

Diabetes can be ameliorated by additive AIRE expression in peripheral DCs
Keywords: mTEC, NOD, type 1 diabetes, Xcr1+ DC
Abstract
Tissue-specific autoimmune diseases are assumed to arise through malfunction of two checkpoints for immune tolerance: defective elimination of autoreactive T cells in the thymus and activation of these T cells by corresponding autoantigens in the periphery. However, evidence for this model and the outcome of such alterations in each or both of the tolerance mechanisms have not been sufficiently investigated. We studied these issues by expressing human AIRE (huAIRE) as a modifier of tolerance function in NOD mice wherein the defects of thymic and peripheral tolerance together cause type I diabetes (T1D). Additive huAIRE expression in the thymic stroma had no major impact on the production of diabetogenic T cells in the thymus. In contrast, huAIRE expression in peripheral antigen-presenting cells (APCs) rendered the mice resistant to T1D, while maintaining other tissue-specific autoimmune responses and antibody production against an exogenous protein antigen, because of the loss of Xcr1+ dendritic cells, an essential component for activating diabetogenic T cells in the periphery. These results contrast with our recent demonstration that huAIRE expression in both the thymic stroma and peripheral APCs resulted in the paradoxical development of muscle-specific autoimmunity. Our results reveal that tissue-specific autoimmunity is differentially controlled by a combination of thymic function and peripheral tolerance, which can be manipulated by expression of huAIRE/Aire in each or both of the tolerance mechanisms.
Introduction
Tissue-specific autoimmune diseases are assumed to arise through malfunction of two immune tolerance checkpoints: defective elimination of autoreactive T cells in the thymus and activation of these T cells by corresponding autoantigens in the periphery. Likewise, two critical steps for the development of type I diabetes (T1D) have been suggested in NOD mice, a murine model of T1D in humans (1–3): generation of T cells reactive against β-islet antigens in the thymus, followed by activation of potentially diabetogenic T cells specific for β-islet antigens in the target organ and/or peripheral lymph nodes (LNs) (4). In the thymus, β-islet antigens are expressed by medullary thymic epithelial cells (mTECs), and presented by mTECs and/or dendritic cells (DCs) to purge autoreactive T cells (5, 6). Several lines of evidence have suggested that the expression level of insulin, a primary autoantigen in the pathogenesis of T1D (7), in mTECs is a critical determinant for elimination of diabetogenic T cells. For example, it has been reported that the T1D protective class III variable number of tandem repeats (VNTRs) in humans is associated with a higher steady-state level of insulin mRNA expression in the thymus (8, 9). Conversely, knockout of insulin 2 (Ins2) in NOD accelerates the development of T1D (10, 11), most likely because of inefficient clonal deletion of Ins2-reactive T cells in the thymus. Aire, the gene responsible for the hereditary autoimmune disease APECED (autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy), is one of the best-characterized factors responsible for controlling the expression levels of tissue-restricted self-antigens (TRAs), such as Ins2, in mTECs, thereby contributing to the establishment of self-tolerance in the thymus (12–14). In the periphery, autoreactive T cells that have escaped negative selection in the thymus subsequently become activated by interaction with corresponding self-antigens to attack target tissues if the peripheral tolerance mechanism does not function appropriately. Indeed, it has been demonstrated that presentation of insulin peptide by antigen-presenting cells (APCs) in pancreatic β-islets and peripheral LNs, not confined to pancreatic LNs (4), is a critical step for diabetogenesis in NOD (15).
Although Aire expression is most prominent in mTECs, it is also expressed by bone marrow (BM)-derived non-conventional APCs that show high MHC-II expression in secondary lymphoid organs, so-called extra-thymic Aire-expressing cells (eTACs) (16, 17). eTACs have been suggested to reinforce immune tolerance by preventing the maturation of autoreactive T cells that have escaped thymic negative selection. B cells in the thymus also express Aire, and their role in thymic tolerance has been suggested (18). Given that Aire exerts its tolerogenic function in both mTECs and BM-derived APCs (i.e. eTACs and B cells), we have recently established a strain of NOD in which human AIRE (huAIRE) is additively expressed under the control of the MHC-II promoter [huAIRE transgenic (huAIRE-Tg)] with the aim of ameliorating the autoimmune pathology in NOD (19). Paradoxically, however, huAIRE-Tg mice developed muscle-specific autoimmunity that was associated with defects in both the thymic and peripheral tolerance mechanisms: mTECs expressing huAIRE showed defective maturation accompanied by reduced TRA expression, including Ins2. In peripheral B cells from huAIRE-Tg mice, expression of the co-stimulatory molecule CD86 was up-regulated. It was noteworthy that muscle-specific autoimmunity developed only when both mTECs and BM-derived APCs expressed huAIRE at high levels, as revealed in BM chimeras (19). Thus, an altered capacity to present self-antigens in thymic and peripheral APCs manipulated through additive huAIRE expression resulted in the development of de novo muscle-specific autoimmunity.
In the present study, we focused on another effect of additive huAIRE expression on the development of T1D in NOD. We found that huAIRE-Tg had defective presentation of β-islet antigens in the periphery because of impaired development and/or function of a particular subset of DCs (i.e. Xcr1+ DCs), as a result of which the mice became resistant to the development of T1D. In contrast to the situation in muscle-specific autoimmunity, mTECs expressing huAIRE had no major impact on the production of diabetogenic T cells revealed by the BM chimeras. Thus, our results suggested that a distinct set of tissue-specific immune responses (i.e. against muscle or against β-islets) is positively or negatively controlled by the altered thymic and/or peripheral tolerance function upon introduction of huAIRE/Aire as a modifier of each tolerance mechanism. These results suggest that control of the tissue-specific immune response may be feasible through manipulation of the thymic and peripheral tolerance mechanisms by expressing huAIRE/Aire as a single factor in each or both of the tolerogenic components.
Methods
Mice
Mice expressing huAIRE under control of the MHC-II promoter were generated as reported previously (19). TCR transgenic (TCR-tg) mice NY8.3 (20) and BDC2.5 (21), and B-cell-deficient NOD mice (22) were purchased from the Jackson Laboratory. NOD/ShiJic-scidJcl mice and Rag2-deficient mice were obtained from CLEA Japan Inc. and Taconic, respectively. Mice were diagnosed as diabetic when blood glucose levels exceeded 250 mg dl−1. The mice were maintained under pathogen-free conditions, and handled in accordance with the Guidelines for Animal Experimentation of Tokushima University School of Medicine.
Pathology
Formalin-fixed tissue sections were subjected to hematoxylin and eosin (H&E) staining, and two pathologists independently evaluated the histology without being informed of the detailed condition of each individual mouse.
TEC preparation and flow cytometric analysis
Preparation of TECs and flow cytometric analysis with a FACS-calibur (BD) and a FACSAria II (BD) were performed as described previously (19). In brief, thymic lobes were isolated from mice and cut into small pieces. The fragments were pipetted to remove the majority of thymocytes in RPMI 1640 medium (Gibco) supplemented with 10% heat-inactivated fetal calf serum (FCS) (Bovogen), 20 mM HEPES, 50 U ml−1 penicillin, 50 μg ml−1 streptomycin and 50 μM 2-mercaptoethanol, hereafter referred to as R10. The resulting thymic fragments were digested with 0.15% collagenase D (Roche) and 10 U ml−1 DNase I (Roche) in R10 at 37°C for 30 min. The supernatants, containing dissociated TECs, were saved, and the remaining thymic fragments were digested with 0.15% collagenase/dispase (Roche) and DNase I at 37°C for 30 min. The supernatants from this digest were combined with the supernatants from the collagenase digests, and the mixture was centrifuged for 5 min at 400 × g. The cells were suspended in phosphate-buffered saline (PBS) containing 5 mM ethylenediaminetetraacetic acid (EDTA) and 1% bovine serum albumin (BSA), and kept on ice until staining. The monoclonal antibodies (mAbs) used were anti-CD45, anti-Ly51, anti-CD80, anti-CD103, anti-CD8a, B220, anti-CD19, anti-CD11b, anti-CD11c and F4/80, all purchased from eBioscience. Anti-EpCAM and anti-Xcr1 mAbs were from BioLegend. Ulex europaeus agglutinin 1 (UEA-1) was from Vector Laboratories.
BM transfer
BM transfer was performed as described previously (19). In brief, BM cells were suspended in R10 medium containing anti-CD90 (Thy1.2) mAb (clone 30-H12; BioLegend) plus low-toxicity rabbit complement (Cedarlane Laboratories). After incubation at 37°C for 45 min, the cells were washed twice and adjusted to 5 × 107 viable cells ml−1 in R10 not containing FCS. Each recipient mouse was then lethally irradiated (9 Gy) and treated with 0.2 ml of donor BM cells i.v. on the same day.
