

Summary
The diploid wild cotton species Gossypium australe possesses excellent traits including resistance to disease and delayed gland morphogenesis, and has been successfully used for distant breeding programmes to incorporate disease resistance traits into domesticated cotton. Here, we sequenced the G. australe genome by integrating PacBio, Illumina short read, BioNano (DLS) and Hi‐C technologies, and acquired a high‐quality reference genome with a contig N50 of 1.83 Mb and a scaffold N50 of 143.60 Mb. We found that 73.5% of the G. australe genome is composed of various repeat sequences, differing from those of G. arboreum (85.39%), G. hirsutum (69.86%) and G. barbadense (69.83%). The G. australe genome showed closer collinear relationships with the genome of G. arboreum than G. raimondii and has undergone less extensive genome reorganization than the G. arboreum genome. Selection signature and transcriptomics analyses implicated multiple genes in disease resistance responses, including GauCCD7 and GauCBP1, and experiments revealed induction of both genes by Verticillium dahliae and by the plant hormones strigolactone (GR24), salicylic acid (SA) and methyl jasmonate (MeJA). Experiments using a Verticillium‐resistant domesticated G. barbadense cultivar confirmed that knockdown of the homologues of these genes caused a significant reduction in resistance against Verticillium dahliae. Moreover, knockdown of a newly identified gland‐associated gene GauGRAS1 caused a glandless phenotype in partial tissues using G. australe. The G. australe genome represents a valuable resource for cotton research and distant relative breeding as well as for understanding the evolutionary history of crop genomes.
Keywords: Gossypium australe, genome sequencing, resistance, Verticillium wilt, delayed gland morphogenesis, gene function
Introduction
In modern agricultural ecosystem, the narrow genetic base of modern crop cultivars, in which diversity has been lost in domestication, is becoming a major bottleneck for crop improvement programmes, especially for cultivated cotton. The use of close relatives of domesticated plants or crop wild relatives (CWRs) is a promising approach to enhance the genetic diversity and resistance to biotic and abiotic stresses of cultivated crops (Mammadov et al., 2018). Genomic analyses of CWRs generate data that support the use of CWRs to expand the genetic diversity of crop plants, which will strongly promote biodiversity, agricultural sustainability and food security (Brozynska et al., 2016). It is becoming increasingly important for genomic studies on CWRs; many such reports published in 2018 and 2019 (Arora et al., 2019; Milner et al., 2019), including wild rice (Zhao et al., 2018), wild wheat (Thind et al., 2018), soya bean wild relatives (Xie et al., 2019), wild tomato (Schmidt et al., 2017), wild peanut (Yin et al., 2018) and so on.
The Gossypium genus is highly diverse and includes the highest economically valuable species among all field crops. This genus is of great significance for plant research studies of plant taxonomy, polyploidization, phylogeny, cytogenetics and genomics (Kunbo and Jonathan, 2018). The wealth of diversity available among wild cotton species is a valuable resource for cotton breeders. There are diverse Gossypium taxa across the world: the A, B, E and F Gossypium genomes are distributed in Asia and Africa, the C, G and K genomes are found in Australia, and the D and AD genomes are distributed in the Americas and Pacific islands (Wendel et al., 2010). To date, cotton genomics researchers have developed high‐quality reference sequences for 2 diploid groups, as well as allotetraploid cottons (‘A‐genome’, ‘D‐genome’ and ‘AD‐genome’ clade) including 3 cultivated species (AA, G. arboreum, AADD, G. hirsutum and G. barbadense) and 1 wild ancestor species (DD, G. raimondii) (Du et al., 2018; Hu et al., 2019; Li et al., 2014, 2015; Paterson et al., 2012; Wang et al., 2012, 2019a; Zhang et al., 2015). Comparatively little research attention has been focused on Australian cotton species such as G. australe (with its ‘GG’ genome in cotton genomics nomenclature) (Chen et al., 2014; Liu et al., 2015; Wang et al., 2018c).
Gossypium species are characterized by their lysigenous glands containing terpenoids, important secondary phytoalexins consisting predominantly of the aldehyde gossypol, which constitute an important plant agent against pests and diseases in cotton (Bell, 1969; Cai et al., 2010; Gao et al., 2013; Tian et al., 2018). However, gossypol deposited in the glands of Gossypium is toxic to nonruminant animals and humans, while glandless cotton varieties (i.e. glandless in both seeds and plants) have no or very low gossypol content, with their resistance to pests and disease being attendantly reduced (Cherry, 1983; Mcmichael, 1959; Vaissayre and Hau, 1985).
Gossypium australe has been an important resource in the era of modern cotton genomics. Specifically, G. australe is highly resistant to Verticillium wilt disease (Benkang and Cun, 1996; Gu et al., 1993; Wang et al., 2018a) and is therefore viewed and has already been used as an important germplasm resource for the genetic improvement of cultivated upland cotton to increase resistance to Verticillium dahliae, seeking the inducement and identification of chromosome introgressions and translocations into G. hirsutum, as well as in the construction of a complete set of alien chromosome addition lines into G. hirsutum that were generated with aim of exploiting G. australe's distinct traits including glanded‐plant and glandless‐seed and resistances to pests and diseases (Benbouza et al., 2009; Chen et al., 2014; Liu et al., 1999; Wang et al., 2018c).
As with other crops, especially polyploid crops, there have been a variety of cotton improvement strategies based on utilization of natural germplasm resources of wild relative and progenitor species to improve plant resistance to diseases such as Verticillium and Fusarium wilt, as well as to pests and abiotic stresses like drought. For example, the Australian wild species Gossypium australe—which exhibits delayed gland morphogenesis wherein the dormant seeds are glandless (low gossypol content) but the germinated cotyledons are glanded (Benkang and Cun, 1996; Gu et al., 1993; Wang et al., 2018a; Wendel et al., 1991)—has been used to generate glandless‐seed but glanded‐plant commercial cotton varieties, thereby providing seeds that lack gossypol and are suitable for food and feed uses but also with strong plant resistance to cotton pests and diseases (Ma et al., 2016; Wendel et al., 1991).
Here, to better enable the continued use of this distinctive cotton wild relative species with its prominent traits like resistance to disease and delayed gland morphogenesis, we performed de novo sequencing of G. australe genome and focused on disease resistance and the G. australe‐specific gland development traits. We used an integrated approach combining four separate sequencing technologies to assemble a high‐quality reference genome sequence. Through genome sequencing of G. australe and transcriptome analysis, we explored the evolution and adaptability of Australian Gossypium species and investigated disease resistance genes and gland formation genes. We hope to provide a theoretical and applied basis for the discovery of the molecular mechanisms underlying the beneficial characteristics of this species and promote cotton breeding for the sustainable development of agriculture, benefiting food security despite the threats of biotic and abiotic stresses.
Results
Genome sequencing and construction of a high‐quality genome assembly for G. australe
Gossypium australe has shown excellent resistance to the fungus disease Verticillium wilt, and the disease had little influence on morphology of G. australe plants (Figure 1a,b); in contrast, the stems and leaves of G. arboreum cultivar plants were greatly damaged after infection with Verticillium wilt. Another prominent trait of G. australe is delayed gland morphogenesis, and the seeds of G. australe (Figure 1c,d,e) have no gland in seed, but were observed during seeds germinate process, differing from that in G. arboretum, which is glanded in whole plants (including seeds) (Figure 1f,g,h). This precious resource could facilitate glandless‐seed and glanded‐plant cotton breeding and provide seeds lacking gossypol for food or feed, as well as maintain the resistance of cotton to pests and diseases.
Figure 1.

The plants of G. australe and G. arboretum, and the forming of new glands during seed germination of G. australe, G. arboretum, G. hirsutum (Xiangmian 18). (a) G. australe plant, resistant or immune to Verticillium wilt. (b) G. arboretum plant, susceptible to Verticillium wilt. (c, f and i) are the delinted seeds of Gossypium australe, G. arboreum and G. hirsutum, respectively. Scale bar, 5 mm. (d and e) are two germination stages of G. australe, early stage before GF (gland formation), beginning stage of GF; (g and h) are the same two germination stages of G. arboreum. (J and k) are stages of Xiangmian 18 (G. hirsutum), scale bars, 1 mm. (l and m) are enlarged versions of the positions indicated by the red box in Figure (e). (n and o) correspond to (h), and (p and q) correspond to (k). The white arrow indicates the location of the glands.
Thus, we sequenced and assembled the G. australe genome with a combination of four technologies: Pacbio single‐molecule real‐time (SMRT) sequencing, paired‐end sequencing, optical mapping (DLS) and Illumina short‐read Hi‐C. Assembly with these complementary data types proceeded in a stepwise fashion, producing progressively improved assemblies (Table 1, Table S1). The initial assembly of the single‐molecule real‐time sequencing data alone resulted in a contig N50 (the minimum length of contigs accounting for half of the haploid genome size) of 2.50 Mb. PacBio contigs were first scaffolded using large‐insert pair‐end library reads, which resulted in a scaffold N50 of 3.59 Mb. The sequences were then scaffolded and corrected using optical mapping data (Figure S1), and the resulting scaffolds were clustered into chromosome‐scale scaffolds using Hi‐C data (FigureS2). With K‐mer distribution analysis, the genome size was estimated to be 1.67 Gb (FigureS3), which is very similar to an earlier prediction (Hendrix and Stewart, 2005). The final assembly comprised 1.75 Gb of sequence with a contig N50 of 1.83 Mb and a scaffold N50 of 143.60 Mb, with only 650 scaffolds covering the 13 haploid G. australe chromosomes (Figure 2, Tables 1, S1).
Table 1.
Summary of genome assembly and annotation for G. australe
| Genomic feature | G. australe |
|---|---|
| Total length of contigs (bp) | 1 729 091 355 |
| Total length of assemblies (bp) | 1 752 741 698 |
| Estimated gap size (bp) | 23 650 343 |
| Percentage of anchoring | 99.08% |
| Percentage of anchoring and ordering | 98.99% |
| Number of contigs | 2598 |
| Contig N50 (bp) | 1 825 353 |
| Contig N90 (bp) | 453 340 |
| Number of scaffolds | 650 |
| Scaffold N50 (bp) | 143 600 552 |
| Scaffold N90 (bp) | 104 992 986 |
| GC content | 36.39% |
| Percentage of repeat sequences | 73.50% |
| Number of genes | 40,694 |
| Number of transcripts | 45 350 |
Figure 2.
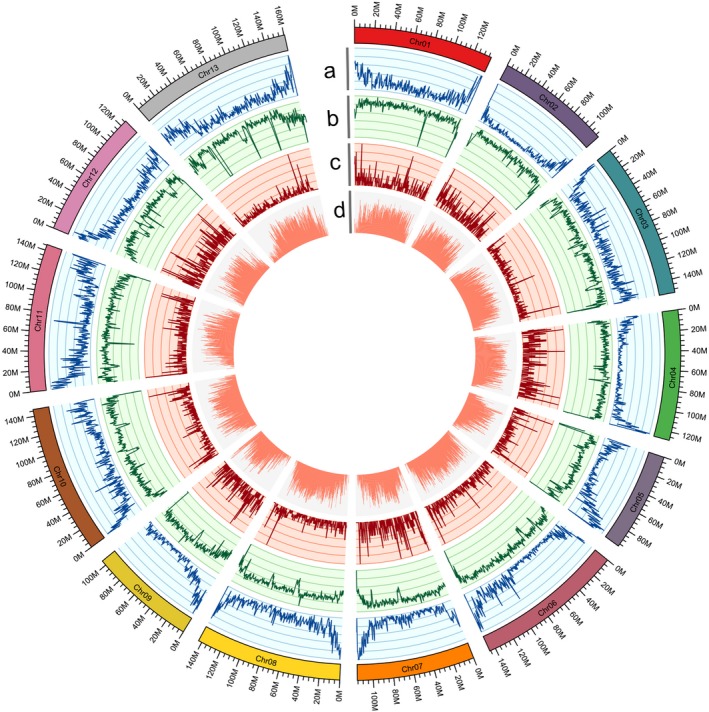
Characterization of the G. australe cotton genome. (a) Gene density in each chromosome; (b) transposable element (TE) density in each chromosome; (c) ncRNA density in each chromosome; (d) GC content in each chromosome.
