

Abstract
Rationale:
Concurrent calreticulin (CALR) mutation and BCR-ABL1 fusion are extremely rare in chronic myelogenous leukemia; to date, only 12 cases have been reported.
Patient concerns:
A 57-year-old male who had an 11-year history of essential thrombocytosis presented to our hospital with leukocytosis and marked splenomegaly for 3 months.
Diagnoses:
Chronic myelogenous leukemia with myeloid fibrosis arising on the background of essential thrombocytosis harboring both BCR-ABL1 fusion and type-1 like CALR mutation.
Interventions:
Imatinib was started at 300 mg daily and increased to 400 mg daily after 3 months; interferon was added after 12 months.
Outcomes:
Partial cytogenetic response was achieved after 3 months of imatinib therapy and complete cytogenetic response was achieved after 1 year of treatment. However, CALR mutation was still present with a stable mutational allele burden.
Lessons:
In this case report and review of additional 12 cases with simultaneous presence of CALR-mutation and BCR-ABL1 fusion, we highlighted the importance of integrating clinical, morphological, and molecular genetic data for classifying atypical myeloid neoplasms.
Keywords: BCR-ABL1 fusion, CALR mutation, chronic myelogenous leukaemia, clonal evolution, cytogenetic response
1. Introduction
Myeloproliferative neoplasms (MPNs) are caused by mutations of various genes, resulting in constitutive activation of their corresponding signaling transduction pathways and aberrant hematopoiesis. Mutation in a gene or a group of genes consequently leads to particular phenotypes and clinical manifestations of MPNs.[1] Key genetic aberrations identified to date include BCR-ABL1 rearrangement in Philadelphia chromosome-positive (Ph+) chronic myelogenous leukemia (CML) and mutations of Janus kinase 2 (JAK2)/myeloproliferative leukemia protein (MPL)/calreticulin (CALR) in Philadelphia chromosome-negative (Ph–) MPNs.[1,2] These gene alterations were initially thought to be mutually exclusive,[3,4] although recent case reports have shown the simultaneous presence of these mutations in MPNs.[5–7]
Mutations of CALR have been reported primarily in wild-type JAK2 and MPL-related essential thrombocytosis (ET) and primary myelofibrosis (PMF). More recently, mutations in CALR exon 9 have been reported to occur in 20% to 25% of ET cases and 25% to 35% of PMF cases,[2,4] but CALR mutations are extremely rare in MPNs with the t(9;22)/BCR-ABL1; to date, only 12 cases have been reported.
In the present case report, we describe a CML patient with an 11-year history of ET who harbored both BCR-ABL1 fusion and type 1-like CALR mutation, a previously unreported atypical MPN case. We then systematically reviewed 12 MPN cases reported in the literature and summarized their characteristics, clonal evolution, treatment options, and responses.
2. Case presentation
In 2006, a 46-year-old male visited our out-patient clinic in The First Hospital of Jilin University (Changchun, China). His laboratory tests showed a platelet count of 800 × 109/L with a normal white blood cell count and hemoglobin level. A bone marrow aspiration smear showed enlarged megakaryocytes with hyperlobulated nuclei and without the classical “dwarf” morphology (Fig. 1A). Bone marrow biopsy further revealed an increase in megakaryocytes, but with normocellular morphology and a normal myeloid-to-erythroid ratio, suggesting a diagnosis of ET without reticulin and collagen fibrosis. The genetic test showed wild-type JAK2 V617F expression at that time. Aspirin was prescribed, and 1 year later, the patient experienced acute myocardial infarction for which 2 stents were placed in his left anterior descending coronary artery. After recovery from the percutaneous coronary intervention, he was given interferon, and his platelet count reached 400 to 600 × 109/L.
Figure 1.
