

Summary
Plant height and branch number are essential components of rapeseed plant architecture and are directly correlated with its yield. Presently, improvement of plant architecture is a major challenge in rapeseed breeding. In this study, we first verified that the two rapeseed BnaMAX1 genes had redundant functions resembling those of Arabidopsis MAX1, which regulates plant height and axillary bud outgrowth. Therefore, we designed two sgRNAs to edit these BnaMAX1 homologs using the CRISPR/Cas9 system. The T0 plants were edited very efficiently (56.30%–67.38%) at the BnaMAX1 target sites resulting in homozygous, heterozygous, bi‐allelic and chimeric mutations. Transmission tests revealed that the mutations were passed on to the T1 and T2 progeny. We also obtained transgene‐free lines created by the CRISPR/Cas9 editing, and no mutations were detected in potential off‐target sites. Notably, simultaneous knockout of all four BnaMAX1 alleles resulted in semi‐dwarf and increased branching phenotypes with more siliques, contributing to increased yield per plant relative to wild type. Therefore, these semi‐dwarf and increased branching characteristics have the potential to help construct a rapeseed ideotype. Significantly, the editing resources obtained in our study provide desirable germplasm for further breeding of high yield in rapeseed.
Keywords: Brassica napus, CRISPR/Cas9, BnaMAX1, plant architecture, semi‐dwarf, increased branching
Introduction
Plant architecture depends greatly on the fates of the axillary meristems, and their regulation by hormonal and environmental factors (Schmitz and Theres, 2005; Sussex and Kerk, 2001). Rapeseed (Brassica napus L., AACC, 2n = 38) is a crucial source of edible oil and biofuel, and its branch number is a major factor affecting its architecture and yield. Plant breeders have been seeking the rapeseed ‘ideotype’ as it directly affects crop yield.
In the vegetative stage, axillary meristems initiate in leaf axils to form axillary buds, which subsequently remain dormant or continue to grow to form branches (Bennett and Leyser, 2006). Levels of auxin and cytokinin hormones have been thought to be the main factors that regulate bud outgrowth for many decades. Auxin synthesized in the primary shoot apex and then transported to the basal regions (basipetally) indirectly inhibits branching, which is termed apical dominance (Ljung et al., 2001; Thimann and Skoog, 1933). In contrast, cytokinin is produced in the root and transported up to the axillary buds promoting their growth (Horvath et al., 2003). Auxin probably partially reduces cytokinin export from roots and synthesis at the node, thus inhibiting bud growth (Chatfield et al., 2000; Nordstrom et al., 2004). Unexpectedly, a study revealed that auxin formed at the apex does not enter the buds, suggesting that another signal is involved in inhibition of bud growth (Morris, 1977).
Studies of several branching mutants, including ramosus (rms) in pea, decreased apical dominance (dad) in petunia, more axillary growth (max) in Arabidopsis and dwarf mutants (d mutants) in rice, revealed that strigolactones (SLs) played a negative role in bud outgrowth and that this function was highly conserved in both monocots and dicots (Gomez‐Roldan et al., 2008; Umehara et al., 2008). The MAX3/RMS5/DAD3/D17 and MAX4/RMS1/DAD1/D10 genes encode carotenoid cleavage dioxygenases 7 (CCD7) and CCD8, respectively, and MAX1 encodes a cytochrome P450 monooxygenase (CYP711A1) that acts downstream of MAX3 and MAX4. All of these enzymes are involved in SL biosynthesis (Alder et al., 2012; Booker et al., 2005; Lazar and Goodman, 2006). MAX1 converts carlactone to carlactonoic acid, and max1 mutants display reduced stature with increased branching and rounder rosette leaves. In contrast, overexpression of MAX1 represses bud outgrowth at the stem base (Abe et al., 2014; Booker et al., 2005; Lazar and Goodman, 2006). The MAX2/RMS4/D3 and DAD2/D14 genes encode an F‐box protein and α/β‐hydrolase, respectively, likely participating in SL signal transduction (Arite et al., 2009; Stirnberg et al., 2007). Recently, the identification of the SL receptor SMAX1‐LIKE/D53 gene in Arabidopsis and rice established an SL biosynthesis–signalling pathway in these plants (Jiang et al., 2013; Soundappan et al., 2015; Wang et al., 2015; Zhou et al., 2013). However, an SL biosynthesis–signalling pathway has not been identified in rapeseed.
Rapeseed is a relatively recent allopolyploid originating from the hybridization of B. rapa (2n = 20, AA) with B. oleracea (2n = 18, CC), and most orthologous genes are duplicated and functionally redundant (Chalhoub et al., 2014). Consequently, characterization of a single gene is complicated, so simultaneous mutation of all members of a multi‐copy gene family is preferable for analysis of gene function. Recently, the CRISPR/CRISPR‐associated 9 (CRISPR/Cas9) gene editing system which can effectively and precisely edit target sites in genomes of all organisms was created (Cong et al., 2013; Gaj et al., 2013; Mali et al., 2013). This system has been successfully used in many plant species (Li et al., 2013; Shan et al., 2013). Moreover, CRISPR/Cas9‐mediated gene editing has been performed in wheat, potato and cotton, showing that CRISPR/Cas9 can be used to edit multi‐gene families in polyploid crops (Andersson et al., 2017; Wang et al., 2014, 2018). To date, several studies have reported using CRISPR/Cas9 for targeted mutagenesis in rapeseed to improve shattering resistance, seed production, plant architecture and disease resistance, respectively (Braatz et al., 2017; Li et al., 2018; Sun et al., 2018; Yang et al., 2017; Zhai et al., 2019).
In this study, we designed sgRNAs to edit the two rapeseed homologs (BnaMAX1) of the Arabidopsis MAX1 gene using the CRISPR/Cas9 system. We obtained plants that contained edited versions of both BnaMAX1 genes and were free of T‐DNA insertions in a spring‐type rapeseed variety. This is the first report of creating semi‐dwarf and increased branching germplasm resources in rapeseed using CRISPR/Cas9 technology and provides a useful germplasm for improving rapeseed plant architecture and yield.
Results and discussion
The 19‐22 mutant is Arabidopsis max1
To investigate the molecular mechanisms controlling plant architecture in rapeseed, we first screened for branching mutants in an Arabidopsis T‐DNA library. We obtained a line (Salk_036990, which we designated 19‐22) that displayed dwarfism with increased branching and rounder rosette leaves (Figure 1a–c). Using Tail‐PCR amplification, we identified one putative insertion site, and linkage analysis showed that this insertion was not responsible for the phenotype of 19‐22 (Figure S1). Further, an F2 segregant from a cross with Col was used to show via genetic experiments that 19‐22 was inherited as a single nuclear recessive mutation (Figure S2a; Table S1). MutMap showed that a 20‐bp deletion in At2g26170 (MAX1) was probably related to the dwarfism and increased branching phenotypes (Table S2). Linkage and sequence analyses confirmed that this deletion was present in 19‐22 and the F2 plants expressing the recessive phenotype (Figure S2b). The 20‐bp deletion caused a frameshift and premature stop in the first exon of MAX1. This represented a novel allele compared with previous studies, and the phenotype of 19‐22 resembled that of the Arabidopsis max1‐3 mutant (Figure 1; Lazar and Goodman, 2006).
Figure 1.

Phenotypes of Arabidopsis wild‐type (WT) and 19‐22 mutant, and alignment of MAX1 protein with its mutations. (a–b) Comparison of WT and 19‐22 mutant vegetative rosettes. The 19‐22 shows rounder leaves. Top is WT and bottom is 19‐22 in (b). (c) 19‐22 mutant showing dwarfism with increased branching phenotype at the flowering stage. (d) Structures of MAX1 and a 20‐bp nucleotide deletion in the first exon that caused a frameshift and premature stop in 19‐22. Lines indicate introns, black boxes indicate exons, and white boxes indicate 5′‐ or 3′‐UTR. Bars = 1 cm.
