

Abstract
Importance:
The recent implication of 108 genomic loci in schizophrenia marked a great advancement in our understanding of the disease. Against the background of its polygenic nature there is a necessity to identify how schizophrenia risk genes interplay. As regulators of gene expression miRNA have repeatedly been implicated in schizophrenia etiology. It is therefore of interest to establish their role in the regulation of schizophrenia risk genes in disease relevant biological processes.
Objective:
To examine the role of miRNA in the context of disease associated genetic variation.
Design, Setting and Participants:
The basis of our study was summary statistics from the largest schizophrenia genome-wide association study to date (83 550 individuals) along with publicly available data for predicted miRNA targets. We examined if schizophrenia risk genes were more likely to be regulated by miRNA. Further, we used gene set analyses to identify miRNA that are regulators of schizophrenia genes.
Main Outcome and Measures:
Results from association tests for miRNA targetomes and related analyses.
Results:
In line with previous studies we found that similarly to other complex traits schizophrenia risk genes were more likely to be regulated by miRNA. Further, the gene set analyses revealed several miRNA regulating schizophrenia risk genes with the strongest enrichment for targets of miR-9-5p. It is further of note that MIR9-2 is located in a genomic region showing strong evidence for association with schizophrenia.
Conclusions and Relevance:
Our study provides evidence for a role of miR-9-5p in the etiology of schizophrenia. Its implication is of particular interest, as the functions of this neurodevelopmental miRNA tie in with established disease biology: it has a regulatory loop with the fragile X mental retardation homolog FXR1, and regulates dopamine D2 receptor density.
Introduction
Schizophrenia is a common psychiatric disorder with considerable morbidity1, high heritability2, and extensive genetic heterogeneity3. Here, we examine the disease from the perspective of a potential influence of miRNA, which are ~22 nucleotide long endogenous RNA molecules that regulate gene expression post-transcriptionally by pairing with the RNA-induced silencing complex, subsequently binding mRNA, and inducing translational repression and/or mRNA degradation. Computer models have been widely used to predict such interactions as detailed in the Supplementary Text.
Accumulating evidence implicate miRNA with schizophrenia: miRNA are known to play important roles in brain development4; miRNAs are found differentially expressed in post-mortem brains of patients with schizophrenia5; miRNA and their targets are found enriched in risk loci from genetic studies at the level of CNVs, as well as common and rare variation6-9. Additionally, much of the signal in genome-wide association studies (GWAS) of schizophrenia is believed to come from variants altering gene expression10,11 thus putting miRNA as regulators of gene expression into the spotlight.
In the present study, the role of miRNA in the etiology of schizophrenia is analyzed at three different levels employing the following approaches: (A) by assessing if schizophrenia risk genes overall are more likely to be regulated by miRNA; (B) by gene set analyses to find conserved miRNA that are regulators of schizophrenia risk genes; and (C) by targeted gene set analyses to systematically characterize the importance of miRNA in previously identified risk loci from GWAS and CNV.
Methods and Materials
The basis for the analyses in this paper (if not stated otherwise) is summary statistics from the most recent schizophrenia GWAS meta-analysis conducted by The Schizophrenia Working Group in the Psychiatric Genomics Consortium (“PGC2”)12. For the purpose of our analyses, only autosomal results were used, and tests excluded the broader MHC-region (chr6:25M-35M).
Part A: Regulation of schizophrenia risk genes by miRNAs
In a first step we aimed to gain a global measure of the magnitude to which schizophrenia genes are regulated by miRNA. We therefore examined whether the degree to which a gene was regulated by miRNA correlated with the gene’s association with schizophrenia by applying a linear model of log-transformed gene p-values with gene and 3'-UTR lengths included as covariates with miRNA target predictions from TargetScan13 (Figure 1a and Supplementary Text). In order to study the specificity of our findings we repeated the analyses with summary statistics from well-powered GWASs for age at menarche, Crohn’s disease, and height14-16.
Figure 1: Illustrations of the analyses in this paper.

a) Flowchart of the linear model for assessing if schizophrenia risk genes are more likely to be targeted by miRNA. b) Flowchart for gene set analyses of all conserved miRNAs and the targeted gene set analyses. More information on the analytical strategies can be found in the main text.
Part B: Gene set enrichment analyses of all conserved miRNAs
A flowchart of the gene set analyses in this paper is presented in Figure 1b. In the following we will provide information on specific aspects of our analyses (see Supplementary Text for more information).
miRNA and their targets
Names and genomic locations of miRNA and stem-loops were taken from mirBase 2017. Based on characteristics of RNA-sequencing experiments, some miRNA are classified as “high confidence” and are considered to have a high probability of representing a bona fide miRNA. For miRNA target sites, TargetScan 6.2 conserved target sites of conserved miRNA-families13 were used unless otherwise stated. Because of its reliance on conservation and the requirement for miRNA to have a seed site, the predicted targets of this algorithm have a higher chance of being functionally important. For the TargetScan miRNA families only names of human miRNA in each family are listed and only gene sets with more than 50 genes were considered. NCBI protein coding genes and their corresponding hg19 positions were used.
