

Abstract
Breast tumor progression is a complex process involving intricate crosstalk between the primary tumor and its microenvironment. In the context of breast tumor-lymphatic interactions, it is unclear how breast cancer cells alter the gene expression of lymphatic endothelial cells and how these transcriptional changes potentiate lymphatic dysfunction. Thus, there is a need for in vitro lymphatic vessel models to study these interactions. In this work, we developed a tumor-lymphatic microfluidic model to study the differential conditioning of lymphatic vessels by estrogen receptor-positive (i.e., MCF7) and triple-negative (i.e., MDA-MB-231) breast cancer cells. The model consisted of a lymphatic endothelial vessel cultured adjacently to either MCF7 or MDA-MB-231 cells. Quantitative transcriptional analysis revealed expression changes in genes related to vessel growth, permeability, metabolism, hypoxia, and apoptosis in lymphatic endothelial cells co-cultured with breast cancer cells. Interestingly, these changes were different in the MCF7-lymphatic co-cultures as compared to the 231-lymphatic co-cultures. Importantly, these changes in gene expression correlated to functional responses, such as endothelial barrier dysfunction. Our results collectively demonstrate the utility of our model for studying breast tumor-lymphatic crosstalk for multiple breast cancer subtypes.
Keywords: microfluidic, lymphatic, breast cancer, estrogen receptor-positive, triple-negative
Introduction
Tumor progression towards metastatic disease involves intricate crosstalk between the primary tumor and its microenvironment. Numerous cellular, molecular, and biophysical factors contribute to this process, including endothelial vessels, stromal fibroblasts and macrophages, growth factors, chemokines, and extracellular matrix proteins [1]. Of these factors, blood and lymphatic vessels provide the underlying physical network of ‘super highways’ for tumor cell dissemination. There is strong clinical evidence demonstrating the initial spread of breast cancer cells to regional lymph nodes prior to distant metastasis, where patients with more metastatic lymph nodes typically have poorer 5-year survival rates [2]. Therefore, lymphatic metastasis is a critical prognostic indicator of patient outcome. In this context, lymphatic vessels are potentially more advantageous routes than blood vessels given their leakier endothelium and natural fluid draining function[3]. Given their role in breast cancer progression, lymphatic vessels are pertinent targets for cancer therapy.
To better understand the biology of lymphatic metastasis in breast cancer and identify potential therapeutics, there has been significant effort in advancing our knowledge of tumor-lymphatic interactions in the breast tumor microenvironment. Multiple studies have established that tumor lymphangiogenesis, in response to growth factors (e.g. VEGF-C and VEGF-D) secreted by cancer cells and stromal cells, provides the physical vessel network for lymphatic spread[3, 4]. Growing evidence also indicates that tumor-associated lymphatic vessels regulate host immunity by suppressing T cell function, helping tumors to escape immune surveillance[5]. Moreover, these vessels play an active role in tumor evolution since they have the potential to enhance tumor invasion by overexpressing chemotactic signals (e.g., CCL21, CXCL12, and CX3CL1) [6]. One recent study further demonstrated that MDA-MB-231 cells condition lymphatic endothelial cells to overexpress CCL5 as a mechanism for tumor invasion[7]. Despite the collective progress in understanding breast tumor-lymphatic interactions, it remains unclear how breast cancer cells alter the gene expression of lymphatic endothelial cells and how these changes contribute to lymphatic dysfunction. Models that enable the investigation of these questions would be instrumental for basic and translational breast cancer research.
Current methods for modeling tumor-lymphatic interactions include in vitro cell culture on plastic surfaces and in 3D gels, or animal models[8]. Animal models provide a more physiological microenvironment, however, their increased complexity reduces experimental tractability. While conventional in vitro cell culture methods offer high-throughput analysis, they commonly fail to capture the in vivo structure-function relationships that influence cell behavior. Recent advances in microfluidic cell culture have enabled the development of in vitro models known as organs-on-a-chip that enable physiological modeling without sacrificing tractability[9, 10]. However, very few microfluidic models available for investigating breast tumor-lymphatic interactions. Only two studies have evaluated the importance of lymphangiogenesis and the invasion of breast tumor cells into lymphatic vessels[11].
Thus, here we developed a tumor-lymphatic microfluidic model to study the crosstalk between estrogen-receptor (ER)-positive (e.g., MCF7) and triple-negative (e.g., MDA-MB-231) breast cancer cells and lymphatic endothelial cells. The model consisted of a tubular lymphatic vessel cultured adjacent to a lumen fill with breast cancer cells in a collagen hydrogel. Gene expression analysis of lymphatic cells co-cultured with MCF7 or MDA-MB-231 cells revealed differential alteration of genes involved in vessel growth, permeability, metabolism, hypoxia, and apoptosis. Importantly, these transcriptional changes correlated to functional changes in endothelial barrier capacity, where MCF7-conditioned vessels had significantly leakier endothelia. Collectively, these results demonstrate the utility of our model for investigating breast tumor-lymphatic crosstalk and offer insight into the conditioning capabilities of different breast cancers.
Results and Discussion
Development of the breast tumor-lymphatic microfluidic model
Currently, there is a lack of physiologically relevant models for studying tumor-lymphatic interactions. Advances in microfluidic cell culture have offered platforms for 3D cell and tissue culture in physiological microenvironments to fill this gap. Specifically, models that recapitulate the tubular structure of endothelial vessels not only provide relevant tissue geometry, but also capture critical structure-function relationships.
