

Graphical abstract
Keywords: Sigma 1 receptor, Amyotrophic lateral sclerosis, Oligomerization, Molecular dynamics simulations, Western blot, Bioluminescence resonance energy transfer
Abstract
The sigma 1 receptor (σ1R) is a unique endoplasmic reticulum membrane protein. Its ligands have been shown to possess therapeutic potential for neurological and substance use disorders among others. The E102Q mutation of σ1R has been found to elicit familial cases of amyotrophic lateral sclerosis (ALS). Despite reports of its downstream signaling consequences, the mechanistic details of the functional impact of E102Q at molecular level are not clear. Here, we investigate the molecular mechanism of the E102Q mutation with a spectrum of biochemical, biophysical, and pharmacological approaches. Our analysis of the interaction network of σ1R indicates that a set of residues near E102 is critical for the integrity of C-terminal ligand-binding domain. However, this integrity is not affected by the E102Q and E102A mutations, which is confirmed by the radioligand binding results. Instead, the E102 mutations disrupt the connection between the C-terminal domain and the N-terminal transmembrane helix (NT-helix). Results from bioluminescence resonance energy transfer and western blot assays demonstrate that these mutations destabilize higher-order σ1R oligomers, while our molecular dynamics simulations based on a σ1R crystal structure reveal a potential mechanism by which the mutations perturb the NT-helix dynamics. Thus, we propose that E102 is at a critical position in propagating the effects of ligand binding from the C-terminal domain to the NT-helix, while the latter may be involved in forming alternative oligomer interfaces, separate from the previously reported trimer interface. Together, these results provide the first account of the molecular mechanism of σ1R dysfunction caused by E102Q.
1. Introduction
The sigma 1 receptor (σ1R) is a unique protein found primarily in the membrane-associated matrix interface of the endoplasmic reticulum (ER) and mitochondrion, although it has been shown to translocate to other membrane environments of the cell [1]. It is diversely expressed in multiple tissues and organs, being abundantly found in the central nervous system, liver, and lung [3]. The primary function of σ1R is most likely as a modulator of intracellular calcium signaling [1], but evidence has also been presented that it may interact as a chaperone or modulator with a panoply of client proteins [2]. σ1R has been implicated in the molecular pathophysiology of several classes of diseases, including substance use disorders [4], depression [5], and neurodegenerative disorders [6], as well as modulating oxidative stress responses [7].
Surprisingly for a protein with such a wide anatomic distribution, σ1R gene (SIGMAR1) knockout in mice is not lethal [8]. Studies with knockout mice have identified deficits in motor control resulting from a loss of motor neurons in the spinal cord [9]. Three distinct phenotypes are associated with reported human SIGMA1R mutations: fronto-temporal lobar degeneration (FTLD) [10], autosomal recessive distal hereditary muscular neuropathy (dHMN) [11], and familial juvenile amyotrophic lateral sclerosis (ALS) [12], [13]. In the latter two pathologies, multiple mutations in SIGMAR1 have been identified. Most of these mutations arise from frameshift or deletion errors in the gene that result in large-scale changes in the σ1R protein sequence. However, ALS can also be caused by a single amino acid residue mutation, E102Q [13]. In in vitro studies, E102Q was found to result in a significant increase in apoptosis in response to oxidative stress, as well as a decrease in the σ1R population at the ER membrane and the aggregation of the mutant protein in the cytoplasm [14], [15]. Thus, in the case of ALS, the E102Q mutation is tractable to in vitro studies and offers a stepping stone for us to understand the molecular mechanisms of σ1R activity.
Until recently, little was known about the molecular structure and functional mechanisms of σ1R. It has been shown that the receptor existed in multiple states of oligomerization, with populations of at least monomer, dimer and tetramer observed [16], [17]. The relative populations of these oligomer states appeared to be affected by ligand binding [17]. The presence of a GxxxG motif, previously observed to be commonly associated with the dimerization of helical protein segments [18], was also noted to be involved in the oligomerization [17]. Recent crystal structures of σ1R were solved by the Kruse group as symmetrical trimers [19], [20]. For each monomer, a single transmembrane helix is located at the N-terminus (NT-helix), while the ligand binding pocket was completely buried in a cupin-like domain at the C terminal end of the protein. Interestingly, this structure places the GxxxG motif not in a helical segment, but still near the trimer interface, while residue E102 is located at the base of the C terminal cupin domain, facing towards the NT-helix near where the helix would emerge from the membrane. The side-chain oxygens of E102 make hydrogen bond contacts with the amide hydrogens in the protein backbone residues of V36 and F37 of the NT-helix, anchoring it in place.
While the σ1R structures offer a structural basis for mechanistic studies, much work remains in understanding its molecular pharmacology. In particular, understanding the mechanisms of how ligand-binding translates into biological activity remains a challenge. Historically, small-molecule σ1R agonists and antagonists were classified by their actions when administered in vivo [6], [21]. In a previous work, we demonstrated that classical σ1R antagonists stabilized higher-order oligomers of σ1R, while classical σ1R agonists did not [22]. In addition, we identified a small side pocket of the main ligand binding site near the trimer interface that was only occupied by ligands that failed to induce oligomerization of σ1R [22]. These observations provide the starting point for additional investigations that aim to elucidate how ligand binding can activate this receptor.
