

Abstract
Background
A causal genetic mutation is found in 40% of families with dilated cardiomyopathy (DCM), leaving a large percentage of families genetically unsolved. This prevents adequate counseling and clear recommendations in these families. We aim to identify novel genes or modifiers associated with DCM.
Methods
We performed computational ranking of human genes based on coexpression with a predefined set of genes known to be associated with DCM, which allowed us to prioritize gene candidates for their likelihood of being involved in DCM. Top candidates will be checked for variants in the available whole‐exome sequencing data of 142 DCM patients. RNA was isolated from cardiac biopsies to investigate gene expression.
Results
PDLIM5 was classified as the top candidate. An interesting heterozygous variant (189_190delinsGG) was found in a DCM patient with a known pathogenic truncating TTN‐variant. The PDLIM5 loss‐of‐function (LoF) variant affected all cardiac‐specific isoforms of PDLIM5 and no LoF variants were detected in the same region in a control cohort of 26,000 individuals. RNA expression of PDLIM5 and its direct interactors (MYOT, LDB3, and MYOZ2) was increased in cardiac tissue of this patient, indicating a possible compensatory mechanism. The PDLIM5 variant cosegregated with the TTN‐variant and the phenotype, leading to a high disease penetrance in this family. A second patient was an infant with a homozygous 10 kb‐deletion of exon 2 in PDLIM5 resulting in early‐onset cardiac disease, showing the importance of PDLIM5 in cardiac function.
Conclusions
Heterozygous PDLIM5 variants are rare and therefore will not have a major contribution in DCM. Although they likely play a role in disease development as this gene plays a major role in contracting cardiomyocytes and homozygous variants lead to early‐onset cardiac disease. Other environmental and/or genetic factors are probably necessary to unveil the cardiac phenotype in PDLIM5 mutation carriers.
Keywords: dilated cardiomyopathy, genetic modifier, genetics, sarcomere
A computational ranking‐based method performed in whole exome sequencing data of dilated cardiomyopathy patient put PDLIM5 forward as novel gene implicated in this cardiac disease.
1. INTRODUCTION
Dilated cardiomyopathy (DCM) is a leading cause of heart failure (HF) and the most frequent cause for cardiac transplantation (Japp, Gulati, Cook, Cowie, & Prasad, 2016). This condition is a complex final phenotype resulting from genetic and environmental triggers (Verdonschot Job, Hazebroek Mark, Ware James, Prasad Sanjay, & Heymans Stephane, 2019). Increasing diagnostic possibilities such as advanced imaging and genetic testing allow better assessment of etiologies contributing to this phenotype in individual patients as a first step toward personalized medicine.
The genetic basis of DCM is an area of great interest as it is likely to explain the observed differences in disease susceptibility between individuals exposed to the same environmental triggers. Currently, over 60 causal genes are described in DCM of which truncating mutations in TTN (OMIM: *188840) are the most prevalent (Japp et al., 2016). Clinical utility of cardio‐genetics is still limited due to incomplete disease penetrance and clinical variability in genetic DCM families. Also, the currently used monogenetic model only explains only up to ~40% in familial cases (Hershberger, Hedges, & Morales, 2013). Therefore, a large percentage of DCM families remain currently unsolved. Interaction between genetic variants can modify the disease, illustrated by a family with a more severe phenotype in the relatives having a mutation in both TTN and LMNA (OMIM: *150330) (Roncarati et al., 2013). This ‘double hit’ phenomenon illustrates the potential of current unknown DCM‐associated genes to modify the disease in patients, explaining the phenotypic variability within families. Large genome‐wide association studies (GWAS) in DCM try to provide further insight in identifying modifying variants without panel restriction. Several interesting single nucleotide polymorphisms (SNP) have been identified in a large variety of genes, potentially modifying the penetrance and susceptibility of DCM (Meder et al., 2014; Villard et al., 2011). However, the large variation among genes with rare variants and their unknown interactions make interpretation and implementation difficult. We therefore believe that “double hit” approach can help us to unravel the gene‐disease interaction network.
This study puts forward new DCM‐associated genes in whole‐exome data using weighted ranking for coexpression with known DCM‐causing genes and careful classification of found variants.
2. METHODS
2.1. DCM cohort
This study consisted of 142 unrelated familial and nonfamilial DCM patients from the Maastricht Cardiomyopathy Registry with inclusion and exclusion criteria as described previously (Verdonschot et al., 2018). In short, both DCM or hypokinetic nondilated cardiomyopathy (HNDC; also called isolated LV dysfunction) according to the latest European Society of Cardiology (ESC) proposal were included (DCM defined as left ventricular ejection fraction (LVEF) <50% with an indexed left ventricular end diastolic diameter (LVEDDi) >33 mm m−2 (men) or >32 mm m−2 (women) measured by echocardiography; and HNDC defined as LVEF <50% with an LVEDDi ≤33 mm m−2 (men) or ≤32 mm m−2 (women) measured by echocardiography) in the absence of a (a) history of myocardial infarction and/or significant coronary artery disease; (b) primary valvular disease; (c) hypertensive or congenital heart disease; (d) acute myocarditis; (e) arrhythmogenic right ventricular dysplasia; (f) hypertrophic or restrictive cardiomyopathy (Pinto et al., 2016). As part of the protocol, patients were referred to the clinical cardio‐genetics department of the Maastricht University Medical Center (MUMC) for genetic counseling and DNA testing (with informed consent) between 2012 and 2018. Genetic analysis was performed using whole‐exome sequencing (WES) in all included patients.
All patients underwent a physical examination, blood sampling, 12‐lead electrocardiogram, 24‐hr Holter monitoring, a complete echocardiographic and Doppler evaluation and coronary angiography (CAG) at baseline. Endomyocardial biopsies (EMB) were performed upon discretion of the treating physician and consent of the patient. The study was performed according to the declaration of Helsinki and was approved by the Medical Ethics Committee of Maastricht University Medical Centre. All patients gave written informed consent.