In this work, we developed a tumor-lymphatic microfluidic device for co-culturing tubular lymphatic vessels and breast cancer cells to study their crosstalk. The design of the device was based on previous methods for making endothelial vessels in natural extracellular matrix (ECM) hydrogel and for co-culturing these vessels with breast cancer cells[10, 12–14]. The device comprised a chamber to inject an unpolymerized collagen type I hydrogel and two adjacent PDMS rods suspended in the chamber (Figure 1A). Empty lumens were formed in the microfluidic device after polymerizing the hydrogel around the lumen rods, followed by removing the rods (Figure 1B). Primary human lymphatic endothelial cells (HLECs) were seeded in one lumen to generate a 3D lymphatic vessel (Figure 1C) and either MCF7 or MDA-MB-231 cells were seeded in the adjacent lumen to form a breast duct filled with cancer cells (Figure 1D–F). The endothelial and lymphatic lineages of the lymphatic vessels were confirmed by immunofluorescence staining for vascular endothelial cadherin and lymphatic vessel endothelial hyaluronan receptor-1 (Figure S1).
Figure 1.
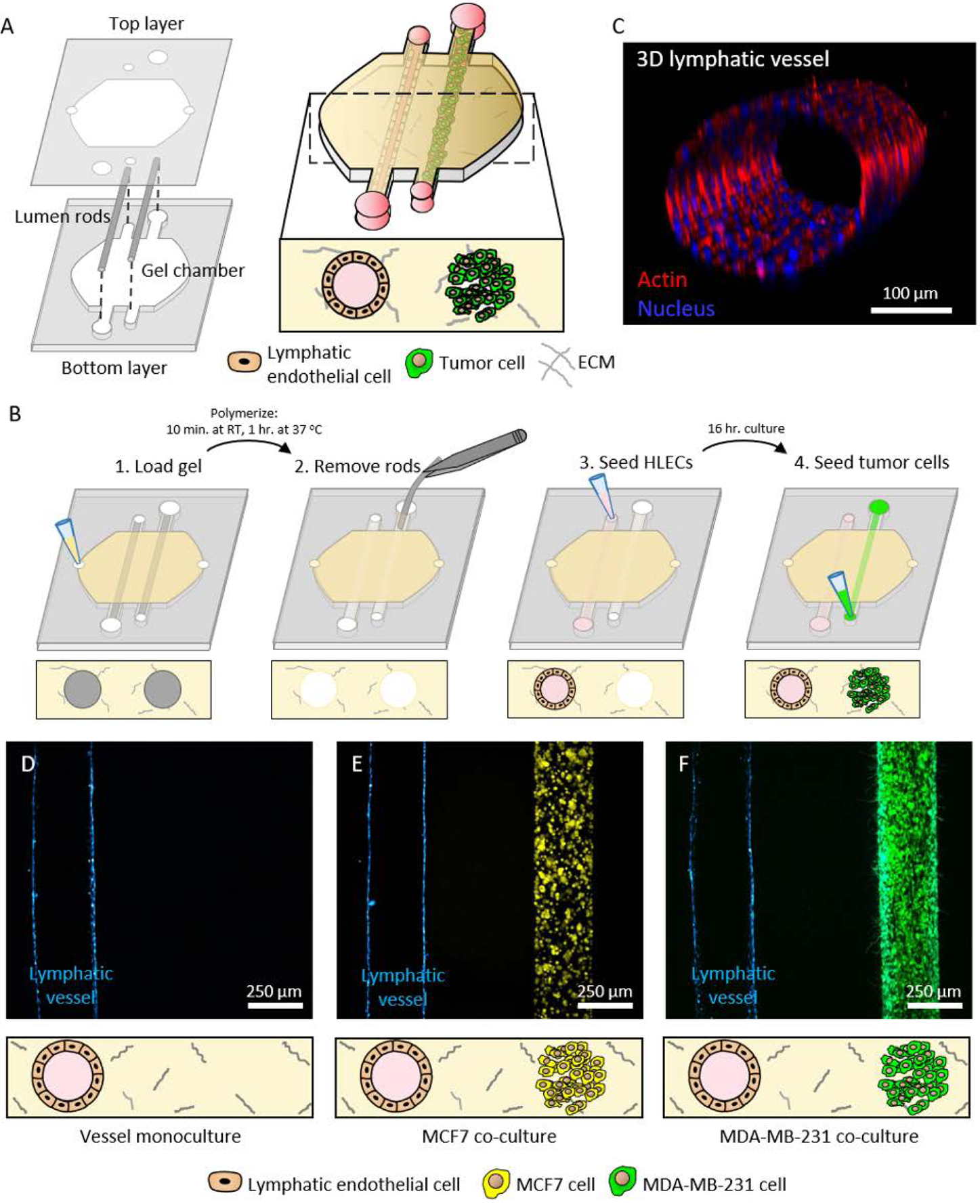
Concept and operation of the breast tumor-lymphatic microfluidic device. A) The device consists of two polydimethylsiloxane (PDMS) layers. The bottom layer comprises a chamber and microchannels for suspending two adjacent lumen rods. The top layer contains the ports for fluid exchange. Cell culture in the device allows the formation of a lumen lined with primary human lymphatic endothelial cells (HLECs) and a second lumen filled with breast cancer cells. B) Illustration of key steps in the operation of the co-culture device. C) Representative confocal image of a 3D lymphatic vessel. D-F) Representative images of vessel monoculture, co-culture with MCF7 cells (in yellow), and co-culture with MDA-MB-231 cells (in green) in the device.