Measurement of proliferation of TCR-Tg T cells specific for β-islet antigens
Spleen cell suspensions prepared from TCR-tg mouse strains NY8.3 and BDC2.5 were depleted of red blood cells by osmotic lysis, and their T cells were purified by depletion with B220+ MicroBeads (Miltenyi Biotec). The resulting preparations contained approximately 95% T cells. The purified NY8.3 CD8+ cells and BDC2.5 CD4+ cells were labeled with 5- (and 6-)carboxyfluorescein diacetate succinimidyl ester (CFSE) (Dojindo), and injected i.v. (6.0–10.0 × 106 cells per mouse) into heterozygous 2m9L-Tg or control mice. Cell proliferation was measured 64 h after T-cell transfer.
Transfer of peripheral T cells into NOD.scid mice
Spleen cell suspensions were depleted of red blood cells by osmotic lysis, and their Thy1+ cells were purified with CD90.2 (Thy1.2) MicroBeads (Miltenyi Biotec). The resulting preparations contained approximately 95% Thy1+ cells. The purified Thy1+ cells were injected i.v. (1.0 × 107 cells per mouse), and development of diabetes was monitored for 20 weeks. Diagnosis of diabetes was performed as described above.
Flow cytometric analysis of BM-APCs from β-islets
β-islets from the pancreas were isolated as described previously (23, 24). Briefly, pancreata were inflated through the common bile duct with 5.0 ml of HBSS supplemented with 380.0 μg ml−1 collagenase. The pancreata were then removed carefully and digested in a 37°C water bath for 13 min. After vigorous shaking for 90 s, the pancreata were washed three times in HBSS and passed through a 70-µm cell strainer to retain the islets. The islets were flushed into a Petri dish and handpicked using a pipette. For flow cytometric analysis, islet cells were dispersed using Cell Dissociation Solution Non-Enzymatic (Sigma) for 3 min at 37°C.
Generation of BM-derived Xcr1+ DCs
BM cells were harvested by flushing the femurs of mice with R10. The cells were cultured in R10 containing 100 ng ml−1 human Flt3L (Peprotech, London, UK) for 8 days as previously described (25). They were then stained with B220, anti-CD11c, anti-CD11b, anti-CD24 and anti-Xcr1 mAbs and B220−CD11c+CD11b+CD24+Xcr1+ cells were sorted by FACSAria II (BD) as Xcr1+ DCs for RNA-seq analysis.
Real-time PCR
RNA was extracted from TECs using RNeasy Mini Kits (Qiagen) and converted to cDNA with SuperScript III or VILO RT Kits (Invitrogen) in accordance with the manufacturer’s instructions. Real-time PCR for quantification of the insulin 2 (Ins2), salivary protein 1 (Sap1) and Hprt genes was performed as described previously (19). Detailed sequence information was provided in our previous paper (19).
RNA-seq analysis
RNA-seq analysis was performed as described previously (19). In brief, total RNAs from sorted DCs were isolated using an RNeasy Mini Kit (Qiagen) in accordance with the manufacturer’s instructions. Eluted RNAs were incubated with RT-grade DNase (Nippon Gene) for 10 min at room temperature, followed by purification on an RNeasy column (Qiagen). RNAs were verified both quantitatively and qualitatively using an Agilent RNA 6000 Pico Kit run on an Agilent 2100 Bioanalyzer (Agilent Technologies). RNA Integrity Numbers were >7. Three independent biological replicates from each genotype were used for RNA-seq analysis. Sequencing libraries were generated from 1.0 ng of total RNA with Ovation SoLo RNA-Seq Systems (NuGEN). The libraries were size-selected with AMPure XP beads (Agilent Technologies) and used for cluster generation on the flow cells. Single-end 150-bp sequencing was performed with an Illumina MiSeq (Illumina), which generated 5.8–20 × 106 sequenced fragments per sample. Prior to alignment, the reads were trimmed to remove adapter sequences. Reads were mapped to the mouse reference genome GRCm38/mm10 using the STAR aligner with the option: -outSAMtype BAM SortedByCoordinate to generate a sorted BAM format. Differential gene expression between the experimental samples was analyzed with Cufflinks. Read count tables and group comparisons for differential expression analysis were performed with Cuffdiff, which is part of the Cufflinks package. The false discovery rate (FDR) for detection of differentially abundant transcripts was set to 5%. The data set associated with this project has been submitted to DDBJ Sequence Reads Archive (DRA accession number: DRA006501).
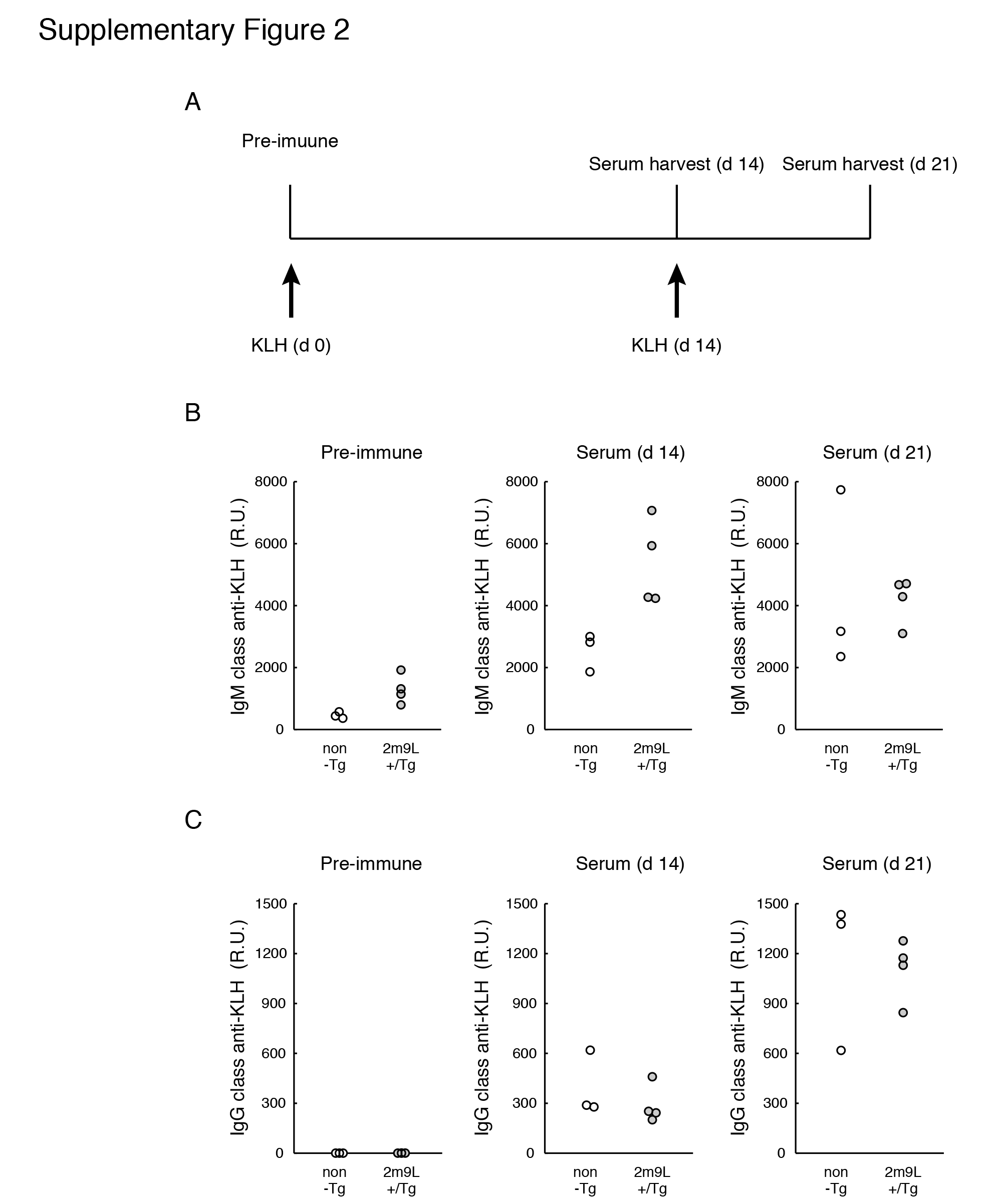
Measurement of anti-keyhole limpet hemocyanin antibody response
Mice were i.p. injected with keyhole limpet hemocyanin (KLH) in complete Freund’s adjuvant, and sera were harvested 14 and 21 days after immunization. Subclass-specific anti-KLH responses were measured by ELISA.