Statistical approach
INRICH18 was used for all gene set analyses. This method tests the overlap between genomic intervals associated with the trait of interest and predefined gene sets. Linkage disequilibrium (LD), variable gene lengths, variable SNP and gene density are taken into account and multiple testing is corrected using a bootstrapping approach. For our analyses, SNPs were filtered for a minor allele frequency (MAF) ≥1% and info score ≥0.8. SNPs were “clumped” with plink 1.9 using all samples of European ancestry from the 1000 genomes project phase 119 with the following settings: for the index SNP, three different significance thresholds were used: 1×10−5, 3.420×10−4, and 0.0110 with the latter two values corresponding to a threshold for the top 1% and top 5% of all SNPs outside the MHC. In all three cases, an r2=0.6 and a window of 500kb was used, i.e. the same parameters that were used to define the associated loci in PGC2.
Scoring our top results
When analyzing the results from INRICH, it was noted that sometimes the p-value for the gene-set would fluctuate when using different p-value thresholds. Furthermore, INRICH gives the same weight to all intervals regardless of how significantly associated they are. This is undesirable, as more significantly associated intervals are more likely to be true risk loci. To circumvent these limitations, a score was assigned to each gene set: , where pi is the p-value corrected for multiple testing of the gene sets based on the ith inclusion threshold and “log” is the natural logarithm. This results in a score that weighs by strength of association and gives higher weight to gene sets that show association across all thresholds.
Characterization of potential confounders
First, we examined whether our top-scoring gene sets were simply those with the highest content of brain-expressed genes. For each gene set, the number of brain-expressed genes was calculated based on information from the “eGenetics/SANBI EST anatomical system data” (Ensembl 75)20. To study the specificity of our findings we repeated our TargetScan based analyses for our top miRNA gene-sets in three unrelated traits14-16 (see above). We also studied the impact of different clumping thresholds on our results through comparison of results from all possible combinations for r2 choices of 0.1 and 0.6 and/or a window size of 500kb and 3000kb. Finally, we studied the impact of target prediction algorithms by using TargetScan predictions filtered with data from 58 AGO CLIP experiments21-23 and the two additional target prediction resources TargetMiner24 and MiRanda25 (see Supplementary Text).
Follow-up of our top finding(s)
To expand on our findings in the gene set analysis we used a framework of different approaches to characterize the relationship of our top miRNA and their targets with schizophrenia. In brief, we checked for association of our top miRNA in the PGC2 GWAS12, analyzed the overlaps in targets of different miRNA, used DAVID26 for functional annotation of the miRNA targets and BrainSpan27 to establish spatiotemporal expression patterns. We also used BrainSpan to identify co-expressed clusters of targeted genes, which we subsequently characterized: We examined their enrichment for common and rare variants using data from PGC2 and a recently published exome sequencing study in schizophrenia28, respectively. Furthermore, we tested for excess in protein-protein interactions within each significantly associated cluster using data from TissueNet29. We also looked at differential expression in data from post-mortem brains of patients with schizophrenia and controls30 in an additional attempt to identify sub-modules of schizophrenia genes targeted by our top ranking miRNA. These analyses are further detailed in the Supplementary Text.
Part C: Targeted gene set analyses
In addition to our analyses in part B we also employed a targeted gene set analysis approach in order to further characterize recent findings from GWAS and CNV analyses12,31. Targetomes of miRNA located in the 108 schizophrenia GWAS loci12 from the PGC2 paper were examined. These “GWAS miRNA” were defined as miRNA whose pri-miRNA genetic sequences overlapped with one of the GWAS loci. In addition to the targetomes of “ GWAS miRNA” we also analyzed targetomes of miRNA located in 10 schizophrenia associated CNVs identified in a recent meta-analysis31. These “CNV miRNA” were defined as those miRNA whose pri-miRNA genetic sequence overlapped with one of the 10 CNVs. All targeted gene set analyses were conducted as described for the analyses in part B with a single exemption: For the TargetScan based analyses, all predicted targets regardless of conservation were used, as only a few of the identified miRNA were conserved.
Results
Part A: Regulation of schizophrenia risk genes by miRNAs
We found a negative correlation between the (log) p-value of a protein coding gene and its corresponding number of predicted miRNA sites (β=−0.016, p<2×10−16), i.e. genes with more predicted miRNA target sites showed on average a stronger association with schizophrenia. However, two out of three additionally tested GWAS traits showed a similar tendency (Supplementary Text). Considering just the 108 genome-wide significant schizophrenia loci, protein coding genes located in these have on average a 21% excess of predicted miRNA-binding sites compared to protein coding genes in general.