In this study, we combine experimental biophysical and pharmacological assays with computational network analysis and all-atom molecular dynamics (MD) simulations to examine the impact of the E102Q mutation on the conformational changes and dynamics of σ1R. In addition, to further characterize the necessary physicochemical properties of the residue at this position, we evaluate two additional mutations, E102D, which has a shorter sidechain, and E102A, which eliminates the H-bonding capability entirely.
2. Materials and methods
2.1. Network analysis
In the network analysis of a protein structure, one common strategy is to reduce each amino acid residue to a single node; two nodes are connected with an edge when the shortest distance between the corresponding two residues is below a defined threshold distance. This representation simplifies identifying key interactions within the protein of interest, and allows network centrality analysis [23]. In this study, we calculated the shortest heavy-atom distance for each residue pair in a σ1R monomer separated by more than 2 residues. For σ1R crystal structures, the distances from three monomers were averaged; for MD simulation results, the distances for each residue pair in all the frames for each given condition were averaged. The program igraph implemented in python (version 0.7.1.post6) [24] was then used to generate an adjacency matrix A, in which each vertex v represents a residue and each edge is a contact. If two vertices i, j form a contact (their distance <5 Å), then Ai,j = 1, otherwise Ai,j = 0. We then calculated the eigenvector centrality score of each vertex, which is proportional to the sum of the centrality scores of its neighbors. To compare across different conditions, we normalized the scores by rescaling, so the maximum score in each condition is 10.
2.2. Molecular dynamics simulations
Based on our previously equilibrated trimeric WT/PD144418 simulation system [22], we built other systems listed in Table S1 by removing the ligand (for the apo conditions), docking and selecting the (+)-pentazocine pose most similar to that in the WT/(+)-pentazocine crystal structure (PDB ID 6DK1 [20]), and/or in silico mutagenesis (for the E102Q and E102A conditions).
The receptor models were immersed in explicit 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine lipid bilayer (POPC). The simple point charge (SPC) water model was used to solvate the system, charges were neutralized, and 0.15 M NaCl was added. The system sizes were ~200000 atoms. Desmond MD systems (D. E. Shaw Research, New York, NY) with the OPLS3 force field were used for the MD simulations. The system was initially minimized and equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by production runs at 310 K with all atoms unrestrained. The NPγT ensemble was used with constant temperature (310 K) maintained with Langevin dynamics. Specifically, 1 atm constant pressure was achieved with the hybrid Nose-Hoover Langevin piston method on an anisotropic flexible periodic cell with a constant surface tension (x-y plane). Overall, 31 trajectories with an aggregated simulated time of ~25 μs were collected (Table S1).
2.3. Conformational analysis
For the analysis of the of the N-terminus distributions along the lipid bilayer plane, we align all the MD frames to chain A of the WT/PD144418 crystal structure. We then calculated the X and Y coordinates for the center of mass (COM) of Cα atoms of the four residues of the first helical turn in the N-terminus (residues 8–11). We calculated the distribution of the NT along the lipid bilayer (XY) plane shown in Fig. S4A. To quantitatively characterize the NT dynamics, we evaluated its distribution along the XY plane in a fixed amount of simulation time. We first selected a representative monomer trajectory for each condition. Using the frames from the last 600 ns for each trajectory, the extents of the COM distributions on the XY plane are evaluated by using a grid with a bin size of 0.25 Å (Fig. S4B) and counting the number of COM points in each bin. These numbers are then sorted in the descending order and plotted in Fig. S4C. Thus, if the NT exhibits more restricted dynamics, it will be more densely populated in fewer bins.
These structural analyses were performed using the MDTraj program [25] and in-house Python scripts.
2.4. DNA constructs, transfection, and cell culture
Δσ1R HEK293T cells were generated using CRISPR-Cas9 gene deletion kit (Santa Cruz). E102Q or E102A single point mutation was made in human σ1R, C-terminally fused σ1R-nanoluciferase, or σ1R-mVenus in pcDNA3.1 plasmid [22]. All constructs were confirmed by sequence analysis. For western blot, 5 µg of σ1R plasmid was transfected in HEK 293T Δσ1R cells using lipofectamine 2000 (Invitrogen) in a 10 cm plate. For radioligand binding, 5 µg of σ1R plasmid was transfected in HEK 293T Δσ1R cells using polyethylenimine (PEI). For acceptor saturating BRET, a constant amount of total plasmid cDNA (5 µg) in varying donor:acceptor ratios for σ1R-nanoluciferase and σ1R-mVenus was transfected using PEI in HEK 293T Δσ1R in 6-well plates. For drug induced BRET, a constant amount of total plasmid cDNA (15 µg) in 1:24 (donor:acceptor ratio for σ1R-nanoluciferase and σ1R-mVenus) was transfected using PEI in a 10 cm plate. Cells were maintained in culture with Dulbecco’s modified Eagle’s medium supplemented with 10% fetal bovine serum and kept in an incubator at 37 °C and 5% CO2. Experiments were performed approximately 48 h post-transfection.