2.2. Genetic analysis
Patients at the cardio‐genetics outpatient clinic in Maastricht received genetic counseling and testing using our 47‐cardiomyopathy gene panel with WES (Table S1). Exome sequencing was performed on a Illumina HiSeq machine with a minimum coverage of 20×. A family history of cardiac‐related disease and sudden cardiac death was obtained by pedigree analysis. Familial inheritance was defined as recommended by the ESC: (a) two or more individuals (first or second degree relatives) have DCM or HNDC fulfilling diagnostic criteria for “definite” disease OR (b) in the presence of an index patient fulfilling diagnostic criteria for DCM/HNDC and a first‐degree relative with autopsy‐proven DCM and sudden death at <50 years of age (Pinto et al., 2016).
Genetic variants were carefully and stringently classified in five different classes: pathogenic, likely pathogenic, variant of clinical unknown significance (VUS), likely benign or benign based on the criteria as proposed by the ACMG guidelines (Richards et al., 2015). Classification of variants was based on the score of in silico prediction software scores (SIFT, MutationTaster, PolyPhen‐2, PhyloP, Align‐GVGD), the frequency in reference population databases (gnomAD, 1,000 genomes, ESP projects), functional studies and previously published variations in NCBI's ClinVar and HGMD. Both pathogenic and likely pathogenic mutations are reported here as pathogenic mutations. All others were considered nonpathogenic. Only truncating mutations with a PSI score >99% in TTN were classified as pathogenic (Ware & Cook, 2018).
The infant carrying a homozygous deletion of exon 2 of PDLIM5 (OMIM: *605904) was treated at Sanford Medical Center Fargo and referred to Rady Children's Institute for Genomic Medicine (RCIGM) for whole‐genome sequencing and variant analysis. Sequencing was performed on a NovaSeq6000 (Illumina) to ~50× coverage, and rapid alignment and nucleotide variant calling was performed using the Dragen (Edico Genome) hardware and software. Structural variants were identified with Manta and CNVnator, then filtered to retain those affecting coding regions of known disease genes and with allele frequencies <2% in the RCIGM database. Multiplex ligation‐dependent probe amplification was used to orthogonally confirm the deletion in the proband (homozygous state) and both parents (heterozygous state). The research groups used GeneMatcher to identify genotypic and phenotypic overlap of their patients (Sobreira, Schiettecatte, Valle, & Hamosh, 2015).
2.3. Control cohort
To compare the frequencies of the genetic variants in candidate genes we collected variant data from ExAC and GnomAD database (Lek et al., 2016). We additionally collected truncating variants from our internal cohort of patients with intellectual disabilities (ID) and their parents (n = 26,000), of whom to a large extend represent similar genetic background as our 142 patients. No cardiomyopathy was reported among ID patients and their parents with a truncating PDLIM5 mutation.
2.4. Network ranking
To support the association of gene candidates with DCM in mammals, we investigated the coexpression of the genes with a predefined set of genes known to be associated with DCM: Z‐disk or cardiac sarcomere cellular component and associated genes, that is genes that might be essential for the initial assembly, stabilization, and functional integrity of the titin filament, but also ion channels, enzymes, nuclear, and desmosomal genes (Table S1). We used expression datasets from total ∼30,000 expression measurements in ~500 murine and ~500 human gene expression datasets collected from the Gene Expression Omnibus (GEO) (Barrett et al., 2013). We selected expression datasets that coregulate with a DCM gene set (query genes) (Szklarczyk et al., 2016). Ranking the coexpression of human genes allowed us to prioritize gene candidates for their likelihood of being involved in DCM. In short, the transcriptome measurements (from GEO) are converted into a correlation matrix. The average correlation with the query set (sgene) is used for gene ranking and the dataset weight calculation. Gene scores sgene from all datasets are combined taking into account the precomputed weights. Subsequently different transcriptome platforms and species data are integrated to arrive at the final ranking. The process is repeated after excluding each query gene to construct a receiver operating characteristic (ROC) curve that visualizes predictive power of the method for a specific query set of genes. All these steps are further elucidated in the methods paper (Szklarczyk et al., 2016).
After the network ranking and prioritization based on known DCM‐associated genes, all previously described genes were excluded, leaving only unreported gene candidates for human DCM. Selection of top gene candidates was based on the combined position on the Z‐disk and sarcomere ranking list. As most top genes were already reported in human DCM, even genes ranked >100 were selected. The created shortlist of genes was further prioritized based on: (a) type of mutation (missense or loss‐of‐function [LoF]; which is defined as any variant which leads to truncation or loss of a protein); (b) location and function of the corresponding protein (potential for DCM development); (c) tissue expression (GTEx: heart/skeletal muscle); and (d) animal models (gene knockout and cardiac disease development). The final list of gene candidates was checked for variants in the WES data of the patients and subsequent variants were analyzed according to the ACMG diagnostic guidelines for variant classification as specified before.
2.5. Gene expression analysis by RT‐PCR
RNA extraction was performed using the miRVana™ miRNA isolation kit (Ambion Life Technologies #AM1560) following the manufacturer's instructions for total RNA isolation. Specifically, EMBs were thawed on ice in 1ml Lysis/Binding buffer followed by homogenization using the TissueLyser II (Qiagen #85300) using the setting 50 oscillations/second for 5 min. The RNA was eluted in 50 µl nuclease free water that had been preheated to 95°C. cDNA synthesis was performed from 100 ng RNA per sample using the Superscript II reverse transcription kit (Thermo Scientific™ #18064014) following the manufacturer's instructions and using a T100™ Thermal Cycler (BIO‐RAD, #1861096). The final cDNA synthesis reaction was diluted 1:10 with nuclease free water prior to qPCR. Primers were ordered from Eurogentec with purification by desalting and designed to measure mature mRNA by spanning exon‐exon boundaries, with a melting temperature (Tm) of between 58°C–62°C and the maximum Tm difference of the primer pair being 0.5°C, a GC content of the primers between 45%–61% and a product size between 90–150 bp. In addition, the absence of primer secondary structure and predicted primer dimers was verified in silico. Furthermore, in silico PCR was performed to confirm absence of nonspecific DNA amplification using the web‐based UCSC In‐Silico PCR programme (http://genome.ucsc.edu/cgi-bin/hgPcr). Each RT‐qPCR reaction was performed in technical triplicate, with the mean Ct value of the triplicates taken forward into subsequent analysis when the values are all within 0.6 Ct. QPCR was performed using a CFX96 Touch™ Real‐Time PCR Detection System (BIO‐RAD #1855195), iQ™ Supermix (BIO‐RAD #1708860), a final primer concentration of 200 nM and PCR programme: 95°C 3 min, (95°C 15 s, 60°C 1 min) × 45 cycles. Expression data of targets were normalized to the geometric mean of three reference genes, GAPDH, SDHA, and TBP, which demonstrated stability between samples from the same amount of starting material.
