

Summary
Chronic exposure of pancreatic β-cells to excess glucose can lead to metabolic acceleration and loss of stimulus-secretion coupling. Here, we examined how exposure to excess glucose (defined here as concentrations above 5 mM) affects mTORC1 signaling and the metabolism of β-cells. Acute exposure to excess glucose stimulated glycolysis-dependent mTORC1 signaling, without changes in the PI3K or AMPK pathways. Prolonged exposure to excess glucose led to hyperactivation of mTORC1 and metabolic acceleration, characterized by higher basal respiration and maximal respiratory capacity, increased energy demand, and enhanced flux through mitochondrial pyruvate metabolism. Inhibition of pyruvate transport to the mitochondria decelerated the metabolism of β-cells chronically exposed to excess glucose and re-established glucose-dependent mTORC1 signaling, disrupting a positive feedback loop for mTORC1 hyperactivation. mTOR inhibition had positive and negative impacts on various metabolic pathways and insulin secretion, demonstrating a role for mTOR signaling in the long-term metabolic adaptation of β-cells to excess glucose.
Subject Areas: Molecular Mechanism of Behavior, Endocrinology, Diabetology, Specialized Functions of Cells
Graphical Abstract
Highlights
-
•
Acute glucose stimulates mTORC1 in β-cells through a glycolysis-dependent mechanism
-
•
Chronic excess glucose promotes mTOR-dependent metabolic acceleration of β-cells
-
•
Metabolic acceleration activates a positive feedback loop for mTORC1 hyperactivation
-
•
mTOR hyperactivation disturbs the metabolism and insulin secretion patterns of β-cells
Molecular Mechanism of Behavior; Endocrinology; Diabetology; Specialized Functions of Cells
Introduction
A tight stimulus-secretion coupling is critical for healthy β-cells to establish glucose homeostasis (Prentki et al., 2013). Obese and/or pre-diabetic patients often present with increased fasting plasma insulin levels and decreased post-prandial insulin secretion, which indicates loss of stimulus-secretion coupling in their β-cells (Corkey, 2012, Pories and Dohm, 2012).
Proper stimulus-secretion coupling in β-cells is attributed to metabolic coupling factors that are generated during the catabolism of glucose and other nutrients (Prentki et al., 2013). Changes in the ATP/ADP ratio, and subsequent closing of ATP-sensitive potassium channels, is one of the metabolic coupling factors that have been identified in β-cells, but other factors also contribute to insulin secretion at various levels of the process, from sensing and amplification of the nutrient signal to triggering insulin granule exocytosis (Lamontagne et al., 2017, Mugabo et al., 2017). Environmental factors that disturb any of these metabolic factors could potentially promote loss of stimulus-secretion coupling (Nolan and Prentki, 2008).
Sugar and fat overload have been linked to loss of stimulus-secretion coupling (Deeney et al., 2000, Erion et al., 2015, Irles et al., 2015, Li et al., 2006); however, the molecular mechanisms or metabolic coupling factors that contribute to increased basal and decreased stimulus-induced insulin secretion are poorly understood. Here, we set up to test the hypothesis that hyperactivation of the nutrient-sensing kinase mTORC1 (mechanistic target of rapamycin complex 1) in β-cells in response to exposure to excess glucose results in metabolic reprogramming and disruption of stimulus-secretion coupling.
mTORC1 signaling is activated in several tissues, either acutely following nutrient stimulation or chronically, as a consequence of obesity (Gonzalez and Hall, 2017). The direct effect of chronic activation of mTORC1 in β-cells was addressed using mice with tissue-specific knockout (KO) of the tuberous sclerosis complex Tsc1/Tsc2 (which are negative regulators of mTORC1). These mice developed progressive hyperinsulinemia, with increased islet β-cell mass (Mori et al., 2009, Rachdi et al., 2008, Shigeyama et al., 2008) and increased mitochondrial biogenesis (Koyanagi et al., 2011). These studies elegantly illustrate the role of mTORC1 in the control of β-cell proliferation, which is an important positive adaptation to compensate for increased insulin demand in response to hyper-caloric diets or obesity-induced insulin resistance. However, these mice also presented with increased fasting insulin levels, a sign of loss of stimulus-secretion coupling that cannot be explained by increased β-cell mass or number of mitochondria, as previously suggested. In one study, β-cell-specific Tsc2 KO mice eventually developed β-cell failure and apoptosis, which culminated in hyperglycemia and diabetes, revealing that prolonged mTORC1 signaling in β-cells can elicit maladaptive responses (Shigeyama et al., 2008). In the present study, we examined how transient exposure of β-cells to excess glucose under well-controlled culture conditions resulted in mTORC1 hyperactivation and mTOR-dependent metabolic reprogramming of β-cells. Unlike most studies looking at the effects of glucose on mTORC1 signaling, which typically involve complete glucose-starvation, we used concentrations of glucose that ranged from normoglycemic (5 mM) to post-prandial levels (11 mM), to completely avoid energy deprivation or glucotoxicity. This approach revealed a mode for acute mTORC1 activation that did not require changes in phosphoinositide 3-kinase (PI3K) or AMP-dependent kinase (AMPK) signaling. Instead, glucose-induced mTORC1 activation was glycolysis dependent and mitochondrial pyruvate metabolism independent. Prolonged mTORC1 activation by excess glucose promoted metabolic acceleration of β-cells, which was characterized by increase in energy demand and flux through mitochondrial pyruvate metabolism. Increased mitochondrial pyruvate metabolism in turn stimulated basal mTORC1 signaling, generating a positive feedback loop for mTORC1 hyperactivation. Furthermore, mTOR inhibition positively and negatively impacted different metabolic pathways and insulin secretion, demonstrating that mTORC1 signaling plays a complex role in the metabolic adaptation of β-cells to excess glucose.
Results
Excess-Glucose Withdrawal Reveals that mTORC1 Signaling in β-Cells Is Glucose Dose Dependent
Insulinoma cells are typically cultured in medium containing 11 mM glucose, which is above the normoglycemic levels in the blood. Previous studies have shown that INS-1 cells chronically cultured under 11 mM glucose have excess fuel, which is evidenced by the accumulation of lipids in the form of lipid droplets (Erion et al., 2015). In contrast, INS-1 cells chronically cultured under 5 mM glucose have fewer lipid droplets and a glucose dose-response curve for insulin secretion that more closely resembles the physiological response, with low secretion at sub-stimulatory glucose concentrations (below 5 mM) and high secretion at stimulatory glucose concentrations (above 8 mM), as compared with cells cultured in 11 mM glucose (Erion et al., 2015). To better understand how culturing INS-1 cells in 11 mM glucose affects nutrient-sensing signaling pathways, we transferred the cells to 5 mM glucose and examined how mTORC1 activity changes over time, as determined by measurements of phospho-S6 and phospho-p70S6K (see Figure 1 A for experimental design). From here on, we will refer to 5 mM glucose as physiological concentration, whereas 11 mM will be referred to as excess glucose, although we acknowledge that this comparison may not be completely appropriate, given that INS-1 are transformed tumoral cells. In cells cultured in physiological glucose for 2 h, basal mTORC1 signaling was elevated and did not significantly change in response to acute glucose stimulation, whereas in cells cultured in physiological glucose for 24 h, basal mTORC1 signaling decreased and sensitivity to glucose improved, with a small but significant increase after acute stimulation with 11 mM glucose (Figure 1B). Remarkably, after 48 h in physiological glucose, mTORC1 signaling became highly sensitive to small changes in glucose, with a significant increase from 5 to 7 mM glucose and a 2-fold increase from 5 to 11 mM (Figure 1B). Thus, excess-glucose withdrawal from the culture media improved glucose sensitivity for mTORC1 activation, with decreased signaling under basal levels of glucose (5 mM) and an increased response to stimulatory glucose (11 mM).