MCF7 and MDA-MB-231 cells differentially alter lymphatic gene expression
Lymphatic endothelial cells were co-cultured with MCF-7 or MDA-MB-231 cells to evaluate the potential alterations in the lymphatic vessel. After 24 hours of co-culture, the hydrogel was sliced in half to remove the lumen with the breast cancer cells and the lymphatic lumen was analyzed by RT-qPCR. The expression of genes related with multiple pathways was analyzed in the co-culture and normalized with the lymphatic lumen monoculture. Given the critical role of tumor-induced lymphangiogenesis in tumor metastasis, we set out to evaluate the expression of lymphangiogenesis-related genes in lymphatic cells (Figure 2A). The results showed the co-culture with MCF7 led to a significant overexpression of several lymphangiogenesis-related genes, including secreted growth factors like fibroblast growth factor 2 (FGF2), vascular endothelial growth factor C (VEGF-C), angiopoietin 2 (ANGPT2), and placental growth factor (PGF)[4]. These factors are amongst the master key regulators of lymphangiogenesis, promoting neo-vascularization, vessel growth and maturation. Altogether these findings suggested that MCF7 cells triggered a lymphangiogenic response in the conditioned lymphatic vessel. We also observed changes in the expression of cell surface receptors in the lymphatic cells, such as VEGF receptor 1 (FLT-1) and 2 (KDR). Interestingly, while FLT-1 was upregulated, KDR was downregulated, suggesting FLT-1 was leading the pro-lymphangiogenic response. On the other hand, the co-culture with MDA-MB-231 cells did not affect the expression of any of the previously described genes, leading only to a minor increase in the expression the chemokine CCL2. Thus, these results demonstrate that breast cancer subtype (e.g. ER-positive vs triple negative) is important to the regulation of lymphangiogenic pathways, and that our model could be used to identify critical pathways as potential therapeutic targets.
Figure 2.

Influence of MCF7 and MDA-MB-231 cells on lymphatic lumen gene expression. A-H) Lymphatic endothelial lumens were co-cultured alone and in the presence of MCF7 or MDA-MB-231 cells for 24 hours. Then, lymphatic endothelial cells were isolated and gene expression was analyzed. The graphs show the genes that were differentially expressed when lymphatic endothelial cells were co-cultured with MCF7 (blue columns) or MDA-MB-231 (red columns). I) Clustergram showing the changes in all the genes analyzed. The clustering algorithm revealed the co-culture with MCF7 led to a more different gene expression signature. J) Radar plot showing the number of genes affected in HLECs by the presence of MCF7 (in blue) and MAD-MB-231 (in red).
Moreover, nutrient starvation and hypoxia are amongst the main inducers of lymphangiogenesis[15]. We analyzed the expression of multiple genes associated with these pathways. The co-culture with MCF7 cells led to changes in genes related with metabolic activity in lymphatic cells (Figure 2B). The ATP Synthase F1 Subunit Alpha (ATP5FA1) and cytochrome C Oxidase Subunit 5A genes were upregulated, these genes are deeply involved in energy production (i.e., ATP synthesis) at the mitochondria. Following a similar trend, glycerol-3-phosphate dehydrogenase 2 (GPD2) was upregulated, which generates glycerol as potential nutrient and controls redox potential. Glycerol production by GPD2 is especially relevant in tissues with high fat content (e.g., breast tissue), since glycerol can be an alternative nutrient source when glucose or other nutrients are depleted. In this context, the observed glucose 6 phosphate dehydrogenase (G6PD) downregulation suggest a glucose-depleted environment, forcing the cells to lymphatic cells to adapt their redox balance through other genes (e.g., GPD2). On the other hand, the co-culture with MDA-MB-231 cells led to different adaptations, showing upregulation of fatty acid metabolism through ATP citrate lyase (ACLY) and carnitine palmitoyl transferase 2 (CPT2). Regarding hypoxia, the co-culture with MCF7 cells induced the overexpression of aryl hydrocarbon receptor nuclear translocator (ARNT, also known as HIF-1β), a key regulator of the hypoxia cellular response, and solute carrier family 2 member 1 (SLC2A1, also known as GLUT1), a canonical glucose transporter that is overexpressed under hypoxic conditions (Figure 2C). Adrenomedullin (ADM) and heme oxygenase 1 (HMOX1) are a vasodilator peptide hormone and an enzyme involved in heme metabolism respectively, and these molecules are commonly overexpressed in blood after a hypoxic event. However, their role in the lymphatic system is poorly understood, highlighting the potential of the model to decipher the specific interaction of these molecules in the lymphatic vessels.
Previous studies have suggested that during the angiogenic process, blood vessel endothelial cells switch from an epithelial phenotype to a more migratory and invasive mesenchymal phenotype (process known as epithelial-mesenchymal transition, EMT)[16]. However, the EMT process has not been well characterized in lymphatic endothelial cells. Here, we observed that the culture with MCF7 cells activated several of the traditionally EMT-associated genes, such as the intermediate filaments desmoplakin (DSP) and keratin 14 (KRT14) (Figure 2D). The presence of MCF-7 also led to an increase in proliferation-associated genes including ki67 (MKI67), a canonical proliferation marker, and aurora kinase A (AURKA) (Figure 2E). In a similar trend, MCF7 cells reduced the expression of apoptosis pathways (e.g., reduced caspase expression), suggested by the downregulation of caspase 7 (CAS7) and Apoptotic Peptidase Activating Factor 1 (APAF1) (Figure 2F). On the other hand, MDA-MB-231 seemed to have little effect on EMT transition and induced a less proliferative state by upregulating the tumor suppressor gene E2F4. Finally, the co-cultures with MCF7 and MDA-MB-231 cells led to dysregulation in the DNA stability and repair of the lymphatic cells (Figure 2G and H).
Taken together, the transcriptional analysis results suggested that the co-culture with MCF7 cells affected the expression of a larger number of genes, leading to a pro-lymphangiogenic and hypoxic phenotype. Additionally, the co-culture with MCF7 increased the expression of genes related with cell proliferation, migratory capacity, and altered metabolism (Figure 2J). Conversely, the co-culture with MDA-MB-231 cells led to different conditioning outcomes, showing effect on a lower number of genes. Furthermore, co-culture with MDA-MB-231 and MCF7 cells lead to different changes, exhibiting unique adaptations (Figure S3 and B). These results highlight how different breast tumor types may condition lymphatic cells through different pathways and the potential utility of the model for identifying new therapeutic targets against the affected pathways (Figure S3C and D).