2.6. Statistical analysis
Statistical analysis was performed in GraphPad Prism v7 by means of a one‐way ANOVA with post hoc Tukey analysis for multiple comparisons for each target gene analyzed. Changes in expression were accepted to be significant when the p‐value was below .05 unless otherwise stated. Statistical stars on data figures represent *p ≤ = .05, **p = ≤ .01, ***p = ≤ .001.
3. RESULTS
3.1. Patient population
The baseline characteristics of our DCM cohort are shown in Table 1. Only patients who had genetic analysis using WES were included, irrespective of the presence of a pathogenic mutation or other etiology. Twenty‐six percent (37/142) of patients had a pathogenic mutation of which 46% (17/37) had at least one relative with DCM. Fifteen percent (22/142) of the total population had familial DCM without a proven mutation.
Table 1.
Baseline characteristics of the included DCM patients
| Parameter | Total cohort (n = 142) |
|---|---|
| Female gender | 45 (32%) |
| Age | 51 ± 12 |
| Etiologies and phenotype | |
| Familial disease | 39 (27%) |
| Muscle involvement | 8 (6%) |
| Genetic mutation | 37 (26%) |
| Electrical disease | 47 (33%) |
| Toxic trigger | 8 (6%) |
| Peripartum | 3 (2%) |
| Systemic | 12 (8%) |
| Inflammation | 37 (26%) |
| Viral | 11 (8%) |
| Viral‐positive inflammation | 8 (6%) |
| Arrhythmias | |
| AV block | 18 (13%) |
| Left bundle branch block | 45 (32%) |
| Nonsustained VT | 44 (31%) |
| Atrial fibrillation | 37 (26%) |
| Outcome | |
| Pacemaker | 7 (5%) |
| ICD | 23 (16%) |
| CRT‐D | 39 (27%) |
| HF rehospitalization | 13 (9%) |
| Death | 11 (8%) |
| Heart transplantation | 2 (1%) |
| Life‐threatening arrhythmia | 23 (16%) |
| Baseline echocardiography | |
| LVEF | 30 ± 11 |
| LVEDD | 62 ± 9 |
| LVESD | 53 ± 11 |
| IVS | 9 ± 2 |
| PW | 9 ± 1 |
| EA ratio | 1.2 ± 0.6 |
Abbreviations: AV, atrioventricular; CRT‐D, cardiac resynchronization therapy device; HF, heart failure; DCM, dilated cardiomyopathy; ICD, implantable cardiac defibrillator; IVS, interventricular septum; LVEDD, left ventricular end‐diastolic diameter; LVEF, left ventricular ejection fraction; LVESD, left ventricular end‐systolic diameter; PW, posterior wall thickness; VT, ventricular tachycardia.
3.2. Ranking of newly discovered candidate genes
After the network ranking and prioritization based on known DCM‐associated genes, we selected 21 genes which were not previously associated with DCM in patients (Table 2). The five most promising genes were selected based on the ranking of coexpression with DCM genes, type of mutation, biological context (location and function), tissue expression of the gene, and potential evidence from animal knockout models (Tables 2 and 3). All variants in these five genes were analyzed according to the ACMG diagnostic guidelines for variant classification, resulting in a list of interesting variants (ACMG class 2/3 or 3): PDLIM5 (n = 2 variants), CMYA5 (n = 2 variants), SPEG (n = 3 variants), SYNM (n = 2 variants), and ALPK3 (n = 1 variant) (Table 3). PDLIM5 was chosen as top candidate considering all collected data (Tables 2 and 3), and an additional case of an infant with a homozygous PDLIM5 deletion.
Table 2.
Information of the top 20 selected genes after ranking all genes based on coexpression with DCM‐associated genes
| Gene | Ranking | Number of variants | Type of mutations | Protein | Protein location/function | Expression | Animal studies | Gene tolerance | |||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Sarcomere | Z‐disk | Heart | Skeletal muscle | pLi | Z‐score | ||||||
| PDLIM5 | 25 | 9 | 5 | 2/5 LoF | PDZ and LIM domain protein 5 | Z‐disk | High | High | KO mouse: DCM | 0.01 | −0.47 |
| MYOM1 | 16 | 37 | 8 | All missense | Myomesin‐1 | Sarcomere | High | High | — | 0 | −0.35 |
| CKM | 28 | 31 | 4 | 2/4 LoF | Creatine kinase, muscle | Cytoplasm | Medium | High | — | 0.01 | 0.95 |
| CMYA5 | 30 | 23 | 14 | 1/14 LoF | Cardiomyopathy associated protein 5 | Anchor protein for PKA | Medium | High | — | 0 | −2.73 |
| PDE4DIP | 32 | 33 | 27 | 1/27 LoF | Phosphodiesterase 4D interacting protein | Anchor protein for cAMP‐dependent pathway | Medium | High | — | — | — |
| TXLNB | 33 | 16 | 5 | All missense | Beta‐taxilin | Promotes motor nerve regeneration | Medium | High | — | 0 | −0.91 |
| MYLK3 | 43 | 92 | 5 | All missense | Myosin light chain kinase 3 | Kinase that phosphorolates MYL2 | High | Medium | KO mouse: DCM | 0.14 | −0.25 |
| LMOD3 | 48 | 26 | 6 | All missense | Leiomodin‐3 | Sarcomere | Low | Low | Only for LMOD2: DCM | 0.01 | −2.05 |
| MYOM2 | 55 | 75 | 16 | All missense | Myomesin‐2 | Sarcomere | High | High | — | 0 | −5.87 |
| MYH11 | 72 | 190 | 7 | All missense | Myosin‐11 | Sarcomere | Absent | Absent | KO mouse: no cardiac phenotype | 1 | 3.15 |
| XIRP1 | 73 | 89 | 10 | All missense | Xin actin binding repeat containing 1 | Sarcomere | Low | Low | KO mouse: myopathy | 0 | −1.7 |
| DMPK | 75 | 131 | 6 | All missense | Dystrophia myotonica protein kinase | Kinase necessary maintaining muscle structure | Medium | Medium | KO mouse: decreased contractility | 0.03 | 1.47 |