Part B: Gene set analyses of all conserved miRNAs
Testing the targetomes of conserved miRNA, several schizophrenia-associated gene sets were found using the predictions of TargetScan (Table 1 and eTable 1). The ten highest scoring miRNAs targetomes are illustrated as a CIRCOS plot32 in Figure 2a and in a cluster plot in Figure 2b. Our top ranking miRNA gene-sets were not simply the largest gene sets or those with the highest number or fraction of brain-expressed genes (eTable 1). Furthermore, our findings were largely consistent under alternative tests conditions. This included additional analyses carried out with less strict thresholds for LD and/or longer windows in clumping (eTables 15-17) and tests with two additional miRNA prediction algorithms (eTables 2,3). Filtering with CLIP data, which on average removed 45% of TargetScan predicted targets, was not found to be better than removing genes at random (Supplementary Text, eTable 4). Further, our top 10 miRNA showed no evidence for association in well-powered studies of unrelated traits (eTable 5). In a first attempt to further characterize our findings (using DAVID26) we found enrichment of genes targeted by two or more miRNA in our top 10 in terms related to transcriptional regulation and neuronal development (eTables 6,7).
Table 1: Top 10 scoring conserved miRNA gene sets.
P-values are corrected for multiple testing within each threshold for all 143 tested gene sets using INRICH’s bootstrapping approach. A Bonferroni corrected alpha level of 0.017 should be applied to correct for all tests performed in our analyses. Please note that due to correlations in the results for the different thresholds a Bonferroni correction seems to be too conservative. The p-values in the table are highlighted as follows: p-values < 0.05 are underlined; < 0.017 bold; < 0.001 bold, and italic. The three different thresholds represent the different significance thresholds for the index-SNP used in clumping. Top-1% of SNPs have p-values less than 3.420×10−4, top 5% of SNPs have p-values less than 0.0110. “Brain” indicates the percentage of the test genes expressed in the brain.
| TargetScan 6.2 miRNA-family |
P-value at different thresholds | Score | Genes | Brain | ||
|---|---|---|---|---|---|---|
| 1E-5 | top-1% | top-5% | ||||
| miR-9-5p | 0.0378 | 0.0056 | 0.0009 | 212.3 | 1237 | 75% |
| miR-485-5p | 0.1474 | 0.1373 | 0.0011 | 68.3 | 379 | 73% |
| miR-137 | 0.0844 | 0.0326 | 0.0319 | 68.3 | 1144 | 77% |
| miR-101-3p | 0.2358 | 0.0135 | 0.0211 | 63.0 | 803 | 78% |
| miR-200bc-3p/429 | 1.0000 | 0.0050 | 0.0011 | 49.5 | 1057 | 77% |
| miR-7-5p | 0.4311 | 0.0555 | 0.0236 | 34.0 | 444 | 73% |
| miR-1/206/613 | 0.2143 | 0.3825 | 0.0052 | 31.1 | 787 | 76% |
| miR-374ab-5p | 0.8970 | 0.0075 | 0.0304 | 29.4 | 678 | 71% |
| miR-28-5p/708-5p/3139 | 0.0716 | 0.0380 | 0.4152 | 29.2 | 209 | 80% |
| miR-34ac-5p/449b-5p/449a | 0.1951 | 0.2136 | 0.0793 | 23.7 | 655 | 78% |
Figure 2: Visualizing the top-10 schizophrenia miRNA gene sets.