MCF7 and MDA-MB-231 cells alter lymphatic vessel physiology
After we analyzed how breast cancer cells modulate the molecular profile of lymphatic endothelial cells, we examined potential associated changes in lymphatic vessel physiology. We assessed changes to the physiological function of the MCF7- and MDA-MB-231-conditioned lymphatic vessels by measuring endothelial cell coverage, lymphangiogenic sprouting, and endothelial barrier capacity. Cell coverage was used as a measure of the physical integrity of the endothelium in the presence of breast cancer cells. Lymphatic vessels in both co-culture conditions exhibited no measurable difference in cell coverage as compared the monoculture control (Figure 3A–C). All conditions showed >99% cell coverage with no observable defects, such as holes, in the endothelium. These data demonstrated that the breast cancer cells do not negatively impact the physical integrity of the vessels nor endothelial cell maintenance. However, the presence of breast cancer cells triggered lymphangiogenic sprouting. Both MCF7 and MDA-MB-231 cells induced sprout formation in the vessels (Figure 3B and D). The monoculture controls had no observable sprouts, indicating that crosstalk with the breast cancer cells was a critical factor for initiating lymphangiogenesis in our model.
Figure 3.
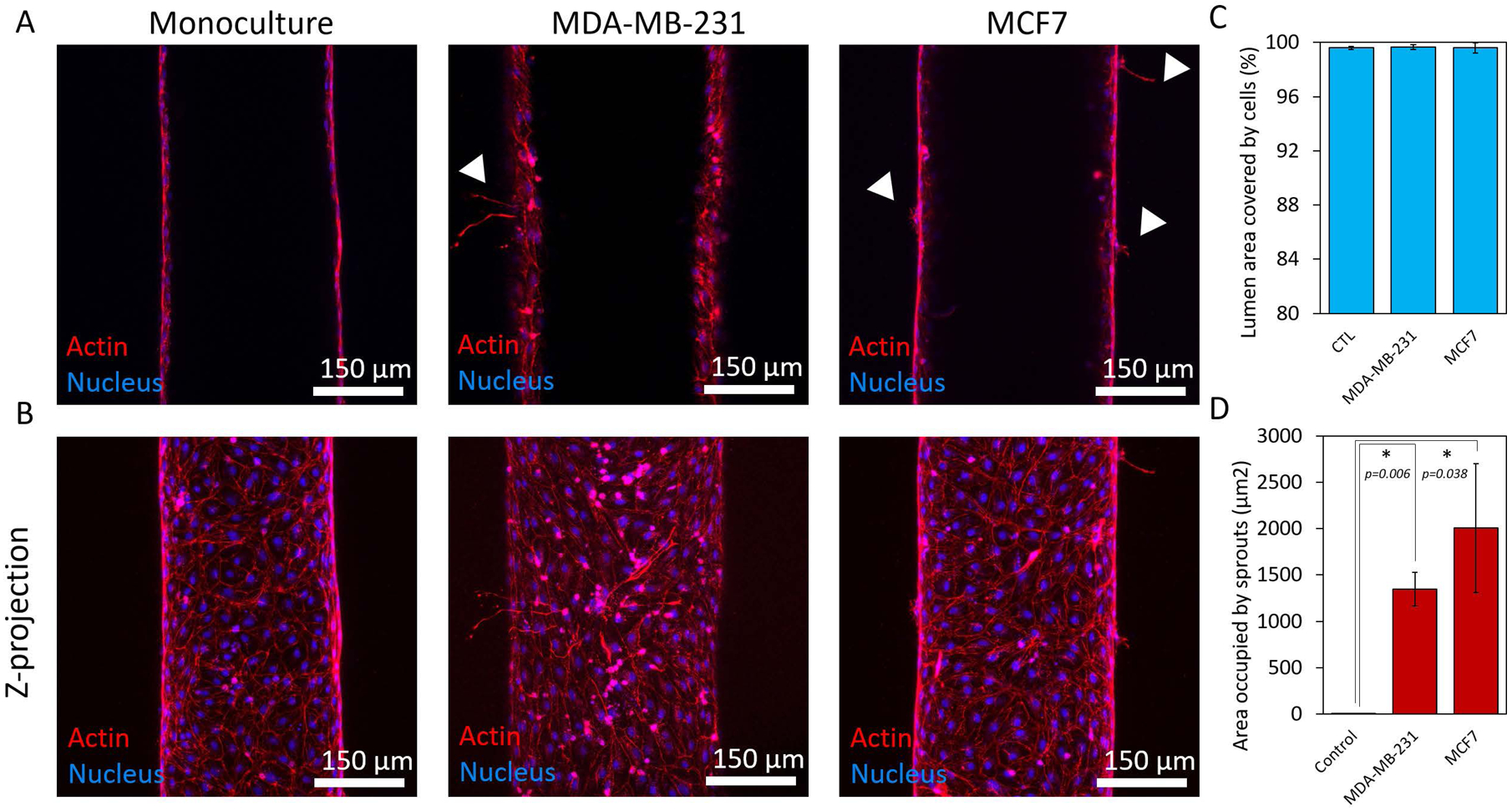
Endothelial cell coverage and lymphangiogenic sprouting. A) Confocal images showing the middle plane of lymphatic vessels in monoculture and co-culture with MCD7 and MDA-MB-231 cells. B) Z-projected images of vessels in monoculture and co-culture with MCF7 and MDA-MB-231 cells. C) Quantification of cell coverage for each culture condition showing an average percentage of cell coverage >99% for all conditions. D) Crosstalk with the breast cancer cells induced lymphangiogenic sprouting in the vessels. There were no observable sprouts in the monoculture control. Three individual vessels (n = 3) were measured for each culture condition to determine the average permeability value (mean ± s.d.).