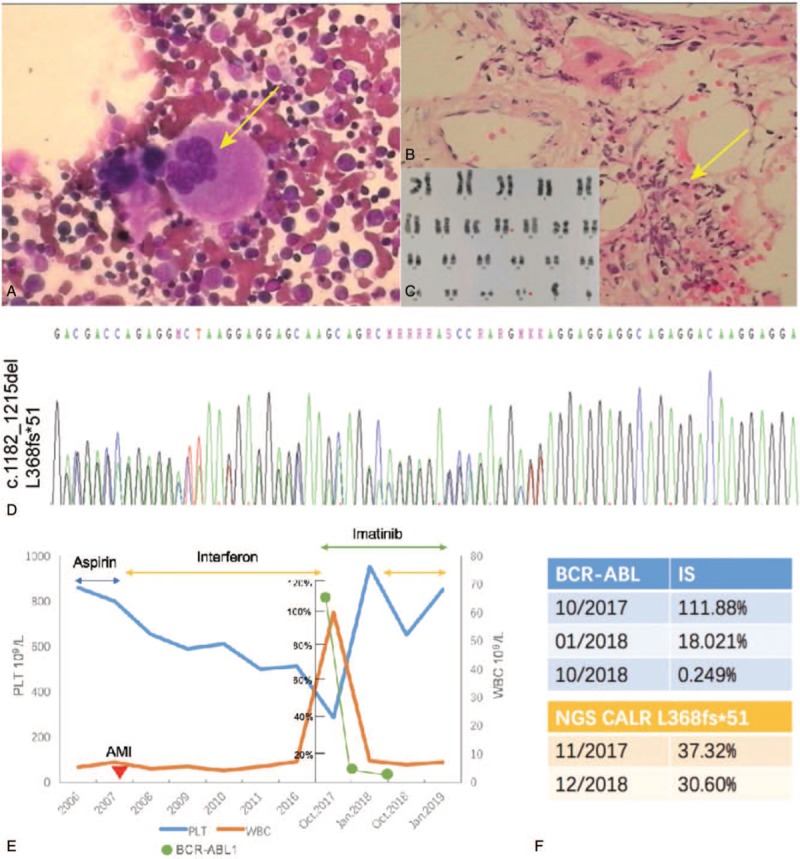
. Clinicopathological features of atypical myeloproliferative neoplasm with BCR-ABL1 fusion and calreticulin (CALR) mutation. (A) Bone marrow aspiration and Wright–Giemsa staining. The patient was admitted into our hospital and underwent bone marrow aspiration with Wright–Giemsa staining. The data show enlarged megakaryocytes and hyperlobulated nuclei, but not classical “dwarf” morphology in megakaryocytes (original magnification, ×400). (B) Hematoxylin and eosin staining of the bone marrow biopsy specimen. The patient was followed up in 2017, and bone marrow biopsy was repeated. Hematoxylin and eosin staining shows leukocytosis and severe marrow fibrosis (original magnification, ×400). (C) Karyotyping of the bone marrow. The data show 46,XX,t[9;22](q34;q11)[10]. (D) Sanger sequencing. The bone marrow was subjected to polymerase chain reaction (PCR) using the primers 5′-GTGGGGCGTAACAAAGGTGA-3′ and 5′-AGAGACATTATTTGGCGCGG-3′ and Sanger sequencing. The data show a frameshift mutation of CALR exon 9 hotspot (c.1182-1215delaaggaggaggaagaagacaagaaacgcaaagagg, p.L368fs∗51). (E) Graphic depiction of peripheral blood counts from 2006 to January 2019. The patient was treated initially with interferon after acute myocardial infarction from 2007 to 2017, and the platelet count was sustained around 400 to 600 × 109/L. In October 2017, he was switched to imatinib due to chronic myeloid leukemia based on leukocytosis, splenomegaly, and BCR-ABL1 fusion. After 3 months of imatinib treatment, thrombocytosis had worsened even though the BCR-ABL1 transcript levels markedly decreased. To control the platelet count, the patient was given a combination of imatinib with interferon. (F) Quantitative reverse-transcriptase (qRT)-PCR. The bone marrow samples were subjected to qRT-PCR (the top table) and next-generation DNA sequencing (the bottom table). The qRT-PCR data show the level of BCR-ABL fusion transcript as an international standard value, whereas the bottom table shows the allele burden of CALR mutation.