2.5. Western blot
Δσ1R HEK293T cells were grown as reported [22] and transiently transfected with WT, E102Q, and E102A σ1R in 10 cm plates. After 48 hr of growth, confluent cells were harvested in Hank’s Balanced Salt Solution (HBSS), centrifuged at 900×g for 8 min, and resuspended in HBSS. The cells were then incubated in 1 µM haloperidol, 1 µM PD144418, 10 µM (+)-pentazocine, or 1% DMSO for 1 h at room temperature. The samples were then centrifuged at 900×g for 4 min and resuspended in lysis buffer (150 mM NaCl, 1.0% triton X-100, 0.5% sodium deoxycholate, Tris 50 mM, pH 7.5, and protease inhibitors (Roche, catalog# 11697498001)). The samples were sonicated, incubated on ice for 30 min, and centrifuged at 20,000×g at 4 °C for 30 min. Supernatants were transferred to new tubes. Protein concentrations of the supernatants were determined with BCA protein assay (Bio-rad, Hercules, CA). Supernatants were mixed with 4x β-mercaptoethanol Laemmli sample buffer to a final 25 µg protein/sample. Samples were electrophoresed on 10% polyacrylamide Tris-glycine gels (Invitrogen) with running buffer (25 mM Tris, 192 mM glycine and 0.1% SDS, pH 8.3, Invitrogen). Proteins were transferred to PVDF membranes (Invitrogen, catalog# IB24002) and immunoblotted with mouse monoclonal α-σ1R antibody (Santa Cruz, B-5) and rabbit polyclonal α-GAPDH antibody (Sant Cruz, FL-335). For secondary antibodies, Donkey α mouse (LI-COR, IRDye® 680RD) and goat α rabbit (LI-COR, IRDye® 800CW). Blots were imaged using an Odyssey LI-COR scanner and analyzed with LI-COR Image Studio™.
2.6. Radioligand binding assay
Membrane fraction of Δσ1R HEK293T cells was prepared as previously described [22]. The radioligand incubation was carried out in 96-well plates with a total volume of 200 µL; containing 60 µL fresh Earle’s Balanced Salts Solution (EBSS) binding buffer (8.7 g/l Earle's Balanced Salts without phenol red (US Biological) and 2.2 g/L sodium bicarbonate, pH to 7.4), 100 µL membranes (10 µg/well for WT, 100 µg/well for E102Q, and 150 µg/well for E102A), 20 µL of radioligand diluted in binding buffer (noted below), and 20 µL of either 10% DMSO for total binding or 100 µM (+)-pentazocine for non-specific binding. Concentrations for [3H]-(+)-pentazocine (American Radiolabeled Chemicals) and [3H]-haloperidol (American Radiolabeled Chemicals) were: 1.5 µM, 1 µM, 500 nM, 100 nM, 50 nM, 10 nM, and 1 nM in EBSS with 10% DMSO. All compound dilutions were tested in triplicate, and samples were incubated for 120 min at room temperature. The reactions were terminated by filtration through Perkin Elmer Uni-Filter-96 GF/B, presoaked in 0.05% PEI for 120 min, and the 96-well filter plates were counted in Perkin Elmer MicroBeta Microplate Counter as described [22] with counter efficiency at 31% for [3H]-(+)-pentazocine. Kd and Bmax values were determined from at least four independent experiments.
2.7. Bioluminescence resonance energy transfer (BRET) assay
Acceptor saturating BRET is performed as described previously [22]. Briefly, cells were distributed in 96-well plates. Expression of venus fusion proteins was estimated by measuring fluorescence at 535 nm with excitation at 485 nm. Expression of NL fusion proteins was estimated by measuring the luminescence of the cells after incubation with 5 µM coelenterazine h (Nanolight) for 30 min. In parallel, BRET was measured as a ratio between measurements at 535 nm for fluorescence and at 485 nm for luminescence using a Pherastar FSX reader (BMG). Results were plotted as fluorescence over luminescence vs. basal-subtracted BRET ratio.
Drug-induced BRET was conducted as reported previously [26]. Briefly, cells were prepared in 96-well plates as in acceptor-saturating BRET. 5 µM coelenterazine h was added to each well. Three minutes after addition of coelenterazine h, ligands [(+)-pentazocine (Sigma), PD144418 (Tocris), and haloperidol (Tocris)] in series of dilution were added to each well. BRET was measured as in acceptor-saturating BRET after 30 min incubation. Results were calculated for the BRET change (BRET ratio for the corresponding drug minus BRET ratio in the absence of the drug). Emax values are expressed as the basal subtracted BRET change in the dose-response graphs.
3. Results
In the E102Q mutation, the sidechain carboxyl group is mutated to an amide group, which alters the charge and hydrogen bonding properties at the junction between the NT-helix and the C-terminal domain and also adjacent to the σ1R ligand binding pocket. To characterize the functional impact of this mutation at the molecular level, we carry out both in silico and in vitro studies, using protein interaction network analysis and molecular dynamics (MD) simulations, as well as biophysical, biochemical, and pharmacological assays.