We used the BUSCO method based on a benchmark of 1440 highly conserved core plant genes to further evaluate assembly quality and completeness (Kriventseva et al., 2019), which revealed that 95.9% of the genes were present in our assembly and indicating that this G. australe, genome assembly is nearly complete (Table S5). Further, the accuracy of the assembly was supported by alignment of the Illumina short‐read data, resulting in a 97.48% mapping ratio. The genome assembly completeness was also validated by aligning full‐length transcripts derived from SMRT sequencing to the assemble genome, which a total of 99% of the 158,566 full‐length transcripts from the G. australe ovules and leaves were detected in our assembly (Table S2).
Annotation of the G. australe genome
We annotated 40 694 gene models in the G. australe genome by combining ab initio gene prediction, homolog protein data searches and the sequences of the aforementioned full‐length transcripts (Figure 2a), and a number similar to the 40 976 and 40 960 consensus protein‐coding‐gene models previously predicted for the G. raimondii and G. arboreum genomes, respectively (Du et al., 2018; Wang et al., 2012). Approximately 97% of the predicted G. australe gene models were annotated by BLAST in four databases, including Nr, Swiss‐prot, KOG and KEGG (Table S3). Additionally, the G. australe genome is predicted to encode 1366 rRNAs, 1292 tRNAs, 339 microRNAs (miRNAs) and 3388 small nuclear RNAs (snRNAs) (S4). Orthologous clustering of the G. australe predicted proteome with 7 closely related plant genomes identified 95 666 gene families in common, with 15 696 gene families that were present specifically in G. australe (FigureS4).
Notably, 73.5% of the G. australe genome is composed of various types of repeat sequences (Figure 2b), a proportion distinct from the reported repeat sequence content of G. arboreum (85.39%), G. hirsutum (69.86%) and G. barbadense (69.83%) (Du et al., 2018; Wang et al., 2019a), and long terminal repeat (LTR) retrotransposons accounted for 92.9% of these sequences in the G. australe genome (Table S6).
Compared with those in the G. raimondii genomes, Gypsy elements showed noticeable proliferation in both the G. australe and G. arboreum genomes, whereas Copia elements have apparently preferentially accumulated in the G. raimondii genome (Table S7). Our results are consistent with previous reports that the detected differences in transposable elements (TEs) between the G. raimondii genome and the other two cotton genomes were established during the divergence of G. raimondii and the common ancestor of G. australe and G. arboreum before the divergence of these two genomes (Hawkins et al., 2006).
Furthermore, a similar proportion of Gypsy subgroup LTR (Llorens et al., 2009) was observed between the G. australe and G. arboreum genomes, with the CRM subgroup being predominant among the Gypsy elements of both genomes. However, a significantly lower proportion of the CRM subgroup than those of G. australe and G. arboretum and different proportions of other subgroups from G. australe and G. arboreum were observed in G. raimondii, which was consistent with the predominance of Copia in the retrotransposons of G. raimondii (Table S8).
Genome evolution in G. australe
The Gossypium genus comprises 8 diploid genome groups (A through G and K), as well as one allopolyploid clade (AD genome) formed from an ancient merger and chromosome doubling from A and D genome ancestors (Kunbo and Jonathan, 2018). Through molecular phylogenetic analyses, we showed that a divergence time for G. australe and G. arboreum of 6.6 (4.1–8.9) million years ago, and their common ancestor had diverged from G. raimondii of 7.7 (4.9–10.1) million years ago (Figure 3a) (Carvalho et al., 2011).
Figure 3.
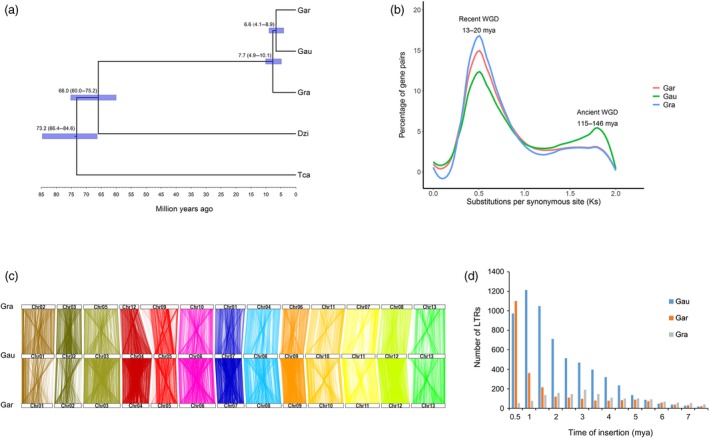
Phylogenetic and evolutionary analysis of the Gossypium genomes. (a) Phylogenetic analysis indicated that G. australe and G. arboreum diverged 6.6 (4.1–8.9) million years ago (mya). Gra: G. raimondii. Gar: G. arboreum; Gau: G. australe; Dzi: Durio zibethinus; Tca, Theobroma cacao. (b) Ks analyses suggested that the Gossypium genomes might have undergone two WGD events. (c) Many collinear blocks were found when comparing either the G. raimondii (Gra) or G. arboreum (Gar) genome with the G. australe (Gau) genome. Numbered rectangles represent the chromosomes. (d) Analysis of the LTR number and insertion time in G. australe (Gau), G. arboreum (Gar) and G. raimondii (Gra).
The G. australe genome was scanned for syntenic gene blocks. We calculated the age distribution for all duplicate gene pairs based on the substitution per synonymous site (Ks) values. We detected a large peak centred around Ks values of approximately 0.5 for G. australe, very similar to the peak detected in the two previously reported diploid cotton genomes (Figure 3b); this apparent whole‐genome duplication (WGD) event has been previously estimated to have occurred 13–20 million years ago (Li et al., 2014; Wang et al., 2012). Our results thus further support that this recent whole‐genome duplication (WGD) event occurred in all of the cotton genomes based on the phylogenetic tree of the Gossypium genus (Hawkins et al., 2006). Notably, a whole‐genome alignment approach revealed that the G. australe genome showed closer collinear relationships with the genome of G. arboreum than of G. raimondii, a finding consistent with multiple reported phylogenetic analyses. Collinear blocks covered 72% of the G. arboreum genome and 71% of the G. australe genome, but covered only 60% of the G. raimondii genome and 60% of the G. australe genome (Figure 3c, Figure S5).
Extensive evidence in plant genomics supports the understanding that the expansion of TE families is one of the major factors that influence genome evolution. Our LTR analysis indicated that the retrotransposition activity of G. australe apparently increased continuously from 7.5 million years ago until about 1 million years ago, before subsequently decreasing (Figure 3d). Notably, with the exception of the most recent 0.5 million years, G. australe apparently had higher retrotransposition activity than did G. arboreum, a situation consistent with the different genome sizes of these three genomes. However, G. australe harbours more than twofold more intact LTRs than G. arboreum, with a similarly sized genome and similar TE ratio between these two genomes, findings which strongly suggest that G. australe has experienced less genome reorganization than the domesticated G. arboreum, which we know has undergone several rounds of artificial selection.
Gene evolution in G. australe
Positive selection plays important roles in plant evolution and adaptation to biotic and abiotic stresses; gene expression and regulation changes have been postulated to be key determinants of the rates of adaptive evolution (Khan et al., 2016; Paape et al., 2018; Seeholzer et al., 2010; Sun et al., 2018a; Zambounis et al., 2016). The positively selected genes (PSGs) between G. australe and G. arboretum remain unknown.
Here, we analysed the PSGs in G. australe to assess how it has adapted to diverse wild environments and to search for loci relating to its strong resistance to Verticillium wilt, a trait for which it is much more resistant than the domesticated G. arboreum. The analysis showed identified selection signatures at 670 and 232 PSGs in the G. australe and G. arboretum genomes, respectively (Figure S6, Data S1). Subsequent KEGG pathway analysis indicated that statistically significant (P < 0.05) enrichment among the G. australe PSGs for the ‘Other types of O‐glycan biosynthesis’ (ko00514) pathway (Figure S7), which was associated with cotton fibre strength in studies of in G. arboreum (Hernandez‐Gomez et al., 2017; Natalio et al., 2017). There was also significant enrichment among the G. australe PSGs for ‘Riboflavin metabolism’ (ko00740) (Asai et al., 2010; Boubakri et al., 2016; Deng et al., 2011) and GPI (Shen et al., 2017) (Figure S7), pathways which have been implicated in plant responses to biotic stresses.
Interestingly, analysis of the PSGs associated with the 12 enriched pathways of (Figure S7, Data S1) identified three genes from the carotenoid biosynthesis pathway, suggesting that these compounds or their downstream metabolites may be involved in G. australe's resistance to fungal pathogens. Carotenoid cleavage dioxygenase 7 (CCD7) is involved in strigolactone biosynthesis by cleaving asymmetrically the 9–10 double bond in various linear and cyclic carotenoids (including branch‐inhibiting hormones and share symbiotic fungi) and showed an elevated Ka/Ks value (Figure S7). Previous reports have shown that CCD7 is involved in disease‐related mechanisms (Decker et al., 2017). Thus, we cloned the GauCCD7 gene, which shared 97.97% identity in amino acid sequence with G. barbadense, and investigated its expression and potential function in disease resistance.
Functional analysis of G. australe genes associated with resistance to Verticillium wilt
The enzyme carotenoid cleavage dioxygenase 7 (CCD7) is involved in strigolactone biosynthesis: it asymmetrically cleaves the 9‐10 double bond in various linear and cyclic carotenoids (including branch‐inhibiting hormones and stimulatory molecules for symbiotic fungi). Given that the G. australe locus encoding CCD7 exhibited an elevated Ka/Ks value (Figure S7), and considering that previous studies have reported a disease‐related function for CCD7 in plants (Decker et al., 2017), we investigated its expression and potential function in disease resistance in G. australe. We conducted a qPCR‐based analysis of GauCCD7 expression in G. australe, G. arboretum and G. raimondii. qPCR analysis of roots, stems and leaves showed that the expression pattern of GauCCD7 in G. australe was different from that in G. arboreum and G. raimondii (Figure S8). Experiments that applied a variety of plant hormones or V. dahliae Kleb isolate to 3‐week‐old G. australe plants revealed that GauCCD7 expression was significantly induced by V. dahliae Kleb and by the plant hormones strigolactone (GR24), salicylic acid (SA) and methyl jasmonate (MeJA), but not by ethylene (applied as ethephon) or abscisic acid (ABA) (FiguresS9 and S10).
We also performed virus‐induced gene silencing (VIGS) experiments to examine the disease resistance‐related phenotypic changes resulting from the knockdown of the GauCCD7 homologue in the Verticillium wilt resistant G. barbadense cultivar Xinhai 15. We first confirmed the ability of a silencing construct targeting GauCCD7 to knockdown the expression of GbCCD7 in Xinhai 15 upon infiltration of tobacco rattle virus (TRV) into the cotyledons of newly germinated seedlings (Figure 4b). Subsequently, 14 days infiltrated control and GbCCD7‐knockdown plants were inoculated with V. dahlia and the disease symptoms were monitored. Compared to controls, the disease index values were significantly increased in GbCCD7‐knockdown plants (Figure 4a,c), thereby experimentally implicating GbCCD7 in G. barbadense defence responses to the fungal pathogen. Upon dissection of these plant materials, we also noted that that the GbCCD7‐knockdown plants exhibited a pronounced vascular browning phenotype (Figure 4d).
Figure 4.