The disease remained stable until 2017 when he displayed marked splenomegaly (19.1 cm in length) and leukocytosis. Thus, he was referred to our in-patient services. Laboratory tests revealed a leukocyte count of 69.88 × 109/L and platelet count of 285 × 109/L. Bone marrow aspiration and biopsy showed a significantly increased myeloid-to-erythroid ratio with 3% myeloblasts and severe marrow fibrosis, respectively (Fig. 1B). Karyotyping showed 46,XX,t[9;22](q34;q11)[20] (Fig. 1C). Quantitative reverse-transcriptase polymerase chain reaction data demonstrated positive BCR-ABL p210 fusion gene, while the quantitative result was 111.88% of an international scale (IS) value. DNA sequencing data also confirmed the presence of type 1-like CALR mutation (c.1182_1215del, L368fs∗51) (NM_004343.3) (Fig. 1D) with a mutational allele burden of 37.32% (Fig. 1F). Thereafter, he was diagnosed with CML in a chronic phase with myeloid fibrosis, probably arising on the background of ET. The treatment regimen was switched from interferon to tyrosine kinase inhibitor (TKI) imatinib at 300 mg daily. Three months later, he achieved a partial cytogenetic response: the Ph+ positive rate decreased to 5% and the BCR-ABL1 fusion gene rate declined to 18.02% (IS) (Fig. 1F). Bone marrow biopsy showed a normal myeloid-to-erythroid ratio, but an increase in large megakaryocytes with grade 3 fibrosis. Imatinib dose was increased to 400 mg daily. One year later, the size of his spleen had obviously shrunk and he achieved a complete cytogenetic response with Ph-negativity and a decrease in the BCR-ABL1 fusion gene rate to 0.249% (IS). However, his platelet count was 900 × 109/L and CALR mutation was still present with a mutational allele burden of 30.60% (Fig. 1F). Bone marrow biopsy still showed grade 3 fibrosis. To further control the platelet count, he was given a combination of imatinib and interferon, and his follow-up showed stable disease. This patient's disease process is summarized and shown in Figure 1E.
3. Discussion
In this article, we reported our own case and then performed literature search to identify additional MPN cases that harbored both CALR mutation and BCR-ABL1 fusion. We identified 12 cases from PubMed. The details, such as clinical characteristics, clonal evolution, treatment options, and responses of our patient and these 12 cases, are summarized in Table 1 and discussed below.
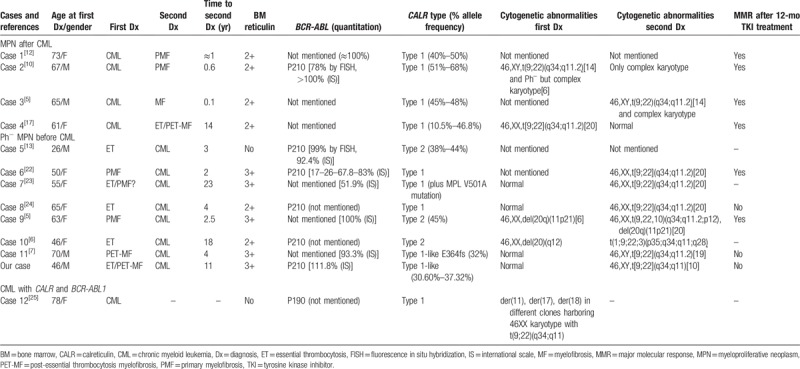
Table 1.
Clinical and molecular characteristics of 13 MPN patients with BCR-ABL1 fusion and CALR mutation.
3.1. Clinical characteristics
Overall, this cohort of 13 patients had a median age of 63 years (range: 26–78 years; Table 1) at the initial diagnosis, and 5 were male for a male/female ratio of 1:1.6 (Table 1). Interestingly, most of the patients were diagnosed with PMF or post-essential thrombocythemia myelofibrosis, either before or after CML diagnosis, suggesting that reticulin fibrosis was the most common phenomenon in patients with concurrent CALR mutation and BCR-ABL1 fusion.