Although there were no substantial differences in endothelial cell coverage or lymphangiogenic sprouting for the MCF7- and MDA-MB-231-conditioned vessels, the molecular analysis by RT-qPCR revealed that the cancer cells altered the expression of growth factors related to vessel permeability, such as VEGF-C. Thus, we assessed the barrier capacity of the lymphatic vessels by quantifying the diffusion of a glucose analog and 70 kDa dextran, which is representative of serum albumin. For both solutes, the conditioned vessels had significantly higher permeability as compared to the monoculture controls, indicating impaired barrier function. Both the MCF7- and MDA-MB-231-conditioned vessels had multiple focal points of leakage in their endothelia (t = 0 min in Figure 4A and 4B), with dispersed diffusion profiles versus the monocultured vessels (Figure 4C). Interestingly, there was a difference in the degree of barrier impairment between the co-culture conditions as indicated by the measured permeability coefficients (Figure 4D). For glucose diffusion, there was a 2.6-fold increase for MCF7-conditioned vessels and a 1.8-fold increase for MDA-MB-231-conditioned vessels against the monoculture control. Similarly for dextran diffusion, there was a higher fold increase in vessel permeability for the MCF7 co-culture versus the MDA-MB-231 co-culture (6.1-fold vs 4.4-fold). Importantly, these data correlated with the increased expression of lymphangiogenic genes related to vessel permeability (e.g. ANGPT2, VEGF-C) for the MCF7 co-culture (Figure 2A). These genes were not upregulated in the MDA-MB-231 co-culture. Our results present a different perspective on the conditioning capabilities of MCF7 cells. In general, MCF7 cells are considered poorly invasive and thus, less of an effector compared with the more invasive MDA-MB-231 cells[7]. The current study suggests that there remains much to elucidate in terms of breast tumor-lymphatic crosstalk to fully understand the conditioning potential of different breast cancer subtypes. Collectively, our results demonstrate that different breast cancer subtypes alter lymphatic vessel physiology to different degrees and that our model can be used to better understand the tumor-lymphatic crosstalk that contributes to lymphatic vessel dysfunction. Previous studies have analyzed the chemokines secreted by breast cancer cells, including MCF7 and MDA-MB-231 cells[12, 17]. These studies have identified multiple chemokines that could be driving the lymphangiogenic process observed in the microfluidic device. Thus, selective inhibitors targeting these chemokines could be used alone or in combination to block the lymphangiogenic process induced by cancer cells. Additionally, future studies could explore the potential conditioning lymphatic vessels exert on breast cancer cells. In this context, previous studies have identified multiple chemokines (e.g., IL-8, G-CSF, follistatin, etc.) produced by lymphatic cells that could impact breast cancer cell biology[18]. In conclusion, these studies could lead to new potential therapies to prevent metastasis
Figure 4.
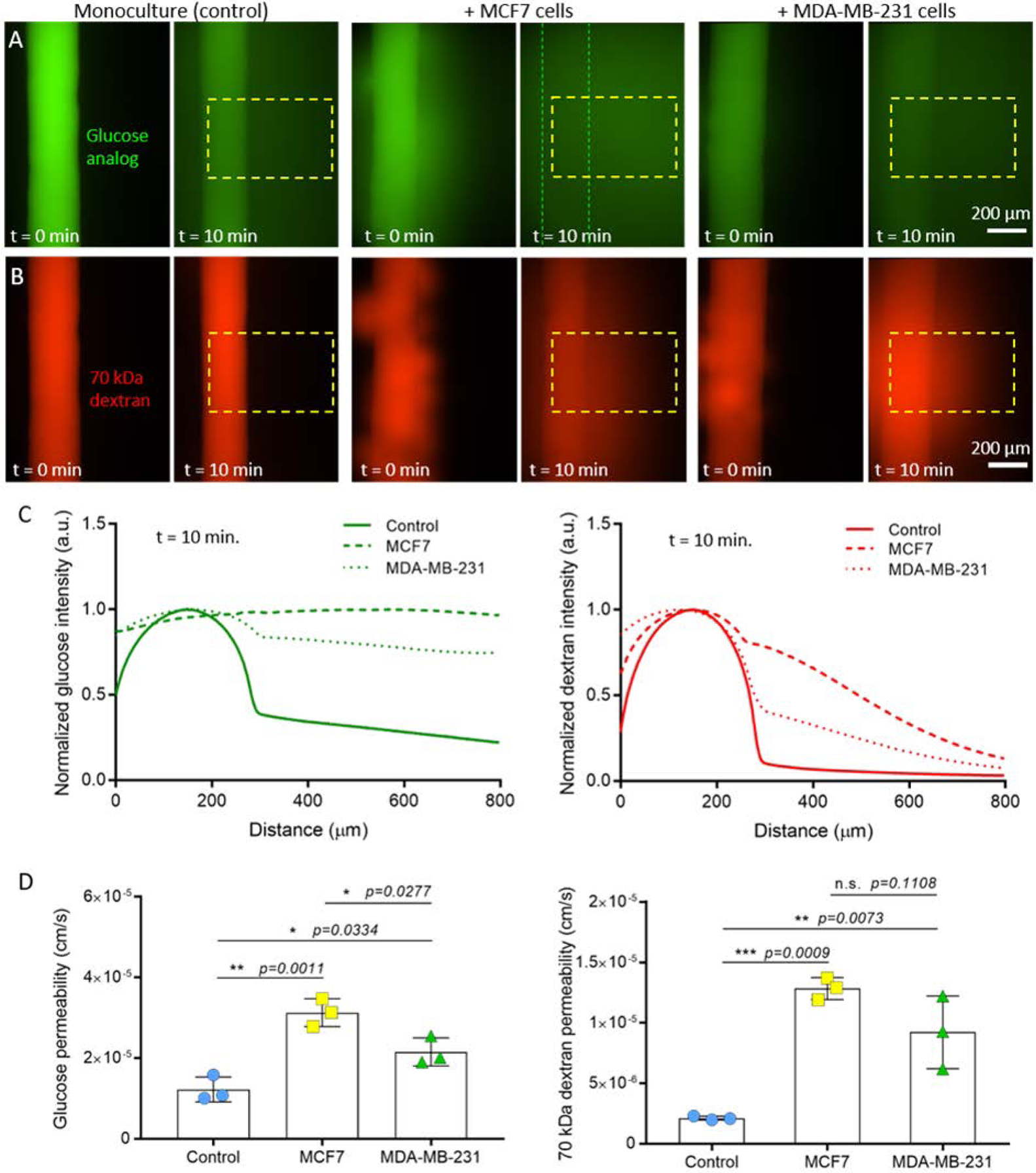
Vessel permeability in monoculture and co-culture. Representative images at t = 0 min. and t = 10 min. for diffusion of A) a glucose analog and B) 70 kDa dextran. Dashed boxes indicate the region of interest used for measuring diffusion profiles. Dashed lines indicate the walls of the vessel. C) Diffusion profiles at t = 10 min. for monoculture and co-culture conditions. D) Glucose and dextran permeability values calculated for monoculture and co-culture conditions. MCF7-conditioned vessels were the leakiest in comparison to MDA-MB-231-conditioned vessels and monoculture controls. Three individual vessels (n = 3) were measured for each culture condition to determine the average permeability value (mean ± s.d.).