The rapeseed genome contains two genes homologous to Arabidopsis MAX1
A BLASTP search identified two close homologs of MAX1 in the rapeseed genome (BnaA03g22900D and BnaC03g26960D). However, BnaA03g22900D only has a partial coding sequence (CDS) in the ‘Darmor‐bzh’ database, and therefore, we obtained the full‐length CDS sequence (BnA03 g0116320.1) from the ‘ZS11’ database (Sun et al., 2017). We named these MAX1 homologs, BnaA03.MAX1 and BnaC03.MAX1. BnaA03.MAX1 shared 88.9% identity and 93.8% similarity with MAX1 at the amino acid level, and BnaC03.MAX1 shared 88.5% and 93.2% similarity with MAX1 at these levels (Figure 2a). These results suggested that the BnaMAX1 homologs might have similar functions as MAX1. In addition, the two BnaMAX1 homologs shared 98.5% identity and 99.4% similarity in their amino acid sequences (Figure 2a, b). Genomic‐synteny analysis showed that the two BnaMAX1s originated from ancestors B. rapa and B. oleracea, respectively (Chalhoub et al., 2014). Therefore, the two BnaMAX1s probably have redundant functions in regulating axillary bud outgrowth.
Figure 2.
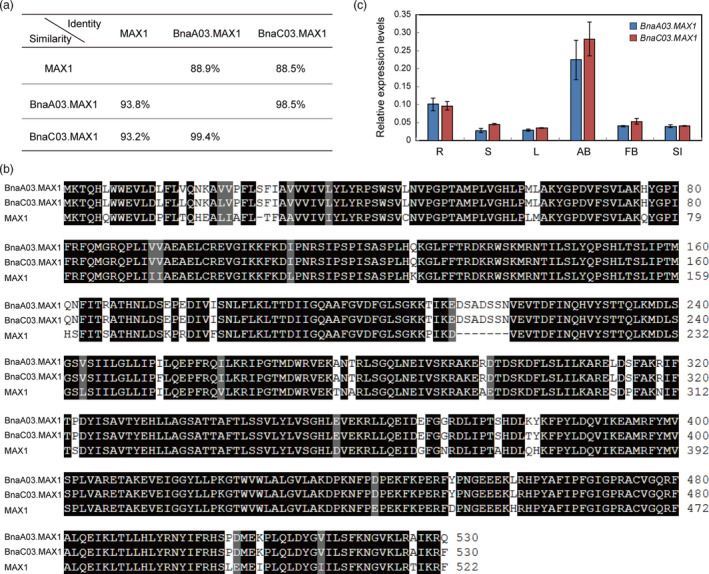
The rapeseed genome contains two BnaMAX1 homologs. (a) Amino acid identities and similarities of MAX1 proteins in Arabidopsis and rapeseed. (b) Alignment of MAX1 and BnaMAX1s amino acid sequences. Black shading indicates identical or similar residues, and numbers indicate amino acid positions. (c) qRT‐PCR analysis of the expression of BnaMAX1s in diverse tissues. Error bars ± standard deviations (n = 3). R, root; S, stem; L, leaf; AB, axillary bud; FB, flower bud; SI, silique.
The SLs are thought to be synthesized in roots and transported to shoots through the xylem to inhibit bud outgrowth (Beveridge, 2006; Kohlen et al., 2011). Our qRT‐PCR analyses showed that the BnaMAX1 transcripts were more abundant in axillary buds, and both were more highly expressed in roots, as was reported for Arabidopsis MAX1 (Figure 2c, Figure S3a), suggesting that both have roles in SL biosynthesis.
BnaMAX1s have similar functions in regulating plant architecture as Arabidopsis MAX1
To further confirm the functions of BnaMAX1s in SL signalling, we constructed vectors expressing BnaA03.MAX1, BnaC03.MAX1 and MAX1 controlled by the 35S promoter and transformed them into the Arabidopsis 19‐22 mutant. All transgenic lines that expressed the three genes exhibited normal Col‐0 leaf phenotypes at the seedling stage (Figure 3a, Figure S3b). Similarly, adult transgenic plants also showed normal plant architecture, including increased plant height (PH) and decreased branch number (BN) relative to the 19‐22 mutant (Figure 3b). Previous studies reported that MAX1 encodes a member of the CYP450 family, CYP711A1, which is a carlactone oxidase that catalyses the last step of SL biosynthesis (Booker et al., 2005). Therefore, the two homologs of BnaMAX1 could have a similar function in SL biosynthesis and repressing the vegetative growth of axillary buds in rapeseed.
Figure 3.

Morphological comparison of wild‐type (WT), 19‐22 and transgenic Arabidopsis plants overexpressing MAX1 or BnaMAX1s in 19‐22 mutant. (a–b) WT, 19‐22 and transgenic plants at vegetative (a) and mature (b) stages. Bars = 1 cm.
Sequence identification for editing BnaMAX1 homologs by CRISPR/Cas9 in rapeseed
In the light of the functional analysis of the two BnaMAX1s in Arabidopsis, we realized that we needed to simultaneously knock out both homologous genes in order to produce the expected phenotypes in rapeseed. We chose the BGK01 vector (Biogle, China) to generate the Cas9‐BnaMAX1 construct, which uses the AtU6‐26 promoter to drive sgRNA expression (Figure 4a). We next designed two sgRNAs to target the first exons of the BnaMAX1s that each had the same target site sequences in their homologous region (Figure 4b). In addition, we analysed the likely off‐target sites in the rapeseed genome using CRISPR‐P (http://crispr.hzau.edu.cn/CRISPR/). The results showed that the two sgRNA sequences had several putative off‐target sites in the rapeseed genome, but all had more than three mismatches compared with the original target sites (Figure S4). This indicated that the two target sites were good choices for specific mutagenesis without off‐target effects.
Figure 4.
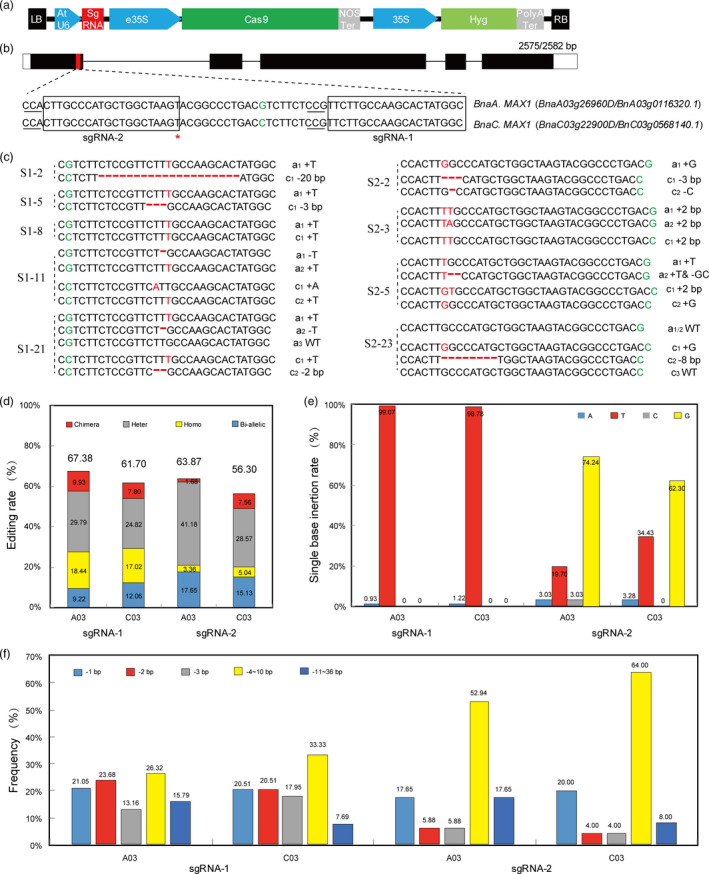
Characterization of editing of BnaMAX1 genes using the CRISPR/Cas9 system in rapeseed. (a) Structures of the BGK01–CRISPR/Cas9 binary vectors. (b) CRISPR/Cas9 sgRNA‐1 targets the first exon of BnaMAX1s. The green colours indicate the SNP upstream sgRNA‐1 target. The protospacer adjacent motif (PAM) is underlined. The box indicates the target sequences. (c) Sequencing of the BnaMAX1 sites targeted by two sgRNAs. Red colours and red hyphens indicate insertions and deletions, respectively. a1, a2, c1 and c2 indicate the four BnaMAX1 alleles, respectively. (d) Editing efficiencies of the two sgRNAs in rapeseed T0 plants. (e) The single nucleotide types and insertion rates in gene editing guided by sgRNAs. (f) Deletion frequencies at the two targeted sites.