In this cohort, patients were all diagnosed with chronic phase CML, and 8 of 13 patients were diagnosed with Ph− MPN (such as PMF, ET, or post-essential thrombocythemia myelofibrosis) before CML diagnosis. There was a shift to the clinical BCR-ABL1+ CML phenotype, with manifestations such as splenomegaly and leukocytosis, between the initial and second diagnoses. However, some typical features of the initial Ph− MPN, for example, large and clustered megakaryocytes and grade 2 to 4 reticulin fibrosis, persisted after TKI treatment. Moreover, there was always a relatively long duration for Ph− MPN transformation to CML with median duration of 4 years (range: 2–23 years). Only 4 patients initially presented with Ph+ CML and without JAK2 V617F and MPL mutations. They were all given TKIs during the course of the diseases and responded well to the treatment with a constant decrease in BCR-ABL1 fusion transcripts, which led to phenotypic changes. In turn, mutation screening showed positivity for CALR mutation and an association with ET or PMF phenotype. Indeed, the retrospective analysis indicated a high CALR mutation burden between initial CML diagnosis and secondary Ph− MPN diagnosis, regardless of the BCR-ABL1 transcript level. Control of the Ph+ clone took no more than 1 year of TKI treatment in these patients showing features of Ph− MPN. In this cohort, only 1 patient (case 12 in Table 1) was diagnosed with CML with concomitantly detectable CALR mutation and BCR-ABL1 fusion transcript.
3.2. Bone morrow features
The simultaneous presence of Ph− MPN/Ph+ CML was evident in bone marrow morphology. For example, granulocytosis is the key CML feature, whereas unusually large and clustered megakaryocytes are characteristic of Ph− MPN (but not CML). Indeed, assessment of megakaryocyte size can differentiate different MPN subtypes; for example, smaller megakaryocytes (micromegakaryocytes) are observed in CML, whereas large atypical forms of megakaryocytes are a typical feature of Ph− MPN, according to a previous study.[8] The presence of large atypical megakaryocyte forms with or without myelofibrosis in the CML bone marrow raises the possibility of simultaneous presence of Ph− MPN and mutated JAK2/CALR/MPL should be searched for.[5,9,10] In contrast, Ph− MPN patients who are morphologically suspected to have CML but without cytogenetic confirmation should undergo further assessments such as fluorescent in situ hybridization and reverse-transcriptase polymerase chain reaction for gene mutation and fusion evolutions.
Moreover, myelofibrosis was a very common phenomenon in the bone marrow biopsy of this atypical MPN, that is, 11 of these 13 cases showing grade 2 to 3 reticulin fibrosis, which is consistent with another case series of concurrent BCR-ABL1 fusion and JAK2 V617F mutation.[11]
3.3. Clonal evolution
Occurrence of CALR driver mutation and BCR-ABL1 fusion in the same patient could indicate the presence of different subclones and clonal evolution during disease progression and remission, mandating further investigation of CALR mutation and BCR-ABL1 fusion in the same or different clones.
From his 11-year history of disease, early diagnostic specimens were no longer available for CALR mutation analysis and clonal evolution. However, based on his thrombocytosis and absence of the Ph chromosome at the first diagnosis, we speculate that our patient initially could have harbored CALR mutation, leading to the clinical and histologic manifestations of a Ph− MPN, while the subsequent development of leukocytosis and acquisition of t(9;22)(q34;q11) suggested that CML evolved at a later point in time. However, we cannot rule out whether he initially harbored the BCR-ABL1 clone with CALR mutation or only the negative CALR mutation.