Statistical analysis
All results are expressed as mean ± SEM. The log-rank test was performed for Kaplan–Meier survival curves. Differences were considered significant at P <0.05.
Results
Acquisition of resistance to T1D in NOD through expression of huAIRE in BM-derived APCs
We have recently reported that transgenic mice additively expressing huAIRE under control of the MHC-II promoter on NOD (huAIRE-Tg) paradoxically developed muscle-specific autoimmunity in two independent lines out of three (i.e. lines 2m9L-Tg and 1m4L-Tg, but not line 8L-Tg) (19): percentages of peripheral B cells expressing huAIRE were, in decreasing order, 2m9L-Tg > 1m4L-Tg > 8L-Tg (Fig. 1A). This corresponded to the order of huAIRE expression levels in mTECs (19). Interestingly, only half of the MHC-II-positive B220+ splenic B cells expressed huAIRE, and the rest did not (Supplementary Figure 1, top). Of note, there was a correlation between the expression of CD86, but not CD80, and huAIRE within B220+ B cells (Supplementary Figure 1, middle and bottom), suggesting that expression of MHC-II may not be sufficient for huAIRE expression. Instead, some other cellular condition such as activation status may control huAIRE expression at the protein level. Although muscle-specific autoimmunity appeared only when the transgene was homozygous, but not heterozygous, for both 2m9L-Tg and 1m4L-Tg, we were able to obtain genetic evidence indicating that this phenotype was attributable to the gene-dosage effect of huAIRE, and not to the inserted mutations. Indeed, 2m9L-Tg (high line) and 1m4L-Tg (intermediate line) developed the disease in 100% and ~20% of individuals, respectively, in the homozygous state (19). In that study, we also confirmed the competence of huAIRE protein in mice by crossing huAIRE-Tg onto Aire-deficient mice (Aire-KO), and observed rescue from the autoimmune lethality caused by Aire deficiency on a NOD background (26). RNA-seq analysis also showed that huAIRE expression in mTECs abolished the differential expression of genes appearing between Aire-sufficient and Aire-deficient mTECs, thus clearly indicating that huAIRE is functional in mice (19).
Fig. 1.

Expression of huAIRE in BM-derived APCs renders NOD resistant to the development of T1D. (A) Detection of peripheral B cells (CD19+CD3−) expressing huAIRE in huAIRE-Tg lines using flow cytometric analysis (left). A summary of the results is shown on the right. One circle corresponds to one mouse analyzed. (B) Female heterozygous 2m9L-Tg showed complete resistance to the development of T1D (n > 50), whereas female mice from two other lines developed T1D when the cumulative incidence of T1D was monitored until 40 weeks of age [55.6% in 1m4L-Tg (n = 9); 75.0% in 8L-Tg (n = 4); 71.7% in non-Tg (n = 46)]. (C) Pancreatic pathology of heterozygous 2m9L-Tg and non-Tg littermates. One representative result from a total of more than five experiments is shown. Scale bar: 100 µm. (D) Development of T1D in reciprocal BM chimeras using heterozygous 2m9L-Tg. Cumulative incidence of T1D was monitored until 40 weeks of age: non-Tg into non-Tg, n = 8; non-Tg into +/Tg, n = 6; +/Tg into non-Tg, n = 7. (E) Resistance to T1D in BM chimeras that had received BM cells from homozygous 2m9L-Tg. non-Tg into non-Tg, n = 4; Tg/Tg into non-Tg, n = 4.
Monitoring of blood glucose levels showed that 2m9L-Tg had complete resistance to the development of T1D when the transgene was heterozygous (+/Tg) (n > 50) (Fig. 1B) (19): these mice showed no pancreatic pathology, in marked contrast to their non-Tg littermates (Fig. 1C). In contrast to 2m9L-Tg (high line), 1m4L-Tg (intermediate line) and 8L-Tg (low line) developed T1D when the transgene was heterozygous [55.6% in 1m4L-Tg (n = 9); 75.0% in 8L-Tg (n = 4); 71.7% in non-Tg (n = 46)] (Fig. 1B). In order to investigate which cell types expressing huAIRE [i.e. stromal component (mTECs) or BM-derived APCs such as B cells (Fig. 1A) and DCs] are responsible for the acquisition of the resistance to T1D in heterozygous 2m9L-Tg, we performed reciprocal BM transfer experiments. Non-Tg (NOD) that received BM cells from heterozygous 2m9L-Tg showed resistance to T1D as untreated heterozygous 2m9L-Tg (Fig. 1D), suggesting that BM-derived cells expressing huAIRE are responsible for the resistance to T1D. Conversely, heterozygous 2m9L-Tg receiving BM cells from non-Tg, albeit with a slightly lower incidence, developed T1D similarly to non-Tg that had received non-Tg BM cells, suggesting that huAIRE expression in mTECs had no major impact on the production of diabetogenic T cells in the thymus (Fig. 1D). The latter finding was rather unexpected because RNA-seq analysis showed that, in heterozygous 2m9L-Tg, the expression level of Ins2, a primary autoantigen in NOD (7), by mTECs was one-third of that in wild-type (WT) NOD [WT = 3.169 reads per kilo base pair of exon per million mapped sequence reads (RPKM) versus +/Tg = 1.003 RPKM]. Because all homozygous 2m9L-Tg die before 25 weeks of age due to severe muscle pathology (19), we were unable to carry out sufficiently long-term observations to determine whether homozygous 2m9L-Tg were also resistant to T1D. However, when BM cells from homozygous 2m9L-Tg were transferred into non-Tg, we found that those chimeras were resistant to T1D (Fig. 1E), strongly suggesting that untreated homozygous 2m9L-Tg might also be resistant to T1D if they were capable of surviving long enough for completion of diabetogenesis. Thus, opposite effects on autoimmunity (i.e. development of muscle-specific autoimmunity versus amelioration of T1D) were observed within the same homozygous 2m9L-Tg individual.
Defective presentation of β-islet antigens by peripheral APCs accounts for resistance to T1D in huAIRE-Tg
During the course of diabetogenesis, T cells generated in the thymus must become activated by recognizing β-islet antigens presented by peripheral APCs for autoimmune attack against β-islets. From the BM transfer experiments (Fig. 1D), we suspected that presentation of β-islet antigens by peripheral BM-derived APCs might be defective in 2m9L-Tg. To test this possibility, we utilized T cells from two types of TCR transgenic mice, NY8.3 (20) and BDC2.5 (21). NY8.3 CD8+ T cells and BDC2.5 CD4+ T cells are specific for islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP) (20) and hybrid peptides resulting from fusion between chromogranin A and insulin (27), respectively. When CFSE-labeled NY8.3 CD8+ T cells were transferred into non-Tg (NOD), we observed active proliferation of NY8.3 CD8+ T cells in the pancreatic LNs and pancreas, but not in inguinal LNs (Fig. 2A, left panels), suggesting that BM-derived APCs presenting the β-islet antigen IGRP were present in the pancreatic LNs and pancreas but not in inguinal LNs. This contrasts with the presentation of insulin peptide that occurs in LNs throughout the body (4). In contrast, NY8.3 CD8+ T cells transferred into heterozygous 2m9L-Tg showed significantly reduced proliferation in the pancreatic LNs and pancreas (Fig. 2A, right panels). Thus, presentation of β-islet antigens (IGRP) coupled with MHC-I to potentially diabetogenic CD8+ T cells was defective in 2m9L-Tg. NY8.3-Tg (NOD) have been reported to develop diabetes relatively earlier than non-Tg NOD mice (28). Indeed, three out of six NY8.3-Tg developed diabetes during the observation period of 8 weeks after birth. In contrast, when NY8.3-Tg were crossed with 2m9L-Tg, none out of seven developed diabetes during the same period (Minoru Matsumoto, Mitsuru Matsumoto and H. Nishijima, unpublished observation), which is consistent with the lack of proliferation of transferred NY8.3 CD8+ T cells in the pancreatic LNs and pancreas in heterozygous 2m9L-Tg (Fig. 2A). Transfer of BDC2.5 CD4+ T cells into heterozygous 2m9L-Tg also reduced the proliferation of BDC2.5 CD4+ T cells in pancreatic LNs relative to that in non-Tg (Fig. 2B). In this case, detection of BDC2.5 CD4+ T cells in the pancreas was difficult in heterozygous 2m9L-Tg, in contrast to non-Tg, possibly because of the poor antigenic stimulation of BDC2.5 CD4+ T cells in the pancreatic LNs. Thus, presentation of β-islet antigens (peptides that are hybrid forms between chromogranin A and insulin) complexed with MHC-II by peripheral APCs was also defective in 2m9L-Tg, together contributing to T1D resistance.
Fig. 2.