| ADCK3 | 84 | 74 | 4 | 1/4 LoF | aarF domain containing kinase 3 | Mitochondrial respiratory chain | Low | High | — | 0 | −1.6 |
| SORBS1 | 87 | 143 | 7 | All missense | Sorbin and SH3 domain containing 1 | Involved in cytoskeletal formation | Low | Low | — | 0 | 0.3 |
| TNXB | 92 | 469 | 34 | 1/34 LoF | Tenascin XB | Extracellular matrix | Low | Low | — | 0.77 | 3 |
| ALPK3 | 92 | 51 | 12 | All missense | Alpha kinase 3 | Cardiomyocyte differentiation | Medium | High | — | 0 | 0.31 |
| OBSCN | 105 | 395 | 200 | 4/200 LoF | Obscurin | Z‐disk | Medium | High | — | −1 | 0 |
| SORBS2 | 109 | 795 | 4 | All missense | Sorbin and SH3 domain containing 2 | Involved in cytoskeletal formation | Medium | Low | — | 0.09 | 0.38 |
| SPEG | 111 | 355 | 22 | All missense | Striated muscle enriched protein kinase | Myocyte cytoskeletal development | Low | Medium | KO mouse: HF | 1 | 6 |
| SYNM | 116 | 84 | 14 | All missense | Synemin | Intermediate filament within the cytoskelet | Low | Medium | KO mouse: HF | 0 | −0.15 |
| XIRP2 | 117 | 128 | 20 | 4/20 LoF | Xin actin binding repeat containing 2 | Sarcomere | Low | High | — | 0 | −5 |
Abbreviations: DCM, dilated cardiomyopathy; KO, knockout; LoF, loss of function; PKA, protein kinase A.
Table 3.
All class 3 variants in the top 5 DCM‐candidate genes
| Gene | Nucleotide change | Amino acid change | PhyloP | CADD_PHRED | Grantham Score | gnoMAD | Classa |
|---|---|---|---|---|---|---|---|
| ALPK3 | c.4447C > T | p.Arg1483Trp | 0.104 | 18.9 | 101 | 7/246240 | 3 |
| CMYA5 | c.8161A > T | p.Lys2721* | 2.513 | 49 | 1,000 | 2/276328 | 3 |
| CMYA5 | c.8885T > G | p.Ile2962Ser | 6.939 | 19.61 | 142 | 0 | 2/3 |
| PDLIM5 | c.189_190delinsGG | p.Leu64Glyfs*15 | 1.665 | 19.85 | 1,000 | 0 | 3 |
| PDLIM5 | c.1333C > T | p.Arg445* | 1.874 | 40 | 1,000 | 1/30906 | 3 |
| SPEG | c.437G > C | p.Arg146Pro | 2.477 | 17.55 | 103 | 0 | 3 |
| SPEG | c.7442C > T | p.Ser2481Leu | 3.364 | 10.74 | 145 | 1/30942 | 2/3 |
| SPEG | c.7442C > T | p.Ser2481Leu | 3.364 | 10.74 | 145 | 1/30942 | 2/3 |
| SYNM | c.1929G > A | p.Val643Ile | 4.667 | 24.9 | 0 | 8/277020 | 2/3 |
| SYNM | c.2838C > T | p.Arg946Trp | 1.89 | 19.32 | 0 | 19/266622 | 2/3 |
Classification according to the ACMG guidelines.
3.3. Cardiac phenotype in an infant with a homozygous PDLIM5 deletion
In a family from native American origin, the firstborn child of healthy consanguineous parents was born at 37 weeks gestation (Figure 1b). The newborn fed poorly. By 3 days old, he was lethargic, cyanotic, and in respiratory distress. Echocardiography revealed a large patent ductus arteriosus, small ventricular septal defect with left to right shunt, small atrial septal defect with bidirectional flow, and cardiac dilatation (initially right‐sided, with subsequent dilatation of all four chambers) with systolic dysfunction. The infant was poorly responsive to treatment with dopamine, ACE inhibitor, and diuretics. He underwent patent ductus arteriosus ligation at 6 weeks old. The cardiac dilatation resolved, and he was weaned off all medications by 4 months old. However, he then experienced growth failure. Family history was negative for sudden death, cardiomyopathies, or congenital heart disease. Echocardiography of the mother revealed normal structural findings.
Figure 1.
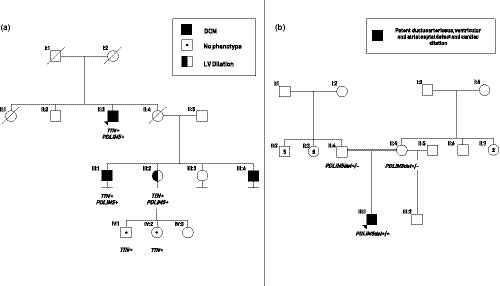
(a) Segregation of the PDLIM5 frameshift variant (p.Leu64Glyfs*15) in a dilated cardiomyopathy (DCM) family with also a truncating TTN variant (p.Thr32477Asnfs*). The index patient (II:3; arrow) has DCM and carries both mutations, just like his cousin (III:1). His niece (III:2) also carries both mutations, but only has left ventricular dilation without a decreased ejection fraction. Both her children had no phenotype at cardiac screening but were only tested for the TTN variant. The other cousin (III:4) is known with DCM but did not consent for genetic analysis. (b) Whole genome sequencing revealed a homozygous deletion in PDLIM5 in a newborn with congenital heart disease (II:1). Both parents were heterozygous for this variant. Echocardiography of the mother revealed no structural abnormalities
Whole‐genome sequencing revealed a homozygous chromosomal deletion of 10.16 kb at the start of PDLIM5 (Hg19: chr4:95,376,436–chr4:95,386,599) which was heterozygous present in both healthy parents. The deletion starts in the second exon and includes nucleotides of the start codon for the heart‐specific isoform (Figure 2). It leaves only the first 39 nucleotides of the exon intact and encompasses deletion of the remaining 259 nucleotides of the second exon, including the start codon. In‐silico analysis predicts loss of the start codon of almost all isoforms. The effect of the homozygous mutation is most likely devastating for the PDLIM5 protein as both the start codon and the donor splice site of the second exon essential for the cardiac expression of the gene are deleted. This loss of PDLIM5 and early‐onset cardiac disease, is a first proof of principle for a connection between the gene and DCM.