Figure 1.

Glucose Stimulates mTORC1 Signaling through Glycolysis Only in β-Cells Cultured in Low Glucose
(A and B) INS-1 cells cultured in excess glucose were transferred to medium containing physiological glucose for 2, 24, or 48 h. Cells were then transferred to serum-free media and stimulated with the indicated glucose dose for 1 h. (A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for (B). (B) Bar graphs and representative western blot (WB) showing glucose dose-dependent activation of mTORC1 signaling after excess glucose withdrawal for 2 (black bars), 24 (dark gray bars), or 48 h (light gray bars).
(C–E) INS-1 cells chronically cultured in physiological glucose were transferred to serum-free media and stimulated with the indicated glucose dose for 1 h, with or without inhibitor. (C) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for Figure 1 (D–E) and Figure 2. (D–E) Bar graphs and representative western blots showing glucose dose-dependent activation of mTORC1 signaling in the presence of DMSO or UK5099 (D) or glucosamine (E). Bar graphs show the average and standard error of at least three separate experiments in which the levels of phospho-S6 were quantified, corrected for loading, and normalized against the 11 mM glucose data point. Statistical significance was calculated by Student's t test. # indicate p values above 0.05, #* indicates p = 0.057, * indicate 0.05 > p > 0.02, ** indicate 0.02 > p > 0.01, and *** indicate 0.01 > p > 0.001.
Glucose Stimulates mTORC1 Signaling through Glycolysis
The mechanisms by which glucose activates mTORC1 are not completely understood and thought to involve changes in energy status (Dibble and Manning, 2013, Herzig and Shaw, 2018). We used INS-1 cells that had been cultured for several days under physiological glucose to investigate whether acute glucose stimulation of mTORC1 depends on glucose catalysis through glycolysis and/or mitochondrial pyruvate metabolism (see Figure 1C for experimental design). Treatment with UK5099, an inhibitor of the mitochondrial pyruvate carrier MPC-1 (Hildyard et al., 2005), reduced basal mTORC1 signaling but did not prevent the glucose dose-dependent stimulation of mTORC1 (Figure 1D). In contrast, treatment with 10 mM glucosamine, which is a competitive inhibitor of hexokinases, completely blocked glucose-dependent mTORC1 signaling (Figures 1E and S1A). To confirm these results, we used freshly isolated mouse islets that were cultured in 6 mM glucose overnight (6 mM glucose was chosen to mimic the normoglycemic levels in mice). Figure 2A shows glucose-dependent mTORC1 stimulation in mouse islets, as measured by phosphorylation of p70S6K and S6 (please note that, despite the low signal-to-noise ratio of the bands for total S6, their levels largely reflected the levels of tubulin, which was used as a loading control). As for INS-1 cells, glucose-dependent stimulation of mTORC1 signaling in islets was sensitive to glucosamine and insensitive to UK5099 (Figure 2A). Glucokinase is the main hexokinase expressed in β-cells and is considered a glucose-sensor owing to its high Km for glucose. The glucose dose-response curve shown in Figure 1D suggested that glucokinase may be involved in the activation of mTORC1 signaling in β-cells. Unlike other hexokinases, glucokinase is insensitive to inhibition by 2-deoxyglucose (2-DG). This is because 2-DG is phosphorylated into 2-deoxyglucose-6-phosphate, which cannot be fully metabolized and accumulates in cells, inhibiting hexokinase. Since glucokinase is not inhibited by glucose-6-phosphate (Matschinsky, 2002), it is insensitive to 2-DG. Interestingly, glucose-dependent mTORC1 activation was not inhibited by 2-DG (Figure S1B). These results are consistent with a role for glucokinase on glucose-dependent mTORC1 activation.
Figure 2.

Up-Regulation of mTORC1 by Glucose Does Not Involve Changes in Insulin/PI3K/Akt or AMPK Signaling
INS-1 cells or freshly isolated mouse islets were chronically cultured in physiological glucose, transferred to serum-free media, and stimulated with the 11 mM glucose (12 mM in the case of islets) for 1 h, with or without inhibitor, as depicted in Figure 1C.
(A and B) Representative western blots showing the effects of acute glucose stimulation on dispersed mouse islets (A) or INS-1 cells (B) treated with or without inhibitors, as indicated. mTORC1 was measured by phosphorylation of p70S6K and S6. PI3K signaling was measured by phosphorylation of Akt. AMPK signaling was measured by phosphorylation of AMPK/Raptor.
(C–E) Glucose-induced mTORC1 signaling, as measured by S6 phosphorylation, in the absence or presence of the mTOR inhibitor, KU, or the PI3K inhibitor, Ly (C); the Akt inhibitor, MK2206 (D); the inhibitor of glucose-stimulated insulin secretion, DZ (E).
(F) Glucose-induced mTORC1 signaling, as measured by S6 phosphorylation, in the absence or presence of exogenous insulin (100 nM). Tables underneath the blots indicate the quantification of the bands (arbitrary units). Bar graphs shown are the average and standard error of at least three separate experiments in which the levels of phospho-S6 were quantified, corrected for loading, and normalized against the 11 mM glucose data point. Statistical significance was calculated by Student's t test. # indicate p values above 0.05, * indicate 0.05 > p > 0.02, ** indicate 0.02 > p > 0.01, and *** indicate 0.01 > p > 0.001.
It is important to point out that glucose stimulation of mTORC1 signaling was observed in cells growing in media containing supra-physiological levels of all amino acids. Therefore, it is unlikely that glucose increased mTORC1 signaling through the biosynthesis of non-essential amino acids. Furthermore, under amino acid deprivation, mTORC1 signaling was dramatically reduced, as expected, and glucose treatment had no effect (Figure S1C), indicating that glucose-derived amino acids were insufficient to activate mTORC1. Together, these results indicate that glucose stimulated mTORC1 signaling through glycolysis and independently of pyruvate oxidative phosphorylation or amino acid biosynthesis.
Glucose Up-Regulates mTORC1 Signaling Independently of Changes in Growth-Factor or Energy-Sensing Pathways
Growth factors activate mTORC1 through PI3K/Akt signaling (Gonzalez and Hall, 2017, Saxton and Sabatini, 2017). Therefore, we examined the contribution of this pathway to the glucose-induced mTORC1 stimulation (see Figure 1C for experimental design). In INS-1 cells and in freshly isolated mouse islets cultured under physiological glucose, phospho-Akt levels were low and, in the case of INS-1, at the threshold of detection (Figures 2A and 2B). Acute exposure to 11 mM glucose stimulated a 2.5-fold increase in phospho-S6 in INS-1 cells (Figures 2B–2F). In mouse islets, 12 mM glucose induced a 5- to 16-fold increase in phospho-S6 (Figures 2A and S6B). These were due to increased mTORC1 signaling, as confirmed by measurements of the mobility shift of p70S6K (Figure S2A) and phosphorylation of p70S6K (Figures 2A and S2B) and 4EBP (Figure S2B), which are direct substrates of mTORC1. Surprisingly, acute exposure to 11–12 mM glucose did not increase phosphorylation of Akt (Figures 2A, 2B, S2B, and S2C) or its substrate GSK3 (Figure S2B), suggesting that glucose simulates mTORC1 without changes in PI3K signaling or mTORC2 activity. Inhibition of mTOR with KU0063794 (KU) or Ly294002 (Ly), which can inhibit mTOR directly or through PI3K inhibition, completely abolished mTORC1 signaling, as expected (Figures 2B, 2C, and S2B). Inhibition of Akt with the pan-inhibitor MK2206 reduced the levels of both basal and glucose-stimulated mTORC1, but it did not prevent the glucose-induced stimulation of mTORC1 signaling in freshly isolated islets or INS-1 (Figure 2A, 2B, and 2D). These results indicated that glucose-dependent stimulation of mTORC1 in β-cells does not involve further activation of Akt.