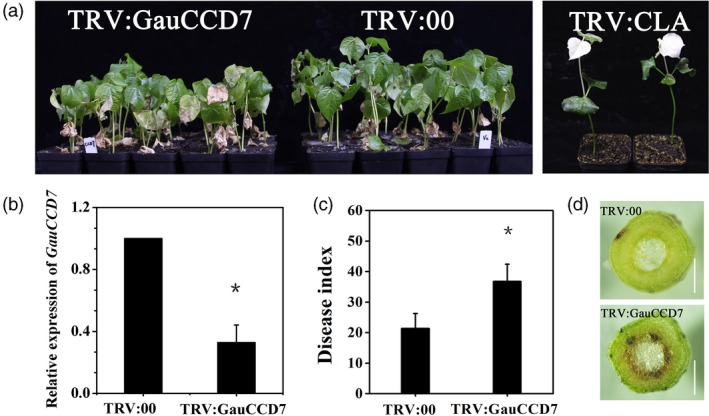
GauCCD7 positively regulates cotton defence against V. dahliae in Xinhai 15. (a) Disease symptoms of TRV:GauCCD7 (left) and TRV:00 plants (centre) inoculated with V. dahliae strain V991, which were photographed 15 days after inoculation, and the albino phenotype of the plants inoculated with TRV:CLA after 15 days (right). (b) (qRT‐PCR) Analysis of the expression of GauCCD7 in TRV:00 and TRV:GauCCD7. Statistical analyses were performed using Student's t test: *P < 0.05. (c) The disease index and incidence rate in TRV:00 and TRV:GauCCD7 were measured at 17 dpi (days postinoculation). Three biological replicates with at least 35 plants per replication. (d) Section anatomy in the stem was observed 17 days after V. dahliae treatment of TRV:00 and TRV:GauCCD7. Bars, 1 mm.
In addition, we conducted an RNA‐seq‐based transcriptomic analysis seeking differentially expressed genes associated with G. australe's resistance responses to Verticillium wilt. Ultimately, we focused on the genes which were simultaneously (1) significantly up‐regulated in G. australe and (2) significantly down‐regulated in G. arboreum during infection with Verticillium wilt, criteria which identified 31 genes, including GAUG00007269 (bHLH19‐like isoform) and GAUG00028019 (calmodulin‐binding protein, CBP) (Figures S11 and S12, Table S9). Among them, GAUG00028019 was selected as a candidate resistance gene of interest because this gene shared 91.86% identity in amino acid sequence with that in G. barbadense (Table S9). Other cotton calmodulin‐binding proteins that was functionally associated with responses to biotic and abiotic stress have been reported (Qin et al., 2018; Sun et al., 2018b; Zheng et al., 2015). We investigated the expression of GauCBP1 in G. australe comparing with G. arboretum and G. raimondii (Figure S13). qPCR analysis of root, stem and leaves showed that GauCBP1 expression was induced by V. dahliae Kleb and by the plant hormones GR24, SA and MeJA, but not by ethylene or ABA (Figures S14 and S15).
We also explored the potential disease‐related functions of the GauCBP1 homologue in the aforementioned Verticillium wilt‐resistant G. barbadense cultivar Xinhai 15. Having confirmed that the silencing construct does indeed knockdown expression of GbCBP1 (Figure S16b), experiments similar to the aforementioned VIGS analysis of GbCCD7 again showed that knockdown of this candidate resistance gene homologue resulted in a significant reduction in Xinhai 15's resistance against Verticillium wilt.
Genes involved in gland formation and function
To explore mechanisms relating to G. australe's delayed gland morphogenesis relative to other cottons, we analysed the embryo and leaf transcriptomes of six cottons, including three Australian diploid G subgroup wild cotton species (G. australe, G. bickii and G. nelsonii) and two different G. hirsutum cultivars (the glandless ZHONG12 and Xiangmian 18) (Figure 1j,k) with few gland in seed but glanded plant. Based on the results of our previous studies, in which 24 differentially expressed cDNAs were identified in the new gland‐forming stage of Xiangmian 18 through suppression subtractive hybridization (SSH) analyses (Cai et al., 2003, 2010), among them we identified a transcription factor GRAS (Belong to GRAS family, GAI, RGA, SCR). GRAS proteins are an important family of plant‐specific proteins named after the first three members: GIBBERELLIC‐ACID INSENSITIVE (GAI), REPRESSOR of GAI (RGA) and SCARECROW (SCR). In this study of the G. australe genome, we cloned the GRAS gene in G. australe based on the GRAS gene cloned from Xiangmian 18 and named it GauGRAS1 (one of GRAS family), and then investigated its expression and function.
The expression levels of GauGRAS1 and GauPGF, a positive regulator of gland formation (Ma et al., 2016), were analysed in embryos of G. australe, glanded G. hirsutum (C5, Jinxianduanguozhi), dominant glandless Zhongmiansuo 12 and recessive glandless Zhongmiansuo 12. The results showed that both GauGRAS1 and GauPGF were highly expressed in glanded G. hirsutum but showed very low expression in G. australe and the two glandless lines (Figure S17). These results indicated that GauGRAS1 is associated with gland formation. We also investigated the expression patterns of GauGRAS1 in the embryos and leaf of G. australe and G. bickii by qRT‐PCR and RNA‐seq (Figures S18 and S19). The results showed that GauGRAS1 has different expression patterns from GauPGF. The relative expression level of GauGRAS1 was significantly lower in the embryos than in the leaves for both G. australe and G. bickii, and the expression level of GauPGF was significantly higher in the embryos than in the leave for both G. australe and G. bickii (Figure S18).
These findings were consistent with the results of the transcriptomic analyses of G. australe, G. bickii and G. nelsonii (Figure S19). In addition, during seed germination of three cotton species, both GauGRAS1 and GauPGF are up‐regulated in the gland‐forming stage compared to early stages before gland formation. The relative expression level of the GauGRAS1 gene showed somewhat differences compared to that of GauPGF by RT‐PCR (Figure S20, Figure 1). The results indicated that GauGRAS1 was associated with gland formation, but its expression pattern was different from that of GauPGF. Thus, the function of GauGRAS1 was further analysed using VIGS technology.
Suppressing GauGRAS1 by VIGS led to glandless stems and petioles in G. australe, but the leaf glands did not change in G. australe (Figure 5a–c), and no glandular cavity was formed in the stems and petioles (Figure 5d). Glands still formed in true leaves, and the number of glands was not different from that in the control plant leaves (TRV:00) (Figure 5a,b). Moreover, the gossypol content in the stem of the GauGRAS1‐silenced plants was significantly reduced, but it remained almost unchanged in the leaves (Figure 5e). The functional results confirmed that the GauGRAS1 gene was responsible for gland formation of partial tissues in G. australe, in contrast to GauPGF, which leads to glandlessness in all tissues, including the leaves and stems, in the GauPGF‐silenced plants of G. australe (Figure S21).
Figure 5.
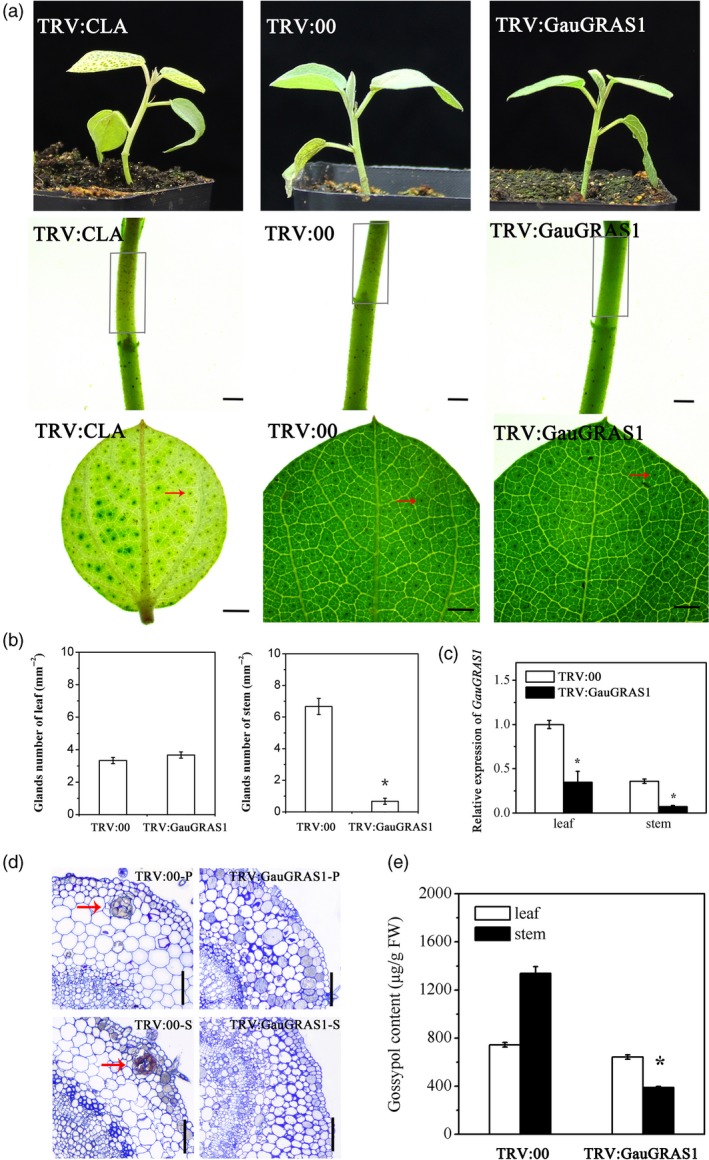
Functional characterization of GauGRAS1 by VIGS. (a) Phenotypes of Gossypium australe after GauGRAS1 silencing by VIGS; TRV:CLA and TRV:00 are the positive control and negative control, respectively. The grey box indicates glands in the stem, and the red arrow indicates glands on the leaf. Scale bars, 1 mm. (b) Statistical chart of the number of glands in the leaves and stems. (c) The silencing efficiency of GauGRAS1. (d) A cavity was observed in the empty vector (TRV:00) leaves and stems but disappeared in the GauGRAS1‐silenced plants. P: petiole, S: stem. Scale bars, 100 μm. (e) Gossypol content in empty vector (TRV:00) and in the GauGRAS1‐silenced leaves of G. australe. Error bars are the SD of three biological repeats. *P < 0.05; Student's t test, n = 3.
To better understand the evolution and function of gossypol/gland formation genes in the botanical system and Gossypium, we performed a comparative transcriptome analysis of the embryos and leaf using several glanded and glandless tetraploid cotton varieties. The differentially expressed genes (DEGs) were identified between glanded and glandless leaves, followed by module partition analysis based on weighted gene co‐expression network analysis (WGCNA). The magenta4 module was positively correlated with the presence/absence of glands (Figure S22, TableS10). Interestingly, the gene with the highest connectivity was glyoxalase I, which responds to stress in higher plants (Espartero et al., 1995; Hasanuzzaman et al., 2017), glutathione S‐transferase (Li et al., 2018) and Laccase 14 (Hu et al., 2018) that were probably associated with disease resistance (Table S10). In addition, 7 genes adjacent to GoPGF were co‐expressed with GoPGF in the magenta4 module (Table S11), indicating that function‐related genes are clustered together.
The pigment gland is a type of glandular trichome that can be found in approximately 30% of all vascular plant species (Huchelmann et al., 2017; Ma et al., 2016). To study the evolution history of gland formation genes, local collinearity was analysed based on forty genes adjacent to the GoPGF and GRAS1 genes to assess the presence/absence in 30 sequenced genomes that represented several orders of plants (Figure S23). We found that the GoPGF gene evolved after the differentiation of dicotyledonous plants and monocotyledonous plants (Figure S24), and GoPGF did not originally function in glandular trichomes but later differentiated in Gossypium and other species. Furthermore, the GRAS1 gene might be present earlier before the differentiation of the glandular trichome formation and regulate tissue‐specific genes’ expression. Later, GRAS1 acquired new regulatory functions for gland information in cotton (Figure S25).
