may be more sensitive than serum lactate level.[3] Furthermore, Lee et al[15] reported that patients with lesions in regions besides the basal ganglia also tended to have an unfavorable functional outcome, such as severe neurologic deterioration during follow-up, which was consistent with other 2 studies.[19,23] Thus, these data demonstrate that the site of lesions may affect clinical manifestations, disease severity, disease progression, and prognosis. Therefore, so we hypothesize that brain MRIs could assist physicians in early diagnosis and in determining the prognosis of Leigh disease.
Furthermore, during disease progression, muscle tissue is also altered because aberrant brain function fails to control muscle contraction, leading to muscle hypotonia, dystonia, and/or ataxia.[3] Thus, muscle biopsy can also help to assess disease severity. However, Lee et al[15] showed that muscle biopsies did not reveal any ragged red fibers, while Sofou et al[11] reported that ragged red fibers occurred in only 51% of muscle biopsies in 104 patients. Thus, early disease might not show abnormalities on muscle biopsies through light-microscopy—a finding that dispels the common misperception among many physicians that muscle biopsy is absolutely required for the diagnosis of mitochondrial abnormalities in patients with Leigh disease. Nonetheless, our current meta-analysis showed that 80% of patients had a deficiency in the mitochondrial respiratory enzyme complex (higher than a previous study of 50%).[2]
In addition, the isolated complex I deficiency was reported to be the most frequent (35%) in Leigh syndrome, followed by combined deficiency (22%), and deficiencies in complex IV (16%) and complex V (11%). This finding suggested that detection of the mitochondrial respiratory enzyme complex activity is a useful diagnostic tool.[24] Furthermore, advances in genetic technologies are applied more broadly and is increasingly used instead of measurements of enzyme activity and muscle biopsy.[25,26] Indeed, mutations of the mitochondrial genome occur in 32% of cases and genomic DNA in 38% of cases. Lee et al[15] reported that frequent mutations of the ND genes are associated with the isolated complex I deficiency, while 2 other studies[17,18] demonstrated that mutations of the MT-ATP6 genes are associated with the isolated complex V deficiency. An additional study showed that up to 75% of the complex IV deficiency resulted from mutations of the SURF1 gene.[27] In addition, the general prognosis of Leigh syndrome is poor, with death often occurring in early childhood,[2] although 1 study[5] showed that 20% of patients could survive to 20 years old of age. Sofou et al reported a favorable survival rate in their study population, with 41% of patients alive after a median follow-up of 11.4 years. So early diagnosis could be the key for treatment and better prognosis.
In conclusion, our study consisted of a meta-analysis to determine the clinical manifestations of Leigh syndrome. We found that upregulated lactate levels in the blood or CSF were the most frequently manifestation. Combined enzymatic and genetic analyses could be used to improve early detection of Leigh disease and possibly lead to gene-specific therapies.
Author contributions
Conceptualization: Wei Zhang, Junhong Guo.
Data curation: Xueli Chang, Yaxin Wu, Jie Zhou.
Formal analysis: Jie Zhou.
Methodology: Huaxing Meng, Wei Zhang.
Software: Huaxing Meng.
Validation: Junhong Guo.
Writing – original draft: Xueli Chang.
Footnotes
Abbreviations: CSF = cerebrospinal fluid, CT = computed tomography, MRI = magnetic resonance imaging, MRS = magnetic resonance spectroscopy, mtDNA = mitochondrial DNA, nDNA = nuclear DNA, SPSS = Statistical Package for the Social Sciences.
How to cite this article: Chang X, Wu Y, Zhou J, Meng H, Zhang W, Guo J. A meta-analysis and systematic review of Leigh syndrome: clinical manifestations, respiratory chain enzyme complex deficiency, and gene mutations. Medicine. 2020;99:5(e18634).
XC and YW contributed equally to this work.
The authors have no conflicts of interest to disclose.