Next, we examined the role of AMPK signaling in the stimulation of mTORC1 by glucose. As a control, INS-1 cells were treated with oligomycin, an inhibitor of ATP synthase, to stop mitochondrial ATP generation. This confirmed that under energy deprivation pAMPK levels increased (indicating activation of AMPK) and mTORC1 signaling decreased, as expected (Figure 2B). In contrast, acute stimulation with 11 mM glucose did not decrease pAMPK levels (Figures 2B and S2D) or phosphorylation of the AMPK substrates Raptor (pS792) and Tsc2 (pT1462) (Figures 2B and S2B), indicating that changes in energy status cannot explain the effect of glucose on mTORC1. These results confirmed that under the conditions used (media containing 5 mM glucose and amino acids and supplemented with 1 mM sodium pyruvate) cells had sufficient nutrients to maintain a positive energy balance.
To test whether the effect of acute exposure to glucose on mTORC1 signaling could be explained by an increase in insulin secreted into the media, we treated INS-1 cells and freshly isolated islets with diazoxide (DZ), an activator of the ATP-dependent potassium channel, to inhibit glucose-stimulated insulin secretion (GSIS). DZ treatment during acute glucose stimulation did not prevent the increase in mTORC1 signaling by 11–12 mM glucose (Figures 2A, 2B, and 2E), demonstrating that autocrine insulin stimulation cannot explain the effects of glucose on mTORC1. This was further confirmed by the addition of exogenous insulin to the media during acute glucose treatment, which stimulated Akt and S6 phosphorylation (Figure 2B), likely through activation of the insulin and/or IGF1 receptors (Rhodes et al., 2013). Remarkably, exogenous insulin synergized with glucose to stimulate mTORC1 (Figures 2B and 2F), suggesting that glucose and insulin activate mTORC1 through independent pathways. Together, these results demonstrate that glucose upregulates mTORC1 independently of changes in insulin secretion, PI3K, or AMPK signaling (energy status).
Prolonged Exposure to Excess Glucose Results in Sustained Activation of mTORC1 Signaling and Metabolic Acceleration
Prolonged exposure of INS-1 cells to excess glucose was previously shown to increase mitochondrial metabolism, as measured by oxygen consumption rate (OCR) (Erion et al., 2015). To understand the role of mTORC1 signaling in the metabolic adaptation of β-cells to prolonged excess glucose, we performed bioenergetic studies in INS-1 cells that were cultured in 5 mM glucose and transiently exposed to 11 mM glucose for 2–24 h. These cells had a gradual and sustained increase in mTORC1 signaling, as measured by phosphorylation of p70S6K, S6, and 4EBP (Figures S3A–S3C), without increases in the levels of the ER stress marker Grp78 (Figure S3A). To measure basal metabolic rate, cells exposed to excess glucose or kept at physiological glucose were transferred to media containing 3 mM glucose and subjected to a mitochondrial stress test (see Figure 3A for experimental design). INS-1 cells exposed to excess glucose had a gradual increase in basal respiration as compared with control cells (Figures 3B and 3C). Since this increase in respiration was maintained even after the excess glucose was removed, we refer to it as metabolic acceleration. Pre-exposure to excess glucose for 2 h was not sufficient to induce changes in respiration, whereas 4-h exposure resulted in a small but significant increase in basal OCR (Figure 3C). Thus, metabolic acceleration is not an acute response, but rather a delayed and gradual adaptation to excess glucose. Increased basal respiration after 20–24 h of excess glucose exposure was due to a substantial increase in ATP-coupled respiration (Figure S4A) and proton leak (Figure S4C), and a small but significant increase in mitochondrial inefficiency, which is measured by the percentage increase in mitochondrial leak over ATP-coupled respiration (Figure S4D). Prolonged exposure to excess glucose also increased the maximal respiratory capacity of INS-1 cells (Figure S4B).
Figure 3.

Exposure to Excess Glucose Accelerates the Metabolism of β-Cells Gradually and Dose-Dependently owing to mTOR Signaling and Increased Energy Demand
INS-1 cells were pre-exposed to excess glucose (7–11 mM) for 2–24 h or kept in physiological glucose and then transferred to complete serum-free Seahorse base-media containing 3 mM glucose for 2 h, after which OCR was measured.
(A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for (B)–(H).
(B) OCR trace along a mitochondrial stress test of cells at 3 mM glucose that had been pre-exposed to excess glucose (11 mM) for the time indicated. Data shown are the average and standard error of four to six replicates and is representative of more than three separate experiments.
(C) Calculated basal mitochondrial OCR (at 3 mM glucose) from cells that were pre-exposed to excess glucose (11 mM) for 2–24 h or kept in physiological glucose (time 0); data are the average and standard error from four to eight experiments with three to six replicates each.
(D) Calculated basal mitochondrial OCR (at 3 mM glucose) from cells that were kept in physiological glucose (5 mM) or pre-exposed to excess glucose (7–11 mM) for 20 h; data shown are the average and standard error of six replicates and is representative of three separate experiments.
(E and F) OCR corresponding to ATP-coupled respiration (E) or maximal respiratory capacity (F) from INS-1 cells, at 3 mM glucose, which had been pre-exposed to excess glucose (dark gray bars) for 20 h or kept in physiological glucose (light gray bars), in the presence of KU (hatched bars) or DMSO (solid bars). Results shown are the average and standard error from three separate experiments with four to six replicates each.
(G and H) OCR corresponding to ATP-coupled respiration (G) or maximal respiratory capacity (H) from INS-1 cells at 3 mM glucose, which had been pre-exposed to excess glucose (dark gray bars) for 20 h or kept in physiological glucose (light gray bars) and treated with cycloheximide (hatched bars) or vehicle (solid bars). Data shown are the average and standard error calculated from four to six replicates and is representative of five separate experiments. # indicate no statistical significance, * indicate 0.05 < p < 0.02, ** indicate 0.02 < p < 0.01, and *** indicate 0.01 < p < 0.003, as determine by Student's t tests. Arrows indicate the times when oligomycin (O), FCCP (f) or antimycin A (AA) were introduced into the cell media.
Next, we exposed INS-1 cells to various doses of glucose for 20 h and measured the effect on basal respiration. Glucose pre-treatment promoted the metabolic acceleration of INS-1 cells in a dose-dependent manner, with significant changes with 2-mM increments (Figures 3D and S4E–S4G), similar to its effects on mTORC1 signaling (Figure 1D). The correlation between excess glucose-induced increase in mTORC1 signaling and in respiration indicates these may have a cause-and-effect relationship.
To demonstrate that mTOR is necessary for the metabolic acceleration of β-cells, we treated cells with KU during exposure to excess glucose. KU treatment inhibited mTORC1 signaling in INS-1 cells kept in physiological glucose or exposed to excess glucose (Figures S3A and S3C) and reduced ATP-coupled respiration and maximal respiratory capacity (Figures 3E and 3F). The basal OCR of cells treated with excess glucose in the presence of KU was similar to the OCR of cells kept under physiological glucose (Figure S5A). Thus, mTOR inhibition prevented the metabolic acceleration by excess glucose. KU treatment decreased ATP-coupled respiration (Figure 3E) and proton leak (Figure S5B) without significantly affecting the efficiency of the mitochondria (Figure S5C). It is important to note that KU was only present at the time of pre-treatment and not at the time of OCR measurements (see Figure 3A), which suggests that the effect of mTOR inhibition on respiration is long-lasting. In fact, acute inhibition of mTOR during OCR measurements had no immediate effect on cellular respiration (Figure S5D). Thus, mTOR activity is necessary for the long-term metabolic adaptation of INS-1 cells to excess glucose.