Defective presentation of β-islet antigens accounts for resistance to T1D in huAIRE-Tg. (A) Reduced proliferation of transferred NY8.3 CD8+ T cells in the pancreatic LNs (pLNs) and pancreas of heterozygous 2m9L-Tg. CFSE-labeled NY8.3 CD8+ T cells isolated from the spleen were i.v.-injected into non-Tg or heterozygous 2m9L-Tg. Sixty-four hours later, division of NY8.3 CD8+ T cells isolated from inguinal LNs (iLN), pLNs and the pancreas was assessed by flow cytometric analysis. One representative experiment from a total of two repeats is shown. (B) Reduced proliferation of transferred BDC2.5 CD4+ T cells in the pLNs, and dramatically reduced numbers of BDC2.5 CD4+ T cells in the pancreas of heterozygous 2m9L-Tg. CFSE-labeled BDC2.5 CD4+ T cells isolated from the spleen were i.v.-injected into non-Tg or heterozygous 2m9L-Tg. Sixty-four hours later, division of BDC2.5 CD4+ T cells isolated from iLNs, pLNs and the pancreas was assessed by flow cytometric analysis. One representative experiment from a total of two repeats is shown. (C) Splenic T cells isolated from pre-diabetic NOD at 15–18 weeks of age were i.v.-injected into Rag2-deficient non-Tg (n = 6), Rag2-deficient heterozygous 2m9L-Tg (+/Tg; n = 5) or Rag2-deficient homozygous 2m9L-Tg (Tg/Tg/; n = 3). Mice were then monitored for the development of T1D.
The defective presentation of β-islet antigens by peripheral APCs in 2m9L-Tg was further confirmed by transferring diabetogenic T cells into lymphopenic hosts. For this purpose, we generated Rag2-deficient heterozygous 2m9L-Tg to allow homeostatic expansion of diabetogenic T cells in the hosts. Diabetogenic T cells were harvested from the spleens of pre-diabetic NOD, and then transferred into Rag2-deficient non-Tg or Rag2-deficient heterozygous 2m9L-Tg recipients both on a NOD background. We found that all the Rag2-deficient non-Tg mice became diabetic after injection of diabetogenic T cells, as expected (Fig. 2C). In contrast, none of the Rag2-deficient heterozygous and homozygous 2m9L-Tg hosts developed T1D after this treatment. These results supported our notion that diabetogenic T cells were not activated by peripheral APCs to cause T1D in 2m9L-Tg.
BM-derived APCs other than B cells from huAIRE-Tg are responsible for defective presentation of β-islet antigens
Because B cells in our huAIRE-Tg mice expressed huAIRE (Fig. 1A), and B cells have been suggested to play a role in diabetogenesis in NOD (22, 29), we examined whether B cells are involved in the acquisition of resistance to T1D by crossing heterozygous 2m9L-Tg onto B-cell-deficient mice (Ighm-KO) on a NOD background (22). We found that none out of 18 female Ighm-KO/heterozygous 2m9L-Tg developed T1D, whereas control Ighm-KO developed T1D (Fig. 3A). The results suggested that B cells were irrelevant to this phenotype, and that BM-derived APCs expressing huAIRE other than B cells were responsible for the T1D resistance in heterozygous 2m9L-Tg.
Fig. 3.

DCs but not B cells from huAIRE-Tg are responsible for defective presentation of β-islet antigens. (A) Development of T1D in female B-cell-deficient NOD (Ighm-KO) (n = 6) and B-cell-deficient heterozygous 2m9L-Tg (Ighm-KO/heterozygous 2m9L-Tg) (n = 18). (B) Expression of huAIRE in CD45+CD11c+F4/80− DCs in spleen, thymus, iLNs and pLNs from heterozygous 2m9L-Tg. One representative experiment from a total of three repeats is shown. (C) Expression levels of huAIRE in DCs were compared with those in mTECs from Aire-reporter mice expressing the fusion gene of huAIRE, GFP and Flag (Aire/AGF-KI) (30)] (left). Expression of endogenous Aire from DCs and mTECs was determined by real-time PCR (right). The expression level of Hprt served as an internal control for RNAs. One representative experiment from a total of two repeats is shown. (D) BM cells isolated from B-cell-deficient heterozygous 2m9L-Tg and from NOD.scid were mixed together at a 1:1 ratio, and then transferred into non-Tg (NOD) (blue circle, n = 6). As a control, non-Tg receiving BM cells from B-cell-deficient heterozygous 2m9L-Tg (red square, n = 8) and non-Tg receiving BM cells from non-Tg (white circle, n = 5) were prepared. Mice were then monitored for the development of T1D.
We evaluated the transgenic huAIRE expression in DCs from heterozygous 2m9L-Tg using flow cytometry, and found that CD45+CD11c+F4/80−MHC-IIhigh DCs isolated from the spleen, thymus, and inguinal and pancreatic LNs expressed huAIRE (Fig. 3B): we noticed that not all of the MHC-IIhigh DCs expressed huAIRE, as we had observed for B cells as described above (Fig. 1A). We assume that some other factors beyond the MHC-II level allow DCs to express huAIRE at the protein level. The expression levels of huAIRE in DCs from heterozygous 2m9L-Tg detected by real-time PCR were higher than those in mTECs from Aire-reporter mice in which a fusion gene of huAIRE and GFP was knocked-in at the Aire locus [Aire/huAIRE-GFP-Flag knock-in (AGF-KI) (30)] (Fig. 3C, left). It has been reported that endogenous Aire expression by DCs is difficult to detect by flow cytometric analysis (31). However, real-time PCR analysis demonstrated Aire expression in DCs from both non-Tg controls and heterozygous 2m9L-Tg, albeit at much lower levels than those in mTECs (Fig. 3C, right). Given that peripheral BM-derived APCs from 2m9L-Tg are defective in the presentation of β-islet antigens, it was assumed that the presence of normal BM-derived APCs other than B cells should cancel the phenotype of resistance to T1D in 2m9L-Tg. We tested this idea by co-transferring BM cells from B-cell-deficient heterozygous 2m9L-Tg together with BM cells from non-Tg NOD.scid into non-Tg (NOD): BM cells from non-Tg NOD.scid provide WT DCs but not any T/B cells. Strikingly, all but one of the six mice receiving this treatment developed T1D (Fig. 3D): the single non-diabetic recipient also showed marked lymphoid cell infiltration into β-islets (Minoru Matsumoto, Mitsuru Matsumoto and H. Nishijima, unpublished observation). In contrast, non-Tg hosts receiving BM cells from B-cell-deficient heterozygous 2m9L-Tg alone showed resistance to the development of T1D (Fig. 3D), further supporting the idea that B cells expressing huAIRE were irrelevant to the T1D resistance revealed by the mouse crossing experiments (Fig. 3A). Non-Tg hosts receiving BM cells from non-Tg developed T1D, as expected. Thus, expression of huAIRE in peripheral BM-derived APCs, most likely DCs, was responsible for the defective presentation of β-islet antigens that is critical for the diabetogenic process in NOD.
Of note, BM-derived APCs from 2m9L-Tg did not show defective antigen presentation overall. Rather, defective presentation was confined to β-islet antigens because homozygous 2m9L-Tg are able to present muscle antigens for the development of muscle-specific autoimmunity (19). Furthermore, immunization with an exogenous antigen of KLH elicited anti-KLH antibody responses equally well in heterozygous 2m9L-Tg, as seen in non-Tg littermates (Supplementary Figure 2).
Effect of huAIRE expression on development of Xcr1+ DCs
It has been reported that pancreatic β-islets in young NOD (i.e. ~ 3 weeks of age) contain two sets of APCs: a major population of macrophages and a minor population of CD8α +CD103+ DCs (equivalent to Xcr1+ DCs) (23, 24), both populations being indispensable for the development of T1D in NOD (15). Interestingly, macrophages residing in β-islets have the capacity to present β-islet antigens, and play an initiating role in communication with diabetogenic T cells (24, 32). To investigate whether APCs in β-islets from 2m9L-Tg show this initial manifestation, we isolated pancreatic islets from 3-week-old heterozygous 2m9L-Tg and examined their APC composition. We found that these β-islets contained CD11cintermediate-highCD11b+F4/80+Xcr1− macrophages similar in percentages to those from non-Tg (Fig. 4A), suggesting that the initial event involving the macrophages was not affected by the expression of huAIRE in MHC-II+ BM-derived APCs.
Fig. 4.