The results suggested that the gland formation pathway might be a branch line of the ancient stress response regulatory network, and this branch line became specialized for gland structure in Malvaceae. The genome of G. australe and its valuable genes and the related regulatory network involved in gossypol/gland formation and disease resistance will be further explored and employed in cotton breeding and sustainable agriculture.
Discussion
Our study was greatly facilitated by the development of SMRT long‐read sequencing technology. Specifically, this technology can dramatically increase the N50 contig lengths of genome assemblies; we used both BioNanooptical and chromatin interaction mapping approaches in combination with paired‐end sequencing, and this combination worked very well in combination with our long‐read assemblies. After our four‐step assembly process, our final G. australe reference genome included 1.75 Gb of sequence, with a scaffold N50 of 143.60 Mb, a contig N50 of 1.83 Mb. Notably, our entire assembly comprised only 650 scaffolds covering the 13 haploid G. australe chromosomes. Our high‐quality assembly enabled comparison with the recently published cotton genomes that have been assembled using PacBio data in combination with multiple scaffolding methods (Du et al., 2018; Wang et al., 2019a).
Based on our new assembly sequencing of G. australe genome, we further explored new genes involved in disease resistance and gland formation. Wilt disease caused by V. dahlia is the most devastating disease of cotton crops in several parts of the world, including China (Zhang et al., 2019). Because the main cultivated upland cotton species (G. hirsutum) lacks genetic resources conferring resistance to Verticillium wilt, researchers have surveyed for such resistance genes from relatives such as, G. barbadense (Gao et al., 2016; Miao et al., 2019; Sun et al., 2013, 2017; Wang et al., 2018a; Xiang et al., 2017; Zhang et al., 2018; Zhou et al., 2018) and other wild species, including G. australe (Benbouza et al., 2009; Tang et al., 2018; Wang et al., 2018a,2018c).
However, the molecular mechanism for cotton resistance to Verticillium wilt remains unclear, which has limited progress in developing cotton varieties with resistance to Verticillium (Han et al., 2019). Exploring the disease resistance of G. australe may facilitate and the genetic improvement of cotton resistance against Verticillium wilt. Our study offers new insights about such molecular mechanisms, in that we empirically demonstrate that expression of the GauCBP1 and GauCCD7 gene is induced by V. dahliae and by treatment with the plant hormones GR24, SA and MeJA (but not by Eth or ABA). GauCBP1 knockdown via VIGS markedly reduced cotton resistance to V. dahliae, implying that GauCBP1 functions in the response processes through which G. australe resists V. dahlia (Figure S16). Calmodulin‐binding proteins (CBPs) are known to transduce calcium signals in response to fungal diseases. The plant‐specific CALMODULIN BINDING PROTEIN 60 (CBP60) protein family includes CBP60a‐g and SYSTEMIC ACQUIRED RESISTANCE DEFICIENT 1 (SARD1) (Bouché et al., 2005; Lu et al., 2018), virus‐induced silencing of GhCBP60b compromised cotton resistance to V. dahliae, revealing that CBP60g, SARD1 and GhCBP60b function in V. dahlia resistance (Qin et al., 2018).
We also found that the carotenoid biosynthesis enzyme CCD7 and the related strigolactone biosynthesis pathway may contribute to Verticillium wilt resistance (Figure S6). Indeed, our VIGS results show that silencing of GauCCD7 compromised resistance to V. dahliae (Figure 4). This is the first report to identify a role for CCD7 against a fungal disease in angiosperms.
Recent studies have reported that genes associated with the strigolactone pathway and with plant architecture function in plant disease resistance (Sun et al., 2019; Wang et al., 2018b). The tomato mutant Slccd8 showed increased susceptibility to both pathogens, indicating a new role for strigolactones in plant defence (Torres‐Vera et al., 2014). The CCD7 and CCD8 enzymes of the strigolactone pathways were also reported to contribute to resistance against the phytopathogenic fungi in the spreading moss Physcomitrella patens (Decker et al., 2017). The F‐box protein MAX2 was confirmed to contribute to strigalactone‐associated resistance to bacterial phytopathogens in Arabidopsis thaliana (Piisilä et al., 2015). Another study reported that a single transcription factor, IPA1 (Ideal Plant Architecture 1), promotes both yield and disease resistance by sustaining a balance between growth and immunity in rice (Wang et al., 2018b). Other work has shown that DWARF14 acts as a receptor for strigolactones in the SL signalling pathway both in rice and cotton (Sun et al., 2016; Wang et al., 2019b). Overexpression of Loose Plant Architecture 1 enabled increased planting densities and resistance to sheath blight disease via activation of PIN‐FORMED 1a in rice (Sun et al., 2019).
In addition, our study identified and cloned a previously unknown gland formation gene: GauGRAS1. VIGS silencing of GauGRAS1 in G. australe resulted in a glandless stem and petiole but a glanded leaf, and significantly reduced the gossypol content in the stem and petiole (Figure 4). The expression pattern of GauGRAS1 was different from that of another known gland development related to gene GauPGF (Figure S21) (Figure 4). These findings indicated that GauGRAS1 may play an important role in delayed morphogenesis of gland morphogenesis in G. australe, a distinct role compared to reported functions from other cotton species (Cheng et al., 2016; Janga et al., 2018; Ma et al., 2016).
In conclusion, our work has generated a high‐quality reference genome assembly for a phenotypically distinct diploid wild relative of tetraploid domesticated cotton. Beyond providing a new genomics‐era tool to help cotton improvement programmes increase disease resistance and potentially develop varieties with new combinations of glandless‐seed and glanded‐plant traits, our study also identified multiple genes which we empirically confirmed to function in increasing G. australe resistance to fungal infection, findings which should help promote the general use of cotton and the efficiency of cotton production as both a fibre and oilseed crop.
Methods
Plant materials and strain selection
DNA samples of G. australe were obtained from the Institute of Cotton Research of the Chinese Academy of Agricultural Sciences (accession G2‐lz); the plants showed genetic homozygosity after 15 successive generations of self‐fertilization and were planted in the nursery of the China National Wild Cotton Plantation in Sanya.
Other Australian wild diploid Gossypium species with glandless‐seed and glanded‐plant traits were obtained from the Institute of Cotton Research of the Chinese Academy of Agricultural Sciences: these included glanded G. australe, G. nelsonii, G. bickii and Zhongya 1 (G. arboreum); few glands in the seeds and glanded plants, such as Xiangmian 18 (G. hirsutum); and with glandless seeds and plants, including dominant and recessive glandless Zhongmiansuo 12, as well as G. raimondii and Xinhai 15 (G. barbadense), C5 (Jinxianduanguozhi, glanded G. hirsutum), were used in this research.
DNA extraction and whole‐genome sequencing
Fresh young leaves of G. australe were collected, immediately frozen in liquid nitrogen and stored at −80 °C until DNA extraction. A standard phenol–chloroform method was used for DNA extraction with RNase A and proteinase K treatment to prevent RNA and protein contamination. Genomic DNA was sheared to a size range of 15–40 kb, enzymatically repaired and converted into SMRTbell template libraries as recommended by Pacific Biosciences. The resulting SMRTbell templates were sequenced on a PacBio Sequel instrument. A total of 18 SMRT cells were sequenced producing 82 Gb SMRT raw data. Genomic DNA was used to construct five paired‐end libraries with insert sizes (in KB) of 0.5, 0.8, 2k, 5k and 10k, using a Paired‐End DNA Sample Prep kit (Illumina). These libraries were sequenced using the Illumina HiSeq Xten platform, producing 151G, 138G, 78G, 75G and 77G raw data, respectively.
De novo assembly of the genome using PacBio and Illumina data
Primary contigs were assembled from PacBio long reads by MECAT (version 1.0) (Xiao et al., 2017). Overlaps of long reads were found using the command mecat2pw (parameters: ‐k 4 –a 2000) and were corrected using the command mecat2cns (parameters: ‐r 0.9 –c 6 –l 5000). The 25× coverage of the longest corrected reads was extracted and assembled using the command mecat2canu (min Overlap Length = 500, min Read Length = 1000). The resulting contigs were polished using more than 100× coverage of Illumina short reads by Pilon (version 1.22) with default parameters (Walker et al., 2014). A total of 32 651 SNPs and 1 029 916 InDels were detected and corrected. SSPACE (version 3.0) was used (with default settings) to join contigs to scaffolds as follows: The large‐insert read pairs are mapped against the pre‐assembled PacBio contigs using Bowtie (version 1.1.1). The position and orientation of each pair that could be mapped is stored in a hash. After removing duplicate read pairs‐pairs, scaffolds are formed by iteratively combining contigs if a minimum number of read pairs (k = 5) support the connection, starting with the largest contig. Scaffolds are extending in the same way direction until either a contig has no links with other contigs.
BioNano Genomics DLS optical maps to improve genome assemblies
Optical maps were de novo assembled into genome maps using BioNano assembler software (Solve System, BioNano Genomics). Single molecules longer than 150 kb with at least 8 fluorescent labels were used to find possible overlaps (P < 1 × 10−10). The BioNano Solve software imports the assembly and identifies putative nick sites in the sequence based on the nicking endonuclease‐specific recognition site. These in silico maps for the sequence contigs were then aligned to the de novo BioNano genome maps. Genome maps orient contigs and size gaps by bridging across repeats and other complex elements that break the NGS/TGS assemblies. A total of 884 conflicts between the two are identified and resolved, and hybrid scaffolds are generated in which sequence maps are used to bridge BioNano maps and vice versa. Finally, the sequence assembly corresponding to this hybrid scaffold was generated.
Hi‐C assembly
We constructed Hi‐C fragment libraries with 300–700 bp insert sizes as described in Rao et al. (2014) and sequenced them with an Illumina platform. The clean Hi‐C reads were first truncated at the putative Hi‐C junctions, and then, the resulting trimmed reads were realigned to the assembly results with a BWA aligner (Li and Durbin, 2009). Only uniquely aligned pair reads whose mapping quality was more than 20 were used for further analysis. Invalid read pairs, including dangling‐end, self‐cycle, relegation and dumped products, were filtered by HiC‐Prov2.8.1 (Servant et al., 2015). The unique mapped read pairs were valid interaction pairs and were used for scaffolds clustered, ordered and orientated onto chromosomes by LACHESIS (Burton et al., 2013). The final pseudo‐chromosomes were constructed after manual adjustment.
Annotation of TEs
Tandem Repeats Finder (Benson, 1999) was used to search the genome for tandem repeats. Both de novo and homology‐based approaches were used to find TEs. Programmes including RepeatProteinMask and RepeatMasker (Tarailo‐Graovac and Chen, 2009) were applied to identify TEs through commonly used databases of known repetitive sequences, and Repbase was used along with a database of plant repeating sequences and our de novo TE library to find repeats with RepeatMasker (Jurka et al., 2005). Intact LTRs were predicted using LTR_STRUC (McCarthy and McDonald, 2003). The insert time of all intact LTRs was calculated with the formula: time = Ks/2r (Ks is synonymous substitutions per synonymous site, and r is the rate of nucleotide substitution, which was set to 7 × 10−9).
Gene prediction
The MAKER pipeline (Campbell et al., 2014) was used to annotate protein‐coding genes, integrating ab initio‐predicted genes including analysis of AUGUSTUS (Stanke et al., 2006), SNAP (Korf, 2004) and GeneMark (Borodovsky and Lomsadze, 2011); 149 916 Gossypium unigenes downloaded from the cottongen website ( https://www.cottongen.org/), de novo assembled transcripts from short‐read mRNA sequencing (mRNA‐seq) in this research, and proteins from A. thaliana, Theobroma cacao, and Durio zibethinus. Transposons and low‐confidence predictions were removed.