The effect of KU on ATP-coupled respiration indicated that mTOR inhibition decreased energy demand in INS-1 cells. Since protein translation is an mTORC1-regulated process that is highly energy demanding (Nicholls, 2016), we examined the effects of cycloheximide, an inhibitor of protein translation, on respiration. Cycloheximide treatment immediately and significantly decreased ATP-coupled respiration of INS-1 cells pre-treated with excess glucose (49%–50% inhibition) or kept in physiological glucose (41%–44% inhibition) and reduced the difference between these two experimental groups (Figures 3G and S5E). Importantly, acute cycloheximide treatment did not significantly decrease maximum respiratory capacity (Figures 3H and S5E), demonstrating that decreased respiration is not due to depletion of enzymes in the metabolic machinery. Together these results show that cells exposed to excess glucose have higher metabolic demand, which is largely due to increased protein translation. Furthermore, inhibiting mTOR activity during glucose exposure completely prevented the metabolic acceleration by excess glucose, which could be due to mTORC1 and/or mTORC2 signaling. Given that excess glucose stimulated mTORC1 signaling without significant changes in mTORC2 (as determined by measurements of pAkt, see above), we suspect that glucose-induced metabolic reprogramming and increased protein translation is mostly regulated by mTORC1 signaling.
Prolonged Exposure to Excess Glucose Results in Enhanced Flux through Pyruvate Metabolism
To examine how prolonged exposure to excess glucose affected fuel utilization and preference, we treated INS-1 cells with UK5099 or etomoxir to inhibit the mitochondrial transport of pyruvate or fatty acids, respectively (see Figure 4A for experimental design). Inhibition of pyruvate transport to the mitochondria with UK5099 significantly reduced ATP-coupled respiration (Figures 4B and 4C) and maximal respiratory capacity (Figures 4B and 4D) of cells pre-exposed to excess glucose. ATP-coupled respiration in cells kept at physiological glucose was also inhibited by UK5099, although to a lesser extent. In contrast, treatment with etomoxir did not decrease respiration of INS-1 cells, whether they were pre-exposed to excess glucose or not (Figures 4B–4D), demonstrating that these cells do not utilize fatty acids as a main source of fuel. These results strongly indicate that pyruvate is the preferred mitochondrial fuel for INS-1 cells and that pyruvate utilization is enhanced after exposure to excess glucose.
Figure 4.

Exposure to Excess Glucose Enhances Mitochondrial Pyruvate Metabolism
(A–F) INS-1 cells were kept in physiological glucose or pre-exposed to excess glucose for 20–24 h, then transferred to complete serum-free Seahorse base-media containing 3 mM glucose for 2 h, after which OCR was measured. (A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for (B)–(F). (B) OCR trace along a mitochondrial stress test at 3 mM glucose with INS-1 cells that were either pre-exposed to excess glucose for 20 h (dark gray squares, triangles, and exes) or kept in physiological glucose (light gray circles, diamonds, and stars). Vehicle (circles and squares), etomoxir (stars and exes), or UK5099 (diamonds and triangles) was injected into the media in between measurements, as indicated by the arrow. Arrows also indicate when oligomycin (O), FCCP (f), and antimycin A (AA) were introduced into the cell media. Data shown are the average and standard error of four to six replicates. (C and D) Calculated OCR corresponding to ATP-coupled respiration (C) or maximal respiratory capacity (D) from the experiment shown in (B). Data shown are the average and standard error from four to six replicates, and is representative of at least three separate experiments. (E) Calculated OCR corresponding to the ATP-coupled respiration of INS-1 cells at 3 mM glucose, which was acutely treated at the time of OCR measurements with various sub-optimum concentrations of UK5099, as indicated. (F) Calculated OCR corresponding to the ATP-coupled respiration of INS-1 cells at 3 mM glucose measured with sodium pyruvate (hatched bars) or without (solid bars) in the media. Cells that had been pre-exposed to excess glucose are shown in dark gray bars, whereas cells that were kept in physiological glucose are shown in light gray bars. (E and F) Data shown are the average and standard error of three separate experiments, with four to six replicates each. # indicate no statistical significance, * indicate 0.05 < p < 0.02, ** indicate 0.02 < p < 0.01, and *** indicate 0.01 < p < 0.003, as determined by Student's t tests.
Next, we examined whether partial inhibition of mitochondrial pyruvate metabolism can reverse the metabolic acceleration of cells exposed to excess glucose. UK5099 dose-dependently inhibited ATP-coupled respiration of INS-1 cells starting at 0.02 μM (100-fold dilution from the suggested dose) (Figure 4E). UK5099 treatment at 0.06 μM completely suppressed the increase in ATP-coupled respiration in response to excess glucose (compare light bars with dark bars). These results demonstrate that mitochondrial pyruvate metabolism is a rate-limiting step in the metabolic acceleration by excess glucose. Furthermore, removal of sodium pyruvate from the cell media right before measurements of OCR decelerated the metabolism of cells exposed to excess glucose but did not affect cells kept at physiological glucose (Figure 4F). These results confirm that mitochondrial pyruvate metabolism is accelerated in cells pre-exposed to excess glucose, possibly due to increased energy demand.
Enhanced Mitochondrial Pyruvate Metabolism Results in a Positive Feedback Loop for mTORC1 Hyperactivation
Next, we examined whether enhanced flux through mitochondrial pyruvate metabolism can explain the elevated basal mTORC1 signaling in cells chronically cultured in excess glucose, as shown in Figure 1. INS-1 cells cultured in physiological glucose were transiently exposed to 11 mM glucose for 20 h, returned to 5 mM glucose for 2 h, and analyzed for glucose-dependent mTORC1 signaling in the presence or absence of UK5099 (see Figure 5A for experimental design). In cells kept under physiological glucose, basal mTORC1 signaling was low and increased upon acute stimulation with 2 mM glucose increments (Figure 5B left panel, and C light bars). In contrast, cells pre-exposed to excess glucose had high basal mTORC1 signaling and loss of glucose dose-dependent stimulation of mTORC1 (Figure 5B, right panel and C, dark bars). In freshly isolated mouse and human islets, basal mTORC1 signaling was also elevated after pre-treatment with excess glucose and fold glucose stimulation of mTORC1 was blunted (Figure S6).
Figure 5.

Exposure to Excess Glucose Results in mTORC1 Hyperactivation owing to Enhanced Mitochondrial Pyruvate Metabolism
(A–E) INS-1 cells were kept in physiological glucose or transiently exposed to excess glucose for 20–24 h. Cells were then returned to 5 mM glucose in serum-free media and stimulated with the indicated glucose dose for 1 h (acute stimulation). (A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for (B)–(E). (B) Representative western blot showing glucose dose-dependent activation of mTORC1 signaling, as measured by S6 phosphorylation, in cells kept in physiological glucose (left lanes) or exposed to excess glucose (right lanes) and stimulated with glucose in the presence or absence of pyruvate or UK5099, as indicated. Blot shown is representative of four separate experiments. (C–E) Bar graphs showing the average and standard error of four independent experiments (identical to the experiment shown in [B]) in which the levels of phospho-S6 were quantified, corrected for loading, and normalized against the 11 mM glucose data point. Cells kept in physiological glucose are shown in light gray bars and cells pre-exposed to excess glucose are shown in dark gray bars. In (D), cells were treated with UK5099 at the time of glucose stimulation. In (E), cells were transferred to serum-free media without pyruvate before treatment with the indicated glucose dose. Statistical significance was calculated by Student's t test. # indicate p values above 0.05, * indicate 0.05 > p > 0.02, ** indicate 0.02 > p > 0.01, and *** indicate 0.01 > p > 0.003.