The first target sequence was CCGTTCTTGCCAAGCACTATGGC (reversed, GCCATAGTGCTTGGCAAGAACGG), with a G/C SNP located 6‐bp upstream of the CGG PAM motif in BnaA03/C03.MAX1, which was used to distinguish the homologous copies by sequencing transgenic plants (Figure 4b). After Agrobacterium tumefaciens‐mediated transformation of variety ‘862’ (a spring‐type rapeseed) hypocotyls, we obtained 4735 calli and 141 positive transgenic plants (141 out of 150 plants) displaying hygromycin (Hyg) resistance. Most transgenic plants had different changes in the sequences of the target sites in the four BnaMAX1 alleles (Figures 4c; S5). In order to distinguish the four BnaMAX1 alleles, we named them a 1 , a 2 , c 1 and c 2 , respectively. We observed that all four BnaMAX1 alleles simultaneously had mutations in 29 T0 lines (Table S4). 9.22%–12.06% of all the loci examined were bi‐allelic mutations, 17.02%–18.44% of the loci were homozygous mutations, and 24.82%–29.79% and 7.80%–9.93% of the loci were heterozygous and chimeric mutations in the A/C genomes, respectively (Figure 4d; Table S4). Notably, in single nucleotide insertion events, 98.78%–99.07% of the loci were insertions of a ‘T’ (Figure 4e), while 57.89%–58.97% of deletions were less than 3 bp (Figure 4f).
The second target sequence was CCACTTGCCCATGCTGGCTAAGT (reversed, ACTTAGCCAGCATGGGCAAGTGG). We changed the first A to G in this sgRNA sequence to suit the AtU6‐26 promoter. A G/C SNP was located 31‐bp downstream of the TGG PAM motif in BnaA03/C03.MAX1 to help identify gene‐edited progeny (Figure 4b). We transformed this construct into hypocotyls of variety ‘862’ and obtained 3382 calli and 119 positive transgenic plants (119 out of 123 plants) showing Hyg resistance. After sequencing the PCR amplicons of the two BnaMAX1 genomic DNAs, we found bi‐allelic mutations in both AC genomes in 7 lines (Figure 4d; Table S4). Similarly, 28.57%–41.18% (34‐49/119) of all loci examined were single heterozygous alleles in the A/C genomes, while the homozygous mutation rate was much lower than that of the sgRNA‐1 edited sites, ranging from 3.36% to 5.04% (Figure 4d; Table S4). Notably, in single nucleotide insertion events, 62.30%–74.24% of the loci were G insertions, whereas T insertions occurred in 19.70%–34.43% of the loci (Figure 4e). As expected, most mutations were adjacent to the PAM sequence in the plants edited with our two sgRNAs. However, three T0 lines contained large deletions (85–165 bp) that lost the entire sgRNA target sites (Figure S5; Table S4). Finally, most of these indels resulted in frameshifts, premature stop codons or amino acid deletions in the first or third exons of the targeted BnaMAX1s.
Previous studies in rapeseed using the CRISPR/Cas9 system selected 20‐nt sequences 5′ to the PAM as target sequences (Braatz et al., 2017; Yang et al., 2017). In our study, we selected 19‐nt targeting sequences and achieved high editing efficiencies. This shows that 19 nt target sequences are also suitable for CRISPR/Cas9 editing in rapeseed, that is the target sequence may be loosened to 18–20 nt (Shan et al., 2013). The sgRNAs targeting‐specific sequences were transcribed by U3 or U6 promoters that initiated with A or G, respectively (Cong et al., 2013; Shan et al., 2013). It was reported that an additional A or G at the 5′ end of sgRNAs that started with a T or C also resulted in genome editing in rice (Ma et al., 2015; Xie and Yang, 2013). In our sgRNA‐2 sequence, the first nucleotide A was an initiation site for U3 promoter transcription, and in subsequent constructs, we changed this A to G to suit the U6 promoter. Intriguingly, the positive transgenic plants expressing sgRNA‐2 also showed high editing levels (56.30%–63.87%, Figure 4e), showing that replacing the transcription start nucleotide to suit the relative promoter functioned in a rapeseed CRISPR/Cas9 system.
1‐bp insertions are typical CRISPR/Cas9 mutations and were found at high rates in both sgRNA‐1 and sgRNA‐2 lines (17.65%–21.05%; Figure 4f). Previous studies showed that single insertions were observed in the 3–4 bp adjacent to the PAM (Cong et al., 2013; Lawrenson et al., 2015). Gs were inserted 4 bp adjacent to the PAM in sgRNA‐2 lines, while Ts were inserted 6 bp adjacent to the PAM in sgRNA‐1 lines, showing that insertions occurred 3–6 bp upstream of the PAM. Previous studies reported that most insertions were As or Ts (Ma et al., 2015; Zhang et al., 2014), whereas most insertions in our sgRNA‐2 plants were Gs. Thus, the base inserted by this type of mutation might be random. In addition, we found that 1‐bp deletions occurred at the lowest rates at both sgRNA target sites, in contrast to previous reports. A possible explanation for these findings is that they were caused by microhomology‐mediated repair (Sanchez‐Leon et al., 2018; Yang et al., 2017). Homozygous mutations occurred at a high rate in the two targeted sites (3.36%–18.44%) in T0 rapeseed plants and occurred simultaneously in both A and C homologs with the same mutation type. It is likely that one chromosome was edited by the Cas9/sgRNA complex, and another homologous chromosome or sub‐homologous chromosome was cleaved by the Cas9/sgRNA complex and repaired using the originally mutated allele as template (Ma et al., 2015). Therefore, both non‐homologous end joining (NHEJ) and homology‐directed repair (HDR) mechanisms might have occurred simultaneously in our edited plants.
Isolation of edited rapeseed BnaMAX1 lines that eliminated the T‐DNA
To assess the inheritance of the CRISPR/Cas9‐induced mutations and obtain stably inherited lines containing mutations in both BnaMAX1 genes that also eliminated the T‐DNA, we self‐pollinated four T0 lines (S1‐5, S1‐8, S1‐11 and S2‐5, which had diverse types of mutations). Real‐time PCR using a Hyg probe in S1‐8 T1 segregants showed that the T‐DNA insertion site was not linked with the plant phenotypes (Table S3). We then screened five independent S1‐8 T1 lines (S1‐8‐8/12/23/36/39) and collected each of their T2 progenies. A quarter of S1‐8‐23 or S1‐8‐39 T2 plants lacking the hygromycin resistance gene, and eight T2 progeny of S1‐8‐8 were homozygous plants contained T‐DNA that corresponded to their T1 parents (Table 1). Notably, in S1‐8 T1 segregants, if one BnaMAX1 allele was normal the plant resembled wild type (Figure S6; Table S3), whereas simultaneous knockout of all four BnaMAX1 alleles resulted in semi‐dwarf and increased branching phenotypes in several T0 plants (e.g. S1‐11/‐12/‐59 and S2‐5/‐41/‐47), indicating that the two BnaMAX1 homologs had functional redundancy.