Reduced numbers and altered function of Xcr1+ DCs in huAIRE-Tg. (A) Macrophages residing within β-islets were examined by flow cytometric analysis after isolating β-islets from heterozygous 2m9L-Tg at 3 weeks of age. Samples were gated for CD45+MHC-II+ cells. One representative experiment from a total of four repeats is shown. (B) Reduced numbers of Xcr1+ DCs in the spleen, thymus and pancreatic LNs (pLN) from heterozygous 2m9L-Tg compared with those from control littermates (left). Cells were gated for the CD45+CD11c+F4/80−MHC-II+ population. A summary of the results is shown on the right. One circle corresponds to one mouse analyzed. **P < 0.01. (C) Reduced percentages of Xcr1+ DCs in pLNs from heterozygous 2m9L-Tg, heterozygous 1m4L-Tg (+/Tg) and homozygous 1m4L-Tg (Tg/Tg) as analyzed in B (left). A summary of the results is shown on the right. One circle corresponds to one mouse analyzed. **P < 0.01; *P < 0.05.
A transcription factor Batf3 controls the differentiation of a subset of Xcr1+ DCs (CD8α +CD103+ DCs) in mice (33) and regulates the development of T1D in NOD (23). We examined Xcr1+ DCs in heterozygous 2m9L-Tg, and found that the percentages of Xcr1+ DCs were significantly reduced in the spleen, thymus and pancreatic LNs compared with those from non-Tg controls (Fig. 4B). Importantly, the degree of reduction of Xcr1+ DCs was correlated with the expression level of huAIRE: heterozygous 1m4L-Tg (intermediate line) demonstrated a milder reduction of Xcr1+ DCs in pancreatic LNs relative to heterozygous 2m9L-Tg (high line) (Fig. 4C). Furthermore, homozygous 1m4L-Tg (Tg/Tg) showed a more severe reduction of Xcr1+ DCs relative to heterozygous 1m4L-Tg (+/Tg), although the degree of reduction evident in homozygous 1m4L-Tg (Tg/Tg) was still milder than that shown by heterozygous 2m9L-Tg (+/Tg) (Fig. 4C).
We confirmed the effect of huAIRE expression in DCs on the production of Xcr1+ DCs by expanding CD11c+ DCs from BM cells in vitro: we cultured BM cells from wild-type WT NOD with FLT3 ligand for 8 days, and classified the viable cells into three major populations (Fig. 5A, top) (25): B220+CD11blow plasmacytoid DCs (pDCs), B220−CD11b+CD24− DCs (CD11b+ DCs) and B220−CD11b+CD24+ DCs: the third population expressed Xcr1 (Xcr1+ DCs) (Fig. 5A, top right). The percentages and absolute numbers of in vitro-expanded Xcr1+ DCs were reduced in heterozygous 2m9L-Tg and, to a lesser extent, in heterozygous 1m4L-Tg compared with those from non-Tg mice (Fig. 5A and B). Expression levels of Xcr1 were also reduced in heterozygous 2m9L-Tg (Fig. 5A, middle right). In contrast, pDCs in huAIRE-Tg were indistinguishable from those in non-Tg mice (Fig. 5B). Percentages of CD11b+ DCs showed a relative increase in huAIRE-Tg because of the reduced percentages of Xcr1+ DCs (Fig. 5B, upper), although the absolute numbers of CD11b+ DCs remained unchanged (Fig. 5B, lower). Thus, Xcr1+ DCs generated from BM cells with FLT3 ligand mirrored the development of Xcr1+ DCs in vivo (Fig. 4C). We evaluated the expression of huAIRE in in vitro-expanded DCs, and found that CD11b+ DCs and Xcr1+ DCs, but not pDCs, expressed huAIRE (Fig. 5C): both CD11b+ DCs and Xcr1+ DCs from heterozygous 2m9L-Tg expressed higher levels of huAIRE than those from heterozygous 1m4L-Tg. Taken together, these results suggested that production of Xcr1+ DCs, a crucial DC population for the development of T1D (23), was negatively regulated by huAIRE in a dose-dependent manner.
Fig. 5.

BM-derived DCs expanded from huAIRE-Tg in vitro reflect the developmental signature of DCs in vivo and diabetogenic pathology. (A) Three populations including pDCs (B220+CD11blow), CD11b+ DCs (B220−CD11b+CD24−) and Xcr1+ DCs (B220−CD11b+CD24+) were induced by treatment with BM cells from non-Tg, heterozygous 2m9L-Tg and heterozygous 1m4L-Tg with Flt3L. Expression of Xcr1 from each population (pDC, green line; CD11b+ DC, blue line; Xcr1+ DC, red line) is shown on the right. (B) A summary of the results in (A) is shown. The percentages (upper) and absolute numbers of Xcr1+ DCs (lower) were reduced in heterozygous 2m9L-Tg and, to a lesser extent, in heterozygous 1m4L-Tg. **P < 0.01; *P < 0.05. (C) Expression of huAIRE from in vitro-expanded DC populations was evaluated by flow cytometric analysis. One representative experiment from a total of two repeats is shown. (D) All the heterozygous 1m4L-Tg (n = 10) but one showed pathological changes in β-islets, whereas homozygous 1m4L-Tg showed significantly milder pathological changes (n = 8) compared with those from heterozygous 1m4L-Tg. Pathological changes in the pancreas were evaluated with H&E staining without being informed of the detailed condition of the individual mice. White column, no pathological change; half-filled column, peri-insulitis and/or peri-vasculitis; full-filled column, massive insulitis. M, male: F, female. All the mice were 7.5–8 months old. Mice shown in (E) are marked with asterisks. (E) Two representative examples (one female and one male) of histology from heterozygous and homozygous 1m4L-Tg summarized in (D) are shown. Scale bar: 100 µm.
Alterations of the production and transcriptome of Xcr1+ DCs account for resistance to T1D
Given that only the 2m9L-Tg line, which showed highest huAIRE expression and most severe reduction of Xcr1+ DCs among the three lines, acquired the resistance to T1D, we suspected that the degree of the reduction of Xcr1+ DCs is crucial for T1D resistance. We tested this idea by monitoring the development of T1D in 1m4L-Tg, heterozygous (+/Tg) individuals of which were susceptible to T1D (Fig. 1B) and homozygous (Tg/Tg) individuals of which showed more severe reduction of Xcr1+ DCs than heterozygous 1m4L-Tg (Fig. 4C). Most of the heterozygous 1m4L-Tg showed pathological changes in β-islets (Fig. 5D and E), and in fact more than half of the heterozygous 1m4L-Tg developed T1D (Fig. 1B). Strikingly, none of the homozygous 1m4L-Tg developed T1D (n > 8), and consequently showed significantly milder pathological changes in the β-islets (Fig. 5D and E). Thus, negative dose-dependent control of the number of Xcr1+ DCs through expression of huAIRE is a critical determinant of T1D development. We suggest that a certain number of Xcr1+ DCs in pancreatic LNs and/or the pancreas is necessary in order to elicit an autoimmune response against β-islets.
Although the reduced numbers of Xcr1+ DCs in pancreatic LNs might largely account for the defect in the presentation of β-islet antigens, and consequently for the resistance to T1D in huAIRE-Tg, we also wanted to evaluate transcriptomic changes in the remaining Xcr1+ DCs in heterozygous 2m9L-Tg. Because the low numbers of Xcr1+ DCs in pancreatic LNs did not allow us to directly examine the transcriptome of this subset, we utilized Xcr1+ DCs expanded from BM cells in vitro as described above. We obtained the data using triplicate samples from heterozygous 2m9L-Tg and control mice, and set FDR q <0.05 as the level of significance. We found that 38 genes were up-regulated whereas 15 genes were down-regulated in BM-derived Xcr1+ DCs from heterozygous 2m9L-Tg (Fig. 6A from DRA006501). The down-regulated genes included C-type lectins such as CD207 (Langerin) and CD209a (DC-SIGN), which are implicated in the development of T1D: NOD showed a decreased number of CD8+CD103+CD207+ DCs in the pancreas (34). It has been reported that CD209a polymorphisms are associated with the development of T1D in humans (35). Because we have performed RNA-seq analysis using in vitro-expanded Xcr1+ DCs from BM, expression levels of Xcr1 and Batf3 remained unchanged in this analysis (Supplementary Figure 3). Of note, key factors that control the development of Xcr1+ DCs such as Irf8, Id2 and Nfil3 (36) also remained unchanged by the expression of huAIRE.
Fig. 6.