Gene family and phylogenetic analysis
All‐versus‐all BLASTP (E value < 1 × 10−7) comparison of all protein sequences for eight species (G. arboreum, G. raimondii, G. australe, Glycine max, Dimocarpus longan, T. cacao, Cucurbita maxima, D. zibethinus) was performed, and orthologous genes were clustered by OrthoMCL (Li et al., 2003). CAFE was applied to identify gene families that had undergone expansion and/or contraction (De Bie et al., 2006).
Single‐copy gene families were used to construct a phylogenetic tree. MUSCLE (Edgar, 2004) was used to generate a multiple sequence alignment of protein sequences for each single‐copy family with default parameters. The alignments of each family were concatenated to a super alignment matrix that was used for phylogenetic tree reconstruction through maximum likelihood (ML) methods. The divergence time between species was estimated using MCMC tree in PAML (Yang, 1997) with the options ‘correlated molecular clock’ and ‘HKY85’ model. A Markov Chain Monte Carlo analysis was run for 1 000 000 generations using a burn‐in of 100 000 iterations. Divergence time for the root node of Malvaceae obtained from the fossil estimate (Carvalho et al., 2011; Grover et al., 2017) and TimeTree database ( http://www.timetree.org/) was used as the calibration point.
Whole‐genome duplication analysis and whole‐genome alignment
We used MCScan (Tang et al., 2008) to identify syntenic blocks and calculate Ks rates for syntenic genes. For analysis of the WGD of G. australe, G. arboreum and G. raimondii, paralogous gene pairs originating from their respective WGDs were identified, and the Ks value of each gene pair was calculated. After the repeat regions were masked, whole‐genome alignment was carried out by LASTZ between G. australe and G. raimondii and between G. australe and G. arboreum.
PSG analysis
Based on the aforementioned phylogenetic tree, the branch‐site model incorporated in the PAML package was used to detect PSGs (Zhang et al., 2005). For the detection of PSGs in G. australe, the branch of G. australe was used as the foreground branch, and all other branches in the phylogenetic tree were used as background branches. Similar approaches were used to detect PSGs in G. arboreum and G. raimondii.
Transcriptome analysis
RNA‐seq reads were mapped to the reference genome using TopHat (Trapnell et al., 2009). To measure the gene expression level in tissues, we calculated the expression of genes using FPKM (fragments per kilobase of exon model per million mapped reads) with Cufflinks (Trapnell et al., 2010). To identify differentially expressed genes across samples or groups, we used the edgeR package ( http://www.rproject.org/). We defined genes with a fold change ≥ 2 and a false discovery rate (FDR) < 0.05 in comparison as significant DEGs. The DEGs were then subjected to enrichment analysis of GO functions and KEGG pathways.
Inoculation of Verticillium dahliae V991
For treatment with V. dahliae, a highly aggressive defoliating fungus, V. dahliae V991, was incubated on a potato‐dextrose agar plate for 1 week and then inoculated into Czapek broth on a shaker at 120 rpm at 25°C for 3–4 days until the concentration of spores reached approximately 108–109 spores (mL−1). The suspension liquid was adjusted to 107 spores (mL−1) with sterile distilled water for inoculation (Xu et al., 2011). The seeds of G. australe, G. arboreum and G. raimondii were grown in commercial sterilized soil at 24°C/20°C day/night temperatures with a photoperiod of 14‐h light and 10‐h dark for 2–3 weeks and a relative humidity of 60%. The cotton seedlings of 2 true leaves were infected with V. dahliae by root dip inoculation into a suspension of fungal spores for 1 min and then returned to their original pots. Control plants were not inoculated but were otherwise treated and were mock inoculated using distilled water in the same way. Whole cotton plants were harvested for sample preparation at 24‐h, 48‐h and 72‐h postinfection time points.
Seed germination experiment
Thirty seeds of G. australe, G. arboreum and G. hirsutum were delinted. The seeds were soaked in distilled water for 5 h, and the outer seed coat was peeled off and then soaked in distilled water for 1 h to remove the inner seed coat. Next, the seeds were covered with moist cotton wool and germinated under dark conditions (28°C). The germinating seeds were collected at 8 h (before gland formation) and 22–46 h for different species (at the beginning stage of gland formation) for transcriptome analysis (Figure 1). Finally, the germinated seeds were taken, photographed under a stereomicroscope, or rapidly frozen with liquid nitrogen prior to extraction of total RNA.
Virus‐induced gene silencing assay
For knockdown of GauPGF, GauGRAS1, GauCCD7 and GauCBP1, approximately 300‐bp fragments of the target genes were PCR‐amplified from G. australe cDNA. The primers used were V‐GauPGF‐F and V‐GauPGF‐R, V‐GauGRAS1‐F and V‐GauGRAS1‐R, V‐GauCCD7‐F and V‐GauCCD7‐R, and V‐GauCBP1‐F and V‐GauCBP1‐R (Table S12). The PCR products were cloned into pTRV2 to produce the VIGS vectors TRV:GauPGF, TRV:GauGRAS1, TRV:GauCCD7 and TRV:GauCBP1. The pTRV1 and recombinant pTRV2 vectors were introduced into the Agrobacterium strain GV3101. Agrobacterium strains harbouring the TRV:GauPGF, TRV:GauGRAS1, TRV:GauCCD7 and TRV:GauCBP1 plasmids combined with strains harbouring the pTRV1 vector were mixed in a 1:1 ratio. Seedlings with mature cotyledons but without a visible rosette leaf (14 days after germination) were infiltrated by inserting the Agrobacterium suspension into the cotyledons via a syringe (the GauCCD7 and GauCBP1 gene silencing using VIGS was done in Xinhai‐15 (G. barbadense), and GauPGF and GauGRAS1 gene silencing was done in G. australe). The plants were grown at 23°C with a 16‐h light and 8‐h dark cycle and 80% humidity. The effectiveness of the VIGS assay was detected by generating the TRV:GbCLA construct using the G. barbadense CLA1 gene as previously described. TRV:00 was used as a control vector.
Identification of Verticillium wilt resistance of VIGS cotton
The VIGS (TRV:GauCCD7, TRV:GauCBP1) plants and the control (TRV:00) plants were inoculated with a V. dahliae conidia suspension by injuring the roots, and the Verticillium wilt symptoms were investigated and compared at 17 days postinfection. The rate of diseased plants was determined from approximately 30 seedlings per treatment, and the assessment was repeated at least three times. The DI was calculated according to the following formula: DI = [(∑disease grades × number of infected plants)/(total examined plants × 4)] × 100%] (Hu et al., 2018). The DI was scored using at least 25 plants per treatment and repeated at least three times.
Quantitative RT‐PCR analysis
Different tissues of the cotton plants, including ovules, roots, stems and leaves, were collected. Total RNA was extracted and then reverse transcribed into cDNA. The GbUBQ7 gene was selected as an internal reference gene, and Primer Premier 6.0 software was used to design specific quantitative primers (q‐UBQ7‐F, q‐UBQ7‐R; q‐GauPGF‐F, q‐GauPGF‐R; q‐GauGRAS1‐F, q‐GauGRAS1‐R; q‐GauCCD7‐F, q‐GauCCD7‐R; q‐GauCBP1‐F, q‐GauCBP1‐R) (Table S12). The experiment was performed using a Roche LightCycler 480 Real‐Time PCR System with Q711‐ChamQ Universal SYBR qPCR Master Mix (Vazyme, Nanjing, China); the reaction procedure was 40 cycles of 95°C for 30 s, 95°C for 10 s and then 60°C for 30 s.
Gossypol content analysis
Leaves and stems were taken separately from the plants to measure the free gossypol content, which was measured by spectrophotometric methods with phloroglucinol, as described previously (Gao et al., 2013). Briefly, each 100 mg plant sample was ground into powder with liquid nitrogen. Then, 0.5 mL extract (acetonitrile/water = 80:20) was added, and the samples were oscillated at 4°C for 45 min. The extract was centrifuged at 12 000 g for 15 min. The supernatant was carefully transferred into a new EP tube. Finally, a double volume of phloroglucinol (Solarbio) chromogenic solution was added, and the samples were incubated in a 55°C water bath for 5 min. The sample was analysed at a wavelength of 550 nm. A gossypol reference standard as purchased from the website http://www.biaowu.com.
Histochemistry and microscopy
A fixative (1 mL 5% glutaraldehyde+4% paraformaldehyde) was added to a 2.0‐mL centrifuge tube. The materials (leaf, stem, petiole) were rapidly cut into 1‐2 mm with a sharp blade, immediately placed into the fixative and incubated overnight at room temperature. Then, the fixative was aspirated, 1 mL 0.1 m phosphate buffer was added, and the sample was rinsed twice (15 min each time). Next, the rinse solution was aspirated, and the samples were dehydrated with 30%, 50%, 70%, 90% and 100% ethanol for 30 min each time. The reagent (100% ethanol: LR White embedding agent = 1:1) was added for 1 h, and the pure embedding agent was added for 5 h. The sample was then soaked in a pure embedding agent overnight. Finally, the sample was embedded in a capsule (polymerization in a 60°C incubator for 12 h). The embedding blocks were formed. Semithin sections (1–2 μm) were cut with a Leica‐UC7 ultrathin microtome and stained with 0.05% crystal violet (a drop of 0.05% crystal violet stain was added to the section, the dye solution was immediately drained, and the excess dye solution was rinsed away with distilled water). The samples were observed and photographed under a microscope.
Author contributions
F.L., Y.F.C., X.Z. and K.B.W. designed and conceived the research programme. F.L., X.Y.C., K.B.W., Z.L.Z., Q.L.W., B.L.Z. and R.H.P. prepared samples of DNA sequencing and RNA‐seq. P.W., C.X.W., S.P.G., B.L., Q.D., Y.Q., J.R.C., Y.Y.Z., Q.S., Y.H.X., Y.C. and X.Y.S. performed gene function analyses. L.Y.M. performed histochemistry and microscopy analyses. Y.C. performed TE insertion time analysis, phylogenetic tree analysis and positively selected genes analysis. S.M.H performed the GO term enrichment and gene family expansion analysis, whole‐genome duplication analysis and collinearity analysis. M.S.S. was involved in the transcriptome analyses. C.W.C., Z.L., F.F.Y., Y.Z. and Y.C.X. involved in bioinformatics analyses. Y.Z. and P.A. directed the genome sequencing and assembly parts of the project. F.F.H. performed genome and transposable elements (TEs) annotation. F.L., Y.F.C., G.Y.A., K.B.W. and X.Z. discussed results. Y.F.C. and F.L. contributed to the writing the main text of the manuscript. F.L., Y.F.C., X.Z., G.Y.A. and K.B.W. reviewed the final version.
Competing Interests
The authors declare there are no competing interests.
Supporting information
Figure S1 Illustration of misassemblies in the genome of G. australe examined using BioNano optical maps.
Figure S2 Interaction frequency distribution of Hi‐C links among chromosomes.
Figure S3 K‐mer analysis for estimating the genome size of G. australe.
Figure S4 Venn diagram analyses of unique and conserved genes or gene families.
Figure S5 Syntenic blocks between G. australe and G. arboreum genome (Left), G. australe and G. raimondii genome (Right).
Figure S6 Venn graph of genes subjected to positive selection in G. australe, G. arboreum and G. raimondii.
Figure S7 Enriched pathway of the PSGs in G. australe and G. arboreum.
Figure S8 Expression levels of CCD7 in different tissues of three diploid cotton species.
Figure S9 Expression levels of GauCCD7 in different tissues of G. australe plant treated with Verticillium dahliae.
Figure S10 Expression levels of GauCCD7 in CSSL‐1 seedlings treated with different hormones.
Figure S11 Trend analysis of differently expressed genes response to Verticillium wilt in G. australe.
Figure S12 Venn diagram analyses of up‐regulated profile genes in G. australe and down‐regulated profile genes in G. arboreum.
Figure S13 Expression levels of CBP1 in different tissues of three diploid cotton species.