Table 1.
Genetic analysis of CRISPR/Cas9‐induced mutations in BnaMAX1s and their transmission to the T1 and T2 generations.
| T0 plants line | T1 plants | T2 plants | ||||
|---|---|---|---|---|---|---|
| No. of tested plants | Line | Chr. A03 | Chr. C03 | No. of homozygous plants | Plants without T‐DNA | |
|
S1‐5 a1 + T a2 WT c1 − CTT c2 WT |
6 | S1‐5‐1 | + T/WT (He) | − CTT (Ho) | 10/24 | None |
| S1‐5‐4 | + T (Ho) | − CTT (Ho) | 8/8 | None | ||
| S1‐5‐6 | + T (Ho) | − CTT (Ho) | 12/12 | S1‐5‐6‐8 | ||
|
S1‐8 a1 + T a2 WT c1 + T c2 WT |
60 | S1‐8‐8 | + T (Ho) | + T (Ho) | 8/8 | None |
| S1‐8‐12 | + T (Ho) | + T (Ho) | 10/10 | None | ||
| S1‐8‐23 | WT | WT | 0/23 | S1‐8‐23‐3/7/10/14/17/18 | ||
| S1‐8‐36 | + T/WT (He) | + T (Ho) | 0/2 | None | ||
| S1‐8‐39 | + T (Ho) | + T (Ho) | 11/11 | S1‐8‐39‐1/4/6/10 | ||
|
S1‐11 a1 − T a2 + T c1 + A c2 + T |
47 | S1‐11‐1 | − T (Ho) | + T (Ho) | 20/20 | S1‐11‐4/7/12/19 |
| S1‐11‐7 | − T/+ T (Bi‐a) | + A (Ho) | 10/22 | All | ||
| S1‐11‐13 | − T/+ T (Bi‐a) | + T (Ho) | 9/22 | All | ||
| S1‐11‐16 | − T (Ho) | + T (Ho) | 22/22 | All | ||
|
S2‐5 a1 + T a2 GC − T c1 + GT c2 + G |
14 | S2‐5‐2 | GC − T (Ho) | + G (Ho) | 16/16 | S2‐5‐2‐6/10/15 |
| S2‐5‐6 | + T/GC − T (Bi‐a) | + GT/+ G (Bi‐a) | 4/16 | S2‐5‐6‐3/5 | ||
| S2‐5‐14 | + T (Ho) | + GT/+ G (Bi‐a) | 6/16 | S2‐5‐14‐2/8 | ||
He, heterozygous; Ho, homozygous; Bi‐a, bio‐allelic. ‘−’ and ‘+’ indicate the deletion and insertion in the sgRNAs target sites, respectively.
Bold indicates the numbers of T0 plants.
To detect the inheritance of bi‐allelic mutations, we sequenced S1‐11 and S2‐5 T1 and T2 plants. We found that the mutations identified in S1‐11 T2 plants matched those observed in the corresponding S1‐11 T0 and T1 lines. We also identified homozygous or bi‐allelic individuals which did not contain T‐DNA in the T2 progeny (Figure 5a; Table 1). Interestingly, a new mutation (+TT) was found in two T2 plants (S2‐5‐14‐6 and S2‐5‐14‐16 of S2‐5‐14), even though it was not detected in S2‐5 T0 and T1 lines. This showed that the sgRNA‐2 site on chromosome A was further edited during the growth of the transgenic plants, even though it has one mismatch with the sgRNA (Figure 5b; Table 1).
Figure 5.

The target site mutations at the T0, T1 and T2 generations in S1‐11 and S2‐5 lines. The green colours indicate the SNP between the two BnaMAX1 genomes, and the bold fonts indicate the PAMs. The red fonts and hyphens indicate insertions and deletions, respectively.
As expected, the plants with homozygous or bi‐allelic mutations resulting in premature stop codons in the two BnaMAX1 genes displayed similar mutant phenotypes. In the S1‐5 line, insertion of a T led to premature stop codons in BnaA03.MAX1 on the A chromosome and an amino acid deletion in BnaC03.MAX1 on the C chromosome. However, homozygous mutant plants displayed normal phenotypes similar to the wild type, suggesting that the single L73 amino acid deletion in BnaC03.MAX1 did not affect its protein function (Figure S7). We also obtained non‐transgenic plants with semi‐dwarf and increased branching phenotypes in S1‐8/S1‐11/S2‐5 segregating populations, suggesting that the Cas9‐induced mutations were stably transmitted to T2 rapeseed plants independently of the T‐DNA construct, and the mutations in BnaMAX1s were heritable (Figure S8; Table 1).
Searching for putative off‐target activities
To determine whether the increased branching phenotype resulted from mutations of off‐target sites in the edited lines, we searched for off‐target effects in our edited plants. We therefore designed PCR primers to detect the possible off‐target sites predicted by CRISPR‐P (9 annotated genes for sgRNA‐1 and only one for sgRNA‐2). After sequencing all PCR products of the putative off‐target genes, no mutations were found in S1‐8 for sgRNA‐1 as well as in S2‐5 for sgRNA‐2. We have also analysed 36 additional independent T0 plants in both sgRNAs, and no off‐target events were found (Figure S9). These results demonstrated that the two sgRNAs had a high specificity for targeting the BnaMAX1 genes in rapeseed, and such high specificity (≥3 mismatches in sgRNA) can avoid off‐targeting (Tang et al., 2018; Yang et al., 2017). In mammals, a single nucleotide mismatch in the 5′ half of an sgRNA sequence showed a high ratio of off‐site targeting, whereas up to five nucleotide mismatches also had low off‐target scores (Doench et al., 2014; Fu et al., 2013; Hsu et al., 2013). In Brassica oleracea, four‐nucleotide mismatches in the sgRNA showed no off‐target effects on the homologs of BolC.GA4.a (BolC.GA4.b), while two mismatches in sgRNA on BolC.GA4.b resulted in off‐target events (Lawrenson et al., 2015). These results suggest that sgRNA with two mismatches or less can simultaneously target multi‐copy genes in plants.
Knockout of two BnaMAX1 homologs can significantly decrease plant height and increase branch number, silique number and yield
We next chose T‐DNA‐free homozygous lines in S1‐5 (S1‐5‐6‐8), S1‐8 (S1‐8‐39‐1) and S1‐11 (S1‐11‐7‐3) for subsequent phenotypic characterization. Five weeks after we sowed seeds in the greenhouse, the WT and all three mutant lines entered the bolting stage. S1‐8 and S1‐11 displayed different phenotypes than WT and S1‐5, with more buds at the stem base. Remarkably, all of the axillary buds were outgrowths in S1‐8 and S1‐11 plants, but these were partially repressed at the bases of leaf petioles of WT and S1‐5 plants (Figure 6a–d). At the mature stage, S1‐5 displayed similar architecture as WT, including PH, BN and yield‐related factors. In contrast, the S1‐8 and S1‐11 lines displayed dramatic semi‐dwarf phenotypes. Height was about 31.9%–36.5% shorter than WT (Figure 6e–i); the total primary branch numbers (axillary and base part) increased from 3 (WT) to 9 (Figure 6i); and the total silique numbers (SN) increased about 62.3%–71.8% relative to WT (Figure 6i). Finally, S1‐8 and S1‐11 had increased yields per plant of about 29.9%–31.3% compared with WT (Figure 6i). We also assessed the yield‐related traits of the two T‐DNA‐free lines (S1‐8 and S1‐11) in the field. Both lines exhibited decreased PH and increased BN and SN as well as yield per plant compared with WT (Figure S10; Table S5).