Gene expression profiles from BM-derived DCs and pancreatic LNs in huAIRE-Tg. (A) Gene expression in in vitro-expanded Xcr1+ DCs from heterozygous 2m9L-Tg versus that from non-Tg, showing the 38 up-regulated genes and 15 down-regulated genes (colored). CD207 (Langerin) and CD209a (DC-SIGN) are highlighted in blue. Data were obtained from triplicate samples from each group of cells, and FDR q <0.05 was defined as significant. (B) Expression of Ins2 and Sap1 in DCs isolated from pancreatic LNs of heterozygous 2m9L-Tg was assessed by real-time PCR. The expression level of Hprt was used as an internal control for RNAs. RNAs from mTECs of non-Tg served as a positive control. One representative experiment from a total of two repeats is shown. N.D., not detected. (C) No expression of Ins2 in in vitro-expanded DCs from heterozygous 2m9L-Tg and 1m4L-Tg revealed by real-time PCR. RNAs from mTECs of non-Tg served as a positive control. One representative experiment from a total of four repeats is shown.
Although enforced expression of Aire in many cell types both in vitro (37, 38) and in vivo (39, 40) has been reported to induce ectopic expression of TRAs, neither B cells (19) nor DCs isolated from pancreatic LNs of heterozygous 2m9L-Tg expressed prototypic Aire-dependent TRAs (e.g. Ins2 and Sap1) in real-time PCR, in contrast to mTECs from non-Tg (Fig. 6B). Similarly, in vitro-expanded Xcr1+ DCs from both heterozygous 2m9L-Tg and 1m4L-Tg showed no Ins2 expression, as was the case for those from non-Tg (Fig. 6C). Taken together, these results suggest that reduced numbers of Xcr1+ DCs together with altered function of this subset caused by additive huAIRE expression are responsible for T1D resistance in huAIRE-Tg.
Maintenance of the diabetogenic T-cell pool requires continuous presentation of β-islet antigens in the periphery
Our data demonstrated that diabetogenic T cells were generated in the thymus from huAIRE-Tg as revealed by BM transfer experiments (Fig. 1D). However, these potentially harmful diabetogenic T cells did not attack β-islets because of the impaired development and/or function of Xcr1+ DCs that act in the periphery. We then investigated whether a diabetogenic T-cell pool is maintained in huAIRE-Tg in the absence of peripheral presentation of β-islet antigens. For this purpose, we transferred splenic T cells isolated from non-Tg (NOD) or heterozygous 2m9L-Tg into NOD.scid. In this case, peripheral APCs in the hosts (NOD.scid) can present β-islet antigens to the transferred diabetogenic T cells. As expected, splenic T cells isolated from non-Tg induced T1D in NOD.scid hosts (Fig. 7). In contrast, splenic T cells isolated from heterozygous 2m9L-Tg did not induce T1D in any hosts. Thus, in order for autoreactive T cells that have escaped thymic selection to induce T1D, they must be maintained in certain numbers and/or in a non-anergic state by continuous interaction with autoantigens presented in the periphery. These results suggest that it may be possible to prevent tissue-specific autoimmunity by attenuating the presentation of the corresponding autoantigens through manipulation of DC development via huAIRE/Aire expression.
Fig. 7.

Autoreactivity is maintained by continuous presentation of autoantigens in the periphery. Splenic T cells isolated from non-Tg, but not from heterozygous 2m9L-Tg, induced T1D when transferred into NOD.scid hosts. T cells from non-Tg into NOD.scid, n = 5; T cells from heterozygous 2m9L-Tg into NOD.scid, n = 5.
Discussion
In the present study, we have demonstrated that additive huAIRE expression in BM-derived APCs, but not in mTECs, rendered NOD mice resistant to T1D. This was because of defective activation of autoreactive T cells specific for β-islet antigens in the periphery caused by altered development and/or function of Xcr1+ DCs. There was an inverse correlation between the level of huAIRE expressed and the Xcr1+ DCs produced: the more huAIRE in BM-derived APCs, the lower the production of Xcr1+ DCs. Accordingly, only 2m9L-Tg, the highest-expressing huAIRE-Tg strain among the three with most severely reduced Xcr1+ DCs, showed resistance to T1D in a heterozygous state. However, when heterozygous 1m4L-Tg were bred onto their homozygous counterparts, mice resistant to T1D were produced, suggesting a negative gene-dosage effect of huAIRE on production of Xcr1+ DCs and T1D susceptibility. In contrast to the diabetogenic process described above, expression of huAIRE both in mTECs and BM-derived APCs was responsible for the development of muscle-specific autoimmunity (19). This process was also dependent on the dose of huAIRE: the more huAIRE in mTECs and BM-derived APCs, the more frequent the appearance of muscle-specific autoimmunity, as reflected in the fact that homozygous 2m9L-Tg and homozygous 1m4L-Tg developed muscle-specific autoimmunity in 100% and ~20% of individuals, respectively (19). Thus, tissue-specific autoimmunity was controlled by the types of APCs that express huAIRE together with the dose of huAIRE expressed in our model.
Development of muscle-specific autoimmunity and prevention of T1D occurred simultaneously in homozygous 2m9L-Tg, since BM-derived APCs from homozygous 2m9L-Tg rendered NOD resistant to T1D (Fig. 1E). Of note, this kind of ambivalence in the tissue-specific immune response is not confined to the transgenic expression of huAIRE demonstrated in the present study. In fact, we have reported previously that deletion of endogenous Aire also resulted in a similar ambivalent outcome: in NOD lacking Aire, autoimmune attack against pancreatic β-islets was completely abrogated, rendering them resistant to T1D, while de novo autoimmune attack against exocrine acinar cells developed (26). Thus, manipulation of Aire’s activity, either by additive huAIRE expression or by disruption of endogenous Aire, elicited similar ambivalence in the tissue-specific immune response. Clearly, therefore, further studies of autoimmunity control through mediation of huAIRE/Aire activity in tolerance mechanisms are warranted.
BM-derived APCs from both heterozygous and homozygous 2m9L-Tg did not present β-islet antigens to elicit T1D because of reduced numbers and/or altered function of Xcr1+ DCs, whereas BM-derived APCs from homozygous 2m9L-Tg presented muscle antigens to induce the muscle-specific immune response. Given that Xcr1+ DCs play an important role in the presentation of antigens transferred from other cells (41) and that transfer of insulin granules from β-islets to Xcr1+ DCs is a critical step for the diabetogenic process in NOD (42), Xcr1+ DCs play an indispensable role in the presentation of β-islet antigens to elicit T1D in the pancreas while other DC subsets can present muscle antigens to induce muscle-specific autoimmunity. Thus, we speculate that DC subsets involved in the induction of T1D and muscle-specific autoimmunity might be distinct in homozygous 2m9L-Tg. The involvement of distinct types of BM-derived APCs in the pathogenesis of tissue-specific autoimmunity was also manifested in the requirement of another BM-derived APC of B cells: B cells were irrelevant to acquisition of T1D resistance in heterozygous 2m9L-Tg, whereas muscle-specific autoimmunity was markedly improved when B cells were depleted from homozygous 2m9L-Tg (19).
Although expression of AIRE/Aire in DCs has been reported in both mice and humans, the precise roles of AIRE/Aire-expressing DCs in the immune response need to be further studied (43, 44). In this regard, one recent report has indicated that AIRE in DCs has no major impact on the expression of TRAs from DCs (45), in contrast to the AIRE in mTECs, suggesting that some other role of AIRE may exist, such as immune defense against Candida albicans (46). So far, to our knowledge, a role of AIRE/Aire in the lineage specification of DCs has not been reported. Interestingly, like Batf3-deficient mice (41), generation of DC subsets other than Xcr1+ DCs (i.e. pDCs and CD11b+ DCs) was not affected in huAIRE-Tg. The mechanism whereby a lack of Batf3 or additive huAIRE expression in DCs results in reduction of a particular subset of Xcr1+ DCs remains unknown. Several other transcription factors beyond Batf3, including Irf8, Id2 and Nfil3, have been reported to control the development of Xcr1+ DCs (36). Our transcriptomic analysis of BM-derived Xcr1+ DCs from heterozygous 2m9L-Tg revealed no major alterations in the expression levels of these key factors (Fig. 6A and Supplementary Figure 3), suggesting that huAIRE expression in DC precursors has an impact distinct from these transcription factors on the developmental program of DCs. The precise mechanism underlying the alteration of Xcr1+ DC development by additive huAIRE expression requires further study.