Figure S14 Expression levels of GauCBP1 in different tissues of G. australe plant treated with Verticillium dahliae.
Figure S15 Expression levels of GauCBP1 in CSSL‐1 seedlings treated with different hormones.
Figure S16 The silencing of GauCBP1 from G. australe compromised cotton resistance to V. dahliae in Xinhai 15.
Figure S17 Expression of GauGRAS1 and GauPGF in ovules of different gland materials.
Figure S18 Relative expression level of GauPGF and GauGRAS1 gene in leaves (adult stage) and ovules (10 dpa) in two diploid G subgroup wild cotton species by qRT‐PCR.
Figure S19 Relative expression level of GoPGF and GRAS1 gene in leaves and ovules in three diploid G subgroup wild cotton species, 16 dpa.
Figure S20 Relative expression level of PGF and GRAS1 gene before and after GF (gland formation) during seed germination of three cotton species.
Figure S21 Functional characterization of GauPGF by VIGS.
Figure S22 Module‐trait relations. Each row corresponds to a module eigengene, column to a transcriptome.
Figure S23 Local collinearity based on forty genes adjacent to GoPGF of two example species.
Figure S24 Local collinearity of GoPGF between G. hirsutum and other genomes of angiosperm selected (blue) from the Angiosperm Phylogeny Group (APG) IV system.
Figure S25 Local collinearity of the GRAS1 gene between G. hirsutum and other genomes of angiosperm selected (blue) from the Angiosperm Phylogeny Group (APG) IV system.
Table S1 Genome assembly statistics for G. australe.
Table S2 Assessment of sequence coverage of the G. australe genome assembly by homologous search using full‐length transcripts.
Table S3 Number of genes with homology or functional classifications by different methods.
Table S4 Analysis of non‐coding RNA genes in the G. australe genome.
Table S5 Evaluation the quality of the annotation using the BUSCO method.
Table S6 Summary and content analysis of different types of TEs in the G. australe genome.
Table S7 Analysis of the content of major TE subfamilies in three Gossypium genomes.
Table S8 Relative distribution (%) of Gypsy retrotransposon subgroups in the genomes of three Gossypium genomes.
Table S9 Annotation of 31 genes that were up‐regulated profile genes in G. australe and down‐regulated profile genes in G. arboreum.
Table S10 Annotation of 10 hub genes with top ten connectivity the magenta4 module of WGCNA analysis.
Table S11 Annotation of genes adjacent to GoPGF co‐expressed in magenta4 module.
Table S12 Primers used in VIGS and qRT‐PCR.
Data S1 PSGs in G. australe, G. arboreum and G. raimondii.
Acknowledgements
We thank Mingcheng Luo in University of California Davis for technical check and assistance. We are grateful to Xiongming Du (Institute of Cotton Research of CAAS) for review of the manuscript. This work was supported by the National Key Research and Development Program of China (NO: 2016YFD0101902) to X. Zhang, the Natural Science Foundation of China (NO: 31530053, U1704104, 31571724) and National Key Research and Development Program of Thirteenth (2016YFD0101413, 2016YFD0100306)
Yingfan Cai, Xiaoyan Cai, Qinglian Wang, Ping Wang, Yu Zhang, Chaowei Cai and Yanchao Xu contributed equally to this work.
Contributor Information
Xiao Zhang, Email: xzhang@henu.edu.cn.
Fang Liu, Email: liufcri@163.com.
Data availability
The raw sequencing data reported in this paper have been deposited at in the NCBI BioProject database under accession number PRJNA513946. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession SMMG00000000. The version described in this paper is version SMMG01000000.
References
- Arora, S. , Steuernagel, B. , Gaurav, K. , Chandramohan, S. , Long, Y. , Matny, O. , Johnson, R. et al (2019) Resistance gene cloning from a wild crop relative by sequence capture and association genetics. Nat. Biotechnol. 37, 139–143. [DOI] [PubMed] [Google Scholar]
- Asai, S. , Mase, K. and Yoshioka, H. (2010) A key enzyme for flavin synthesis is required for nitric oxide and reactive oxygen species production in disease resistance. Plant J. Cell Mol. Biol. 62, 911–924. [DOI] [PubMed] [Google Scholar]
- Bell, A.A. (1969) Phytoalexin production and Verticillium wilt resistance in cotton. Tech. Rep. Arch. Image Lib. 2013, 1–8. [Google Scholar]
- Benbouza, H. , Lognay, G. , Scheffler, J. , Baudoin, J.P. and Mergeai, G. (2009) Expression of the ‘glanded‐plant and glandless‐seed’ trait of Australian diploid cottons in different genetic backgrounds. Euphytica, 165, 211–221. [Google Scholar]
- Benkang, G. and Cun, M. (1996) China Cotton Breeding resistant to Disease. Nanjing: Jiangsu Science and Technology Publishing Press. [Google Scholar]
- Benson, G. (1999) Tandem repeats finder: a program to analyze DNA sequences. Nucleic Acids Res. 27, 573–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borodovsky, M. and Lomsadze, A. (2011) Eukaryotic gene prediction using GeneMark.hmm‐E and GeneMark‐ES. Curr. Protoc. Bioinformatics, Chapter 4, Unit 4.6.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boubakri, H. , Gargouri, M. , Mliki, A. , Brini, F. , Chong, J. and Jbara, M. (2016) Vitamins for enhancing plant resistance. Planta, 244, 529–543. [DOI] [PubMed] [Google Scholar]
- Bouché, N. , Yellin, A. , Snedden, W.A. and Fromm, H. (2005) Plant‐specific calmodulin‐binding proteins. Annu. Rev. Plant Biol. 56, 435–466. [DOI] [PubMed] [Google Scholar]
- Brozynska, M. , Furtado, A. and Henry, R.J. (2016) Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol. J. 14, 1070–1085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton, J.N. , Adey, A. , Patwardhan, R.P. , Qiu, R. , Kitzman, J.O. and Shendure, J. (2013) Chromosome‐scale scaffolding of de novo genome assemblies based on chromatin interactions. Nat. Biotechnol. 31, 1119–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai, Y. , Mo, J. , Zeng, Y. , Ren, W. , Xu, Y. , Wang, S. and Chen, F. (2003) Cloning of cDNAs of differentially expressed genes in the development of special pigment gland of cotton by suppression subtractive hybridization. J. Beijing Univ. Forest, 25, 6–10. [Google Scholar]
- Cai, Y. , Xie, Y. and Liu, J. (2010) Glandless seed and glanded plant research in cotton. A review. Agron. Sustain. Dev. 30, 181–190. [Google Scholar]
- Campbell, M.S. , Holt, C. , Moore, B. and Yandell, M. (2014) Genome annotation and curation using MAKER and MAKER‐P. Curr. Protoc. Bioinformatics, 48, 4.11.11–14.11.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho, M.R. , Herrera, F.A. , Jaramillo, C.A. , Wing, S.L. and Callejas, R. (2011) Paleocene Malvaceae from Northern South America and their biogeographical implications. Am. J. Bot. 98, 1337–1355. [DOI] [PubMed] [Google Scholar]
- Chen, Y. , Wang, Y. , Wang, K. , Zhu, X. , Guo, W. , Zhang, T. and Zhou, B. (2014) Construction of a complete set of alien chromosome addition lines from Gossypium australe in Gossypium hirsutum: morphological, cytological, and genotypic characterization. Theor. Appl. Genet. 127, 1105–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Lu, C. , John, Z.Y. , Zou, C. , Zhang, Y. , Wang, Q. , Huang, J. et al (2016) Fine mapping and candidate gene analysis of the dominant glandless gene Gl2e in cotton (Gossypium spp.). Theor. Appl. Genet. 129, 1347–1355. [DOI] [PubMed] [Google Scholar]
- Cherry, J.P. (1983) Cottonseed oil. J. Am. Oil Chem. Soc. 60, 360–367. [Google Scholar]
- De Bie, T. , Cristianini, N. , Demuth, J.P. and Hahn, M.W. (2006) CAFE: a computational tool for the study of gene family evolution. Bioinformatics, 22, 1269–1271. [DOI] [PubMed] [Google Scholar]
- Decker, E.L. , Alder, A. , Hunn, S. , Ferguson, J. , Lehtonen, M.T. , Scheler, B. , Kerres, K.L. et al (2017) Strigolactone biosynthesis is evolutionarily conserved, regulated by phosphate starvation and contributes to resistance against phytopathogenic fungi in a moss, Physcomitrella patens. New Phytol. 216, 455–468. [DOI] [PubMed] [Google Scholar]
- Deng, B. , Deng, S. , Sun, F. , Zhang, S. and Dong, H. (2011) Down‐regulation of free riboflavin content induces hydrogen peroxide and a pathogen defense in Arabidopsis . Plant Mol. Biol. 77, 185–201. [DOI] [PubMed] [Google Scholar]
- Du, X. , Huang, G. , He, S. , Yang, Z. , Sun, G. , Ma, X. , Li, N. et al (2018) Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50, 796–802. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2004) MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Espartero, J. , Sanchez‐Aguayo, I. and Pardo, J.M. (1995) Molecular characterization of glyoxalase‐I from a higher plant; upregulation by stress. Plant Mol. Biol. 29, 1223–1233. [DOI] [PubMed] [Google Scholar]
- Gao, W. , Long, L. , Zhu, L.F. , Xu, L. , Gao, W.H. , Sun, L.Q. , Liu, L.L. et al (2013) Proteomic and virus‐induced gene silencing (VIGS) Analyses reveal that gossypol, brassinosteroids, and jasmonic acid contribute to the resistance of cotton to Verticillium dahliae . Mol. Cell Proteomics, 12, 3690–3703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, W. , Long, L. , Xu, L. , Lindsey, K. , Zhang, X. and Zhu, L. (2016) Suppression of the homeobox gene HDTF1 enhances resistance to Verticillium dahliae and Botrytis cinerea in cotton. J. Integr. Plant Biol. 58, 503–513. [DOI] [PubMed] [Google Scholar]
- Grover, C.E. , Arick, M.A. , Conover, J.L. , Thrash, A. , Hu, G. , Sanders, W.S. , Hsu, C.‐Y. et al (2017) Comparative genomics of an unusual biogeographic disjunction in the cotton tribe (Gossypieae) yields insights into genome downsizing. Genome Biol. Evol. 9, 3328–3344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, B. , Li, J. , Gu, P. , Qian, S. , Huang, J. , Zhou, B. , Peng, Y. et al (1993) The identification of resistance to Verticillium and Fusarium wlt in Gossypium genus wild species. Jiangsu Agricul. Sci. 5, 36–37. [Google Scholar]
- Han, L.B. , Li, Y.B. , Wang, F.X. , Wang, W.Y. , Liu, J. , Wu, J.H. , Zhong, N.Q. et al (2019) The cotton apoplastic protein CRR1 stabilizes chitinase 28 to facilitate defense against the fungal pathogen Verticillium dahliae . Plant Cell, 31, 520–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasanuzzaman, M. , Nahar, K. , Anee, T.I. and Fujita, M. (2017) Glutathione in plants: biosynthesis and physiological role in environmental stress tolerance. Physiol. Mol. Biol. Plants, 23, 249–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawkins, J.S. , Kim, H. , Nason, J.D. , Wing, R.A. and Wendel, J.F. (2006) Differential lineage‐specific amplification of transposable elements is responsible for genome size variation in Gossypium . Genome Res. 16, 1252–1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hendrix, B. and Stewart, J.M. (2005) Estimation of the nuclear DNA content of Gossypium species. Ann. Bot. 95, 789–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez‐Gomez, M.C. , Runavot, J.L. , Meulewaeter, F. and Knox, J.P. (2017) Developmental features of cotton fibre middle lamellae in relation to cell adhesion and cell detachment in cultivars with distinct fibre qualities. BMC Plant Biol. 17, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Q. , Min, L. , Yang, X. , Jin, S. , Zhang, L. , Li, Y. , Ma, Y. et al (2018) Laccase GhLac1 modulates broad‐spectrum biotic stress tolerance via manipulating phenylpropanoid pathway and jasmonic acid synthesis. Plant Physiol. 176, 1808–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu, Y. , Chen, J. , Fang, L. , Zhang, Z. , Ma, W. , Niu, Y. , Ju, L. et al (2019) Gossypium barbadense and Gossypium hirsutum genomes provide insights into the origin and evolution of allotetraploid cotton. Nat. Genet. 51, 739–748. [DOI] [PubMed] [Google Scholar]