Conclusion
Lymphatic vessels are intimately involved in breast cancer progression. Improved understanding of how breast cancer cells condition lymphatic vessels towards metastasis could, ultimately, advance therapy options and patient outcomes. We developed a breast tumor-lymphatic model to examine the crosstalk between lymphatic vessels, MCF7 cells, and MDA-MB-231 cells. This model showed the profound conditioning that breast cancer cells can exert on the lymphatic vasculature, affecting multiple pathways such as angiogenesis, hypoxia response, metabolism and metabolite transport, EMT or even DNA stability and repair. The presence of breast cancer cells also triggered different functional responses in the lymphatic vasculature, leading to the generation of new lymphatic sprouts from the pre-existing vessel and increasing vascular permeability. Additionally, breast cancer is a highly heterogenous disease (e.g., ER+, PR+, HER+, TNBC) and tumor evolution, treatment and even patient outcome is highly dependent on the specific breast cancer subtype considered. Using this model, we observed that different breast cancer cell types led to different adaptations in the lymphatic vasculature, highlighting again the heterogeneity of this disease. Thus, this model could be used to decipher the role of different breast cancer subtypes on lymphatic vessel dysfunction, helping to determine the optimal treatment for individual patients.
Methods
Cell culture
MCF7 (ATCC) and MDA-MB-231 (ATCC) cells were cultured in standard cell culture flasks and maintained in RPMI 1640 (BE12–702F, Lonza) supplemented with 10% FBS (97068–085, VWR). Human lymphatic endothelial cells (HLECs) isolated from the lymph node (2500, ScienCell) were cultured in standard cell culture flasks coated with fibronectin (5μg/cm2, F1141–5MG, Sigma Aldrich) at a starting cell concentration of 5 × 105 as per supplier instructions. HLECs were maintained with Endothelial Basal Medium-2 (CC-3156, Lonza) supplemented with EGM-2 MV SingleQuot Kit (CC-4147, Lonza) and used at 95% confluency at passages 3 and 4 for all experiments.
Microfluidic device concept and fabrication
The tumor-lymphatic microfluidic model is a two-layer polydimethylsiloxane (PDMS)-based device fabricated using soft lithography (Fig. 1A). Its design is based on previously developed methods for generating lumens in ECM hydrogel and co-culturing these lumens with tumor cells[12, 14]. The basic device concept consists of two adjacent PDMS rods suspended in a gel chamber, such that they can be encapsulated in ECM gel and removed after gel polymerization to form separate hollow lumens. Ports on either side of the gel chamber enable gel loading into the device. Ports flanking the lumen rods allow fluid exchange through the hollow lumens after the rods are removed. Lumen rods were made by curing PDMS in 25-gauge hypodermic needles (inner diameter ~250 μm, 14-840-84, Fisher Scientific). Fully-assembled devices were bonded to glass-bottom culture dishes (P50G-1.5–30-F, MaTek Corporation) and stored until use.
Co-culture in the microfluidic device
All microfluidic devices were UV sterilized for 15 min. and used in a sterile biosafety hood. Figure 1C illustrates the key steps for co-culturing lymphatic vessels and tumor cells in the device. Prior to gel loading, devices were 1) incubated with 2% polyethyleneimine solution (03880, Sigma-Aldrich) at room temperature for 10 min, 2) incubated with 0.1% glutaraldehyde solution (G6257, Sigma-Aldrich) at room temperature for 30 min, and 3) aspirated and washed with sterile deionized water (5 times per device). These solutions were added to the devices to minimize collagen delamination. Collagen type I gel solution was loaded into the gel chamber of each device (6 μL per device), incubated at room temperature for 10 min. to initiate polymerization, and then transferred to 37 °C for 1 hour. The collagen solution was prepared on ice by diluting stock rat-tail collagen type I gel solution (354249, BD Biosciences) in sterile PBS to a final concentration of 3 mg/mL and neutralized using 0.5 M NaOH.
Following collagen gel polymerization, lumen rods were manually removed using a tweezer to generate the empty lumens. One empty lumen was incubated with fibronectin solution (33 μg/mL, 10 μL per lumen) at room temperature for 30 min. to support cell adhesion, followed by the addition of HLECs at a concentration of 20,000 cells/μL (4 μL per lumen). This lymphatic lumen was cultured overnight at 37 °C. After overnight culture, the remaining empty lumen was seeded with either MCF7 or MDA-MB-231 cells at 40,000 cells/μL (4 μL per lumen) depending on the co-culture condition. Cultures were maintained in HLEC media with two media exchanges on a daily basis for up to five days.