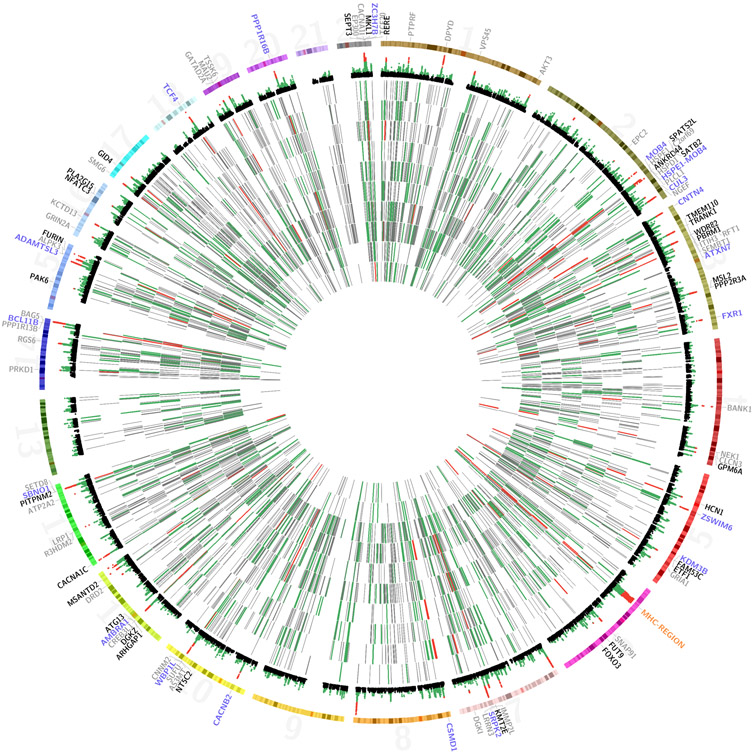

a) CIRCOS-plot of the top-10 scoring miRNA gene sets. The innermost 10 tracks illustrate the targets of each miRNA. The targets are color-coded based on their gene p-values. The miRNA were ordered by their correlational clustering. Peripherally to this, a Manhattan plot is shown (only SNPs with a p-value <0.02 located in protein coding genes are included). At the edge, the genome-wide significant genes targeted by the top-10 miRNA are shown. They are color-coded based on the number of miRNA in the top-10 list that target them. Note that for illustrative purposes the MHC-region is included here although it wasn't part of the gene set tests and that p-values from PGC2 are without replication. In eFigure 1, a zoomed in view of this region is presented. b) Legend for the figure and the clustering of miRNA based on the Jaccard distance between the targets of each miRNA. In eFigure 2 this clustering is repeated considering only the targets showing increasing degrees of association with schizophrenia. “Height” is the dissimilarity measure in the clustering. GW: Genome-wide.
miR-9-5p and related follow-up analyses
In our analysis of conserved miRNA, the targetome of miR-9-5p showed the strongest association with schizophrenia (Table 1). Based on these results we attempted to evaluate its role in schizophrenia: We examined the GWAS signal at the three genes encoding miR-9-5p (eFigure 3). MIR9-2, the most highly expressed gene in BrainSpan of the three, and the only one expressed in neuronal progenitor cells33, is contained within the r2=0.6 clump of rs181900, a SNP just shy of significance in PGC2 (p=7.1×10−8 including replication). For the 50 most schizophrenia-associated genes targeted by miR-9-5p we provide an overview in eTable 8. Amongst those are 21 genes residing in regions with a genome-wide significant signal in the PGC2 GWAS12. Functional annotation of the miR-9-5p gene set using DAVID showed enrichment in regulatory function, brain development and various transcription factors with FOXO3 having the highest fold enrichment (eTables 9-11) In our analyses of spatiotemporal expression patterns miR-9-5p showed a distinct peak in expression around the 16th post-conception week across all brain regions (eFigure 4).
To find a more homogeneous subset of miR-9-5p targets we performed cluster analyses using brain expression data from BrainSpan27. We identified a cluster of 497 genes (cluster 4, Supplementary Text and eTable 12) that subsequently was shown to be enriched for protein-protein interactions (p=3×10−5 for enrichment; Supplementary Text and eFigure 5). More importantly, however, it was enriched for schizophrenia risk genes compared to randomly drawn subsets of the original set of miR-9-5p targets (p=5×10−3 for enrichment; Supplementary Text). Cluster 4 was also enriched in rare variant analyses using summary statistics from a recently published schizophrenia exome sequencing study28 (one-sided binominal test, MAF<0.1%, disruptive mutations; p=0.013, 194 vs. 153 mutations). This was not the case for the full miR-9-5p targetome (p=0.10, 524 vs. 487 mutations). We also examined the miR-9-5p targetome expression in post-mortem brains30, but the targets in the full set and cluster 4 showed only nominally significant module enrichments (Supplementary Text).
Overlap in targetomes of miR-9 and miR-137
Consistent with its previous implication in schizophrenia we identified miR-137 among our top ranking miRNA (Table 1). During our cluster analysis for the top 10 ranking miRNA we found that the targetomes of miR-137 and miR-9-5p clustered together and shared 231 predicted target genes (eFigure 2). This overlap may a priori seem to be larger than what can be expected by chance. However, miRNA have a markedly skewed distribution of number of genes they target, and correcting for this reveals the overlap to be non-significant (p=0.28, Supplementary Text and eFigure 6).
Part C: Targeted gene set analyses
A total of 43 different mature miRNA were found to be located in schizophrenia GWAS loci (Table 2). However, the targetomes of these miRNA did not show consistent association with schizophrenia (eTable 13). Compared to our analysis for conserved miRNAs (part B) the TargetScan gene set for miR-137 tested here, which included all targets regardless of conservation, shows a less significant association. A total of 17 different mature miRNA were found to be located in schizophrenia associated CNVs (Table 2). For the targetomes of these miRNA, miR-185-5p, located in the 22q11.21 deletion, showed the most consistent association with schizophrenia (eTable 14).