Figure 2.
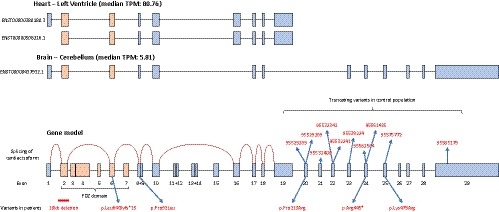
Cardiac isoforms of PDLIM5 in comparison with the longer isoform in the brain. Note the low expression of the transcript in the brain compared to the heart (TPM = transcripts per million). Exon skipping of the cardiac isoform is shown by the red line in the gene model. Position of truncating variants in the control population correspond to their respective genomic location using the GRCh38 human genome assembly
3.4. PDLIM5 variants in a DCM cohort
The next step was to investigate the heterozygous PDLIM5 variants identified in the DCM cohort. In total, we detected five variants in PDLIM5: one frameshift, one nonsense, and three nonsynonymous missense variants (Table S2). Both LoF variants were extremely rare in reference database, located in functional domains of the protein and predicted to have functional consequences, and therefore classified as class 3. In contrast, the missense mutations were less rare in the gnomAD database.
The human PDLIM5 gene constitutes of 625 amino acids and is divided in 29 exons. The gene is highly complex as it undergoes extensive splicing; currently 23 splice variants have been described that have tissue‐specific expression. Using the available expression data of the PDLIM5 isoforms in the GTEx portal, we could identify ENST00000508216.1 and ENST00000380180.3 as almost exclusively expressed in the heart and therefore representing the cardiac isoforms (Figure 2). After mapping of our variants to the cardiac isoforms, one LoF and one missense variant were in an exon included in the cardiac isoform (Figure 2). Since the other three found variants are absent or only very low expressed in the heart, they are probably not contributing to the cardiac phenotype. The clinical characteristics of both patients who carry a variant in the cardiac isoform can be found in Table S3.
3.5. Burden of PDLIM5 loss‐of‐function variants in a control cohort
PDLIM5 has a pLi‐score of 0.01, suggesting its tolerance for LoF variants. However, the pLi‐score does not make a distinction among different isoforms. We used available WES data of our control cohort of 26,000 individuals to extract a list of LoF variants in PDLIM5. We identified 12 truncating variants (10 unique variants; prevalence of 0.05%) and mapped them along the gene. All the LoF variants identified were localized in the 3' ending of the gene and not expressed in the cardiac isoforms near the PDZ‐domain at the 5' ending (Figure 2). This highlights the scarcity of LoF mutations, and in particular the variants in cardiac isoforms we detected in our DCM cohort (2/142; prevalence of 1.4%), depicting the frameshift in the PDZ‐domain of PDLIM5 (p.Leu64Glyfs*15).
3.6. Clinical characteristics associated with a heterozygous frameshift PDLIM5 variant
We focused on the family with the most interesting frameshift mutation (p.Leu64Glyfs*15), as the other LoF variant was not in the cardiac isoform and the missense variant in the cardiac isoform was too prevalent in gnomAD (Table S2). The frameshift variant was found in a male patient of 67 years old who presented with acute heart failure (LVEF 10%) and atrial fibrillation (AF). He rapidly normalized after initial medical treatment with standard heart failure therapy (LVEF 53%). Diagnostic work‐up revealed a pathogenic truncating TTN mutation (c.97427dup; p.Thr32477Asnfs*13) which segregated with the phenotype among the relatives (Figure 1a; Table 4). In all tested affected family members the PDLIM5 variant was detected (c.189_190delinsGG; p.Leu64Glyfs*15).
Table 4.
Clinical features of affected patients with PDLIM5 mutations
| Individual | Sex | Origin | Age at diagnosis | Presenting symptoms | Outcome | Genotype |
|---|---|---|---|---|---|---|
| A‐II:3 | M | Dutch | 67 | Acute heart failure | Normalized LVEF | Heterozygous TTN: p.Thr32477Asnfs* + heterozygous PDLIM5: p.Leu64Glyfs*15 |
| A‐III:1 | M | Dutch | 26 | Severe biventricular DCM | Normalized LVEF | Heterozygous TTN: p.Thr32477Asnfs* + heterozygous PDLIM5: p.Leu64Glyfs*15 |
| A‐III:2 | F | Dutch | 48 | LV dilatation at echocardiography | Stable cardiac function | Heterozygous TTN: p.Thr32477Asnfs* + heterozygous PDLIM5: p.Leu64Glyfs*15 |
| A‐III:4 | M | Dutch | 26 | Severe DCM with NSVTs | LVEF 30%, multiple appropriate ICD shocks | NA |
| B‐III:1 | M | Native American | At birth | Patent ductus arteriosus, ventricular and atrial septal defect, and cardiac dilation | Normalized cardiac dimensions | Homozygous PDLIM5 deletion of 10.16 kb (Hg19: chr4:95,376,436–chr4:95,386,599) |
Abbreviations: DCM, dilated cardiomyopathy; F, female; ICD, implantable cardiac defibrillator; LV, left ventricular; LVEF, left ventricular ejection fraction; M, male; NSVT, nonsustained ventricular tachycardia.
3.7. Cardiac PDLIM5 expression in DCM patients
RNA was isolated from cardiac tissue of the patient with the PDLIM5 frameshift variant and eight control DCM patients. The short isoform of PDLIM5 was detected in all cardiac tissues, in contrast to the longer isoforms which could not be detected. Instead, the longer isoform could be detected in cDNA of a mixed HEK/HeLa cell line, confirming tissue‐specificity of the different isoforms of PDLIM5.