3.1. E102 is closely associated with the structural motif connecting the N-terminal transmembrane and C-terminal domains
Network analysis has been adopted to understand the structure and dynamics of proteins [27]. In this family of methods, a protein structure (or an ensemble of its closely related structures, such as frames from an MD trajectory) is converted into a graph consisting of nodes and edges, then graph theory is applied to characterize the mechanistic properties of the protein (see Materials and Methods). In the present study, we first built the interaction network for the crystal structures of σ1R at the residue level, by calculating the interacting partners for each residue (Fig. 1A). As expected, we found that the C-terminal domain is in general populated with residues with larger numbers of interactions, while residues in the NT-helix have relatively fewer interactions (Fig. S1A).
Fig. 1.

E102 is closely associated with the structural motif connecting the NT-helix and the C-terminal domain. (A) Contacts in the σ1R WT/PD144418 crystal structure (PDB ID 5HK1) are represented as lines connecting the contacting residue pairs. (B) The eigenvector centrality score of each residue (see Materials and Methods) in the σ1R WT/PD144418 crystal structure C-terminal domain is represented by the radius of the sphere. The residues with the highest centrality scores (Fig. S1B) are colored in cyan. (C) A zoom-in view of the ligand binding pocket showing the interactions between the sidechain of E102 and the loop connecting the N-terminal transmembrane helix with the C-terminal domain. The nearby residues essential to ligand binding are shown as sticks as well. PD144418 is represented in green spheres. The positions of the high-centrality residues shown in panel B are colored on the backbone ribbons. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further characterize the relative importance of the residues, we used network centrality analysis [23] to identify the residues that are well connected and thus are hypothesized to be more influential on the network (see Materials and Methods). We found that the residues with higher centrality scores, F58, L61, V104, L106, F107, T127, I128, I129, W169, M170, V171 are located in four regions of the C-terminal domain (Fig. 1B and Fig. S1B). Except for F58 and L61, the others are in the β-sheet enclosing the ligand binding pocket on the intracellular side, namely [19] in the β3, β5, and β10 strands, while the α3 helix, in which F58 and L61 are located, interacts with all these three β strands (Fig. 1B,C).
The high-centrality residues in the β-sheet are in close proximity to residues that are known to be essential to ligand binding. Specifically, residues W169, M170, and V171 are in the β10 strand, as is E172, the residue forming a key salt bridge with the charged nitrogen of bound σ1R ligands. Similarly, V104, L106, and F107 are in β3 along with Y103, which interacts with E172. T127, I128, and I129 are in β5 as is D126, the protonated form of which interacts with and stabilizes the aforementioned E172 in the crystal structures. Interestingly, E102, the putative causal mutation for ALS, is located in β3 and forms a hydrogen bond with Y173 in β10. Thus, E102 is in a position that may indirectly affect ligand binding or facilitate propagating its impact.
3.2. The E102 mutations disrupt the connection between the NT-helix and the C-terminal domain
Starting from the crystal structures of σ1R, we then investigated the impact of the bound ligands ((+)-pentazocine and PD144418) and the mutations (E102Q, E102A, and E102D) on the conformation of σ1R, using all-atom molecular dynamics (MD) simulations (Table S1).
To compare the residue interactions in simulations to those in the crystal structures, we calculated the residue-residue distances, and averaged them over all frames for each condition. Using these averaged distances, we repeated the network analysis described above and found that the residues with higher centrality scores in all WT simulations are identical to those in the WT crystal structures, i.e., the residues shown in Fig. 1B are consistently top ranked. Thus, the consistency of these residues being important across all simulated conditions, as well as the crystal structures, suggests that they are important for the integrity and functionality of σ1R. Indeed, when we examined the root-mean-square fluctuation (RMSF) of the individual residues in the simulations, the results show that these highly connected residues all fall into low-RMSF regions (Fig. S2), indicating that these residues form a critical part of the core of the C-terminal domain that is less flexible.
Interestingly, the network analysis of our E102 simulation results show that the mutations did not change the centrality score ranking of the residues shown in Fig. 1B. Thus, the mutations appeared to have minimal impact on the integrity of this core and the C-terminal domain in general. We also compared the contact frequencies between PD144418 and its interacting residues in WT and mutant simulations, which also showed that the mutations have minimal impact on the ligand binding site located in the C-terminal domain (Table S2). These conclusions are consistent with our radioligand binding results (see below).