Figure 6.
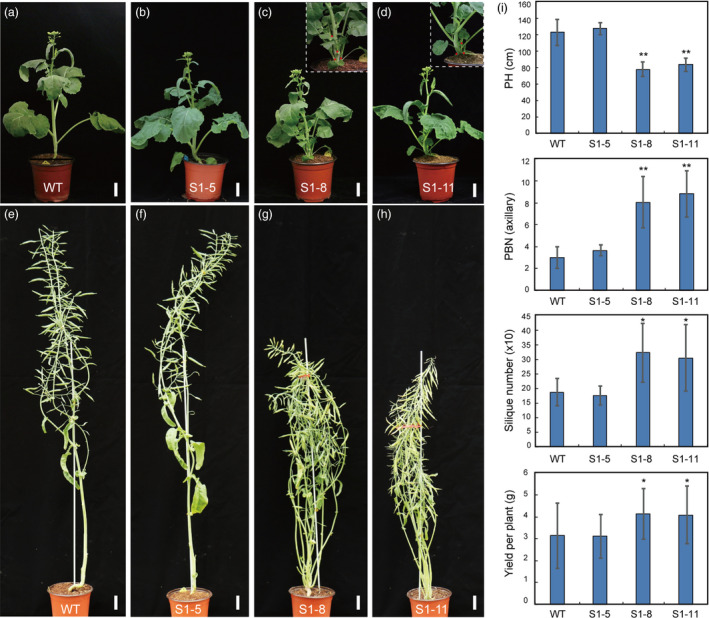
Morphological comparison of wild‐type (WT) and BnaMAX1 homozygous mutant plants at various developmental stages. (a–d) The S1‐8‐39‐1 and S1‐11‐7‐3 homozygous plants show more axillary buds than WT and S1‐5‐6‐8 at the bolting stage. (e–h) The S1‐8‐39‐1 and S1‐11‐7‐3 homozygous plants show dwarfism and higher branch numbers than WT and S1‐5‐6‐8 at the mature stage. (i) Statistical analysis of plant height (PH), branching number (BN), silique number (SN), and yield per plant in WT, S1‐5‐6‐8, S1‐8‐39‐1 and S1‐11‐7‐3 homozygous plants in the greenhouse. Error bars ± standard deviation (n = 15 for PH, BN and yield; 10 for SN in WT, S1‐5‐6‐8, S1‐8‐39‐1 and S1‐11‐7‐3). Student's t‐test was used for statistical analysis (*P ≤ 0.05; **P ≤ 0.01). Bars = 5 cm.
The ‘Green revolution gene’ was successfully adopted in cereal crops in the late 1960s (Peng et al., 1999; Evenson and Gollin, 2003). Presently, rice breeding has moved from ‘breeding for dwarfs’ and ‘using hybrid heterosis’ into ‘super rice’ to obtain greater yield. It was reported that selecting for reasonable PH is an effective method for breeding rapeseed for high yield (Fu and Zhou, 2013). Although dwarf types are adapted to mechanical harvesting, this is sometimes accompanied by decreased biomass (Salas Fernandez et al., 2009). Decreasing PH and increasing BN would benefit the stabilization of biomass. However, the lack of special semi‐dwarf germplasm resources and their biomass constraints has limited rapeseed breeding. So far, increased branching germplasms have not been identified in rapeseed. In a previous study, Cardoso et al. (2014) found that Bala (a rice indica cultivar) displayed more tillers than Azucena (a japonica cultivar) due to lack of two MAX1 homologs in the indica genome, indicating that abolishing MAX1 could benefit production. We analysed the resequencing data of 991 worldwide rapeseed germplasm accessions from Wu et al. (2019), and no insertion or deletion sites were found in each BnaMAX1 exon on Chr. A03 or C03, indicating that natural mutations in both BnaMAX1s are difficult to obtain. In our study, we found that knockout of the two BnaMAX1 homologs mediated by CRISPR/Cas9‐targeted mutagenesis resulted in semi‐dwarf and increased branching phenotypes and also increased SN and yield per plant. Significantly, the semi‐dwarf habit can reduce the risk of lodging and facilitate mechanical harvesting, and more branching could maintain stable biomass and increase SN. In the future, it will be necessary to continue aggregating other traits (smaller branch angle, erect leaf, shattering resistance, etc.) in order to create an ‘ideotype’ that can be used for breeding high yield in rapeseed.
Experimental procedures
Plant materials
We used ‘862’ (wild‐type, a spring‐type rapeseed variety) as the donor plants for transformation in our study. All wild‐type and transgenic plants were grown in a greenhouse (16/8 h of light/dark at 20–23 °C). The Arabidopsis 19‐22 mutant was obtained from TAIR (https://www.arabidopsis.org/, Salk_036990). All Col, 19‐22 and transgenic plants were grown in a greenhouse (16/8 h of light/dark at 20 °C).
MutMap
One bulked DNA sample was prepared by mixing an equal ratio of DNA of 30 plants that displayed an increased branching phenotype from an F2 population. The libraries for Illumina sequencing were constructed from Arabidopsis Col (wild type) and the bulked pool. The clean data (1.7 Gb) from Col were first reconstructed to determine the new reference genome, and then, the clean data (3.5 Gb) from bulked samples were BLASTed with the new reference genome to find mutation sites (Indel or SNP) using Coval and selected Indel or SNP index > 0.8 as candidate sites (Abe et al., 2012).
qRT‐PCR
Total RNA was extracted using the RNA Prep Pure Plant Kit (Tiangen), and cDNA was synthesized using Oligo (dT) 18 primer and the First cDNA transcriptase kit (Takara). qRT‐PCR was performed using a Fast Start Universal Probe Master Mix (Roche) in an ABI 7500 Fast PCR system, and the rapeseed TMA7 gene (BnaC05g11560D) was used as an endogenous control (Zhu et al., 2017). Primers for qRT‐PCR are listed in Table S6. Data from three biological replicates were analysed following the relative quantification method (2−ΔΔCT).
Complementary and CRISPR/Cas9 plasmid construction, transformation and positive transgenic‐plant identification
We performed a BLASTP search of the MAX1 protein against the rapeseed reference genome (http://www.genoscope.cns.fr/brassicanapus/, Sun et al., 2018) and identified two MAX1 homologs (BnaA03g22900D and BnaC03g26960D or BnA03g0116320.1 and BnC03g0568140.1). The full‐length CDS of MAX1 and the two BnaMAX1s were cloned into gateway 100 vectors controlled by the 35S promoter, and the three plasmids were transformed into the Arabidopsis 19‐22 mutant using the floral dip method mediated by Agrobacterium tumefaciens (GV3101). To select positive lines, 250 mg/L phosphinothricin was sprayed on Arabidopsis T1 transgenic seedlings.
We designed the two sgRNA target sequences using the online tool CRISPR‐P1.0 (http://crispr.hzau.edu.cn/CRISPR/, Lei et al., 2014). Two target sequences that each had high similarity with both BnaMAX1 genes, and few putative off‐target sites were selected as candidate sgRNAs. The sgRNA‐1 target sequence initiated with G, and the sgRNA‐2 initiated with A which we replaced with G. The two sgRNA sequences were cloned into BGK01 vector (Biogle, China; http://www.biogle.cn/) to complete the constructs and then transformed into 862 hypocotyls mediated by Agrobacterium tumefaciens (GV3101).