Table 2: miRNA from genes in GWAS loci and CNVs associated with Schizophrenia.
The following schizophrenia CNVs didn’t contain any miRNA genes: NEDD4L-exonic duplication, 3q26.1 deletion, VIPR2 exonic duplication, C16orf72 exonic duplication, 3q29 deletion. The miRNA in bold are conserved and/or from a highly confident transcript. Names of miRNA are underlined when the targetome for this miRNA showed a nominal significant association with schizophrenia using the top 1% of SNPs and target predictions from TargetScan including non-conserved targets.
| Schizophrenia GWAS loci |
|---|
| miR-29b-2-5p, miR-29b-3p, miR-29c-3p, miR-29c-5p, miR-33a-3p, miR-33a-5p, miR-33b-3p, miR-33b-5p, miR-130a-3p, miR-130a-5p, miR-137, miR-378i, miR-640, miR-1228-3p, miR-1228-5p, miR-1281, miR-1307-3p, miR-1307-5p, miR-2682-3p, miR-2682-5p, miR-3160-3p, miR-3160-3p, miR-3160-5p, miR-3160-5p, miR-3655, miR-4301, miR-4304, miR-4529-3p, miR-4529-5p, miR-4655-3p, miR-4655-5p, miR-4677-3p, miR-4677-5p, miR-4688, miR-6773-3p, miR-6773-5p, miR-6777-3p, miR-6777-5p, miR-6843-3p, miR-6889-3p, miR-6889-5p, miR-8064, miR-8072 |
| Schizophrenia CNVs |
| 1q21.1-Deletions: miR-6736-5p, miR-6736-3p |
| 3q29-Deletions: miR-922 |
| 15q13.3-Deletions: miR-4509 |
| 16p11.2-Duplications: miR-3680-5p, miR-3680-3p |
| 22q11.21-Deletions: miR-185-3p, miR-185-5p, miR-648, miR-1306-3p, miR-1306-5p, miR-3198, miR-3618, miR-4761-5p, miR-4761-3p, miR-6816-5p, miR-6816-3p |
miR-2682-5p and miR-137
The miR-137 locus on chromosome 1 also contains the high confidence miRNA-gene MIR2682 just 719bp downstream of MIR137. The targetome of miR-2682-5p was nominally significant at the lowest threshold using target predictions from TargetScan (eTable 13). It has been shown that miRNA closely located together often are co-expressed34 and co-target the same genes35. Analysis of MIR137 and MIR2682 expression using BrainSpan27 shows that both miRNA have similar spatiotemporal expression patterns with a peak in expression in early childhood (eFigure 4). In addition, MIR2682 is the gene that shows the highest degree of expressional correlation with MIR137 among all genes in BrainSpan (r=0.679). However, the overlap (n=225) in targetomes of miR-2682-5p with miR-137 is not significantly larger than that of a random miRNA (p=0.31, Supplementary Text and eFigure 6).
Discussion
We found evidence for an overall involvement of miRNA in the etiology of multiple traits including schizophrenia. This finding is in line with results from a previous report that using GWAS data found enrichments of risk variants in miRNA genes and binding sites across multiple traits7. In contrast to this unspecific association, results of our gene set analyses in targetomes of conserved miRNAs revealed a more differentiated picture. Our results suggest the existence of several schizophrenia-associated miRNA targetomes, for which no evidence of association was found in additionally tested unrelated traits. In line with this, many of our top miRNA are known to be brain specific and/or have known regulatory functions in the brain36-41.
The association of both miR-9-5p and its targetome marks our strongest finding. Intriguingly, a recent study identified miR-9-5p as the highest abundance miRNA with significant differential expression (of 800 queried miRNAs). This result was found studying neuronal progenitor cells differentiated from human induced pluripotent stem cells from patients with schizophrenia. (Kristen Brennand and Gang Fang, written communication, October 2015).
Experimental evidence has implicated miR-9-5p as an important regulator of neuronal differentiation4,36 and it is predicted13 and experimentally validated42 that miR-9-5p targets the dopamine D2 receptor (DRD2), the predominant drug-target in schizophrenia43. Further of interest are the functional correlations with an additional target of miR-9-5p, the genome-wide significant gene FXR1. Along with FXR2, FXR1 is a homolog of the fragile X mental retardation 1 gene (FMR1), which itself is targeted by miR-9-5p. It has been shown that FXR1 regulates the level of miR-9-5p and is necessary for efficient processing of pre-miR-9-5p44. Moreover, FMR1 gene sets have shown association with schizophrenia in PGC2, an exome sequencing study, and a CNV study12,28,45. Additionally, we used a framework of different approaches to expand our knowledge about the role of miR-9-5p and its targetome in the etiology of schizophrenia. The expression pattern for MIR9-2 identified in our analyses is in agreement with the suggested neurodevelopmental role of this miRNA36. Despite strong evidence for an importance of an identified subset of 497 genes in the miR-9-5p targetome we were not successful in identifying a specific biological process that is connected to these genes. However, our identification of a co-regulating function with FOXO3 (a member of this cluster) is of interest as this gene falls just shy of genome-wide significance in PGC2 and plays a critical role in oxidative stress-induced neuronal cell death46.