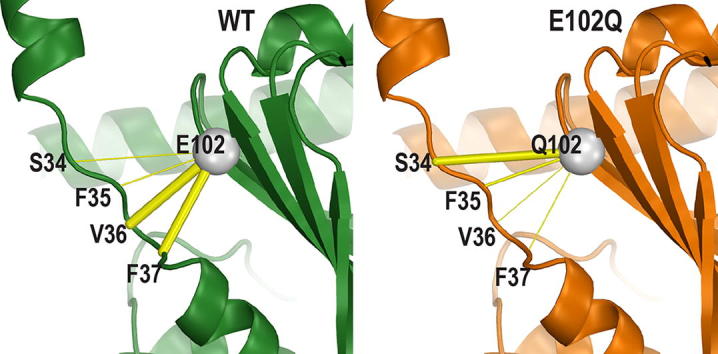
However, the results of MD simulations show that in the E102Q and E102A mutants the interactions between residue 102 and the loop region connecting the NT-helix and C-terminal domain are distorted or weakened. When we specifically calculated the frequencies of the polar interactions between residue 102 and the loop region (residues 34–37), we found the interactions to V36 and F37 are significantly weakened but that to S34 is strengthened in E102Q mutant, while E102A does not form any polar interaction with the loop (Fig. 2A–C). In comparison, E102D largely retains the interaction pattern to the loop as that of E102, but the interaction to F37 was weakened (Fig. 2D). These results suggest that the E102 mutations disrupt the interaction network that coordinates the NT-helix and the C-terminal domain, while both the length and charge of the sidechain of E102 matter for the association with the loop, especially the charge.
Fig. 2.

The E102 mutations disrupt the connection between the NT-helix and the C-terminal domain. The Cα atom of residue 102 is shown as a gray sphere and its contacts are shown in yellow sticks with varied thickness proportional to their frequency of the polar interactions to the loop region (residues 34–37) in the simulated conditions. Protein backbone ribbons are colored in green, orange, red, and light green for WT/PD144418 (PD) (A), E102Q/PD (B), E102A/PD (C), and E102D/PD (D) conditions, respectively. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
3.3. The E102 mutations disrupt S1R oligomerization
To characterize the potential impact of the E102 mutations on the oligomerization state of σ1R, we carried out western blot analysis. In this study, we used a rescue expression approach, in which WT or mutant human σ1R is expressed in a σ1R knock-out (Δσ1R) HEK293T cell line. As we described recently [28], when anti-σ1R antibodies are used to visualize unmodified WT σ1R on a western blot, multiple bands of protein are detected, each band being at a multiple of the monomeric molecular weight of 25 kDa. These bands clearly correspond to monomer through hexamer of σ1R. However, for the E102Q mutant, the ladder-like pattern of the higher-order oligomer bands disappears and only the bands corresponding to the monomer and dimer remain. For the E102A mutant, only the monomer band of σ1R remains (Fig. 3A). The anti-σ1R antibody did not detect any protein bands in the Δσ1R cells, confirming the specificity of the antibody (Fig. 3A). The lack of higher-order oligomer bands beyond the dimer in the western blots of the E102Q and E102A σ1R mutants suggests that the mutations disrupt intermolecular interactions necessary for oligomerization.
Fig. 3.

WT σ1R forms high-order oligomers while E102Q and E102A do not. (A) Representative western blot from five experiments are shown to visualize σ1R (red) and GAPDH (green) bands for wildtype (WT), E102Q, and E102A constructs expressed in Δσ1R HEK293T cells. Protein extract from Δσ1R cells without σ1R rescue expression is run in the last lane. (B) Molecular interactions between σ1R monomers are measured by BRET. Δσ1R HEK 293T cells were transfected with a constant amount of the RLuc-fusion construct and increasing amounts of the Venus-fusion construct (green-WT, orange-E102Q, and red-E102A). All data points were performed in triplicate (S.E.M. shown as error bars). The BRETmax and BRET50 values were calculated by nonlinear regression using a single-site saturation binding model. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To further probe this hypothesis, we generated σ1R fusion constructs with Renilla reniformis luciferase (Rluc) and Venus (yellow fluorescent protein) to allow for bioluminescence resonance energy transfer (BRET) studies (see Materials and Methods). BRET signals for varied expression ratios of donor and acceptor constructs were measured in saturation BRET to assess the specificity of protein-protein interactions (Fig. 3B). When compared to WT, the E102Q mutant showed decreases in both BRETmax and BRET50, indicating a significant reduction in intermolecular interactions between σ1R monomers. The E102A mutant showed a largely linear relationship between the ratio of Rluc to Venus and the net BRET ratio, which is characteristic of non-saturating kinetics. Such kinetics suggest that very few, if any, stable oligomers containing both Rluc and Venus-tagged σ1R monomers were formed. These results support the hypothesis that there is a loss of oligomerization capacity in σ1R monomers bearing the E102 mutations.
3.4. The E102 mutations abrogate ligand-induced changes in the S1R oligomerization states
Next, we examined the influence of E102 mutations in ligand-induced σ1R oligomerization. Western blots showed significant intensity changes induced by σ1R ligands in the higher-order bands corresponding to pentamer, hexamer, and higher molecular weight states for WT: compared to the DMSO control, (+)-pentazocine increased the intensity of the higher-order bands, while haloperidol and PD144418 decreased their intensity [28]. Strikingly, both the E102Q and E102A mutations abrogated any ligand-induced changes in band intensity (Fig. 4A,B).
Fig. 4.