Diffusion assays
Vessel permeability was assessed by measuring the diffusion of a glucose analog, 2-NBDG (2-(N-(7-Nitrobenz-2-oxa-1,3-diazol-4-yl)Amino)-2-Deoxyglucose) (NBDG) (N13195, Thermo Fisher), and 70 kDa Texas Red-dextran (D1830, ThermoFisher) across the lymphatic vessel endothelium. A mixed solution of glucose analog and dextran was prepared at 200 and 100 μM respectively. A volume of 3 μL of the solution was added to each vessel such that fluid was flush with the ports to minimize flow from a pressure head. Diffusion was imaged with the Nikon TI® Eclipse inverted microscope every minute over 10 minutes. The permeability coefficient was calculated using the following relationship between fluorescence intensity and the diameter of the vessel, which is an adaptation of a previously defined equation[19]:
where If-out is the intensity of solute outside the vessel at 10 minutes, If-in is the intensity of solute inside the vessel at 10 minutes, to is the initial time point, tf is the final time point of 10 minutes, and D is vessel diameter.
Microscopy and image analysis
Cells were visualized with a Leica SP8 3X STED super-resolution confocal microscope equipped with a super-continuum white-light laser for fluorescent excitation from 470nm to 670nm, and a separate 405nm diode laser. The unit is equipped with 3 PMTs and 2 high-sensitive HyD detectors for image collection. Z-stacks of the lymphatic vessels were acquired by taking images every 10 microns at 20X magnification. Diffusion assays were performed in a Nikon TI® Eclipse fluorescence microscope equipped with a stage top incubator set at 37 °C and 5% CO2. Diffusion profiles and lymphatic vessel immunofluorescence images were analyzed with Fiji (https://fiji.sc/).
RT-qPCR
RT-qPCR was used to analyze the expression of multiple lymphatic cell genes altered by co-culture with breast cancer cells. Briefly, after 48 hours in the microfluidic device, the hydrogel was cut in half to separate the lymphatic vessel from the cancer cells. Lymphatic cells were lysated and mRNA was isolated using the Dynabeads™ mRNA DIRECT™ Purification Kit (61011, ThermoFisher). Isolated mRNA was quantified using a Qubit fluorometer (ThermoFisher) and a Qubit™ RNA BR Assay Kit (Q10210, ThermoFisher). mRNA was retrotranscribed to cDNA using the RT2 PreAMP cDNA Synthesis Kit (330451, Qiagen). This kit included a preamplification step that amplifies the amount of cDNA available for downstream RT-qPCR analysis. cDNA was analyzed by RT-qPCR using a Qiagen RT2 profiler custom panel (CLAH25337, Qiagen) and data was analyzed using the Qiagen online software (http://pcrdataanalysis.sabiosciences.com/pcr/arrayanalysis.php). More than 95% of the genes analyzed were detectable in the RT-qPCR experiment. Only genes that exhibited statistically significant differences were discussed in the results section.
Statistical Analysis
RT-qPCR data was analyzed using the online Qiagen Data analysis center (https://www.qiagen.com/us/shop/genes-and-pathways/data-analysis-center-overview-page/). Statistical analysis was conducted using GraphPad Prism 7 (GraphPad Software). Significance tests for the glucose and dextran diffusion data were performed using ordinary one-way ANOVA with Tukey’s multiple comparisons test. The normality of the data was first checked with the Shapiro-Wilk test. Tests were considered significant for p ≤ 0.05. All experiments were repeated at least three times.
Supplementary Material
Figure S1. Immunofluorescent staining of vascular endothelial cadherin (VE-cadherin) and lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1) for the primary human lymphatic endothelial cells in 2D.
Figure S2. Example of a standard glass-bottom culture dish containing six devices used for experiments. Schematic of device layers and components. Fluid handling in the devices is accomplished by using standard micropipettes.
Figure S3. Breast cancer cells condition lymphatic endothelial cell gene expression. A) Waterfall plot showing genes differentially expressed in lymphatic endothelial cells co-cultured with MCF7 cells. B) Similar waterfall plot showing the changes induced by MDA-MB-231 in lymphatic endothelial cells. C) Molecular network highlighting the molecular interactions affected in the MCF7 co-culture. FDA-approved drugs are also shown as yellow hexagons. D) Molecular networks illustrating the interactions affected in the MDA-MB-231 co-culture.
Acknowledgements
The authors acknowledge funding support from the University of Wisconsin Carbone Cancer Center (AAB7173), Morgridge Research Institute, Wisconsin Head and Neck Cancer SPORE (P50DE026787), National Institutes of Health (R01 CA164492; R01 CA185747; and R01 CA205101), Mary Kay Foundation (067-16), Department of Defense Breast Cancer Research Program (W81XWH-13-1-0194), and National Science Foundation (CBET-1642287).
Footnotes
Conflict of Interest
David J. Beebe is a board member and stockowner of Tasso, Inc. and a stockowner of Bellbrook Labs, LLC. David J. Beebe is a founder, stockowner, and consultant of Salus Discovery LLC. David J. Beebe is an advisor and stockowner of Lynx Biosciences, LLC, Onexio Biosystems, LLC, and Stacks to the Future, LLC. David J. Beebe holds equity in Bellbrook Labs, LLC, Tasso Inc., Salus Discovery LLC, Stacks to the Future, LLC and Onexio Biosystems, LLC.