- Huchelmann, A. , Boutry, M. and Hachez, C. (2017) Plant glandular trichomes: natural cell factories of high biotechnological interest. Plant Physiol. 175, 6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janga, M.R. , Pandeya, D. , Campbell, L.M. , Konganti, K. , Villafuerte, S.T. , Puckhaber, L. , Pepper, A. et al (2018) Genes regulating gland development in the cotton plant. Plant Biotechnol. J. 17, 1142–1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurka, J. , Kapitonov, V.V. , Pavlicek, A. , Klonowski, P. , Kohany, O. and Walichiewicz, J. (2005) Repbase update, a database of eukaryotic repetitive elements. Cytogenet Genome Res. 110, 462–467. [DOI] [PubMed] [Google Scholar]
- Khan, A.M. , Khan, A.A. , Azhar, M.T. , Amrao, L. and Cheema, H.M. (2016) Comparative analysis of resistance gene analogues encoding NBS‐LRR domains in cotton. J. Sci. Food Agric. 96, 530–538. [DOI] [PubMed] [Google Scholar]
- Korf, I. (2004) Gene finding in novel genomes. BMC Bioinformatics, 5, 59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kriventseva, E.V. , Kuznetsov, D. , Tegenfeldt, F. , Manni, M. , Dias, R. , Simao, F.A. and Zdobnov, E.M. (2019) OrthoDB v10: sampling the diversity of animal, plant, fungal, protist, bacterial and viral genomes for evolutionary and functional annotations of orthologs. Nucleic Acids Res. 47, D807–D811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kunbo, W. and Jonathan, W. (2018) Designations for individual genomes and chromosomes in Gossypium. J. Cotton Res. 1, 3. [Google Scholar]
- Li, H. and Durbin, R. (2009) Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, L. , Stoeckert, C.J. and Roos, D.S. (2003) OrthoMCL: identification of ortholog groups for eukaryotic genomes. Genome Res. 13, 2178–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, F. , Fan, G. , Wang, K. , Sun, F. , Yuan, Y. , Song, G. , Li, Q. et al (2014) Genome sequence of the cultivated cotton Gossypium arboreum . Nat. Genet. 46, 567–572. [DOI] [PubMed] [Google Scholar]
- Li, F. , Fan, G. , Lu, C. , Xiao, G. , Zou, C. , Kohel, R.J. , Ma, Z. et al (2015) Genome sequence of cultivated upland cotton (Gossypium hirsutum TM‐1) provides insights into genome evolution. Nat. Biotechnol. 33, 524–530. [DOI] [PubMed] [Google Scholar]
- Li, Z.K. , Chen, B. , Li, X.X. , Wang, J.P. , Zhang, Y. , Wang, X.F. , Yan, Y.Y. et al (2018) A newly identified cluster of glutathione S‐transferase genes provides Verticillium wilt resistance in cotton. Plant J. 98, 213–227. [DOI] [PubMed] [Google Scholar]
- Liu, C.J. , Heinstein, P. and Chen, X.Y. (1999) Expression pattern of genes encoding farnesyl diphosphate synthase and sesquiterpene cyclase in cotton suspension‐cultured cells treated with fungal elicitors. Mol. Plant Microbe Interact. 12, 1095–1104. [DOI] [PubMed] [Google Scholar]
- Liu, Q. , Chen, Y. , Chen, Y. , Wang, Y. , Chen, J. , Zhang, T. and Zhou, B. (2015) A new synthetic allotetraploid (A1A1G2G2) between Gossypium herbaceum and G. australe: bridging for simultaneously transferring favorable genes from these two diploid species into upland cotton. PLoS ONE, 10, e0123209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llorens, C. , Munoz‐Pomer, A. , Bernad, L. , Botella, H. and Moya, A. (2009) Network dynamics of eukaryotic LTR retroelements beyond phylogenetic trees. Biol. Direct, 4, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu, Y. , Truman, W. , Liu, X. , Bethke, G. , Zhou, M. , Myers, C.L. , Katagiri, F. et al (2018) Different modes of negative regulation of plant immunity by calmodulin‐related genes. Plant Physiol. 176, 3046–3061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma, D. , Hu, Y. , Yang, C. , Liu, B. , Fang, L. , Wan, Q. , Liang, W. et al (2016) Genetic basis for glandular trichome formation in cotton. Nat. Commun. 7, 10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammadov, J. , Buyyarapu, R. , Guttikonda, S.K. , Parliament, K. , Abdurakhmonov, I.Y. and Kumpatla, S.P. (2018) Wild relatives of maize, rice, cotton, and soybean: treasure troves for tolerance to biotic and abiotic stresses. Front. Plant. Sci. 9, 886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy, E.M. and McDonald, J.F. (2003) LTR_STRUC: a novel search and identification program for LTR retrotransposons. Bioinformatics, 19, 362–367. [DOI] [PubMed] [Google Scholar]
- Mcmichael, S.C. (1959) Hopi cotton, a source of cottonseed free of gossypol pigments. Agron. J. 51, 630. [Google Scholar]
- Miao, Y. , Xu, L. , He, X. , Zhang, L. , Shaban, M. , Zhang, X. and Zhu, L. (2019) Suppression of tryptophan synthase activates cotton immunity by triggering cell death via promoting SA synthesis. Plant J. 98, 329–345. [DOI] [PubMed] [Google Scholar]
- Milner, S.G. , Jost, M. , Taketa, S. , Mazon, E.R. , Himmelbach, A. , Oppermann, M. , Weise, S. et al (2019) Genebank genomics highlights the diversity of a global barley collection. Nat. Genet. 51, 319–326. [DOI] [PubMed] [Google Scholar]
- Natalio, F. , Fuchs, R. , Cohen, S.R. , Leitus, G. , Fritz‐Popovski, G. , Paris, O. , Kappl, M. et al (2017) Biological fabrication of cellulose fibers with tailored properties. Science, 357, 1118–1122. [DOI] [PubMed] [Google Scholar]
- Paape, T. , Briskine, R.V. , Lischer, H.E.L. , Halsteadnussloch, G. , Shimizuinatsugi, R. , Hatekayama, M. , Tanaka, K. et al (2018) Patterns of polymorphism, selection and linkage disequilibrium in the subgenomes of the allopolyploid Arabidopsis kamchatica . Nat. Commun. 9, 3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paterson, A.H. , Wendel, J.F. , Gundlach, H. , Guo, H. , Jenkins, J. , Jin, D. , Llewellyn, D. et al (2012) Repeated polyploidization of Gossypium genomes and the evolution of spinnable cotton fibres. Nature, 492, 423–427. [DOI] [PubMed] [Google Scholar]
- Piisilä, M. , Keceli, M.A. , Brader, G. , Jakobson, L. , Jõesaar, I. , Sipari, N. , Kollist, H. et al (2015) The F‐box protein MAX2 contributes to resistance to bacterial phytopathogens in Arabidopsis thaliana . BMC Plant Biol. 15, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin, J. , Wang, K. , Sun, L. , Xing, H. , Wang, S. , Li, L. , Chen, S. et al (2018) The plant‐specific transcription factors CBP60g and SARD1 are targeted by a Verticillium secretory protein VdSCP41 to modulate immunity. Elife, 7, e34902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao, S.S. , Huntley, M.H. , Durand, N.C. , Stamenova, E.K. , Bochkov, I.D. , Robinson, J.T. , Sanborn, A.L. et al (2014) A 3D map of the human genome at kilobase resolution reveals principles of chromatin looping. Cell, 159, 1665–1680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidt, M.H. , Vogel, A. , Denton, A.K. , Istace, B. , Wormit, A. , van de Geest, H. , Bolger, M.E. et al (2017) De novo assembly of a new Solanum pennellii accession using nanopore sequencing. Plant Cell, 29, 2336–2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeholzer, S. , Tsuchimatsu, T. , Jordan, T. , Bieri, S. , Pajonk, S. , Yang, W.X. , Jahoor, A. et al (2010) Diversity at the Mla powdery mildew resistance locus from cultivated barley reveals sites of positive selection. Mol. Plant Microbe Interact. 23, 497–509. [DOI] [PubMed] [Google Scholar]
- Servant, N. , Varoquaux, N. , Lajoie, B.R. , Viara, E. , Chen, C.J. , Vert, J.P. , Heard, E. et al (2015) HiC‐Pro: an optimized and flexible pipeline for Hi‐C data processing. Genome Biol. 16, 259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, Q. , Bourdais, G. , Pan, H. , Robatzek, S. and Tang, D. (2017) Arabidopsis glycosylphosphatidylinositol‐anchored protein LLG1 associates with and modulates FLS2 to regulate innate immunity. Proc. Natl Acad. Sci. USA, 114, 5749–5754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanke, M. , Keller, O. , Gunduz, I. , Hayes, A. , Waack, S. and Morgenstern, B. (2006) AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res. 34, W435–W439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Q. , Jiang, H. , Zhu, X. , Wang, W. , He, X. , Shi, Y. , Yuan, Y. et al (2013) Analysis of sea‐island cotton and upland cotton in response to Verticillium dahliae infection by RNA sequencing. BMC Genom. 14, 852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Q. , Du, X.M. , Cai, C.W. , Long, L. , Zhang, S. , Qiao, P. , Wang, W.N. et al (2016) To be a flower or fruiting branch: insights revealed by mRNA and small RNA transcriptomes from different cotton developmental stages. Sci. Rep. 6, 23212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, Q. , Wang, G.H. , Zhang, X. , Zhang, X.R. , Qiao, P. , Long, L. , Yuan, Y.L. et al (2017) Genome‐wide identification of the TIFY gene family in three cultivated Gossypium species and the expression of JAZ genes. Sci. Rep. 7, 42418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, S. , Wang, T. , Wang, L. , Li, X. , Jia, Y. , Liu, C. , Huang, X. et al (2018a) Natural selection of a GSK3 determines rice mesocotyl domestication by coordinating strigolactone and brassinosteroid signaling. Nat. Commun. 9, 2523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, T. , Busta, L. , Zhang, Q. , Ding, P. , Jetter, R. and Zhang, Y. (2018b) TGACG‐binding factor 1 (TGA1) and TGA4 regulate salicylic acid and pipecolic acid biosynthesis by modulating the expression of systemic acquired resistance deficient 1 (SARD1) and calmodulin‐binding protein 60g (CBP60g). New Phytol. 217, 344–354. [DOI] [PubMed] [Google Scholar]
- Sun, Q. , Li, T.Y. , Li, D.D. , Wang, Z.Y. , Li, S. , Li, D.P. , Han, X. et al (2019) Overexpression of loose plant architecture 1 increases planting density and resistance to sheath blight disease via activation of PIN‐FORMED 1a in rice. Plant Biotechnol. J. 17, 855–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang, H. , Bowers, J.E. , Wang, X. , Ming, R. , Alam, M. and Paterson, A.H. (2008) Synteny and collinearity in plant genomes. Science, 320, 486–488. [DOI] [PubMed] [Google Scholar]
- Tang, D. , Feng, S. , Li, S. , Chen, Y. and Zhou, B. (2018) Ten alien chromosome additions of Gossypium hirsutum–Gossypium bickii developed by integrative uses of GISH and species‐specific SSR markers. Mol. Genet. Genomics, 293, 945–955. [DOI] [PubMed] [Google Scholar]
- Tarailo‐Graovac, M. and Chen, N. (2009) Using repeatmasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinformatics, 4, 1–4. 10 [DOI] [PubMed] [Google Scholar]