E102 mutations abrogated drug response in σ1R oligomerization. (A-B) Representative western blots from five experiments are shown to visualize σ1R (red) and GAPDH (green) bands after drug treatment for E102Q (A) and E102A (B) constructs expressed in Δσ1R HEK293T cells. (C-D) Drug-induced changes in σ1R homomer BRET ratios are shown for the E102Q (C) and E102A (D) constructs expressed in Δσ1R HEK293T cells. None of (+)-pentazocine (red), PD144418 (cyan), or haloperidol (blue) induced significant changes in either of the mutants. Data represent mean ± S.E.M. (n = 5 or more). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
To verify this absence of pharmacological effects in cells expressing the E102 mutants and any possible concentration dependence, we utilized a BRET assay. As reported previously [22], for the WT fusion construct, a concentration-dependent increase in BRET activity was observed for haloperidol-like compounds (i.e., haloperidol and PD144418), while (+)-pentazocine failed to induce any change in the expression rescue approach with Δσ1R cells (Fig. S3). However, consistent to the western blot results, the E102Q and E102A mutations largely abrogated ligand effects on σ1R oligomerization (Fig. 4C,D).
To evaluate whether the lack of pharmacological response in both mutants is due to disrupted ligand binding, we studied the ligand binding affinity with radioligand binding assays. Using the expression rescue approach, specific binding activities of the mutants can be studied effectively. Both [3H](+)-pentazocine and [3H]haloperidol showed a significantly decreased Bmax in E102Q while the extent of decrease was even greater for E102A (Table S3), which may indicate that the mutants were not inserted or translocated to membranes properly. However, only moderate increases in the Kd values were observed for E102Q and E102A mutants for both classes of σ1R ligands (Table S3). The presence of significant binding activities indicates that the ligand binding site is largely intact, therefore the lack of pharmacological response in western blot and BRET is likely due to the disrupted propagation of ligand binding effects within σ1R.
3.5. The E102 mutations disrupt NT-helix dynamics that may be functionally important
By reconstructing the neighboring asymmetric units of the σ1R crystal structures, we found that in addition to the originally reported trimer interface, the NT-helices from six monomers cluster together (Fig. 5A). In this cluster, two helices are in inverted orientation from the other four, which is unlikely to happen under physiological conditions. While the overall structure of this cluster is an artifact of crystallography, it may contain oligomer interface(s) that are potentially functionally relevant – the four helices in the same orientation form two asymmetrical dimers, while the other two helices form a symmetrical dimer. It has been found that Trp, Tyr, and Arg are enriched in protein-protein interaction interfaces, and are called “hot spot” residues [29]. Interestingly, the presence of a large number of hot-spot residues in the NT-helix, i.e., in total two Arg and five Trp residues out of the first 30 residues of σ1R, suggests that this transmembrane helix has significant potential to form oligomer interface(s). In particular, four out of five Trp are involved in forming the symmetrical dimer interface, which includes very tightly packed residues 3–11 (Fig. 5B). In addition, one N-terminus from each of the asymmetric dimers interacts with another in a similar manner as the symmetric dimer. Such close associations of N-termini in dimers are consistent with our recent results showing that the modification of the N-terminus disrupts the changes of the oligomer states in response to different bound ligands [28].
Fig. 5.

The crystal structures of σ1R may include other oligomer interface(s). (A) In addition to the trimer interface reported previously [19], analysis of the asymmetric units in a representative crystal structure of σ1R (PDB ID 5HK1) shows that the NT-helices from 6 monomers cluster together (in the dotted box), with two of them (cyan) in inverted orientation from the other four (gray). Although overall this cluster of NT-helices is an artifact of crystallography, it may include other oligomeric interface(s) that are potentially functionally relevant, such as that in panel B. The backbone ribbons of three monomers in a trimer are colored in green, cyan, and magenta. (B) In this symmetric dimer interface (dotted box in panel A), three Trp and one Val residues from each monomer are tightly packed together at their N-termini, while several other residues face each other within an interacting range at the bottom of these two NT-helices. E102 interacts with the backbone amine groups in the loop connecting the NT-helix and C-terminal domain. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Because E102 makes extensive interactions with the loop connecting the NT-helix and C-terminal domain (Fig. 2), we hypothesized that E102 is at a critical position to propagate the impact of ligand binding to the NT-helix. Thus, to understand the potential mechanisms of how ligands with different pharmacological profiles may differentially affect the σ1R oligomerization and how the E102 mutations disrupt the oligomerization at the molecular level, we characterized the dynamics of the NT-helix by calculating the center-of-mass distributions of residues 8–11 along the lipid bilayer plane (see Materials and Methods, Fig. S4).
Interestingly, the N-termini in E102Q has significantly restricted dynamics in all simulated conditions, regardless of the bound ligands. To quantitatively characterize the altered N-terminus dynamics, we calculated the distribution for the same amount of simulation time for each condition (Fig. S4A,B). We found that in WT conditions the N-terminus can visit more area in the WT than in mutants, i.e., the most densely populated areas in mutants are denser than those in WT (Fig. S4C). Previously, we have observed a similar phenomenon in dopamine transporter where a mutation at a key position disrupted functionally important conformational dynamics and trapped the protein in a specific conformation [30]. Thus, we propose that the E102 mutations may cause σ1R to lose dynamics that are likely functionally important, i.e., disrupting the involvement of the N-terminus in oligomerization.