Kulshrestha and colleagues have reported that NOD mice that express mouse Aire under the control of the CD11c promoter (CD11c-Aire Tg) exhibited delayed onset and reduced incidence of T1D in comparison with non-Tg littermates (40). However, the degree of resistance to T1D in CD11c-Aire Tg was less prominent than that shown by our huAIRE-Tg: the cumulative incidence of diabetes was reported to be approximately 80% in non-Tg versus 50% in CD11c-Aire Tg when assessed at 40 weeks of age. Although the studies reported by Kulshrestha and our group both concluded that DCs transgenically expressing mouse Aire and huAIRE, respectively, were responsible for the acquisition of T1D resistance, there was a striking difference in the mechanisms to which those transgenic DCs contributed. Kulshrestha and colleagues found that DCs from CD11c-Aire Tg acquired the ability to express β-islet antigens including both Aire-dependent (i.e. Ins2) and Aire-independent (i.e. Gad67) TRAs. They reported that those ectopically expressed β-islet antigens in DCs actively induced ‘exhausted’ populations of CD4+ and CD8+ T cells in an antigen-specific manner (40). In marked contrast to these findings, our studies demonstrated that expression of huAIRE resulted in reduction of Xcr1+ DCs, thereby rendering them unable to present β-islet antigens to activate diabetogenic T cells in the pancreas and/or pancreatic LNs. This idea was confirmed by the fact that a co-population of BM-derived APCs from heterozygous 2m9L-Tg with WT DCs from NOD.scid in non-Tg recipients induced T1D. Furthermore, we did not observe any expression of Ins2 by DCs expressing huAIRE (Fig. 6B and C). The use of different species of Aire (mouse Aire versus huAIRE) and/or promoter (CD11c versus MHC-II promoter) for transgenic expression might have accounted for the mechanistic difference between the two studies. Because the mode of action of huAIRE/Aire in peripheral APCs is a critical issue when using huAIRE/Aire as a tolerance modifier for self-antigen-specific immune responses, precise elucidation of the mechanisms awaits further studies.
It is interesting to note that expression of IGRP (16) and BDC antigen (p31) (17), both under control of the Aire promoter, induced resistance to T1D in NOD when they were subjected to MHC-I-restricted NY8.3 TCR-Tg and MHC-II-restricted BDC2.5 TCR-Tg, respectively. In contrast to these studies, our huAIRE-Tg was not designed to express any particular β-islet antigens. Instead, huAIRE itself was expressed in thymic and peripheral APCs. Furthermore, the resistance to T1D in our huAIRE-Tg was observed in a polyclonal setting without any crossing with TCR-Tg specific for β-islet antigens. In the reports discussed above (16, 17), the authors demonstrated that IGRP and p31 expressed by BM-derived eTACs, which were distinct from conventional DCs, were responsible for the deletional tolerance and dominant tolerance for diabetogenic T cells in the periphery, respectively (16, 17). Clearly, the resistance to T1D in huAIRE-Tg is not caused by the manipulation of eTACs because the originally reported eTACs belong to a non-DC subset: CD45lowEpCAMhighCD11c−CD80lowCD86low (16, 17). Together, these results suggest that there are several different approaches for amelioration of T1D using huAIRE/Aire as a transgene itself and/or as a promoter to express β-islet-related antigens.
It has been clearly demonstrated that insulin plays a primary role in diabetogenesis in NOD: deletion of immunologically relevant insulin molecules completely abrogates the development of T1D (7). Conversely, overexpression of preproinsulin 2 in NOD using the MHC-II promoter, as employed in the present study, substantially reduces the onset and severity of T1D (47). In this case, tolerance was induced not only by the BM-derived APCs but also by radioresistant thymic epithelial cells (TECs) in these animals (47). The authors further demonstrated that overexpression of preproinsulin 2 from TECs did not induce the production of Tregs specific for preproinsulin 2 (i.e. dominant tolerance). Instead, they suggested that overexpression of preproinsulin 2 ensured recessive tolerance (i.e. clonal deletion) against preproinsulin 2 (47). Clearly, a difference in the transgenes (i.e. preproinsulin 2 versus huAIRE) rendered NOD resistant to T1D through different mechanisms, despite the fact that the same MHC-II promoter was used.
Given that huAIRE expression in mTECs resulted in reduced expression of Ins2 (19), the fact that mTECs from huAIRE-Tg had no major impact on the production of diabetogenic T cells revealed by BM chimeras (Fig. 1D) was somewhat unexpected. However, there is a precedent for Aire-deficient NOD in which reduced expression of Ins2 by mTECs conferred complete resistance to T1D (26, 48). One possible explanation for the discrepancy between the reduced expression of Ins2 from mTECs and the resistance to T1D in Aire-deficient NOD might be the production of anti-IFN-α autoantibody that could be protective against the diabetogenic process, as reported for patients with AIRE deficiency (49). However, production of anti-IFN-α autoantibody has not been observed in Aire-deficient mice, at least on C57BL/6 (50) and BALB/c (51) backgrounds. Furthermore, a link between resistance to T1D and production of anti-IFN-α autoantibody in APECED patients has been recently challenged (52). Thus, further studies are required to clarify the exact role of endogenous Aire in T1D pathogenesis.
Finally, mTECs expressing huAIRE were differentially involved in the production of autoreactive T cells with distinct tissue specificity. Muscle-specific T cells were generated only when mTECs expressed a high level of huAIRE, whereas diabetogenic T cells were generated irrespective of huAIRE expression in mTECs. Because huAIRE expression in BM-derived APCs also showed a contrasting outcome in terms of the tissue-specific immune response [i.e. amelioration of T1D (demonstrated in the present study) and a co-operative role in the induction of muscle-specific autoimmunity (19)], it might be possible to control the tissue-specific immune responses positively or negatively by manipulating the activity of huAIRE/Aire in either the thymic or peripheral tolerance mechanism selectively, or both simultaneously.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
Acknowledgements
We thank Profs T. Egawa and T. Kaisho for helpful discussions. We thank Ms F. Hirota and Mr R. Shinohara for technical assistance. We also thank members from Prof. Unanue’s laboratory at Washington University School of Medicine for their kind instruction how to isolate β-islets.
Funding
This work was supported in part by Japan Society for the Promotion of Science grant numbers JP16K21731, JP16H06496 and JP16H05342, by the Core Research for Evolutional Science and Technology (to Mitsuru Matsumoto), by Japan Society for the Promotion of Science grant number 19K16613 (to Minoru Matsumoto) and by Japan Society for the Promotion of Science grant number 17K08885 (to H.N.). The data set associated with this project has been submitted to DDBJ Sequence Reads Archive (DRA accession number: DRA006501).
Conflicts of interest statement: the authors declared no conflicts of interest.
References
- 1. Anderson M. S. and Bluestone J. A. 2005. The NOD mouse: a model of immune dysregulation. Annu. Rev. Immunol. 23:447. [DOI] [PubMed] [Google Scholar]
- 2. Unanue E. R. 2014. Antigen presentation in the autoimmune diabetes of the NOD mouse. Annu. Rev. Immunol. 32:579. [DOI] [PubMed] [Google Scholar]
- 3. Pearson J. A., Wong F. S. and Wen L. 2016. The importance of the non obese diabetic (NOD) mouse model in autoimmune diabetes. J. Autoimmun. 66:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wan X., Zinselmeyer B. H., Zakharov P. N. et al. 2018. Pancreatic islets communicate with lymphoid tissues via exocytosis of insulin peptides. Nature 560:107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Klein L., Kyewski B., Allen P. M. and Hogquist K. A. 2014. Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 14:377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Abramson J. and Anderson G. 2017. Thymic epithelial cells. Annu. Rev. Immunol. 35:85. [DOI] [PubMed] [Google Scholar]
- 7. Nakayama M., Abiru N., Moriyama H. et al. 2005. Prime role for an insulin epitope in the development of type 1 diabetes in NOD mice. Nature 435:220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bennett S. T., Wilson A. J., Esposito L. et al. 1997. Insulin VNTR allele-specific effect in type 1 diabetes depends on identity of untransmitted paternal allele. The IMDIAB Group. Nat. Genet. 17:350. [DOI] [PubMed] [Google Scholar]
- 9. Pugliese A., Zeller M., Fernandez A. Jr et al. 1997. The insulin gene is transcribed in the human thymus and transcription levels correlated with allelic variation at the INS VNTR-IDDM2 susceptibility locus for type 1 diabetes. Nat. Genet. 15:293. [DOI] [PubMed] [Google Scholar]