References
- [1].Leigh D. Subacute necrotizing encephalomyelopathy in an infant. J Neurol Neurosurg Psychiatry 1951;14:216–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Finsterer J. Leigh and Leigh-like syndrome in children and adults. Pediatr Neurol 2008;39:223–35. [DOI] [PubMed] [Google Scholar]
- [3].Baertling F, Rodenburg RJ, Schaper J, et al. A guide to diagnosis and treatment of Leigh syndrome. J Neurol Neurosurg Psychiatry 2014;85:257–65. [DOI] [PubMed] [Google Scholar]
- [4].Montpetit VJ, Andermann F, Carpenter S, et al. Subacute necrotizing encephalomyelopathy. A review and a study of two families. Brain 1971;94:1–30. [DOI] [PubMed] [Google Scholar]
- [5].Rahman S, Blok RB, Dahl HH, et al. Leigh syndrome: clinical features and biochemical and DNA abnormalities. Ann Neurol 1996;39:343–51. [DOI] [PubMed] [Google Scholar]
- [6].Saneto RP, Friedman SD, Shaw DW. Neuroimaging of mitochondrial disease. Mitochondrion 2008;8:396–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sofou K, Steneryd K, Wiklund LM, et al. MRI of the brain in childhood-onset mitochondrial disorders with central nervous system involvement. Mitochondrion 2013;13:364–71. [DOI] [PubMed] [Google Scholar]
- [8].Dahl HH. Getting to the nucleus of mitochondrial disorders: identification of respiratory chain-enzyme genes causing Leigh syndrome. Am J Hum Genet 1998;63:1594–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Goldenberg PC, Steiner RD, Merkens LS, et al. Remarkable improvement in adult Leigh syndrome with partial cytochrome c oxidase deficiency. Neurology 2003;60:865–8. [DOI] [PubMed] [Google Scholar]
- [10].Huntsman RJ, Sinclair DB, Bhargava R, et al. Atypical presentations of leigh syndrome: a case series and review. Pediatr Neurol 2005;32:334–40. [DOI] [PubMed] [Google Scholar]
- [11].Sofou K, De Coo IF, Isohanni P, et al. A multicenter study on Leigh syndrome: disease course and predictors of survival. Orphanet J Rare Dis 2014;9:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Richardson WS, Polashenski WA, Robbins BW. Could our pretest probabilities become evidence based? A prospective survey of hospital practice. J Gen Intern Med 2003;18:203–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Loney PL, Chambers LW, Bennett KJ, et al. Critical appraisal of the health research literature: prevalence or incidence of a health problem. Chronic Dis Canada 1998;19:170–6. [PubMed] [Google Scholar]
- [14].Juni P, Witschi A, Bloch R, et al. The hazards of scoring the quality of clinical trials for meta-analysis. JAMA 1999;282:1054–60. [DOI] [PubMed] [Google Scholar]
- [15].Lee JS, Kim H, Lim BC, et al. Leigh syndrome in childhood: neurologic progression and functional outcome. J Clin Neurol 2016;12:181–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ma YY, Wu TF, Liu YP, et al. Genetic and biochemical findings in Chinese children with Leigh syndrome. J Clin Neurosci 2013;20:1591–4. [DOI] [PubMed] [Google Scholar]
- [17].Ogawa E, Shimura M, Fushimi T, et al. Clinical validity of biochemical and molecular analysis in diagnosing Leigh syndrome: a study of 106 Japanese patients. J Inherit Metab Dis 2017;40:685–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Fang F, Shen Y, Shen DM, et al. Clinical and genetic characteristics of children with Leigh syndrome. Zhonghua Er Ke Za Zhi 2017;55:205–9. [DOI] [PubMed] [Google Scholar]
- [19].Lee HF, Tsai CR, Chi CS, et al. Leigh syndrome: clinical and neuroimaging follow-up. Pediatr Neurol 2009;40:88–93. [DOI] [PubMed] [Google Scholar]
- [20].Yang YL, Sun F, Zhang Y, et al. Clinical and laboratory survey of 65 Chinese patients with Leigh syndrome. Chin Med J 2006;119:373–7. [PubMed] [Google Scholar]
- [21].Mak SC, Chi CS, Tsai CR. Mitochondrial DNA 8993 T > C mutation presenting as juvenile Leigh syndrome with respiratory failure. J Child Neurol 1998;13:349–51. [DOI] [PubMed] [Google Scholar]
- [22].Sobreira C, Marques W, Jr, Pontes Neto OM, et al. Leigh-like syndrome with the T8993G mutation in the mitochondrial ATPase 6 gene: long-term follow-up discloses a slowly progressive course. J Neurol Sci 2009;278:132–4. [DOI] [PubMed] [Google Scholar]
- [23].Jin T, Shen H, Zhao Z, et al. Clinical, pathological, and neuroimaging analyses of two cases of Leigh syndrome in a Chinese family. J Child Neurol 2014;29:N143–8. [DOI] [PubMed] [Google Scholar]
- [24].Koenig MK. Presentation and diagnosis of mitochondrial disorders in children. Pediatr Neurol 2008;38:305–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lake NJ, Compton AG, Rahman S, et al. Leigh syndrome: one disorder, more than 75 monogenic causes. Ann Neurol 2016;79:190–203. [DOI] [PubMed] [Google Scholar]
- [26].Taylor RW, Pyle A, Griffin H, et al. Use of whole-exome sequencing to determine the genetic basis of multiple mitochondrial respiratory chain complex deficiencies. JAMA 2014;312:68–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Gerards M, Sallevelt SC, Smeets HJ. Leigh syndrome: resolving the clinical and genetic heterogeneity paves the way for treatment options. Mol Genet Metab 2016;117:300–12. [DOI] [PubMed] [Google Scholar]