To date, clonal analysis has been performed in only 1 case. Cabagnols et al[12] assessed clonal hierarchy during evolution of the disease and showed that some of the original del52CALR clone was subjected to homozygosity transition, whereas the other clone led to BCR-ABL1 fusion, suggesting that del52CALR and BCR-ABL1 fusion were from the same clone and del52CALR was acquired before BCR-ABL1 fusion. Although other cases did not have data from clonal analysis, they also discussed possible clonal heterogeneity and evolution in their patients.[5,6,10–17] For instance, a case presented by Loghavi et al[10] had fluorescence in situ hybridization data showing BCR-ABL1 fusion signals in 78% of interphased cells in the initial sample, whereas the CALR-mutation allele occurred in 51% cells. Thus, they speculated that the BCR-ABL1-positive subclone could have arisen from a dominant clone with a heterozygous CALR mutation. Moreover, in case 5, the high CALR mutation level, concurrent with BCR-ABL fusion in almost 100% of the bone marrow cells, could be another strong argument in favor of the same clone that harbors both genetic alterations, which helped to shift the ET morphology to that of CML.[13]
However, several studies presented an opposing view, showing presence of 2 different clones or subclones, which can grow independently from each other.[6,14–18] For example, a recent MPN study[11] of concurrent BCR-ABL1 fusion and JAK2 mutation indicated that some JAK2+ MPN cells acquired BCR-ABL1 fusions, whereas others had both genetic drivers at the initial diagnosis, suggesting that 2 independent clones arose from genetically unstable hematopoietic stem and progenitor cells and then competed with each other.
In summary, the majority of previously reported cases of Ph+ and CALR-mutated MPNs showed that BCR-ABL1 fusion emerged from a previously CALR-mutated clone, and throughout the course of TKI treatment, the mutant CALR allele burden remained stable or progressively increased as the BCR-ABL1 transcript declined, indicating that the Ph+/CALR-mutated clones were sensitive to TKIs, whereas Ph−/CALR-mutated clones were resistant to TKIs.
3.4. CALR subtype
Two main types of CALR mutations have been described in the literature to date: the 52-bp deletion (p.L367fs∗46, type 1 mutation) and the 5-bp insertion (p.K385fs∗47, type 2 mutation) in more than 80% of CALR-mutant MPNs.[19] Since the discovery of these mutations, CALR mutations could be grouped into type 1 and type 2 CALR mutations in MPNs according to their predicted effects on the C-terminus of CALR.[20] Pietra et al[20] associated CALR type 1 mutation with a significant increase in the risk of myelofibrotic transformation among ET patients, whereas CALR type 2 mutations are preferentially associated with the ET phenotype, such as a low risk of thrombosis despite a very high platelet count and indolent disease progression, suggesting that these CALR-mutant subtypes correspond to different clinical phenotypes and outcomes. However, it will be interesting to know whether MPNs with concurrent CALR mutation and BCR-ABL1 fusion show the same trend. Indeed, 10 of the 13 cases (76.9%) discussed here had type 1 or type 1-like CALR mutations and exhibited moderate myelofibrosis, whereas 2 of the other 3 cases harbored type 2 CALR mutation and showed an ET phenotype.[6,13] Altogether, these 13 cases of MPNs more frequently had type 1 and type 1-like CALR mutations in PMF, which led to the evolution of a CALR-mutant MPN to Ph+ CML.
3.5. Treatment and response
The 13 patients were all treated with TKIs. After treatment, the BCR-ABL1 transcript levels declined, although the mutant CALR allele burden remained stable or progressively increased. These data indicated that Ph+/CALR-mutated clones were sensitive to TKIs whereas the Ph−/CALR-mutated clones were not, suggesting that a combination of TKIs with cytoreductive medications, such as hydroxyurea and interferon, or with JAK inhibitor ruxolitinib, could effectively control disease progression. However, data concerning the timing, efficacy, and toxicity of TKI use in combination with other cytoreductive medications have not been presented previously, and more data are needed to confirm this notion. Moreover, case 9 received allogeneic hematopoietic stem cell transplantation and achieved complete remission with undetectable CALR mutation and BCR-ABL1 fusion gene, suggesting that hematopoietic stem cell therapy is a useful option to cure this disease, especially in patients with treatment-related refractory disease.