- 10. Thébault-Baumont K., Dubois-Laforgue D., Krief P. et al. 2003. Acceleration of type 1 diabetes mellitus in proinsulin 2-deficient NOD mice. J. Clin. Invest. 111:851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Moriyama H., Abiru N., Paronen J. et al. 2003. Evidence for a primary islet autoantigen (preproinsulin 1) for insulitis and diabetes in the nonobese diabetic mouse. Proc. Natl Acad. Sci. USA 100:10376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Mathis D. and Benoist C. 2009. Aire. Annu. Rev. Immunol. 27:287. [DOI] [PubMed] [Google Scholar]
- 13. Anderson M. S. and Su M. A. 2016. AIRE expands: new roles in immune tolerance and beyond. Nat. Rev. Immunol. 16:247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Constantine G. M. and Lionakis M. S. 2019. Lessons from primary immunodeficiencies: autoimmune regulator and autoimmune polyendocrinopathy-candidiasis-ectodermal dystrophy. Immunol. Rev. 287:103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Unanue E. R., Ferris S. T. and Carrero J. A. 2016. The role of islet antigen presenting cells and the presentation of insulin in the initiation of autoimmune diabetes in the NOD mouse. Immunol. Rev. 272:183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gardner J. M., Devoss J. J., Friedman R. S. et al. 2008. Deletional tolerance mediated by extrathymic Aire-expressing cells. Science 321:843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gardner J. M., Metzger T. C., McMahon E. J. et al. 2013. Extrathymic Aire-expressing cells are a distinct bone marrow-derived population that induce functional inactivation of CD4⁺ T cells. Immunity 39:560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yamano T., Nedjic J., Hinterberger M. et al. 2015. Thymic B cells are licensed to present self antigens for central T cell tolerance induction. Immunity 42:1048. [DOI] [PubMed] [Google Scholar]
- 19. Nishijima H., Kajimoto T., Matsuoka Y. et al. 2018. Paradoxical development of polymyositis-like autoimmunity through augmented expression of autoimmune regulator (AIRE). J. Autoimmun. 86:75. [DOI] [PubMed] [Google Scholar]
- 20. Anderson B., Park B. J., Verdaguer J., Amrani A. and Santamaria P. 1999. Prevalent CD8(+) T cell response against one peptide/MHC complex in autoimmune diabetes. Proc. Natl Acad. Sci. USA 96:9311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tourne S., Nakano N., Viville S., Benoist C. and Mathis D. 1995. The influence of invariant chain on the positive selection of single T cell receptor specificities. Eur. J. Immunol. 25:1851. [DOI] [PubMed] [Google Scholar]
- 22. Noorchashm H., Lieu Y. K., Noorchashm N. et al. 1999. I-Ag7-mediated antigen presentation by B lymphocytes is critical in overcoming a checkpoint in T cell tolerance to islet beta cells of nonobese diabetic mice. J. Immunol. 163:743. [PubMed] [Google Scholar]
- 23. Ferris S. T., Carrero J. A., Mohan J. F., Calderon B., Murphy K. M. and Unanue E. R. 2014. A minor subset of Batf3-dependent antigen-presenting cells in islets of Langerhans is essential for the development of autoimmune diabetes. Immunity 41:657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Carrero J. A., McCarthy D. P., Ferris S. T. et al. 2017. Resident macrophages of pancreatic islets have a seminal role in the initiation of autoimmune diabetes of NOD mice. Proc. Natl Acad. Sci. USA 114:E10418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hemmi H., Hoshino K. and Kaisho T. 2016. In vivo ablation of a dendritic cell subset expressing the chemokine receptor XCR1. Methods Mol. Biol. 1423:247. [DOI] [PubMed] [Google Scholar]
- 26. Niki S., Oshikawa K., Mouri Y. et al. 2006. Alteration of intra-pancreatic target-organ specificity by abrogation of Aire in NOD mice. J. Clin. Invest. 116:1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Delong T., Wiles T. A., Baker R. L. et al. 2016. Pathogenic CD4 T cells in type 1 diabetes recognize epitopes formed by peptide fusion. Science 351:711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Verdaguer J., Schmidt D., Amrani A., Anderson B., Averill N. and Santamaria P. 1997. Spontaneous autoimmune diabetes in monoclonal T cell nonobese diabetic mice. J. Exp. Med. 186:1663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Wong F. S., Wen L., Tang M. et al. 2004. Investigation of the role of B-cells in type 1 diabetes in the NOD mouse. Diabetes 53:2581. [DOI] [PubMed] [Google Scholar]
- 30. Kawano H., Nishijima H., Morimoto J. et al. 2015. Aire expression is inherent to most medullary thymic epithelial cells during their differentiation program. J. Immunol. 195:5149. [DOI] [PubMed] [Google Scholar]
- 31. Hubert F. X., Kinkel S. A., Webster K. E. et al. 2008. A specific anti-Aire antibody reveals Aire expression is restricted to medullary thymic epithelial cells and not expressed in periphery. J. Immunol. 180:3824. [DOI] [PubMed] [Google Scholar]
- 32. Ferris S. T., Zakharov P. N., Wan X. et al. 2017. The islet-resident macrophage is in an inflammatory state and senses microbial products in blood. J. Exp. Med. 214:2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Edelson B. T., Wumesh K. C., Juang R. et al. 2010. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J. Exp. Med. 207:823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Welzen-Coppens J. M., van Helden-Meeuwsen C. G., Leenen P. J., Drexhage H. A. and Versnel M. A. 2012. Reduced numbers of dendritic cells with a tolerogenic phenotype in the prediabetic pancreas of NOD mice. J. Leukoc. Biol. 92:1207. [DOI] [PubMed] [Google Scholar]
- 35. da Silva R. C., Cunha Tavares N. A., Moura R. et al. 2014. DC-SIGN polymorphisms are associated to type 1 diabetes mellitus. Immunobiology 219:859. [DOI] [PubMed] [Google Scholar]
- 36. Murphy T. L., Grajales-Reyes G. E., Wu X. et al. 2016. Transcriptional control of dendritic cell development. Annu. Rev. Immunol. 34:93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Abramson J., Giraud M., Benoist C. and Mathis D. 2010. Aire’s partners in the molecular control of immunological tolerance. Cell 140:123. [DOI] [PubMed] [Google Scholar]
- 38. Kont V., Laan M., Kisand K., Merits A., Scott H. S. and Peterson P. 2008. Modulation of Aire regulates the expression of tissue-restricted antigens. Mol. Immunol. 45:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Guerau-de-Arellano M., Mathis D. and Benoist C. 2008. Transcriptional impact of Aire varies with cell type. Proc. Natl Acad. Sci. USA 105:14011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kulshrestha D., Yeh L. T., Chien M. W., Chou F. C. and Sytwu H. K. 2017. Peripheral autoimmune regulator induces exhaustion of CD4+ and CD8+ effector T cells to attenuate autoimmune diabetes in non-obese diabetic mice. Front. Immunol. 8:1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hildner K., Edelson B. T., Purtha W. E. et al. 2008. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science 322:1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vomund A. N., Zinselmeyer B. H., Hughes J. et al. 2015. Beta cells transfer vesicles containing insulin to phagocytes for presentation to T cells. Proc. Natl Acad. Sci. USA 112:E5496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ryan K. R., Hong M., Arkwright P. D. et al. 2008. Impaired dendritic cell maturation and cytokine production in patients with chronic mucocutaneous candidiasis with or without APECED. Clin. Exp. Immunol. 154:406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindmark E., Chen Y., Georgoudaki A. M. et al. 2013. AIRE expressing marginal zone dendritic cells balances adaptive immunity and T-follicular helper cell recruitment. J. Autoimmun. 42:62. [DOI] [PubMed] [Google Scholar]
- 45. Fergusson J. R., Morgan M. D., Bruchard M. et al. 2018. Maturing human CD127+ CCR7+ PDL1+ dendritic cells express AIRE in the absence of tissue restricted antigens. Front. Immunol. 9:2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. de Albuquerque J. A. T., Banerjee P. P., Castoldi A. et al. 2018. The role of AIRE in the immunity against Candida albicans in a model of human macrophages. Front. Immunol. 9:567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jaeckel E., Lipes M. A. and von Boehmer H. 2004. Recessive tolerance to preproinsulin 2 reduces but does not abolish type 1 diabetes. Nat. Immunol. 5:1028. [DOI] [PubMed] [Google Scholar]
- 48. Jiang W., Anderson M. S., Bronson R., Mathis D. and Benoist C. 2005. Modifier loci condition autoimmunity provoked by Aire deficiency. J. Exp. Med. 202:805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Meyer S., Woodward M., Hertel C. et al. ; APECED Patient Collaborative 2016. AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies. Cell 166:582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Hubert F. X., Kinkel S. A., Crewther P. E. et al. 2009. Aire-deficient C57BL/6 mice mimicking the common human 13-base pair deletion mutation present with only a mild autoimmune phenotype. J. Immunol. 182:3902. [DOI] [PubMed] [Google Scholar]
- 51. Kärner J., Meager A., Laan M. et al. 2013. Anti-cytokine autoantibodies suggest pathogenetic links with autoimmune regulator deficiency in humans and mice. Clin. Exp. Immunol. 171:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Landegren N., Rosen L. B., Freyhult E. et al. 2019. Comment on ‘AIRE-deficient patients harbor unique high-affinity disease-ameliorating autoantibodies’. Elife. 8. pii: e43578. doi: 10.7554/eLife.43578 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.