Treatment with UK5099 decreased mTORC1 signaling by about 40%–80%, as expected, and restored glucose sensitivity in cells that had been pre-exposed to excess glucose (Figures 5B and 5D). Likewise, removal of sodium pyruvate from the cell media during acute glucose stimulation improved sensitivity to glucose, mostly due to a decrease in mTORC1 signaling at 5 mM glucose (Figures 5B and 5E), confirming that pyruvate metabolism plays a significant role in the hyperactivation of basal mTORC1. Thus, mTORC1 hyperactivation by excess glucose was bimodal, with an initial pyruvate-independent component that is activated during acute exposure (Figure 1C) followed by a pyruvate-dependent component that is activated after prolonged exposure (Figure 5D). Furthermore, pyruvate-dependent mTORC1 activation masked the glucose dose-dependent mTORC1 activation in cells pre-exposed to excess glucose.
Exposure to Excess Glucose Stimulates mTOR-Sensitive and mTOR-Insensitive Metabolic Changes
To better characterize the impact of prolonged excess glucose and mTOR hyperactivation in the metabolism of β-cells, we used metabolomic analysis. Cells were either pre-exposed to excess glucose for 20 h or kept at physiological glucose, with or without KU, then switched to media containing 3 mM glucose before metabolite extraction and analysis (see Figure 6A for experimental design). Since neither excess glucose nor KU was present in the media at the time of metabolite extraction, only sustained metabolic alterations were expected.
Figure 6.

mTOR Inhibition Further Increased the Levels of a Subset of Metabolites that Were Induced by Exposure to Excess Glucose.
(A–M) INS-1 cells were kept in physiological glucose or pre-exposed to excess glucose for 20–24 h, transferred to complete serum-free media containing 3 mM glucose for 2 h, and lysed for metabolite extraction. (A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for Figures 6B–6M and 7A–7O. (B)–(M) Bar graphs depicting the levels (arbitrary units) of various metabolites extracted from INS-1 cells at 3 mM glucose. Cells that had been pre-exposed to excess glucose without KU are shown in solid dark gray bars, with KU in hatched dark gray bars. Cells that were kept in physiological glucose without KU are shown in solid light gray bars and with KU in hatched light gray bars. Data shown are the average and standard error from three independently generated samples. # indicate no statistical significance, * indicate 0.05 < p < 0.02, ** indicate 0.02 < p < 0.01, *** indicate 0.01 < p < 0.001, and **** indicate p ≤ 0.001, as determine by Student’s t tests.
As compared with cells kept at physiological glucose, cells pre-exposed to excess glucose had increased levels of all the glycolytic intermediates (excluding pyruvate, which was added as a supplement in the media), TCA cycle intermediates, and NADH/NAD (Figures 6B–6G and 7A–7D). These strongly suggest increased flux through glycolysis, anaplerosis, and oxidative phosphorylation, which is consistent with the metabolic acceleration characterized through measurements of oxygen consumption (Figure 3). Nonetheless, the relative levels of ATP/ADP and the levels of AMP were similar when these two experimental groups were compared (Figures 7E and 7F), confirming that there were no major differences in energy status between cells kept under physiological glucose or pre-exposed to excess glucose.
Figure 7.

mTOR Inhibition Prevented/Reduced the Changes Caused by Exposure to Excess Glucose on a Subset of Metabolites
(A–O) INS-1 cells were kept in physiological glucose or pre-exposed to excess glucose for 20–24 h, transferred to complete serum-free media containing 3 mM glucose for 2 h, and lysed for metabolite extraction (as described in Figure 6A). (A-O) Bar graphs depicting the levels (arbitrary units) of various metabolites extracted from INS-1 cells at 3 mM glucose. Cells that had been pre-exposed to excess glucose without KU are shown in solid dark gray bars and with KU in hatched dark gray bars. Cells that were kept in physiological glucose without KU are shown in solid light gray bars and with KU in hatched light gray bars. Data shown are the average and standard error from three independently generated samples. # indicate no statistical significance, * indicate 0.05 < p < 0.02, ** indicate 0.02 < p < 0.01, *** indicate 0.01 < p < 0.001, and **** indicate p ≤ 0.001, as determined by Student’s t tests.
Consistent with the observation that INS-1 cells exposed to excess glucose accumulate fat in the form of lipid droplets (Erion et al., 2015, Mugabo et al., 2017), these cells had a significant increase in glycerol 3-phosphate (Figure 6K), which is a product of glycolysis and an intermediate in glycerolipid synthesis. Likewise, cells pre-exposed to excess glucose had significantly higher levels of many lipid species (Figures 6L, 6M, and 7G–7I).
The levels of several metabolites that increased in cells pre-exposed to excess glucose were KU sensitive, including intermediates of the pentose-phosphate pathway (Figures 7J–7L), confirming previous findings observed in cancer cells with Tsc1/2 knockdown (Duvel et al., 2010). Dihydroxyacetone phosphate (DHAP) and phosphoenolpyruvate (PEP) were the only glycolysis intermediates that decreased with KU treatment, although statistical significance was only reached for DHAP (Figures 7A and 7B).
Surprisingly, most of the metabolites that were enhanced by exposure to excess glucose increased even further with mTOR inhibition. For example, the glycolysis and TCA cycle intermediates, glucose-6-phosphate/fructose-6-phosphate, malate, and succinate, further increased with KU treatment (Figures 6B–6G). Lactate production was also significantly enhanced by mTOR inhibition (Figure 6I), reflecting increased anaerobic glycolysis in KU-treated cells. Despite that, cells treated with KU had slightly higher AMP levels (Figure 7F), which indicates that they had some degree of energy deficit, likely owing to decreased mitochondrial respiration (see Figure 3E). We suspect that enhanced anaerobic glycolysis and anaplerosis in KU-treated cells are secondary effects of mTOR inhibition, triggered by the need of cells to compensate for lower mitochondrial respiration.
The intracellular content of 12 amino acids decreased slightly in cells pre-exposed to excess glucose (statistical significance was reached for 6 of them) and were all restored upon mTOR inhibition (statistical significance was reached for all of them) (Figures 7M–7O and Table S1). Since these amino acids were supplied in the media, we suggest that this decrease could be due to a higher demand for amino acids in cells pre-exposed to excess nutrients, as a result of increased mTORC1-dependent protein translation (assuming that the transport of these amino acids into the cells can be rate limiting). Alanine is the only amino acid that increased upon pre-exposure to excess glucose and further increased upon mTOR inhibition (Figure 6J). Since alanine can be synthesized from pyruvate through alanine aminotransferase, we suspect that these increases reflect high levels of cytosolic pyruvate.
Several of the lipid species that accumulated owing to exposure to excess glucose further increased upon mTOR inhibition (Figures 6L and 6M and Table S1), which is consistent with increased fuel availability and decreased energy demand. Increased fatty acid accumulation can also be explained by an increase in lipolysis, which has been previously linked to mTORC1 inhibition (Ben-Sahra and Manning, 2017), without concomitant increase in β-oxidation, which seems to be suppressed in these cells (see Figures 4B–4D). However, a few lipid species, such as eicosenoic acid, linoleic acid, arachidic acid, and cholesterol (Figures 7G–7I and Table S1), were reduced in KU-treated cells. Most likely, these lipids are imported into the cells from the serum (which is present in the growth medium during glucose exposure). Thus, it is unclear whether mTOR inhibition increased the metabolism of these particular lipids or whether it prevented their uptake through the membrane. However, we cannot rule out that some of them may be de novo synthesized by the β-cell. In fact, mTORC1 has been previously shown to regulate SREBP-2, a transcription factor that controls the expression of enzymes in the cholesterol synthesis pathway (Ben-Sahra and Manning, 2017).