4. Discussion
The ALS causing mutation of σ1R, E102Q, is located at an “anchor” region between the NT-helix and C-terminal domain. This region is on the opposite side of the C-terminal domain that forms the previously reported trimer interface [19]. However, both of our western blot and BRET results indicate that the E102 mutations surprisingly and drastically impair the oligomerization of σ1R. On the other hand, even though many of the residues in the C-terminal domain with high centrality scores were found to be in close proximity to E102, our MD simulations of the E102 mutant constructs, followed by network analysis, showed that the C-terminal domain was minimally affected by the mutations, which is consistent with our radioligand binding results. As E102 and the trimer interface are separated by the ligand binding pocket, the relatively small changes in ligand binding affinities for the mutant constructs also suggest that the E102 mutations may have limited perturbations on the trimer interface. Instead, these mutations disrupt the interactions between E102 and the loop connecting the NT-helix and C-terminal domain, which confer rigidity of the NT-helix.
Based on these findings and our analysis of the asymmetric units in the crystal structures of σ1R, we propose that it is possible that NT-helix contributes to form an oligomer interface that is different from the trimer interface. Indeed, in our recent study, we found the fusion modifications on the N-terminus by the Myc tag or nanoluciferase disrupt the changes of the σ1R oligomerization state in response to different bound ligands, suggesting that the intact N-terminus structure and dynamics are necessary in forming higher-order oligomers [28]. Intriguingly, a missense mutation at the residue position 2 from glutamine to proline resulted in significantly disrupted sensory function in neuropathic pain patients [31]. Thus, the very end of N-terminus, even though in a flexible extended conformation in the crystal structures, is functionally relevant and may be potentially involved in oligomerization (Fig. S5). In addition, the capability of the NT-helix to form dimers is not only consistent with the conclusions deduced from our in vitro investigations of the E102 mutations, but can also be easily reconciled with the commonly observed dimer formations in previous biochemical studies of σ1R [16], [17], [32]. However, the NT-helix and the trimer interface may coordinate to form higher-order oligomers.
Together, it is tempting to speculate that the destabilization of the “anchor” region by the E102 mutations leads to disrupted propagation of conformational changes from the ligand binding pocket in the C-terminal domain to the N-terminus on the other side of the membrane. Our results provide the first account of the molecular mechanism of the σ1R dysfunction induced by E102Q (and E102A) mutations and characterize the general importance of this “anchor” region in the receptor’s function. Indeed, as many σ1R’s client proteins are membrane proteins, the revealed functional significance of the “anchor” region, which is closely associated with membrane and the transmembrane segment of σ1R, provides an important context for future mechanistic studies of the interactions between σ1R and its client proteins.
Author contribution
A.M.A., M.X., H.Y., and L.S. designed the study. A.M.A, M.X., A.D.F., and L.S. carried out computational modeling, simulations and analysis, L.L., S.N., and H.Y. performed the experiments. All authors took part in interpreting the results. A.M.A., H.Y., A.D.F., and L.S. wrote the manuscript.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
We would like to thank Drs. Yoki Nakamura and Tsung-Ping Su at NIDA for collaborating with us to develop σ1R KO HEK cells used in this assay and sharing their image detection instrument (LI-COR). Support for this research was provided by the National Institute on Drug Abuse–Intramural Research Program, Z1A DA000606-03 (L.S.). This work utilized the computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2019.12.012.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hayashi T., Su T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca(2+) signaling and cell survival. Cell. 2007;131(3):596–610. doi: 10.1016/j.cell.2007.08.036. [DOI] [PubMed] [Google Scholar]
- 2.Su T.P. The Sigma-1 receptor as a pluripotent modulator in living systems. Trends Pharmacol Sci. 2016;37(4):262–278. doi: 10.1016/j.tips.2016.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stone J.M. [123I]TPCNE–a novel SPET tracer for the sigma-1 receptor: first human studies and in vivo haloperidol challenge. Synapse. 2006;60(2):109–117. doi: 10.1002/syn.20281. [DOI] [PubMed] [Google Scholar]
- 4.Katz J.L. A role for sigma receptors in stimulant self-administration and addiction. Behav Pharmacol. 2016;27(2-3 Spec Issue):100–115. doi: 10.1097/FBP.0000000000000209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hashimoto K. Sigma-1 receptor chaperone and brain-derived neurotrophic factor: emerging links between cardiovascular disease and depression. Prog Neurobiol. 2013;100:15–29. doi: 10.1016/j.pneurobio.2012.09.001. [DOI] [PubMed] [Google Scholar]