However, given the paucity of data, it is still debatable whether the concurrent presence of CALR mutation indicates a better response to TKIs. Lewandowski et al[21] speculated that simultaneous presence of different molecular alterations could complicate interpretation of the response of CML patients to TKIs. In their recent study, they demonstrated that TKI treatment was effective in CML patients with JAK2 V617 and CALR mutations, and patients achieved major molecular response; however, 3 of 4 such patients still showed thrombocytosis and/or splenomegaly, indicating that TKIs could not control all clinical symptoms of this type of atypical MPN. In other studies, Fava and colleagues[17] reported that 3 CML patients with simultaneous JAK2 mutation and BCR-ABL1 fusion only reached suboptimal responses to TKIs, whereas Boddu et al[5] showed that 2 patients with JAK-V617-mutated CML rapidly progressed to a blast crisis after treatment with TKIs. In another study of 8 MPN patients with concurrent BCR-ABL1 fusion and JAK2 V617F/CALR mutations,[6] only 4 patients achieved an optimal response after imatinib treatment, whereas 3 developed imatinib resistance, 2 of whom had BCR-ABL1 kinase domain mutations. Taken together, these findings suggest that multiple gene alterations could complicate treatment selections and responses.
In the current case series, the response to TKIs among patients with a concurrent CALR mutation depended on the sequential appearance of CML and Ph− MPN. For example, 4 patients with Ph− MPN subsequent to CML all achieved major molecular response within 12 months of TKI treatment, whereas only 1 of the 5 patients with Ph− MPN before CML achieved an optimal response within 12 months of TKI treatment. Ph+ clone could be the predominant clone and sensitive to TKI treatment. However, if BCR-ABL1 fusion occurred after a long period of Ph− MPN phase, such a clone might harbor other genetic aberrations, due to selective pressure from other drug treatments, for example, hydroxyurea, leading to an unfavorable treatment response. However, this interpretation needs to be confirmed by DNA sequencing of each clonal sample in the future.
4. Conclusion
In this case report and review of 12 additional cases with concurrent CALR-mutation and BCR-ABL1 fusion, we highlighted the importance of integrated clinical, morphologic, and molecular genetic data for classifying atypical myeloid neoplasms for treatment and responses of patients. The coexistence of these gene alterations should be thoroughly investigated in cases of CML with persistent thrombocytosis, uncharacteristically advanced myelofibrosis, unusual megakaryocyte morphology, or failure to achieve an optimal response to primary therapy. So far, most studies have reported that BCR-ABL1 fusion emerged from a previous CALR-mutated clone and that the Ph+/CALR-mutated clone, but not the Ph−/CALR-mutated clone, was sensitive to TKI treatment, suggesting that a combination of the TKIs with cytoreductive medications may be effective. Our current study lacks survival data; thus, there is a need for further investigation of the effects of these gene alterations and treatment options on the survival of MPN patients.
Author contributions
CL designed the study and was involved in revising the manuscript. CW performed the data collection and was involved in writing the manuscript. RH performed all the molecular analysis for this patient. ZD performed the bone marrow aspiration and biopsy examination. MA provided more detailed information on case 4, respectively. All authors reviewed and approved the final version of the manuscript.
Data curation: Chunshui Liu, Manuel Abecasis, Cong Wang.
Formal analysis: Chunshui Liu, Cong Wang.
Funding acquisition: Cong Wang.
Investigation: Chunshui Liu, Ruiping Hu, Zhonghua Du, Cong Wang.
Methodology: Chunshui Liu, Ruiping Hu, Zhonghua Du, Cong Wang.
Project administration: Chunshui Liu, Cong Wang.
Resources: Cong Wang.
Software: Chunshui Liu, Ruiping Hu, Zhonghua Du, Manuel Abecasis, Cong Wang.
Supervision: Ruiping Hu, Cong Wang.
Validation: Cong Wang.