Together, the results shown revealed that INS-1 cells under excess glucose and mTOR inhibition have increased anaplerosis, flux through glycolysis, and glycerolipid synthesis, all indicative of excess fuel. Yet, they also have signs of decreased mitochondrial pyruvate metabolism (higher lactate production) and decreased anabolic processes (decreased flux through the pentose phosphate pathway and decreased demand for amino acids). Thus, metabolic deceleration by mTOR inhibition can be explained by a decrease in both energy demand and aerobic metabolism.
mTOR Inhibition in β-Cells Exposed to Excess Glucose Results in Altered Insulin Secretion
Several of the metabolic changes described above can potentially interfere with stimulus-secretion coupling in β-cells (Nicholls, 2016). Thus, we examined the effect of excess glucose and KU treatment on insulin secretion, with special emphasis on secretion at sub-stimulatory glucose doses. INS-1 cells were pre-exposed to physiological or excess glucose for 20 h and then transferred to 2 mM glucose for a resting period of 2 h, after which insulin secretion was measured (see Figure 8A for experimental design). The data were normalized by secretion at 2 mM glucose to exclude any effects on proinsulin processing, which has been previously linked to mTORC1 signaling (Alejandro et al., 2017, Blandino-Rosano et al., 2017).
Figure 8.

mTOR Inhibition Affects Stimulus-Secretion Coupling in Cells Exposed to Excess Glucose
(A–D) INS-1 cells or intact mouse islets cultured in physiological glucose were exposed to excess glucose in the presence or absence of KU, transferred to serum-free media, and stimulated with the indicated glucose dose for assessment of insulin secretion. (A) Diagram depicting the media composition and glucose concentrations throughout the different stages of the experimental timeline for (A)–(D). (B) Insulin secretion of INS-1 cells under sub-stimulatory (2 and 4 mM) and stimulatory (8 mM) glucose. (C) Same data shown in (B) but highlighting the effect on secretion at sub-stimulatory glucose. (D) Insulin secretion of freshly isolated mouse islets under sub-stimulatory (3 and 5 mM) and stimulatory (8 mM) glucose. Islets were treated as described in (A), with small adjustments in the glucose concentrations used, which were 6 mM for physiological, 12 mM for excess, and 3 mM for resting glucose. Cells kept in physiological glucose are shown in light gray bars, and cells pre-exposed to excess glucose are shown in dark gray bars, with KU (hatched bars) or without (solid bars). (B,C) Data shown are the secretion normalized by the 2 mM glucose dose and is the average and standard error of four independent experiments with three replicates each. (D) Data shown are the average and standard error from three separate replicas and are representative of three separate experiments.
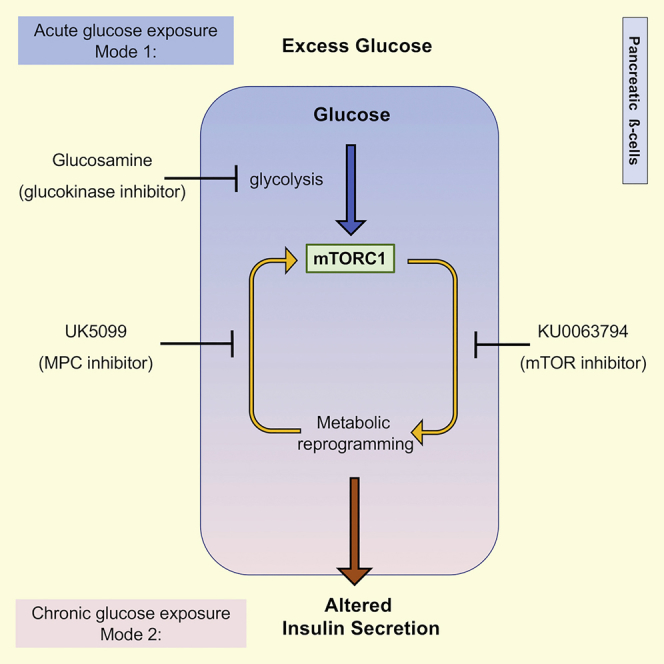
(E) Potential model to explain the role of mTORC1 hyperactivation in metabolic reprograming due to exposure to excess glucose.
Figure 8B shows that exposure of INS-1 cells to excess glucose caused a left-shift in the glucose dose-response curve for insulin secretion, as previously reported (Erion et al., 2015). INS-1 cells chronically cultured at physiological glucose had no basal insulin secretion (the amount of insulin secreted at 4 mM glucose relative to the amount secreted at 2 mM equaled 1), whereas cells pre-exposed to excess glucose had a significant increase in basal secretion (secretion at 4 mM glucose was more than 2-fold the secretion at 2 mM) (Figure 8C). When cells were pre-exposed to excess glucose in the presence of KU, insulin secretion was reduced and the glucose dose-response was similar to the response obtained from cells kept in physiological glucose (Figures 8B and 8C).
The same experimental design was used to assess secretion in freshly isolated mouse islets, with a slight adjustment in the concentrations of glucose used to better reflect the higher normoglycemic levels in the mouse circulation. Paradoxically, we observed an increase in insulin secretion in islets co-treated with excess glucose and KU (Figure 8D). These revealed that the net result of mTOR inhibition in islets is different from that in INS-1 cells. It is possible that the compensatory increase in anaerobic glycolysis and enhanced lipid accumulation seen with mTOR inhibition (Figure 6) promotes insulin secretion in islets, but not in INS-1 cells, masking any potential effect of a lower pyruvate metabolism. In summary, our results demonstrate that excess glucose-induced mTORC1 signaling has a complex and significant impact on insulin secretion, likely due to its primary and secondary effects on the metabolism of β-cells.
Discussion
mTORC1 is a major energy-sensing and signaling hub that regulates anabolic processes and allows cells to adapt to an environment of nutrient surplus (Saxton and Sabatini, 2017). In the context of cancer, mTORC1 signaling is considered an important therapeutic target to slow down cell proliferation (Gomes et al., 2017). In the context of diabetes, on the other hand, the use of mTORC1 inhibitors has generated mixed outcomes (Ardestani et al., 2018). This is likely because glucose homeostasis requires the concerted action of various tissues, which may differ in the way they regulate and respond to mTORC1 signaling. Even within one cell type, mTORC1 activation may have positive or negative implications for cell function, depending on the intensity and duration of the signal. Transient mTORC1 activation signals for proliferation and survival, which are positive adaptations; on the other hand, prolonged mTORC1 activation leads to ER stress and apoptosis, which are negative adaptations (Ardestani et al., 2018). Here we showed that exposure of β-cells to excess glucose leads to hyperactivation of mTORC1 and mTOR-dependent metabolic reprogramming that severely affected stimulus-secretion coupling.
Unlike most published studies on the effect of glucose in β-cells, our work was performed within a short time frame (up to 24 h) and a normal range for glucose variation in vivo, from normoglycemic (5 mM) to post-prandial (11 mM). Thus, our experimental approach better mimics the environment that β-cells from overfed, obese, or pre-diabetic patients may be subjected to, thereby avoiding more extreme mTORC1-mediated responses such as glucotoxicity-induced ER stress or glucose starvation-induced autophagy. In a recent study, INS-1 cells exposed to 25 mM glucose for 48 h had increased anaplerosis and flux through the pentose phosphate pathway (Haythorne et al., 2019), which are consistent with our findings. In contrast, oxidative metabolism was reduced and pyruvate metabolism was blocked in INS-1 cells exposed to 25 mM glucose for 48 h, which is the opposite result from what we obtained using 11 mM glucose and shorter exposure. Given that mTORC1-mediated metabolic acceleration reported here was an early response to excess glucose (within 4–20 h exposure), it most likely precedes and may even contribute to other delayed events.