- 6.Nguyen L. Sigma-1 receptors and neurodegenerative diseases: towards a hypothesis of sigma-1 receptors as amplifiers of neurodegeneration and neuroprotection. Adv Exp Med Biol. 2017;964:133–152. doi: 10.1007/978-3-319-50174-1_10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pal A. The sigma-1 receptor protects against cellular oxidative stress and activates antioxidant response elements. Eur J Pharmacol. 2012;682(1–3):12–20. doi: 10.1016/j.ejphar.2012.01.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sabino V. Sigma-1 receptor knockout mice display a depressive-like phenotype. Behav Brain Res. 2009;198(2):472–476. doi: 10.1016/j.bbr.2008.11.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mavlyutov T.A. The sigma-1 receptor is enriched in postsynaptic sites of C-terminals in mouse motoneurons. An anatomical and behavioral study. Neuroscience. 2010;167(2):247–255. doi: 10.1016/j.neuroscience.2010.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Luty A.A. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann Neurol. 2010;68(5):639–649. doi: 10.1002/ana.22274. [DOI] [PubMed] [Google Scholar]
- 11.Li X. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology. 2015;84(24):2430–2437. doi: 10.1212/WNL.0000000000001680. [DOI] [PubMed] [Google Scholar]
- 12.Watanabe S. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol Med. 2016;8(12):1421–1437. doi: 10.15252/emmm.201606403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Saif A., Al-Mohanna F., Bohlega S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann Neurol. 2011;70(6):913–919. doi: 10.1002/ana.22534. [DOI] [PubMed] [Google Scholar]
- 14.Wong A.Y. Aberrant subcellular dynamics of sigma-1 receptor mutants underlying neuromuscular diseases. Mol Pharmacol. 2016;90(3):238–253. doi: 10.1124/mol.116.104018. [DOI] [PubMed] [Google Scholar]
- 15.Dreser A. The ALS-linked E102Q mutation in Sigma receptor-1 leads to ER stress-mediated defects in protein homeostasis and dysregulation of RNA-binding proteins. Cell Death Differ. 2017;24(10):1655–1671. doi: 10.1038/cdd.2017.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mishra A.K. The sigma-1 receptors are present in monomeric and oligomeric forms in living cells in the presence and absence of ligands. Biochem J. 2015;466(2):263–271. doi: 10.1042/BJ20141321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gromek K.A. The oligomeric states of the purified sigma-1 receptor are stabilized by ligands. J Biol Chem. 2014;289(29):20333–20344. doi: 10.1074/jbc.M113.537993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Russ W.P., Engelman D.M. The GxxxG motif: a framework for transmembrane helix-helix association. J Mol Biol. 2000;296(3):911–919. doi: 10.1006/jmbi.1999.3489. [DOI] [PubMed] [Google Scholar]
- 19.Schmidt H.R. Crystal structure of the human sigma1 receptor. Nature. 2016;532(7600):527–530. doi: 10.1038/nature17391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Schmidt H.R. Structural basis for sigma1 receptor ligand recognition. Nat Struct Mol Biol. 2018;25(10):981–987. doi: 10.1038/s41594-018-0137-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Romieu P. Involvement of the sigma(1) receptor in cocaine-induced conditioned place preference: possible dependence on dopamine uptake blockade. Neuropsychopharmacology. 2002;26(4):444–455. doi: 10.1016/S0893-133X(01)00391-8. [DOI] [PubMed] [Google Scholar]
- 22.Yano H. Pharmacological profiling of sigma 1 receptor ligands by novel receptor homomer assays. Neuropharmacology. 2018;133:264–275. doi: 10.1016/j.neuropharm.2018.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Doshi U. Dynamical network of residue-residue contacts reveals coupled allosteric effects in recognition, catalysis, and mutation. Proc Natl Acad Sci USA. 2016;113(17):4735–4740. doi: 10.1073/pnas.1523573113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Csardi G., Nepusz T. The igraph software package for complex network research. InterJournal, Complex Syst. 2006;1695(5):1–9. [Google Scholar]
- 25.McGibbon R.T. MDTraj: a modern open library for the analysis of molecular dynamics trajectories. Biophys J. 2015;109(8):1528–1532. doi: 10.1016/j.bpj.2015.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Urizar E. CODA-RET reveals functional selectivity as a result of GPCR heteromerization. Nat Chem Biol. 2011;7(9):624–630. doi: 10.1038/nchembio.623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.O'Rourke K.F., Gorman S.D., Boehr D.D. Biophysical and computational methods to analyze amino acid interaction networks in proteins. Comput Struct Biotechnol J. 2016;14:245–251. doi: 10.1016/j.csbj.2016.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yano H. The effects of terminal tagging on homomeric interactions of the sigma 1 receptor. Front Neurosci. 2019;13:1356. doi: 10.3389/fnins.2019.01356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bogan A.A., Thorn K.S. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280(1):1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- 30.Abramyan A.M. The isomeric preference of an atypical dopamine transporter inhibitor contributes to its selection of the transporter conformation. ACS Chem Neurosci. 2017;8(8):1735–1746. doi: 10.1021/acschemneuro.7b00094. [DOI] [PubMed] [Google Scholar]
- 31.Sachau J. SIGMA-1 receptor gene variants affect the somatosensory phenotype in neuropathic pain patients. J Pain. 2019;20(2):201–214. doi: 10.1016/j.jpain.2018.08.011. [DOI] [PubMed] [Google Scholar]
- 32.Hong W.C. The sigma-1 receptor modulates dopamine transporter conformation and cocaine binding and may thereby potentiate cocaine self-administration in rats. J Biol Chem. 2017;292(27):11250–11261. doi: 10.1074/jbc.M116.774075. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.