- Thind, A.K. , Wicker, T. , Mueller, T. , Ackermann, P.M. , Steuernagel, B. , Wulff, B.B. , Spannagl, M. et al (2018) Chromosome‐scale comparative sequence analysis unravels molecular mechanisms of genome evolution between two wheat cultivars. Genome Biol. 19, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian, X. , Ruan, J.X. , Huang, J.Q. , Yang, C.Q. , Fang, X. , Chen, Z.W. , Hong, H. et al (2018) Characterization of gossypol biosynthetic pathway. Proc. Natl Acad. Sci. USA, 115, E5410–E5418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres‐Vera, R. , García, J.M. , Pozo, M.J. and López‐Ráez, J.A. (2014) Do strigolactones contribute to plant defence? Mol. Plant Pathol. 15, 211–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Pachter, L. and Salzberg, S.L. (2009) TopHat: discovering splice junctions with RNA‐Seq. Bioinformatics, 25, 1105–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trapnell, C. , Williams, B.A. , Pertea, G. , Mortazavi, A. , Kwan, G. , van Baren, M.J. , Salzberg, S.L. et al (2010) Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 28, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaissayre, M. and Hau, B. (1985) New results on the susceptibility of glandless cotton varieties to phyllophagous insects. Coton Fibres Tropic. 40, 159–168. [Google Scholar]
- Walker, B.J. , Abeel, T. , Shea, T. , Priest, M. , Abouelliel, A. , Sakthikumar, S. , Cuomo, C.A. et al (2014) Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS ONE, 9, e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, K. , Wang, Z. , Li, F. , Ye, W. , Wang, J. , Song, G. , Yue, Z. et al (2012) The draft genome of a diploid cotton Gossypium raimondii . Nat. Genet. 44, 1098–1103. [DOI] [PubMed] [Google Scholar]
- Wang, C. , Ulloa, M. , Duong, T. and Roberts, P.A. (2018a) Quantitative trait loci mapping of multiple independent loci for resistance to Fusarium oxysporum f. sp. vasinfectum races 1 and 4 in an interspecific cotton population. Phytopathology, 108, 759–767. [DOI] [PubMed] [Google Scholar]
- Wang, J. , Zhou, L. , Shi, H. , Chern, M. , Yu, H. , Yi, H. , He, M. et al (2018b) A single transcription factor promotes both yield and immunity in rice. Science, 361, 1026–1028. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Feng, S. , Li, S. , Tang, D. , Chen, Y. , Chen, Y. and Zhou, B. (2018c) Inducement and identification of chromosome introgression and translocation of Gossypium australe on Gossypium hirsutum . BMC Genom. 19, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, M. , Tu, L. , Yuan, D. , Zhu, Shen.C. , Li, J. , Liu, F. , Pei, L. et al (2019a) Reference genome sequences of two cultivated allotetraploid cottons, Gossypium hirsutum and Gossypium barbadense . Nat. Genet. 51, 224–229. [DOI] [PubMed] [Google Scholar]
- Wang, P. , Zhang, S. , Qiao, J. , Sun, Q. , Shi, Q. , Cai, C. , Mo, J. et al (2019b) Functional analysis of the GbDWARF14 gene associated with branching development in cotton. PeerJ, 7, e6901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendel, J.F. , Stewart, J.M. and Rettig, J.H. (1991) Molecular evidence for homoploid reticulate evolution in Australian species of Gossypium . Evolution, 45, 694–711. [DOI] [PubMed] [Google Scholar]
- Wendel, J.F. , Brubaker, C.L. and Seelanan, T. (2010) The Origin and Evolution of Gossypium In Physiology of Cotton (Stewart J.M., Oosterhuis D.M., Heitholt J.J. & Mauney J.R. eds), pp. 1–18. Netherlands, Dordrecht: Springer. [Google Scholar]
- Xiang, L. , Liu, J. , Wu, C. , Deng, Y. , Cai, C. , Zhang, X. and Cai, Y. (2017) Genome‐wide comparative analysis of NBS‐encoding genes in four Gossypium species. BMC Genom. 18, 292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao, C.L. , Chen, Y. , Xie, S.Q. , Chen, K.N. , Wang, Y. , Han, Y. , Luo, F. et al (2017) MECAT: fast mapping, error correction, and de novo assembly for single‐molecule sequencing reads. Nat. Methods, 14, 1072–1074. [DOI] [PubMed] [Google Scholar]
- Xie, M. , Chung, C.Y. , Li, M.W. , Wong, F.L. , Wang, X. , Liu, A. , Wang, Z. et al (2019) A reference‐grade wild soybean genome. Nat. Commun. 10, 1216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, L. , Zhu, L. , Tu, L. , Liu, L. , Yuan, D. , Jin, L. , Long, L. et al (2011) Lignin metabolism has a central role in the resistance of cotton to the wilt fungus Verticillium dahliae as revealed by RNA‐Seq‐dependent transcriptional analysis and histochemistry. J. Exp. Bot. 62, 5607–5621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Z. (1997) PAML: a program package for phylogenetic analysis by maximum likelihood. Comput. Appl. Biosci. 13, 555–556. [DOI] [PubMed] [Google Scholar]
- Yin, D. , Ji, C. , Ma, X. , Li, H. , Zhang, W. , Li, S. , Liu, F. et al (2018) Genome of an allotetraploid wild peanut Arachis monticola: a de novo assembly. Gigascience, 7, giy066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zambounis, A. , Ganopoulos, I. , Kalivas, A. , Tsaftaris, A. and Madesis, P. (2016) Identification and evidence of positive selection upon resistance gene analogs in cotton (Gossypium hirsutum L.). Physiol. Mol. Biol. Plants, 22, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, J. , Nielsen, R. and Yang, Z. (2005) Evaluation of an improved branch‐site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. [DOI] [PubMed] [Google Scholar]
- Zhang, T. , Hu, Y. , Jiang, W. , Fang, L. , Guan, X. , Chen, J. , Zhang, J. et al (2015) Sequencing of allotetraploid cotton (Gossypium hirsutum L. acc. TM‐1) provides a resource for fiber improvement. Nat. Biotechnol. 33, 531–537. [DOI] [PubMed] [Google Scholar]
- Zhang, L. , Wang, M. , Li, N. , Wang, H. , Qiu, P. , Pei, L. , Xu, Z. et al (2018) Long noncoding RNA s involve in resistance to Verticillium dahliae, a fungal disease in cotton. Plant Biotechnol. J. 16, 1172–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, D.D. , Wang, J. , Wang, D. , Kong, Z.Q. , Zhou, L. , Zhang, G.Y. , Gui, Y.J. et al (2019) Population genomics demystifies the defoliation phenotype in the plant pathogen Verticillium dahliae . New Phytol. 222, 1012–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, Q. , Feng, Q. , Lu, H. , Li, Y. , Wang, A. , Tian, Q. , Zhan, Q. et al (2018) Pan‐genome analysis highlights the extent of genomic variation in cultivated and wild rice. Nat. Genet. 50, 278. [DOI] [PubMed] [Google Scholar]
- Zheng, X.Y. , Zhou, M. , Yoo, H. , Prunedapaz, J.L. , Spivey, N.W. , Kay, S.A. and Dong, X. (2015) Inaugural article by a recently elected academy member: spatial and temporal regulation of biosynthesis of the plant immune signal salicylic acid. Proc. Natl Acad. Sci. USA, 112, 9166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, Y. , Sun, L. , Wassan, G.M. , He, X. , Shaban, M. , Zhang, L. , Zhu, L. et al (2018) Gb SOBIR 1 confers Verticillium wilt resistance by phosphorylating the transcriptional factor Gbb HLH 171 in Gossypium barbadense . Plant Biotechnol. J. 17, 152–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Illustration of misassemblies in the genome of G. australe examined using BioNano optical maps.
Figure S2 Interaction frequency distribution of Hi‐C links among chromosomes.
Figure S3 K‐mer analysis for estimating the genome size of G. australe.
Figure S4 Venn diagram analyses of unique and conserved genes or gene families.
Figure S5 Syntenic blocks between G. australe and G. arboreum genome (Left), G. australe and G. raimondii genome (Right).
Figure S6 Venn graph of genes subjected to positive selection in G. australe, G. arboreum and G. raimondii.
Figure S7 Enriched pathway of the PSGs in G. australe and G. arboreum.
Figure S8 Expression levels of CCD7 in different tissues of three diploid cotton species.
Figure S9 Expression levels of GauCCD7 in different tissues of G. australe plant treated with Verticillium dahliae.
Figure S10 Expression levels of GauCCD7 in CSSL‐1 seedlings treated with different hormones.
Figure S11 Trend analysis of differently expressed genes response to Verticillium wilt in G. australe.
Figure S12 Venn diagram analyses of up‐regulated profile genes in G. australe and down‐regulated profile genes in G. arboreum.
Figure S13 Expression levels of CBP1 in different tissues of three diploid cotton species.
Figure S14 Expression levels of GauCBP1 in different tissues of G. australe plant treated with Verticillium dahliae.
Figure S15 Expression levels of GauCBP1 in CSSL‐1 seedlings treated with different hormones.
Figure S16 The silencing of GauCBP1 from G. australe compromised cotton resistance to V. dahliae in Xinhai 15.
Figure S17 Expression of GauGRAS1 and GauPGF in ovules of different gland materials.
Figure S18 Relative expression level of GauPGF and GauGRAS1 gene in leaves (adult stage) and ovules (10 dpa) in two diploid G subgroup wild cotton species by qRT‐PCR.
Figure S19 Relative expression level of GoPGF and GRAS1 gene in leaves and ovules in three diploid G subgroup wild cotton species, 16 dpa.
Figure S20 Relative expression level of PGF and GRAS1 gene before and after GF (gland formation) during seed germination of three cotton species.
Figure S21 Functional characterization of GauPGF by VIGS.
Figure S22 Module‐trait relations. Each row corresponds to a module eigengene, column to a transcriptome.
Figure S23 Local collinearity based on forty genes adjacent to GoPGF of two example species.
Figure S24 Local collinearity of GoPGF between G. hirsutum and other genomes of angiosperm selected (blue) from the Angiosperm Phylogeny Group (APG) IV system.
Figure S25 Local collinearity of the GRAS1 gene between G. hirsutum and other genomes of angiosperm selected (blue) from the Angiosperm Phylogeny Group (APG) IV system.
Table S1 Genome assembly statistics for G. australe.
Table S2 Assessment of sequence coverage of the G. australe genome assembly by homologous search using full‐length transcripts.
Table S3 Number of genes with homology or functional classifications by different methods.
Table S4 Analysis of non‐coding RNA genes in the G. australe genome.
Table S5 Evaluation the quality of the annotation using the BUSCO method.
Table S6 Summary and content analysis of different types of TEs in the G. australe genome.
Table S7 Analysis of the content of major TE subfamilies in three Gossypium genomes.
Table S8 Relative distribution (%) of Gypsy retrotransposon subgroups in the genomes of three Gossypium genomes.
Table S9 Annotation of 31 genes that were up‐regulated profile genes in G. australe and down‐regulated profile genes in G. arboreum.
Table S10 Annotation of 10 hub genes with top ten connectivity the magenta4 module of WGCNA analysis.
Table S11 Annotation of genes adjacent to GoPGF co‐expressed in magenta4 module.
Table S12 Primers used in VIGS and qRT‐PCR.
Data S1 PSGs in G. australe, G. arboreum and G. raimondii.
Data Availability Statement
The raw sequencing data reported in this paper have been deposited at in the NCBI BioProject database under accession number PRJNA513946. This Whole Genome Shotgun project has been deposited at DDBJ/ENA/GenBank under the accession SMMG00000000. The version described in this paper is version SMMG01000000.