Our studies revealed that excess glucose-induced mTORC1 activation is bimodal. By keeping β-cells under a normoglycemic environment, we uncovered a mode for mTORC1 activation that is glycolysis dependent but mitochondrial pyruvate metabolism independent (Figure 8E). This mechanism for mTORC1 activation does not involve modulation by AMPK, as had been previously described in studies where the glucose effect was measured in cells subjected to complete glucose starvation. Instead, it is possible that excess glucose stimulated mTORC1 signaling in β-cells by increasing the levels of phosphatidic acid, a known mTORC1 activator and intermediate in the glycerolipid synthetic pathway (Foster et al., 2014, Menon et al., 2017). Another possibility is that glucokinase (hexokinase IV), which has a high Km for glucose and thus functions as a major glucose sensor in β-cells, may directly bind to and sequester Raptor, the regulatory subunit of mTORC1, in a manner similar to the one that was described for hexokinase II in cardiomyocytes (Roberts et al., 2014). The exact mechanism that links glycolysis to mTORC1 activation will be addressed in future work. Regardless of the mechanism, we predict that excess glucose-dependent mTORC1 signaling will be more significant in β-cells, which respond poorly to insulin (Rhodes et al., 2013), than in liver or muscle, which are insulin-responsive tissues.
Chronic exposure of β-cells to excess glucose triggered a second mode for mTORC1 activation that was dependent on mitochondrial pyruvate metabolism. Pyruvate-dependent mTORC1 activation is likely to involve direct sensing of ATP turnover by the mTORC1 complex (McDaniel et al., 2002). Since accelerated metabolism of pyruvate in the mitochondria was mTORC1 dependent, our results revealed a positive feedback loop for activation of mTORC1 in β-cells under energy surplus (Figure 8E). Positive feedback loops are often associated with pathological states and are critical steps for therapeutic intervention. In future research, we hope to identify better drug targets for the treatment of β-cell dysfunction by further dissecting the signals downstream of mTORC1 that elicit this positive feedback loop.
Excess glucose increased ATP-coupled respiration in a mTORC1-dependent manner (Figure 3). Interestingly, this increase in ATP-coupled respiration was cycloheximide sensitive and glucose dose dependent, with a significant increase from 5 (non-secretory dose) to 7 mM (secretory dose) glucose. Therefore, we suspect that newly synthesized ATP is largely being used to translate more proinsulin to replenish insulin content and granules lost (Boland et al., 2017).
There are several metabolic coupling factors that can interfere with normal insulin secretion by the β-cell. Based on our metabolomic analysis, we can identify several mTOR-dependent metabolites that could affect stimulus-secretion coupling after exposure to excess glucose. For example, cholesterol, eicosenoic acid, and arachidic acid are lipid species that were reduced by the treatment with KU and therefore could potentially contribute to elevated basal secretion in INS-1 (Yaney and Corkey, 2003). Other lipid species that further accumulated in KU-treated cells, such as myristate and omega-hydroxy tetradecanoate, could potentially explain the effect of KU on potentiating islet secretion. In addition, accelerated flux through mitochondrial metabolism per se could induce a shift in the glucose threshold for insulin secretion, given the remarkable effect of KU in preventing the excess glucose-induced metabolic acceleration. Future research will focus on dissecting the downstream effectors of mTORC1 that mediate specific metabolic changes and their effect on secretion, to distinguish which mTOR responses are beneficial and which are detrimental to proper β-cell function.
Limitations of the Study
Bioenergetics and metabolomic experiments were performed using a β-cell line, INS-1. The use of primary islets in these analyses is complicated by the fact that primary islets contain alpha and delta cells in addition to β-cells, which can affect the outcome of the analysis. Therefore, the findings from these analyses are suggestive but cannot be conclusive in the absence of primary cell data. Our studies on glucose-dependent mTORC1 signaling in mouse islets are limited by low signal-to-noise ratio of the total S6 blots. Our studies on glucose-dependent mTORC1 signaling in human islets are limited by lack of additional phenotypic information on donors. Although the results obtained with human islets are consistent with those of mice islets and INS-1 cells, in the absence of donor information the potential impact of other confounding factors cannot be conclusively ruled out. Animal studies will be necessary to confirm the in vitro/ex vivo findings. The exact mechanism that links glycolysis to mTORC1 activation has not been uncovered and will be addressed in future work.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We would like to thank Drs. Keith Tornheim and Hartmut Wohlrab for insightful discussions and Eleni Ritou for help with Seahorse experiments. This work was supported by start-up funds from Vanderbilt University School of Medicine, Department of Biochemistry, USA, the Evans Foundation funds from Boston University, USA, and the National Institute of Health (NIH), USA, grants 1UL1TR001430 (CTSI), DK0007201 (T32), DK46200 (BNORC), DK74778. We thank the Islet Procurement and Analysis (IPA) core from Vanderbilt University Medical Center (VUMC), USA, for mouse islet isolation and purification and analysis and the Hormone Assay and Analytical Service (HAAS) core from Vanderbilt University Medical Center (VUMC), USA, for insulin assays. The HAAS core is supported by the National Institute of Health (NIH), USA, grants DK059637 and DK020593, and the IPA core is supported by the Vanderbilt Diabetes Research and Training Center (DRTC), NIH, USA, grant P60 DK020593.
Authors Contributions
C.Z.R. designed, performed, and analyzed the experiments; J.L. and J.W.L. designed and analyzed metabolomic experiments; B.E.C. provided expertise and feedback; J.T.D. conceived and provided reagents, expertise, and feedback; L.E.R. conceived, designed, performed, and analyzed the experiments and wrote the manuscript.
Declaration of Interests
The Authors declare no competing interests.
Published: February 21, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.100858.
Supplemental Information
INS-1 cells were pre-exposed to excess glucose for 20 h, with KU or without, or kept in physiological glucose, with KU or without, as indicated. Included in the spreadsheet are the names of the metabolites, chemical formulas, M/Z, and relative abundance (arbitrary units).