References
- [1].Joyce JA, Pollard JW, Nat Rev Cancer 2009, 9, 239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tang C, Wang P, Li X, Zhao B, Yang H, Yu H, Li C, Plos One 2017, 12, e0182953; [DOI] [PMC free article] [PubMed] [Google Scholar]; Truong PT, Vinh-Hung V, Cserni G, Woodward WA, Tai P, Vlastos G, Eur J Cancer 2008, 44, 1670; [DOI] [PubMed] [Google Scholar]; Shen J, Hunt KK, Mirza NQ, Krishnamurthy S, Singletary SE, Kuerer HM, Meric-Bernstam F, Feig B, Ross MI, Ames FC, Babiera GV, Cancer 2004, 101, 1330. [DOI] [PubMed] [Google Scholar]
- [3].Ran S, Volk L, Hall K, Flister MJ, Pathophysiology : the official journal of the International Society for Pathophysiology 2010, 17, 229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Alitalo A, Detmar M, Oncogene 2012, 31, 4499. [DOI] [PubMed] [Google Scholar]
- [5].Dieterich LC, Ikenberg K, Cetintas T, Kapaklikaya K, Hutmacher C, Detmar M, Frontiers in immunology 2017, 8, 66; [DOI] [PMC free article] [PubMed] [Google Scholar]; Swartz MA, Cancer Immunol Res 2014, 2, 701. [DOI] [PubMed] [Google Scholar]
- [6].Roussos ET, Condeelis JS, Patsialou A, Nat Rev Cancer 2011, 11, 573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lee E, Fertig EJ, Jin K, Sukumar S, Pandey NB, Popel AS, Nat Commun 2014, 5, 4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Servais EL, Colovos C, Bograd AJ, White J, Sadelain M, Adusumilli PS, J Mol Med (Berl) 2011, 89, 753; [DOI] [PMC free article] [PubMed] [Google Scholar]; Shields JD, Fleury ME, Yong C, Tomei AA, Randolph GJ, Swartz MA, Cancer cell 2007, 11, 526; [DOI] [PubMed] [Google Scholar]; Harris AR, Perez MJ, Munson JM, BMC cancer 2018, 18, 718; [DOI] [PMC free article] [PubMed] [Google Scholar]; Katt ME, Placone AL, Wong AD, Xu ZS, Searson PC, Frontiers in bioengineering and biotechnology 2016, 4, 12; [DOI] [PMC free article] [PubMed] [Google Scholar]; Asghar W, El Assal R, Shafiee H, Pitteri S, Paulmurugan R, Demirci U, Mater Today (Kidlington) 2015, 18, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Rothbauer M, Rosser JM, Zirath H, Ertl P, Current opinion in biotechnology 2019, 55, 81; [DOI] [PubMed] [Google Scholar]; Ashammakhi N, Elkhammas E, Hasan A, Journal of biomedical materials research. Part B, Applied biomaterials 2018; [DOI] [PubMed] [Google Scholar]; Bhatia SN, Ingber DE, Nat Biotechnol 2014, 32, 760; [DOI] [PubMed] [Google Scholar]; Huh D, Torisawa YS, Hamilton GA, Kim HJ, Ingber DE, Lab on a chip 2012, 12, 2156. [DOI] [PubMed] [Google Scholar]
- [10].Ayuso JM, Gillette A, Lugo-Cintron K, Acevedo-Acevedo S, Gomez I, Morgan M, Heaster T, Wisinski KB, Palecek SP, Skala MC, Beebe DJ, EBioMedicine 2018, 37, 144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Pisano M, Triacca V, Barbee KA, Swartz MA, Integrative biology : quantitative biosciences from nano to macro 2015, 7, 525; [DOI] [PubMed] [Google Scholar]; Kim S, Chung M, Jeon NL, Biomaterials 2016, 78, 115; [DOI] [PubMed] [Google Scholar]; Shim S, Belanger MC, Harris AR, Munson JM, Pompano RR, Lab on a chip 2019, 19, 1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Ayuso JM, Truttschel R, Gong MM, Humayun M, Virumbrales-Munoz M, Vitek R, Felder M, Gillies SD, Sondel P, Wisinski KB, Patankar M, Beebe DJ, Skala MC, Oncoimmunology 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ingram PN, Hind LE, Jiminez-Torres JA, Huttenlocher A, Beebe DJ, Advanced healthcare materials 2018, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Jimenez-Torres JA, Peery SL, Sung KE, Beebe DJ, Advanced healthcare materials 2016, 5, 198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ji RC, Cancer Lett 2014, 346, 6. [DOI] [PubMed] [Google Scholar]
- [16].Cho JG, Lee A, Chang W, Lee MS, Kim J, Frontiers in immunology 2018, 9, 294; [DOI] [PMC free article] [PubMed] [Google Scholar]; Li Y, Lui KO, Zhou B, Nature reviews. Cardiology 2018, 15, 445. [DOI] [PubMed] [Google Scholar]
- [17].Hernandez-Bedolla MA, Carretero-Ortega J, Valadez-Sanchez M, Vazquez-Prado J, Reyes-Cruz G, Biochimica et biophysica acta 2015, 1853, 166; [DOI] [PubMed] [Google Scholar]; Subramaniyan A, Maddaly R, J Cell Biochem 2018, 119, 1309; [DOI] [PubMed] [Google Scholar]; Bravata V, Minafra L, Forte GI, Cammarata FP, Russo G, Di Maggio FM, Augello G, Lio D, Gilardi MC, International journal of radiation biology 2017, 93, 1217. [DOI] [PubMed] [Google Scholar]
- [18].Osaki T, Serrano JC, Kamm RD, Regenerative engineering and translational medicine 2018, 4, 120; [DOI] [PMC free article] [PubMed] [Google Scholar]; Gong MM, Lugo-Cintron KM, White BR, Kerr SC, Harari PM, Beebe DJ, Biomaterials 2019, 214, 119225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Huxley VH, Curry FE, Adamson RH, The American journal of physiology 1987, 252, H188. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Immunofluorescent staining of vascular endothelial cadherin (VE-cadherin) and lymphatic vessel endothelial hyaluronan receptor 1 (LYVE1) for the primary human lymphatic endothelial cells in 2D.
Figure S2. Example of a standard glass-bottom culture dish containing six devices used for experiments. Schematic of device layers and components. Fluid handling in the devices is accomplished by using standard micropipettes.
Figure S3. Breast cancer cells condition lymphatic endothelial cell gene expression. A) Waterfall plot showing genes differentially expressed in lymphatic endothelial cells co-cultured with MCF7 cells. B) Similar waterfall plot showing the changes induced by MDA-MB-231 in lymphatic endothelial cells. C) Molecular network highlighting the molecular interactions affected in the MCF7 co-culture. FDA-approved drugs are also shown as yellow hexagons. D) Molecular networks illustrating the interactions affected in the MDA-MB-231 co-culture.