PDLIM5 expression is correlated to LVEF: a higher expression was observed in the patients with a higher LVEF (p < .001, R 2: .91; Figure 3a). In contrast, PDLIM5 RNA was highly expressed in the heart of the patient with the PDLIM5 frameshift variants although he had a LVEF of 10% (Figure 3b). In line with previous research regarding a PDLIM5‐knockout mouse developing DCM (Cheng et al., 2010), we investigated the most important interactors of PDLIM5 which showed transcriptional upregulation after loss of PDLIM5: LDB3 and MYOZ2 which form a complex with PDLIM5 at the Z‐line, in close interaction with MYOT (Figures 4 and 5). All the PDLIM5‐interactors were also upregulated in the myocardium of our patient (Figure 4).
Figure 3.
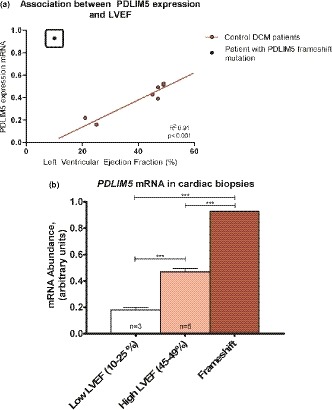
(a) Association between PDLIM5 expression in the heart and left ventricular ejection fraction (LVEF) in the patient with the PDLIM5 frameshift mutation and eight control dilated cardiomyopathy (DCM) patients. (b) Expression of PDLIM5 mRNA in the cardiac biopsy of the patient with the PDLIM5 frameshift mutation and two DCM control groups with high and low LVEF, respectively. Statistical stars on data figures represent ***p ≤ .001
Figure 4.
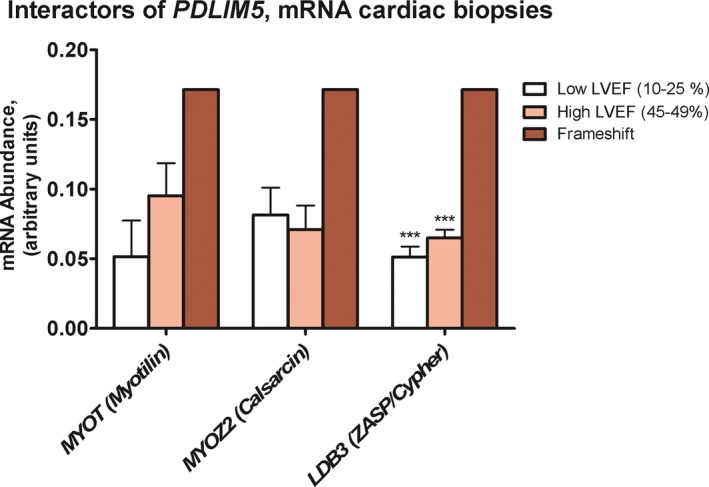
Expression of known interactors with PDLIM5 in the cardiac biopsy of the patient with the PDLIM5 frameshift mutation and two dilated cardiomyopathy (DCM) control groups with high and low left ventricular ejection fraction (LVEF), respectively. Statistical stars on data figures represent ***p ≤ .001
Figure 5.
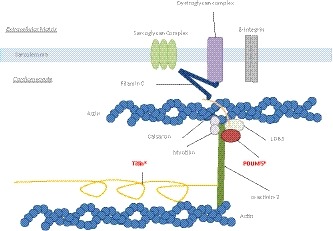
Schematic figure of the Z‐line in the cardiomyocyte. PDLIM5 has a direct interaction with LDB3 and forms a complex together with calsarcin. They bind to α‐actinin‐2 on one side and to the filamin C‐complex at the other side. The large protein titin also binds to α‐actinin‐2. The patient described in our manuscript has a loss of function mutation in both TTN and PDLIM5 (as indicated by an asterisk)
4. DISCUSSION
This study used an innovative method to discover new DCM‐associated genes in WES data. We ranked variants in PDLIM5, CMYA5, SPEG, SYNM, and ALPK3 as the most promising disease candidate genes using computational ranking based on coexpression followed by strict variant selection. The interesting complexity and tissue‐specificity of PDLIM5 isoforms called for further investigation of this gene, although the other genes also require additional research. Based on the computational, literature and molecular indications we show altered RNA expression of PDLIM5 in a patient carrying a heterozygous frameshift mutation in the short cardiac isoform. Moreover, the patient also harbors a pathogenic TTN mutation that segregates with the disease in the family. All affected family members that were tested also harbored the PDLIM5 variant. Heterozygous mutations in PDLIM5 do not seem to play a major role in DCM, but it can be speculated that myocardial PDLIM5 dysfunction leads to increased susceptibility of the heart to develop DCM. The familial segregation of both TTN and PDLIM5 and high disease penetrance is suggestive for an additive effect of both mutations in the development of DCM. In addition, a neonatal cardiac patient carrying a homozygous PDLIM5 deletion strongly suggests the association between PDLIM5 dysfunction and cardiac failure.
4.1. Genetic complexity of PDLIM5
Four major isoforms of PDLIM5 have been identified: one long isoform (ENH1) expressed in all tissues, and three shorter isoforms (ENH2‐4) that are mainly expressed in cardiac and skeletal tissues (Niederlander, Fayein, Auffray, & Pomies, 2004). ENH1 appears to be the embryonic isoform in the heart, whereas ENH2‐4 are found in the adult heart (Yamazaki et al., 2010). Subsequent research showed that PDLIM5 undergoes even more extensive splicing and that there are multiple isoforms in the mouse, and 23 transcripts in the human; although only 14 are shown to be protein coding (Cheng et al., 2010; GTEx Consortium, 2013). We showed that only the short isoform could be found in the human myocardium, and that the longer isoforms were undetectable. This strongly indicates tissue specificity of the short form of PDLIM5 in the heart. The heterozygous frameshift mutation we found in exon 6 is present in almost all isoforms of PDLIM5. Remarkably, the LoF mutations detected in a large control cohort were all located in exons not present in the cardiac isoforms of PDLIM5. In addition, the homozygous deletion started in exon 2 including the start codon which probably prevents the transcription of PDLIM5 including the cardiac isoform. Therefore, it is unlikely that variants occurring in exons not included in the cardiac isoform will have any effect on cardiac function.