Visualization: Cong Wang.
Writing – original draft: Chunshui Liu.
Writing – review & editing: Cong Wang.
Footnotes
Abbreviations: CALR = calreticulin, ET = essential thrombocytosis, IS = international scale, JAK2 = Janus kinase 2, MPL = myeloproliferative leukemia protein, MPNs = myeloproliferative neoplasms, PMF = primary myelofibrosis, TKI = tyrosine kinase inhibitor.
How to cite this article: Liu C, Hu R, Du Z, Abecasis M, Wang C. Atypical myeloproliferative neoplasm with concurrent BCR-ABL1 fusion and CALR mutation: a case report and literature review. Medicine. 2020;99:5(e18811).
Funding: This work was supported by grants obtained from research program from the Education Department of Jilin Province (grant no. JJKH20180208KJ), the Natural Science Foundation of Jilin Province (grant no. 20190201041JC), and the Health Foundation of Jilin Province (grant no. 2018J065).
Ethical approval and consent to participate: This study was approved by the ethics committee of The First Hospital of Jilin University. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki Declaration and its subsequent amendments or comparable ethical standards. Written informed consent was obtained from individual participants.
Patient consent for publication: All data published here are under the consent for publication. Written informed consent was obtained from all individual participants included in the study.
Data availability: The datasets generated and analyzed during the present study are available from the corresponding author on reasonable request.
The authors report no conflicts of interest.
References
- [1].Nangalia J, Green AR. Myeloproliferative neoplasms: from origins to outcomes. Blood 2017;130:2475–83. [DOI] [PubMed] [Google Scholar]
- [2].Tefferi A, Thiele J, Vannucchi AM, et al. An overview on CALR and CSF3R mutations and a proposal for revision of WHO diagnostic criteria for myeloproliferative neoplasms. Leukemia 2014;28:1407–13. [DOI] [PubMed] [Google Scholar]
- [3].Nangalia J, Massie CE, Baxter EJ, et al. Somatic CALR mutations in myeloproliferative neoplasms with nonmutated JAK2. N Engl J Med 2013;369:2391–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Klampfl T, Gisslinger H, Harutyunyan AS, et al. Somatic mutations of calreticulin in myeloproliferative neoplasms. N Engl J Med 2013;369:2379–90. [DOI] [PubMed] [Google Scholar]
- [5].Boddu P, Chihara D, Masarova L, et al. The co-occurrence of driver mutations in chronic myeloproliferative neoplasms. Ann Hematol 2018;97:2071–80. [DOI] [PubMed] [Google Scholar]
- [6].De Roeck L, Michaux L, Debackere K, et al. Coexisting driver mutations in MPN: clinical and molecular characteristics of a series of 11 patients. Hematology 2018;23:785–92. [DOI] [PubMed] [Google Scholar]
- [7].Kandarpa M, Wu YM, Robinson D, et al. Clinical characteristics and whole exome/transcriptome sequencing of coexisting chronic myeloid leukemia and myelofibrosis. Am J Hematol 2017;92:555–61. [DOI] [PubMed] [Google Scholar]
- [8].Nafe R, Holgado de Colombo S, Choritz H, et al. Morphometry of megakaryocytes for supporting the histologic diagnosis of chronic myeloproliferative diseases [in German]. Pathologe 1995;16:34–40. [DOI] [PubMed] [Google Scholar]
- [9].Vardiman J. Chronic myelogenous leukemia, BCR-ABL1+. Am J Clin Pathol 2009;132:250–60. [DOI] [PubMed] [Google Scholar]
- [10].Loghavi S, Pemmaraju N, Kanagal-Shamanna R, et al. Insights from response to tyrosine kinase inhibitor therapy in a rare myeloproliferative neoplasm with CALR mutation and BCR-ABL1. Blood 2015;125:3360–3. [DOI] [PubMed] [Google Scholar]