For rapeseed hypocotyl transformation, plump seeds were sterilized using 75% ethyl alcohol, 1.5% mercuric chloride and ddH2O in turn. We placed them on filter paper to dry, then placed them on M0 plates (2.2 g/L MS; 7.5 g/L agar; pH 6.0). After growth for about 1 week at 24 °C in the dark, the elongated hypocotyls were cut into 1 cm pieces in DM medium (4.4 g/L MS; 30 g/L sucrose; 0.1 mm AS; pH 6.0). These pieces were then soaked in resuspended Agrobacterium (harbouring the Cas9 plasmid) for about 15 min. Filter paper was then used to dry the hypocotyl pieces, which were placed on M1 medium (2.2 g/L MS; 7.5 g/L agar; pH 6.0) and left for 2 days at 24 °C in the dark. The non‐dehydrated hypocotyl pieces were selected on M2 medium (4.4 g/L MS; 30 g/L sucrose; 18 g/L Mannitol; 1 mg/L 2,4‐D; 0.3 mg/L KT; 7 g/L agar; 15 mg/L Hyg B; pH 6.0) for about 3 weeks at 24 °C under light. The putative transgenic pieces were placed on M3 medium (4.4 g/L MS; 10 g/L glucose; 0.25 g/L xylose; 0.6 g/L MES; 10−4 mg/L IAA; 7 g/L agar; 300 mg/L TMT; 1 mg/L zeatin; 15 mg/L Hyg B; pH 6.0) for about 2 weeks at 24 °C under light, which was repeated at 2 week intervals. Finally, the regenerated shoots were allowed to take root in B5 culture medium.
The positive T2 rapeseed plants were identified by PCR using the special Hyg TaqMan probe (Table S6). The T0 seeds were identified after growing on MS medium containing 30 mg/L Hyg B in darkness for about 1 week, and seedlings with elongated hypocotyls were putative positive plants.
Mutant identification and off‐target analysis
Genomic DNA of the T0 and T1 lines was isolated from the leaf samples by a CTAB extraction method. The specific primers that bracketed the CRISPR/Cas9 target sites were used to perform PCR amplification (Table S6), and the PCR products were directly sequenced or cloned into the pEASY‐T vector (TransGen Biotech) and then sequenced using the Sanger method.
Putative off‐target sites (9 annotated genes for sgRNA‐1 and only one for sgRNA‐2) were identified by CRISPR‐P1.0 (http://crispr.hzau.edu.cn/CRISPR/) against the reference (Brassica napus v4.1). The specific primers that surrounded the putative off‐target sites were used to perform PCR amplification (Table S6), and 37 independent T0 plants in both sgRNAs were used for off‐target analysis. The products were sequenced using the Sanger method.
Phenotypic observations
The wild‐type and T0 and T1 transgenic lines were grown in a greenhouse. Whole plants were photographed at the bolting stage, especially at the branching part. At the mature stage, 15 each of wild‐type, S1‐5, S1‐8 and S1‐11 homozygous plants were selected to determine PH, BN and yield per plant, and then, ten plants of each were selected to determine SN per plant. For the field experiment, the wild‐type and all T‐DNA‐free lines were grown in plots of 2 rows, with 10–11 plants in each row. Ten plants were selected from the centre of each row to measure the following four traits at maturity: PH, BN, SN and yield per plant.
Author contributions
M.Z. and W.H. designed the experiments; M.Z., L.Z., M.T., J.L. and H.Y. performed the experiments; H.L. and L.Z. analysed the genome and sequencing data; L.Z. and S.F. characterized the agronomic traits; M.Z. wrote the manuscript, and W.T., H.W. and W.H. revised the manuscript. All authors read and approved the final manuscript.
Conflict of interest
The authors declare no competing financial interest.
Supporting information
Figure S1 Identification of insert site in 19‐22 mutant.
Figure S2 Phenotype observation and linkage analysis of 19‐22 mutant.
Figure S3 The expression pattern of MAX1 in various Arabidopsis tissues (a) and the transgenic plants (b).
Figure S4 The candidate off‐targets of two sgRNAs sites according to the CRISPR‐P.
Figure S5 Alignment of the different insertion and deletion types found at the target sites of two sgRNAs.
Figure S6 Phenotypes of BnaMAX1 allele plants.
Figure S7 Amino acid sequence alignment of BnaMAX1s among WT, S1‐5, S1‐8 and S1‐11 T0 plants.
Figure S8 Real‐time PCR amplification of S1‐8 and S2‐5T2 plants using Hyg probe.
Figure S9 PCR identification of the off‐targets on two sgRNAs (a, sgRNA‐1; b, sgRNA‐2).
Figure S10 Phenotypes of WT, S1‐8 and S1‐11 plants in the field.
Table S1 Genetic analysis between wild‐type (WT) and 19‐22 mutant.
Table S2 Twenty‐three Indel between WT and 19‐22 by Mutmap.
Table S3 The target site mutations in T1 generation from S1‐8 line.
Table S4 Genotypes at S1 and S2 target site of Cas9‐BnaMAX1s T0 plants.
Table S5 Yield related traits of WT, S1‐8 and S1‐11 T‐DNA‐free plants in the field.
Table S6 Primers used in this study.
Acknowledgements
This study was supported from the National Key Research and Development Program of China (2016YFD0101007), the National Key Basic Research Program of China (2015CB150203) and the National Natural Science Foundation of China (31801402).