Previously, MIR137 has seen the highest degree of interest resulting from GWASes of schizophrenia. In this article we demonstrated that another miRNA located in this schizophrenia hit region, MIR2682, is co-expressed with MIR137 and shares part of its targetome. Further, the implication of miR-185-5p (22q11 microdeletion locus) and its targetome in schizophrenia is in line with results from a previous study that used an earlier, overlapping version of the PGC GWAS47.
Our study is not without limitations. Of particular concern are limitations pertaining to miRNA target prediction methods (see Supplementary Text and eTables 1-3). Additionally, the sample size of the PGC2 study is still insufficient to detect all disease-associated variants at reasonable significance levels6. An additional limitation is our exclusion of the broad MHC-region, which has potentially impacted the identification of miRNA mainly targeting genes in this region. Finally, our scoring based approach, which was meant to rank the miRNA targetomes based on likelihood for their involvement in schizophrenia, could have prevented us from focusing on miRNAs with an important role in the etiology of schizophrenia. However, additional analyzes with rank sum or log sum based scoring procedures revealed that miR-9-5p’s leading position in our study was independent of the utilized scoring function (data not shown). Moreover, our scoring approach was not intended to exclude other miRNAs from downstream analyses but to assist in the interpretation of our results (Table 1 and eTable 1). Future studies are warranted to illustrate the schizophrenia related role of miRNAs in general and miR-9-5p’s role in particular.
In conclusion, we used an analytical framework that, broadly studied the role of miRNAs in common variant schizophrenia susceptibility and found further evidence for their involvement. In particular, we identified a tripartite correlation between schizophrenia, miR-9-5p, and FMR1/FXR1 with the corollary that establishing the functional overlaps and differences between the FMR1 and its homologues could potentially shed light on both the function of miR-9-5p and the etiology of schizophrenia.
Supplementary Material
At a glance:
Using common genetic risk variants the role of miRNA in schizophrenia is examined.
Several miRNA regulate schizophrenia risk genes with the strongest associations for miR-9-5p, miR-485-5p, and miR-137.
miR-9-5p is itself a risk gene and its function ties in with established disease biology.
Acknowledgements
This study was funded by The Lundbeck Foundation; iSEQ (Centre for Integrative Sequencing), Aarhus University; and The Faculty of Health, Aarhus University.
Footnotes
Financial Disclosures:
The authors declare no conflict of interest. The Lundbeck Foundation had no involvement in any aspect of the study.
A list of all members is available in the supplementary Text.
URLs
Schizophrenia GWAS:
http://www.med.unc.edu/pgc/downloads
Age at Menarche GWAS:
http://www.reprogen.org/data_download.html
Crohn’s Disease GWAS:
http://www.ibdgenetics.org/downloads.html
Height GWAS:
http://www.broadinstitute.org/collaboration/giant/
INRICH
https://atgu.mgh.harvard.edu/inrich/
PLINK
Contributor Information
Mads Engel Hauberg, Department of Biomedicine, Aarhus University, Aarhus, Denmark. The Lundbeck Foundation Initiative of Integrative Psychiatric Research (iPSYCH), Denmark. Centre for Integrative Sequencing (iSEQ), Aarhus University, Aarhus, Denmark, Address: Wilhelm Meyers Allé 4, Building 1242, Room 129, 8000 Aarhus C, Denmark.
Panos Roussos, Department of Psychiatry, Icahn School of Medicine at Mount Sinai, New York, NY 10029, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY 10029, Institute for Genomics and Multiscale Biology, Icahn School of Medicine at Mount Sinai, New York, NY 10029, Friedman Brain Institute, Icahn School of Medicine at Mount Sinai, New York, NY 10029, James J. Peters VA Medical Center, Mental Illness Research Education and Clinical Center (MIRECC), 130 West Kingsbridge Road, Bronx, NY 10468.
Jakob Grove, Department of Biomedicine, Aarhus University, Aarhus, Denmark. The Lundbeck Foundation Initiative of Integrative Psychiatric Research (iPSYCH), Denmark, Centre for Integrative Sequencing (iSEQ), Aarhus University, Aarhus, Denmark, Bioinformatics Research Centre (BiRC), Aarhus University, Aarhus, Denmark..