4.2. Homology within the PDLIM protein family
All PDLIM proteins can be divided into two subfamilies: the ALP and Enigma family (Zheng, Cheng, Banerjee, & Chen, 2010). PDLIM5 is located at the Z‐line of the sarcomere, in a complex formed by the Enigma family (Sequeira, Nijenkamp, Regan, & van der Velden, 2014). All members of the Enigma family interact directly with α‐actinin‐2, and constitute of PDLIM7 (also called LMP), LDB3 (also called ZASP; and PDLIM6 in the past), and PDLIM5 (also called ENH). They contain a N‐terminal PDZ domain and 3 C‐terminal LIM‐domains which are used for binding to α‐actinin and multiple signaling proteins. Variants in LDB3 are well‐known and investigated as cause for dilated and hypertrophic cardiomyopathies in human and mice (Vatta et al., 2003; Zheng et al., 2009). Variants in PDLIM7 have not been described in humans. However, a PDLIM7 knockout mouse developed mild cardiac dysfunction mainly due to structural aberrations of the cardiac valves, despite the main tissue expression of human PDLIM7 is in the vascular and gastrointestinal system (Krcmery et al., 2013). This contrasts with PDLIM5 and LDB3 which are mainly expressed in the heart.
Next to the Enigma‐family of PDLIM‐proteins, the ALP‐family constituting of PDLIM1‐4, all contain 1 C‐terminal LIM‐domain (Zheng et al., 2010). Although all members of the Enigma‐family have been linked to a cardiac phenotype in animal models, only a PDLIM3 knockout mouse is related to isolated right ventricular DCM (Pashmforoush et al., 2001). Selective genotyping of PDLIM3 in 185 DCM patients yielded one suspicious variant in a 33‐year‐old woman who developed DCM during her pregnancy (Arola et al., 2007). Transfection experiments with the specific variant showed no exogeneous PDLIM3 expression. They speculated that myocardial PDLIM3 deficiency could have increased the susceptibility of the maladaptive responses to the hemodynamic and hormonal burdens of pregnancy. Variants in PDLIM3 do not seem to play a causal role in DCM, however, due to their multiple significant interactions at the Z‐line, they may play a role in the disease pathogenesis by unknown mechanisms. In line with this observation, the same might be true for PDLIM5 in DCM, suggesting a contributing role for PDLIM‐variants in the pathogenesis of DCM.
4.3. PDLIM5 and its interactors in cardiac disease
The finding of LDB3 variants DCM is a strong indication that additional members of the Enigma subfamily could be involved in DCM, corroborated by the presence of cardiac‐specific isoforms of human PDLIM5. Indeed, a cardiac‐specific ENH (PDLIM5) knockout mouse model developed DCM within 3 months with progressive deterioration (Cheng et al., 2010). Recently, it was shown that novel polymorphisms in PDLIM5 are associated with an increased risk for developing DCM in a Chinese Han population (Wang et al., 2019). In addition, the congenital heart disease in an infant with a homozygous PDLIM5 deletion strengthens the association between PDLIM5 and cardiac (dys)function. We observed a correlation between LVEF and PDLIM5 expression, with a decrease in PDLIM5 expression with decreasing LVEF in patients without a PDLIM5 LoF variant. In contrast, our patient with a frameshift mutation had an increased expression of PDLIM5 despite of a low LVEF of 10%. This could indicate a strong compensatory effect by the wild‐type allele due to the frameshift mutation in PDLIM5, irrespective of the functional status of the cardiomyocyte. However, this finding needs to be validated and investigated in more detail.
It is important to realize the complexity of the Z‐line which constitutes of many different protein complexes involved in the signal transduction, myocyte stability, and force transmission (Sequeira et al., 2014). Besides the Enigma family, myotilin (MYOT) and calsarcin (MYOZ2) also bind to α‐actinin‐2, and a previous pull‐down assay showed complex formation between PDLIM5, LDB3 and MYOZ2 (Cheng et al., 2010). The cardiac‐specific knockout of PDLIM5 subsequently led to a significant decrease in MYOZ2 and LDB3 and a strong increase in MYOT protein expression over time, probably to compensate for the loss of the PDLIM5‐containing complex. This was accompanied by an increase in the RNA expression of MYOZ2 and LDB3, suggesting a posttranslational mechanism for the decreased protein expression and not due to decreased mRNA synthesis. In our experiments, we also observed an increase in LDB3, MYOZ2, and MYOT RNA expression in our patient, although we could not verify their expression at protein level. MYOZ2 has been shown to interact with filamin C (FLNC), which directly interacts with β1 integrin, and is associated with the sarcoglycan and dystroglycan complexes at the sarcolemma (Gontier et al., 2005). In the knockout model, these filamin C‐associated complexes were upregulated, suggesting a compensatory mechanism to strengthen the disruption of the Z‐line stability to the extracellular matrix (Cheng et al., 2010).
4.4. Potential link between PDLIM5 and TTN
Besides these complexes, titin (TTN) is also an important protein which binds directly to α‐actinin‐2 (Ware & Cook, 2018). The fact that both PDLIM5 and TTN bind to the same protein in the Z‐line, makes our patient with a LoF mutation in both genes very interesting. Evidence is accumulating that truncating variants in TTN (TTNtv) are likely to be susceptibility variants needing a second hit to develop a pronounced phenotype (Ware & Cook, 2018). In the general population, ~0.4% carries a TTNtv with an estimated penetrance of 12% (Ware & Cook, 2018). This would mean that only a minority of the TTNtv carriers will develop a phenotype. Apparently, the heart can compensate adequately for the physiological consequences of TTN haploinsufficiency (Schafer et al., 2017; Verdonschot et al., 2018). Multiple studies have shown an increased burden of TTNtv in populations with acquired causes for DCM, such as chemotherapy, pregnancy and alcohol (Linschoten et al., 2017; Ware et al., 2018, 2016). It is likely that, besides acquired factors, also multiple genetic modifiers can influence the penetrance of TTNtv. PDLIM5 is a good candidate to be such modifier as it is physically involved in the Z‐line in approximation of titin attached to α‐actinin‐2. Two patients with a pronounced DCM and one with LV dilation in our family all carried the mutation in PDLIM5 and TTN. This high penetrance of disease could be due to the combination of both mutations, as disease penetrance in TTN‐associated DCM is relatively low, however further investigations are necessary to better dissect this protein–protein interaction. The concept of mutations as susceptibility factor was elegantly shown in hypertrophic cardiomyopathy (HCM) patients with a MYL2 mutation (Claes et al., 2016). Carriers of the mutation only developed HCM when hypertension was present, clearly showing a gene–environment interaction for disease development.