References
- Abe, A. , Kosugi, S. , Yoshida, K. , Natsume, S. , Takagi, H. , Kanzaki, H. , Matsumura, H. et al. (2012) Genome sequencing reveals agronomically important loci in rice using MutMap. Nat. Biotechnol. 30, 174–178. [DOI] [PubMed] [Google Scholar]
- Abe, S. , Sado, A. , Tanaka, K. , Kisugi, T. , Asami, K. , Ota, S. , Il Kim, H. et al. (2014) Carlactone is converted to carlactonoic acid by MAX1 in Arabidopsis and its methyl ester can directly interact with AtD14 in vitro. Proc. Natl. Acad. Sci. USA, 111, 18084–18089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alder, A. , Jamil, M. , Marzorati, M. , Bruno, M. , Vermathen, M. , Bigler, P. , Ghisla, S. et al. (2012) The path from beta‐carotene to carlactone, a strigolactone‐like plant hormone. Science, 335, 1348–1351. [DOI] [PubMed] [Google Scholar]
- Andersson, M. , Turesson, H. , Nicolia, A. , Falt, A.S. , Samuelsson, M. and Hofvander, P. (2017) Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR‐Cas9 expression in protoplasts. Plant Cell Rep. 36, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arite, T. , Umehara, M. , Ishikawa, S. , Hanada, A. , Maekawa, M. , Yamaguchi, S. and Kyozuka, J. (2009) d14, a strigolactone‐insensitive mutant of rice, shows an accelerated outgrowth of tillers. Plant Cell Physiol. 50, 1416–1424. [DOI] [PubMed] [Google Scholar]
- Bennett, T. and Leyser, O. (2006) Something on the side: axillary meristems and plant development. Plant Mol. Biol. 60, 843–854. [DOI] [PubMed] [Google Scholar]
- Beveridge, C.A. (2006) Axillary bud outgrowth: sending a message. Curr. Opin. Plant Biol. 9, 35–40. [DOI] [PubMed] [Google Scholar]
- Booker, J. , Sieberer, T. , Wright, W. , Williamson, L. , Willett, B. , Stirnberg, P. , Turnbull, C. et al. (2005) MAX1 encodes a cytochrome P450 family member that acts downstream of MAX3/4 to produce a carotenoid‐derived branch‐inhibiting hormone. Dev. Cell, 8, 443–449. [DOI] [PubMed] [Google Scholar]
- Braatz, J. , Harloff, H.J. , Mascher, M. , Stein, N. , Himmelbach, A. and Jung, C. (2017) CRISPR‐Cas9 targeted mutagenesis leads to simultaneous modification of different homoeologous gene copies in polyploid oilseed rape (Brassica napus). Plant Physiol. 174, 935–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardoso, C. , Zhang, Y.X. , Jamil, M. , Hepworth, J. , Charnikhova, T. , Dimkpa, S.O.N. , Meharg, C. , et al. (2014) Natural variation of rice strigolactone biosynthesis is associated with the deletion of two MAX1 orthologs. P Natl Acad Sci USA 111, 2379–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalhoub, B. , Denoeud, F. , Liu, S. , Parkin, I.A. , Tang, H. , Wang, X. , Chiquet, J. , et al. (2014) Plant genetics. Early allopolyploid evolution in the post-Neolithic Brassica napus oilseed genome. Science 345, 950–953. [DOI] [PubMed] [Google Scholar]
- Chatfield, S.P. , Stirnberg, P. , Forde, B.G. and Leyser, O. (2000) The hormonal regulation of axillary bud growth in Arabidopsis. Plant J. 24, 159–169. [DOI] [PubMed] [Google Scholar]
- Cong, L. , Ran, F.A. , Cox, D. , Lin, S.L. , Barretto, R. , Habib, N. , Hsu, P.D. et al. (2013) Multiplex genome engineering using CRISPR/Cas systems. Science, 339, 819–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doench, J.G. , Hartenian, E. , Graham, D.B. , Tothova, Z. , Hegde, M. , Smith, I. , Sullender, M. et al. (2014) Rational design of highly active sgRNAs for CRISPR‐Cas9‐mediated gene inactivation. Nat. Biotechnol. 32, 1262–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evenson, R.E. and Gollin, D. (2003) Assessing the impact of the green revolution, 1960 to 2000. Science 300, 758–762. [DOI] [PubMed] [Google Scholar]
- Fu, T. and Zhou, Y. (2013) Progress and future development of hybrid rapeseed in China. Eng. Sci. 11, 13–18. [Google Scholar]
- Fu, Y.F. , Foden, J.A. , Khayter, C. , Maeder, M.L. , Reyon, D. , Joung, J.K. and Sander, J.D. (2013) High‐frequency off‐target mutagenesis induced by CRISPR‐Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaj, T. , Gersbach, C.A. and Barbas, C.F. (2013) ZFN, TALEN, and CRISPR/Cas‐based methods for genome engineering. Trends Biotechnol. 31, 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez‐Roldan, V. , Fermas, S. , Brewer, P.B. , Puech‐Pages, V. , Dun, E.A. , Pillot, J.P. , Letisse, F. et al. (2008) Strigolactone inhibition of shoot branching. Nature, 455, 189–194. [DOI] [PubMed] [Google Scholar]
- Horvath, D.P. , Anderson, J.V. , Chao, W.S. and Foley, M.E. (2003) Knowing when to grow: signals regulating bud dormancy. Trends Plant Sci. 8, 534–540. [DOI] [PubMed] [Google Scholar]
- Hsu, P.D. , Scott, D.A. , Weinstein, J.A. , Ran, F.A. , Konermann, S. , Agarwala, V. , Li, Y.Q. et al. (2013) DNA targeting specificity of RNA‐guided Cas9 nucleases. Nat. Biotechnol. 31, 827–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, L. , Liu, X. , Xiong, G. , Liu, H. , Chen, F. , Wang, L. , Meng, X. et al. (2013) DWARF 53 acts as a repressor of strigolactone signalling in rice. Nature, 504, 401–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlen, W. , Charnikhova, T. , Liu, Q. , Bours, R. , Domagalska, M.A. , Beguerie, S. , Verstappen, F. et al. (2011) Strigolactones are transported through the xylem and play a key role in shoot architectural response to phosphate deficiency in Nonarbuscular Mycorrhizal host Arabidopsis. Plant Physiol. 155, 974–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrenson, T. , Shorinola, O. , Stacey, N. , Li, C.D. , Ostergaard, L. , Patron, N. , Uauy, C. et al. (2015) Induction of targeted, heritable mutations in barley and Brassica oleracea using RNA‐guided Cas9 nuclease. Genome Biol. 16, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar, G. and Goodman, H.M. (2006) MAX1, a regulator of the flavonoid pathway, controls vegetative axillary bud outgrowth in Arabidopsis. Proc. Natl. Acad. Sci. USA, 103, 472–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei, Y. , Lu, L. , Liu, H.Y. , Li, S. , Xing, F. and Chen, L.L. (2014) CRISPR‐P: a web tool for synthetic single‐guide RNA design of CRISPR‐system in plants. Mol. Plant. 7, 1494–1496. [DOI] [PubMed] [Google Scholar]
- Li, J.F. , Norville, J.E. , Aach, J. , McCormack, M. , Zhang, D.D. , Bush, J. , Church, G.M. et al. (2013) Multiplex and homologous recombination‐mediated genome editing in Arabidopsis and Nicotiana benthamiana using guide RNA and Cas9. Nat. Biotechnol. 31, 688–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, C. , Hao, M. , Wang, W. , Wang, H. , Chen, F. , Chu, W. , Zhang, B. et al. (2018) An efficient CRISPR/Cas9 platform for rapidly generating simultaneous mutagenesis of multiple gene homoeologs in allotetraploid oilseed rape. Front. Plant Sci. 9, 442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljung, K. , Bhalerao, R.P. and Sandberg, G. (2001) Sites and homeostatic control of auxin biosynthesis in Arabidopsis during vegetative growth. Plant J. 28, 465–474. [DOI] [PubMed] [Google Scholar]
- Ma, X. , Zhang, Q. , Zhu, Q. , Liu, W. , Chen, Y. , Qiu, R. , Wang, B. et al. (2015) A robust CRISPR/Cas9 system for convenient, high‐efficiency multiplex genome editing in monocot and dicot plants. Mol Plant. 8, 1274–1284. [DOI] [PubMed] [Google Scholar]