Anders Dupont Børglum, Department of Biomedicine, Aarhus University, Aarhus, Denmark. The Lundbeck Foundation Initiative of Integrative Psychiatric Research (iPSYCH), Denmark. Centre for Integrative Sequencing (iSEQ), Aarhus University, Aarhus, Denmark, Research Department P, Aarhus University Hospital, Risskov, Denmark. Translational Neuropsychiatry Unit, Department of Clinical Medicine, Aarhus University, Denmark..
Manuel Mattheisen, Department of Biomedicine, Aarhus University, Aarhus, Denmark. The Lundbeck Foundation Initiative of Integrative Psychiatric Research (iPSYCH), Denmark. Centre for Integrative Sequencing (iSEQ), Aarhus University, Aarhus, Denmark, Address: Wilhelm Meyers Allé 4, Building 1243, Room 213, 8000 Aarhus C, Denmark.
References
- 1.Millier A, Schmidt U, Angermeyer MC, et al. Humanistic burden in schizophrenia: a literature review. J Psychiatr Res. 2014;54:85–93. [DOI] [PubMed] [Google Scholar]
- 2.Jv Os, Kapur S. Schizophrenia. Lancet. 2009;374(9690):635–645. [DOI] [PubMed] [Google Scholar]
- 3.International Schizophrenia Consortium, Purcell SM, Wray NR, et al. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature. 2009;460(7256):748–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sun AX, Crabtree GR, Yoo AS. MicroRNAs: regulators of neuronal fate. Current opinion in cell biology. 2013;25(2):215–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chao Y-L, Chen C-H. An introduction to microRNAs and their dysregulation in psychiatric disorders. Tzu Chi Medical Journal. 2013;25(1):1–7. [Google Scholar]
- 6.Ripke S, O'Dushlaine C, Chambert K, et al. Genome-wide association analysis identifies 13 new risk loci for schizophrenia. Nature genetics. 2013;45(10):1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Goulart LF, Bettella F, Sønderby IE, et al. MicroRNAs enrichment in GWAS of complex human phenotypes. BMC genomics. 2015;16(1):304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Earls LR, Fricke RG, Yu J, Berry RB, Baldwin LT, Zakharenko SS. Age-dependent microRNA control of synaptic plasticity in 22q11 deletion syndrome and schizophrenia. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2012;32(41):14132–14144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Warnica W, Merico D, Costain G, et al. Copy Number Variable MicroRNAs in Schizophrenia and Their Neurodevelopmental Gene Targets. Biological psychiatry. 2015;77(2):158–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maurano MT, Humbert R, Rynes E, et al. Systematic localization of common disease-associated variation in regulatory DNA. Science. 2012;337(6099):1190–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Richards AL, Jones L, Moskvina V, et al. Schizophrenia susceptibility alleles are enriched for alleles that affect gene expression in adult human brain. Molecular psychiatry. 2012;17(2):193–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature. 2014;511(7510):421–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Garcia DM, Baek D, Shin C, Bell GW, Grimson A, Bartel DP. Weak seed-pairing stability and high target-site abundance decrease the proficiency of lsy-6 and other microRNAs. Nature structural & molecular biology. 2011;18(10):1139–1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perry JR, Day F, Elks CE, et al. Parent-of-origin-specific allelic associations among 106 genomic loci for age at menarche. Nature. 2014;514(7520):92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491(7422):119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wood AR, Esko T, Yang J, et al. Defining the role of common variation in the genomic and biological architecture of adult human height. Nature genetics. 2014;46(11):1173–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research. 2013:gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee PH, O'Dushlaine C, Thomas B, Purcell SM. INRICH: interval-based enrichment analysis for genome-wide association studies. Bioinformatics. 2012;28(13):1797–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.The 1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature. 2012;491(7422):56–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Flicek P, Amode MR, Barrell D, et al. Ensembl 2014. Nucleic acids research. 2013:gkt1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li JH, Liu S, Zhou H, Qu LH, Yang JH. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-Seq data. Nucleic acids research. 2014;42(Database issue):D92–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Balakrishnan I, Yang X, Brown J, et al. Genome-Wide Analysis of miRNA-mRNA Interactions in Marrow Stromal Cells. Stem Cells. 2014;32(3):662–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Boudreau RL, Jiang P, Gilmore BL, et al. Transcriptome-wide discovery of microRNA binding sites in human brain. Neuron. 2014;81(2):294–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bandyopadhyay S, Mitra R. TargetMiner: microRNA target prediction with systematic identification of tissue-specific negative examples. Bioinformatics. 2009;25(20):2625–2631. [DOI] [PubMed] [Google Scholar]