References
- Alejandro E.U., Bozadjieva N., Blandino-Rosano M., Wasan M.A., Elghazi L., Vadrevu S., Satin L., Bernal-Mizrachi E. Overexpression of kinase-dead mTOR impairs glucose homeostasis by regulating insulin secretion and not beta-cell mass. Diabetes. 2017;66:2150–2162. doi: 10.2337/db16-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardestani A., Lupse B., Kido Y., Leibowitz G., Maedler K. mTORC1 signaling: a double-edged sword in diabetic beta cells. Cell Metab. 2018;27:314–331. doi: 10.1016/j.cmet.2017.11.004. [DOI] [PubMed] [Google Scholar]
- Ben-Sahra I., Manning B.D. mTORC1 signaling and the metabolic control of cell growth. Curr. Opin. Cell Biol. 2017;45:72–82. doi: 10.1016/j.ceb.2017.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blandino-Rosano M., Barbaresso R., Jimenez-Palomares M., Bozadjieva N., Werneck-de-Castro J.P., Hatanaka M., Mirmira R.G., Sonenberg N., Liu M., Ruegg M.A. Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat. Commun. 2017;8:16014. doi: 10.1038/ncomms16014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland B.B., Rhodes C.J., Grimsby J.S. The dynamic plasticity of insulin production in beta-cells. Mol. Metab. 2017;6:958–973. doi: 10.1016/j.molmet.2017.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corkey B.E. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes. 2012;61:4–13. doi: 10.2337/db11-1483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeney J.T., Gromada J., Hoy M., Olsen H.L., Rhodes C.J., Prentki M., Berggren P.O., Corkey B.E. Acute stimulation with long chain acyl-CoA enhances exocytosis in insulin-secreting cells (HIT T-15 and NMRI beta-cells) J. Biol. Chem. 2000;275:9363–9368. doi: 10.1074/jbc.275.13.9363. [DOI] [PubMed] [Google Scholar]
- Dibble C.C., Manning B.D. Signal integration by mTORC1 coordinates nutrient input with biosynthetic output. Nat. Cell Biol. 2013;15:555–564. doi: 10.1038/ncb2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvel K., Yecies J.L., Menon S., Raman P., Lipovsky A.I., Souza A.L., Triantafellow E., Ma Q., Gorski R., Cleaver S. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell. 2010;39:171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erion K.A., Berdan C.A., Burritt N.E., Corkey B.E., Deeney J.T. Chronic exposure to excess nutrients left-shifts the concentration dependence of glucose-stimulated insulin secretion in pancreatic beta-cells. J. Biol. Chem. 2015;290:16191–16201. doi: 10.1074/jbc.M114.620351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster D.A., Salloum D., Menon D., Frias M.A. Phospholipase D and the maintenance of phosphatidic acid levels for regulation of mammalian target of rapamycin (mTOR) J. Biol. Chem. 2014;289:22583–22588. doi: 10.1074/jbc.R114.566091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes A.P., Schild T., Blenis J. Adding polyamine metabolism to the mTORC1 toolkit in cell growth and cancer. Dev. Cell. 2017;42:112–114. doi: 10.1016/j.devcel.2017.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez A., Hall M.N. Nutrient sensing and TOR signaling in yeast and mammals. EMBO J. 2017;36:397–408. doi: 10.15252/embj.201696010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haythorne E., Rohm M., van de Bunt M., Brereton M.F., Tarasov A.I., Blacker T.S., Sachse G., Silva Dos Santos M., Terron Exposito R., Davis S. Diabetes causes marked inhibition of mitochondrial metabolism in pancreatic beta-cells. Nat. Commun. 2019;10:2474. doi: 10.1038/s41467-019-10189-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzig S., Shaw R.J. AMPK: guardian of metabolism and mitochondrial homeostasis. Nat. Rev. Mol. Cell Biol. 2018;19:121–135. doi: 10.1038/nrm.2017.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildyard J.C., Ammala C., Dukes I.D., Thomson S.A., Halestrap A.P. Identification and characterisation of a new class of highly specific and potent inhibitors of the mitochondrial pyruvate carrier. Biochim. Biophys. Acta. 2005;1707:221–230. doi: 10.1016/j.bbabio.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Irles E., Neco P., Lluesma M., Villar-Pazos S., Santos-Silva J.C., Vettorazzi J.F., Alonso-Magdalena P., Carneiro E.M., Boschero A.C., Nadal A. Enhanced glucose-induced intracellular signaling promotes insulin hypersecretion: pancreatic beta-cell functional adaptations in a model of genetic obesity and prediabetes. Mol. Cell. Endocrinol. 2015;404:46–55. doi: 10.1016/j.mce.2015.01.033. [DOI] [PubMed] [Google Scholar]
- Koyanagi M., Asahara S., Matsuda T., Hashimoto N., Shigeyama Y., Shibutani Y., Kanno A., Fuchita M., Mikami T., Hosooka T. Ablation of TSC2 enhances insulin secretion by increasing the number of mitochondria through activation of mTORC1. PLoS One. 2011;6:e23238. doi: 10.1371/journal.pone.0023238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamontagne J., Al-Mass A., Nolan C.J., Corkey B.E., Madiraju S.R.M., Joly E., Prentki M. Identification of the signals for glucose-induced insulin secretion in INS1 (832/13) beta-cells using metformin-induced metabolic deceleration as a model. J. Biol. Chem. 2017;292:19458–19468. doi: 10.1074/jbc.M117.808105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C., Ford E.S., McGuire L.C., Mokdad A.H., Little R.R., Reaven G.M. Trends in hyperinsulinemia among nondiabetic adults in the U.S. Diabetes Care. 2006;29:2396–2402. doi: 10.2337/dc06-0289. [DOI] [PubMed] [Google Scholar]
- Matschinsky F.M. Regulation of pancreatic beta-cell glucokinase: from basics to therapeutics. Diabetes. 2002;51(Suppl 3):S394–S404. doi: 10.2337/diabetes.51.2007.s394. [DOI] [PubMed] [Google Scholar]
- McDaniel M.L., Marshall C.A., Pappan K.L., Kwon G. Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes. 2002;51:2877–2885. doi: 10.2337/diabetes.51.10.2877. [DOI] [PubMed] [Google Scholar]
- Menon D., Salloum D., Bernfeld E., Gorodetsky E., Akselrod A., Frias M.A., Sudderth J., Chen P.H., DeBerardinis R., Foster D.A. Lipid sensing by mTOR complexes via de novo synthesis of phosphatidic acid. J. Biol. Chem. 2017;292:6303–6311. doi: 10.1074/jbc.M116.772988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori H., Inoki K., Opland D., Munzberg H., Villanueva E.C., Faouzi M., Ikenoue T., Kwiatkowski D.J., Macdougald O.A., Myers M.G., Jr. Critical roles for the TSC-mTOR pathway in beta-cell function. American journal of physiology. Endocrinol. Metab. 2009;297:E1013–E1022. doi: 10.1152/ajpendo.00262.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mugabo Y., Zhao S., Lamontagne J., Al-Mass A., Peyot M.L., Corkey B.E., Joly E., Madiraju S.R.M., Prentki M. Metabolic fate of glucose and candidate signaling and excess-fuel detoxification pathways in pancreatic beta-cells. J. Biol. Chem. 2017;292:7407–7422. doi: 10.1074/jbc.M116.763060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D.G. The pancreatic beta-cell: a bioenergetic perspective. Physiol. Rev. 2016;96:1385–1447. doi: 10.1152/physrev.00009.2016. [DOI] [PubMed] [Google Scholar]
- Nolan C.J., Prentki M. The islet beta-cell: fuel responsive and vulnerable. Trends Endocrinol. Metab. 2008;19:285–291. doi: 10.1016/j.tem.2008.07.006. [DOI] [PubMed] [Google Scholar]
- Pories W.J., Dohm G.L. Diabetes: have we got it all wrong? Hyperinsulinism as the culprit: surgery provides the evidence. Diabetes Care. 2012;35:2438–2442. doi: 10.2337/dc12-0684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prentki M., Matschinsky F.M., Madiraju S.R. Metabolic signaling in fuel-induced insulin secretion. Cell Metab. 2013;18:162–185. doi: 10.1016/j.cmet.2013.05.018. [DOI] [PubMed] [Google Scholar]
- Rachdi L., Balcazar N., Osorio-Duque F., Elghazi L., Weiss A., Gould A., Chang-Chen K.J., Gambello M.J., Bernal-Mizrachi E. Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc. Natl. Acad. Sci. U S A. 2008;105:9250–9255. doi: 10.1073/pnas.0803047105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhodes C.J., White M.F., Leahy J.L., Kahn S.E. Direct autocrine action of insulin on beta-cells: does it make physiological sense? Diabetes. 2013;62:2157–2163. doi: 10.2337/db13-0246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts D.J., Tan-Sah V.P., Ding E.Y., Smith J.M., Miyamoto S. Hexokinase-II positively regulates glucose starvation-induced autophagy through TORC1 inhibition. Mol. Cell. 2014;53:521–533. doi: 10.1016/j.molcel.2013.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxton R.A., Sabatini D.M. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168:960–976. doi: 10.1016/j.cell.2017.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shigeyama Y., Kobayashi T., Kido Y., Hashimoto N., Asahara S., Matsuda T., Takeda A., Inoue T., Shibutani Y., Koyanagi M. Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol. Cell. Biol. 2008;28:2971–2979. doi: 10.1128/MCB.01695-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaney G.C., Corkey B.E. Fatty acid metabolism and insulin secretion in pancreatic beta cells. Diabetologia. 2003;46:1297–1312. doi: 10.1007/s00125-003-1207-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
INS-1 cells were pre-exposed to excess glucose for 20 h, with KU or without, or kept in physiological glucose, with KU or without, as indicated. Included in the spreadsheet are the names of the metabolites, chemical formulas, M/Z, and relative abundance (arbitrary units).