- Mali, P. , Yang, L.H. , Esvelt, K.M. , Aach, J. , Guell, M. , DiCarlo, J.E. , Norville, J.E. et al. (2013) RNA‐guided human genome engineering via Cas9. Science, 339, 823–826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris, D.A. (1977) Transport of exogenous auxin in two‐branched dwarf pea seedlings (Pisum sativum L.): some implications for polarity and apical dominance. Planta, 136, 91–96. [DOI] [PubMed] [Google Scholar]
- Nordstrom, A. , Tarkowski, P. , Tarkowska, D. , Norbaek, R. , Astot, C. , Dolezal, K. and Sandberg, G. (2004) Auxin regulation of cytokinin biosynthesis in Arabidopsis thaliana: a factor of potential importance for auxin‐cytokinin‐regulated development. Proc. Natl. Acad. Sci. USA, 101, 8039–8044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng, J. , Richards, D.E. , Hartley, N.M. , Murphy, G.P. , Devos, K.M. , Flintham, J.E. , Beales, J. , et al.(1999) 'Green revolution' genes encode mutant gibberellin response modulators. Nature 400, 256–261. [DOI] [PubMed] [Google Scholar]
- Salas Fernandez, M.G. , Becraft, P.W. , Yin, Y. and Lubberstedt, T. (2009) From dwarves to giants? Plant height manipulation for biomass yield. Trends Plant Sci. 14, 454–461. [DOI] [PubMed] [Google Scholar]
- Sanchez‐Leon, S. , Gil‐Humanes, J. , Ozuna, C.V. , Gimenez, M.J. , Sousa, C. , Voytas, D.F. and Barro, F. (2018) Low‐gluten, nontransgenic wheat engineered with CRISPR/Cas9. Plant Biotechnol. J. 16, 902–910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz, G. and Theres, K. (2005) Shoot and inflorescence branching. Curr. Opin. Plant Biol. 8, 506–511. [DOI] [PubMed] [Google Scholar]
- Shan, Q.W. , Wang, Y.P. , Li, J. , Zhang, Y. , Chen, K.L. , Liang, Z. , Zhang, K. et al. (2013) Targeted genome modification of crop plants using a CRISPR‐Cas system. Nat. Biotechnol. 31, 686–688. [DOI] [PubMed] [Google Scholar]
- Soundappan, I. , Bennett, T. , Morffy, N. , Liang, Y. , Stanga, J.P. , Abbas, A. , Leyser, O. et al. (2015) SMAX1‐LIKE/D53 family members enable distinct MAX2‐dependent responses to Strigolactones and Karrikins in Arabidopsis. Plant Cell. 27, 3143–3159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, F. , Fan, G. , Hu, Q. , Zhou, Y. , Guan, M. , Tong, C. , et al. (2017) The high-quality genome of Brassica napus cultivar 'ZS11' reveals the introgression history in semi-winter morphotype. Plant J 92, 452–468. [DOI] [PubMed] [Google Scholar]
- Stirnberg, P. , Furner, I.J. and Ottoline Leyser, H.M. (2007) MAX2 participates in an SCF complex which acts locally at the node to suppress shoot branching. Plant J. 50, 80–94. [DOI] [PubMed] [Google Scholar]
- Sun, Q. , Lin, L. , Liu., D. , Wu, D. , Fang, Y. , Wu, J. and Wang, Y. (2018) CRISPR/Cas9‐mediated multiplex genome editing of the BnWRKY11 and BnWRKY70 genes in Brassica napus L. Int. J. Mol. Sci., 19, E2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussex, I.M. and Kerk, N.M. (2001) The evolution of plant architecture. Curr. Opin. Plant Biol. 4, 33–37. [DOI] [PubMed] [Google Scholar]
- Tang, X. , Liu, G. , Zhou, J. , Ren, Q. , You, Q. , Tian, L. , Xin, X. et al. (2018) A large‐scale whole‐genome sequencing analysis reveals highly specific genome editing by both Cas9 and Cpf1 (Cas12a) nucleases in rice. Genome Biol. 19, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thimann, K.V. and Skoog, F. (1933) Studies on the growth hormone of plants: III. The inhibiting action of the growth substance on bud development. Proc. Natl. Acad. Sci. USA, 19, 714–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umehara, M. , Hanada, A. , Yoshida, S. , Akiyama, K. , Arite, T. , Takeda‐Kamiya, N. , Magome, H. et al. (2008) Inhibition of shoot branching by new terpenoid plant hormones. Nature, 455, 195–200. [DOI] [PubMed] [Google Scholar]
- Wang, Y.P. , Cheng, X. , Shan, Q.W. , Zhang, Y. , Liu, J.X. , Gao, C.X. and Qiu, J.L. (2014) Simultaneous editing of three homoeoalleles in hexaploid bread wheat confers heritable resistance to powdery mildew. Nat. Biotechnol. 32, 947–951. [DOI] [PubMed] [Google Scholar]
- Wang, L. , Wang, B. , Jiang, L. , Liu, X. , Li, X. , Lu, Z. , Meng, X. et al. (2015) Strigolactone signaling in Arabidopsis regulates shoot development by targeting D53‐Like SMXL repressor proteins for ubiquitination and degradation. Plant Cell. 27, 3128–3142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, P.C. , Zhang, J. , Sun, L. , Ma, Y.Z. , Xu, J. , Liang, S.J. , Deng, J.W. et al. (2018) High efficient multisites genome editing in allotetraploid cotton (Gossypium hirsutum) using CRISPR/Cas9 system. Plant Biotechnol. J. 16, 137–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu, D.Z. , Liang, Z. , Yan, T. , Xu, Y. , Xuan, L.J. , Tang, J. , Zhou, G. et al. (2019) Whole‐genome resequencing of a worldwide collection of rapeseed accessions reveals the genetic basis of ecotype divergence. Mol Plant. 12, 30–43. [DOI] [PubMed] [Google Scholar]
- Xie, K.B. and Yang, Y.N. (2013) RNA‐guided genome editing in plants using a CRISPRCas system. Molecular Plant. 6, 1975–1983. [DOI] [PubMed] [Google Scholar]
- Yang, Y. , Zhu, K. , Li, H. , Han, S. , Meng, Q. , Khan, S.U. , Fan, C. et al. (2017) Precise editing of CLAVATA genes in Brassica napus L. regulates multilocular silique development. Plant Biotechnol. J. 16, 1322–1335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhai, Y. , Cai, S. , Hu, L. , Yang, Y. , Amoo, O. , Fan, C. and Zhou, Y. (2019) CRISPR/Cas9‐mediated genome editing reveals differences in the contribution of INDEHISCENT homologues to pod shatter resistance in Brassica napus L. Theor. Appl. Genet. 132, 2111–2123. [DOI] [PubMed] [Google Scholar]
- Zhang, H. , Zhang, J. , Wei, P. , Zhang, B. , Gou, F. , Feng, Z. , Mao, Y. et al. (2014) The CRISPR/Cas9 system produces specific and homozygous targeted gene editing in rice in one generation. Plant Biotechnol. J. 12, 797–807. [DOI] [PubMed] [Google Scholar]
- Zhou, F. , Lin, Q. , Zhu, L. , Ren, Y. , Zhou, K. , Shabek, N. , Wu, F. et al. (2013) D14‐SCF(D3)‐dependent degradation of D53 regulates strigolactone signalling. Nature, 504, 406–410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, X.Y. , Huang, C.Q. , Zhang, L. , Liu, H.F. , Yu, J.H. , Hu, Z.Y. and Hua, W. (2017) Systematic analysis of Hsf family genes in the Brassica napus genome reveals novel responses to heat, drought and high CO2 stresses. Front. Plant Sci. 8, 1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 Identification of insert site in 19‐22 mutant.
Figure S2 Phenotype observation and linkage analysis of 19‐22 mutant.
Figure S3 The expression pattern of MAX1 in various Arabidopsis tissues (a) and the transgenic plants (b).
Figure S4 The candidate off‐targets of two sgRNAs sites according to the CRISPR‐P.
Figure S5 Alignment of the different insertion and deletion types found at the target sites of two sgRNAs.
Figure S6 Phenotypes of BnaMAX1 allele plants.
Figure S7 Amino acid sequence alignment of BnaMAX1s among WT, S1‐5, S1‐8 and S1‐11 T0 plants.
Figure S8 Real‐time PCR amplification of S1‐8 and S2‐5T2 plants using Hyg probe.
Figure S9 PCR identification of the off‐targets on two sgRNAs (a, sgRNA‐1; b, sgRNA‐2).
Figure S10 Phenotypes of WT, S1‐8 and S1‐11 plants in the field.
Table S1 Genetic analysis between wild‐type (WT) and 19‐22 mutant.
Table S2 Twenty‐three Indel between WT and 19‐22 by Mutmap.
Table S3 The target site mutations in T1 generation from S1‐8 line.
Table S4 Genotypes at S1 and S2 target site of Cas9‐BnaMAX1s T0 plants.
Table S5 Yield related traits of WT, S1‐8 and S1‐11 T‐DNA‐free plants in the field.
Table S6 Primers used in this study.