- 25.Betel D, Koppal A, Agius P, Sander C, Leslie C. Comprehensive modeling of microRNA targets predicts functional non-conserved and non-canonical sites. Genome biology. 2010;11(8):R90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang da W, Sherman BT, Tan Q, et al. DAVID Bioinformatics Resources: expanded annotation database and novel algorithms to better extract biology from large gene lists. Nucleic acids research. 2007;35(Web Server issue):W169–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.BrainSpan: Atlas of the Developing Human Brain. 2011; Funded by ARRA Awards 1RC2MH089921-089901, 089921RC089922MH090047-089901, and 089921RC089922MH089929-089901. Available at: http://brainspan.org. Accessed 17 Jun, 2014. [Google Scholar]
- 28.Purcell SM, Moran JL, Fromer M, et al. A polygenic burden of rare disruptive mutations in schizophrenia. Nature. 2014;506(7487):185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barshir R, Basha O, Eluk A, Smoly IY, Lan A, Yeger-Lotem E. The TissueNet database of human tissue protein–protein interactions. Nucleic acids research. 2013;41(D1):D841–D844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Roussos P, Katsel P, Davis KL, Siever LJ, Haroutunian V. A system-level transcriptomic analysis of schizophrenia using postmortem brain tissue samples. Archives of general psychiatry. 2012;69(12):1205–1213. [DOI] [PubMed] [Google Scholar]
- 31.Levinson DF, Duan J, Oh S, et al. Copy number variants in schizophrenia: confirmation of five previous findings and new evidence for 3q29 microdeletions and VIPR2 duplications. The American journal of psychiatry. 2011;168(3):302–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krzywinski M, Schein J, Birol I, et al. Circos: an information aesthetic for comparative genomics. Genome research. 2009;19(9):1639–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delaloy C, Liu L, Lee J-A, et al. MicroRNA-9 coordinates proliferation and migration of human embryonic stem cell-derived neural progenitors. Cell stem cell. 2010;6(4):323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baskerville S, Bartel DP. Microarray profiling of microRNAs reveals frequent coexpression with neighboring miRNAs and host genes. Rna. 2005;11(3):241–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wang Y, Li X, Hu H. Transcriptional regulation of co-expressed microRNA target genes. Genomics. 2011;98(6):445–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Coolen M, Katz S, Bally-Cuif L. miR-9: a versatile regulator of neurogenesis. Front Cell Neurosci. 2013;7:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cohen JE, Lee PR, Chen S, Li W, Fields RD. MicroRNA regulation of homeostatic synaptic plasticity. Proceedings of the National Academy of Sciences. 2011;108(28):11650–11655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Siegert S, Seo J, Kwon EJ, et al. The schizophrenia risk gene product miR-137 alters presynaptic plasticity. Nature neuroscience. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Choi PS, Zakhary L, Choi W-Y, et al. Members of the miRNA-200 family regulate olfactory neurogenesis. Neuron. 2008;57(1):41–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.De Chevigny A, Coré N, Follert P, et al. miR-7a regulation of Pax6 controls spatial origin of forebrain dopaminergic neurons. Nature neuroscience. 2012;15(8):1120–1126. [DOI] [PubMed] [Google Scholar]
- 41.Chang S-J, Weng S-L, Hsieh J-Y, Wang T-Y, Chang MD, Wang H-W. MicroRNA-34a modulates genes involved in cellular motility and oxidative phosphorylation in neural precursors derived from human umbilical cord mesenchymal stem cells. BMC medical genomics. 2011;4(1):65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shi S, Leites C, He DL, et al. MicroRNA-9 and MicroRNA-326 Regulate Human Dopamine D2 Receptor Expression, and the MicroRNA-mediated Expression Regulation Is Altered by a Genetic Variant. Journal of Biological Chemistry. 2014;289(19):13434–13444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kapur S, Mamo D. Half a century of antipsychotics and still a central role for dopamine D 2 receptors. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2003;27(7):1081–1090. [DOI] [PubMed] [Google Scholar]
- 44.Xu XL, Zong R, Li Z, et al. FXR1P but not FMRP regulates the levels of mammalian brain-specific microRNA-9 and microRNA-124. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2011;31(39):13705–13709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Szatkiewicz JP, O'Dushlaine C, Chen G, et al. Copy number variation in schizophrenia in Sweden. Molecular psychiatry. 2014;19(7):762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xie Q, Hao Y, Tao L, et al. Lysine methylation of FOXO3 regulates oxidative stress-induced neuronal cell death. EMBO reports. 2012;13(4):371–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Forstner AJ, Basmanav FB, Mattheisen M, et al. Investigation of the involvement of MIR185 and its target genes in the development of schizophrenia. J Psychiatry Neurosci. 2014;39(4):130189. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.