- [11].Soderquist CR, Ewalt MD, Czuchlewski DR, et al. Myeloproliferative neoplasms with concurrent BCR-ABL1 translocation and JAK2 V617F mutation: a multi-institutional study from the bone marrow pathology group. Mod Pathol 2018;31:690–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cabagnols X, Cayuela J, Vainchenker W. A CALR mutation preceding BCR-ABL1 in an atypical myeloproliferative neoplasm. N Engl J Med 2015;372:688–90. [DOI] [PubMed] [Google Scholar]
- [13].Bonzheim I, Mankel B, Klapthor P, et al. CALR-mutated essential thrombocythemia evolving to chronic myeloid leukemia with coexistent CALR mutation and BCR-ABL translocation. Blood 2015;125:2309–11. [DOI] [PubMed] [Google Scholar]
- [14].Zhou A, Knoche EM, Engle EK, et al. Concomitant JAK2 V617F-positive polycythemia vera and BCR-ABL-positive chronic myelogenous leukemia treated with ruxolitinib and dasatinib. Blood Cancer J 2015;5:e351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Hussein K, Bock O, Seegers A, et al. Myelofibrosis evolving during imatinib treatment of a chronic myeloproliferative disease with coexisting BCR-ABL translocation and JAK2V617F mutation. Blood 2007;109:4106–7. [DOI] [PubMed] [Google Scholar]
- [16].Cambier N, Renneville A, Cazaentre T, et al. JAK2V617F-positive polycythemia vera and Philadelphia chromosome-positive chronic myeloid leukemia: one patient with two distinct myeloproliferative disorders. Leukemia 2008;22:1454–5. [DOI] [PubMed] [Google Scholar]
- [17].Dogliotti I, Fava C, Serra A, et al. CALR-positive myeloproliferative disorder in a patient with Ph-positive chronic myeloid leukemia in durable treatment-free remission: a case report. Stem Cell Investig 2017;4:57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Martin-Cabrera P, Haferlach C, Kern W, et al. BCR-ABL1-positive and JAK2 V617F-positive clones in 23 patients with both aberrations reveal biologic and clinical importance. Br J Haematol 2017;176:135–9. [DOI] [PubMed] [Google Scholar]
- [19].Merlinsky TR, Levine RL, Pronier E. Unfolding the role of calreticulin in myeloproliferative neoplasm pathogenesis. Clin Cancer Res 2019;25:2956–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Pietra D, Rumi E, Ferretti VV, et al. Differential clinical effects of different mutation subtypes in CALR-mutant myeloproliferative neoplasms. Leukemia 2016;30:431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Lewandowski K, Gniot M, Wojtaszewska M, et al. Coexistence of JAK2 or CALR mutation is a rare but clinically important event in chronic myeloid leukemia patients treated with tyrosine kinase inhibitors. Int J Lab Hematol 2018;40:366–71. [DOI] [PubMed] [Google Scholar]
- [22].Diamond JM, de Almeida AM, Belo HJ, et al. CALR-mutated primary myelofibrosis evolving to chronic myeloid leukemia with both CALR mutation and BCR-ABL1 fusion gene. Ann Hematol 2016;95:2101–4. [DOI] [PubMed] [Google Scholar]
- [23].Klairmont MM, Cheng J, Schwartzberg L, et al. Chronic myeloid leukemia, BCR-ABL1-positive with CALR and MPL mutations. Int J Lab Hematol 2018;40:e41–2. [DOI] [PubMed] [Google Scholar]
- [24].Xia D, Hsi ED, Dal Cin P, et al. Composite chronic myeloid leukemia and essential thrombocythemia with BCR-ABL1 fusion and CALR mutation. Am J Hematol 2019;94:504–5. [DOI] [PubMed] [Google Scholar]
- [25].Seghatoleslami M, Ketabchi N, Ordo A, et al. Coexistence of p190 BCR/ABL transcript and CALR 52-bp deletion in chronic myeloid leukemia blast crisis: a case report. Mediterr J Hematol Infect Dis 2016;8:e2016002. [DOI] [PMC free article] [PubMed] [Google Scholar]