4.5. Limitations
This study describes only two families; therefore, the results should be interpreted with caution. However, data from our families and the previous animal studies suggest that PDLIM5 is an interesting candidate to investigate further in relation to DCM. Functional studies are necessary to estimate any consequences related to the specific described heterozygous mutations, in absence and presence of any additional mutation in the Z‐line proteins. As cardiac tissue obtained during life is quite limited, we did not have enough tissue from the patient with the frameshift mutation to study PDLIM5 at the protein level. Therefore, we were limited to the data from RNA expression. Also, as cardiac tissue is gathered during a diagnostic procedure, we do not have any available tissue from healthy control subjects for measuring RNA expression. Some individuals that tested mutation‐negative in our study may have other monogenic causes that are not covered by the diagnostic 47 gene DCM panel. As an extension of this, in an ideal situation we would have the WES data of a DCM cohort constituting of familial disease without proven pathogenic mutation in the absence of an underlying, as this will increase the likelihood of identifying novel genetic variants. However, due to our extensive diagnostic trajectory we already identified a contributing etiology in most DCM patients. Although this does not necessary exclude a genetic predisposition.
5. CONCLUSION
Computational ranking based on coexpression is a valid tool to create a shortlist of interesting DCM‐associated candidate genes. Our study suggests a role for PDLIM5 in DCM, although heterozygous mutations are not a major cause of DCM in contrast to a homozygous LoF mutation which leads to early onset cardiac disease. However, as PDLIM5 plays a major role in contracting cardiomyocytes, a heterozygous LoF is likely to play a role in disease development. Other environmental and/or genetic factors are probably necessary to unveil the cardiac phenotype in heterozygous PDLIM5 mutation carriers.
CONFLICT OF INTEREST
None of the authors has any conflict of interest to declare.
AUTHORS CONTRIBUTION
All listed authors contributed to either the conceptual design of the study, in drafting and/or revising the manuscript. All listed authors gave final approval of the version to be published. Each author participated sufficiently in the work to take public responsibility for appropriate portions of the content; and agreed to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Supporting information
Verdonschot JAJ, Robinson EL, James KN, et al. Mutations in PDLIM5 are rare in dilated cardiomyopathy but are emerging as potential disease modifiers. Mol Genet Genomic Med. 2020;8:e1049 10.1002/mgg3.1049
REFERENCES
- Arola, A. M. , Sanchez, X. , Murphy, R. T. , Hasle, E. , Li, H. , Elliott, P. M. , … Bowles, N. E. (2007). Mutations in PDLIM3 and MYOZ1 encoding myocyte Z line proteins are infrequently found in idiopathic dilated cardiomyopathy. Molecular Genetics and Metabolism, 90(4), 435–440. 10.1016/j.ymgme.2006.12.008 [DOI] [PubMed] [Google Scholar]
- Barrett, T. , Wilhite, S. E. , Ledoux, P. , Evangelista, C. , Kim, I. F. , Tomashevsky, M. , … Soboleva, A. (2013). NCBI GEO: Archive for functional genomics data sets–update. Nucleic Acids Research, 41(D1), D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, H. , Kimura, K. , Peter, A. K. , Cui, L. I. , Ouyang, K. , Shen, T. , … Chen, J. U. (2010). Loss of enigma homolog protein results in dilated cardiomyopathy. Circulation Research, 107(3), 348–356. 10.1161/CIRCRESAHA.110.218735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claes, G. R. , van Tienen, F. H. , Lindsey, P. , Krapels, I. P. , Helderman‐van den Enden, A. T. , Hoos, M. B. , … van den Wijngaard, A. (2016). Hypertrophic remodelling in cardiac regulatory myosin light chain (MYL2) founder mutation carriers. European Heart Journal, 37(23), 1815–1822. 10.1093/eurheartj/ehv522 [DOI] [PubMed] [Google Scholar]
- Gontier, Y. , Taivainen, A. , Fontao, L. , Sonnenberg, A. , van der Flier, A. , Carpen, O. , … Borradori, L. (2005). The Z‐disc proteins myotilin and FATZ‐1 interact with each other and are connected to the sarcolemma via muscle‐specific filamins. Journal of Cell Science, 118(Pt 16), 3739–3749. 10.1242/jcs.02484 [DOI] [PubMed] [Google Scholar]
- GTEx Consortium . (2013). The Genotype‐Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hershberger, R. E. , Hedges, D. J. , & Morales, A. (2013). Dilated cardiomyopathy: The complexity of a diverse genetic architecture. Nature Reviews Cardiology, 10(9), 531–547. 10.1038/nrcardio.2013.105 [DOI] [PubMed] [Google Scholar]
- Japp, A. G. , Gulati, A. , Cook, S. A. , Cowie, M. R. , & Prasad, S. K. (2016). The diagnosis and evaluation of dilated cardiomyopathy. Journal of the American College of Cardiology, 67(25), 2996–3010. 10.1016/j.jacc.2016.03.590 [DOI] [PubMed] [Google Scholar]
- Krcmery, J. , Gupta, R. , Sadleir, R. W. , Ahrens, M. J. , Misener, S. , Kamide, C. , … Simon, H.‐G. (2013). Loss of the cytoskeletal protein PDLIM7 predisposes mice to heart defects and hemostatic dysfunction. PLoS ONE, 8(11), e80809 10.1371/journal.pone.0080809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lek, M. , Karczewski, K. J. , Minikel, E. V. , Samocha, K. E. , Banks, E. , Fennell, T. , … MacArthur, D. G. (2016). Analysis of protein‐coding genetic variation in 60,706 humans. Nature, 536(7616), 285–291. 10.1038/nature19057 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linschoten, M. , Teske, A. J. , Baas, A. F. , Vink, A. , Dooijes, D. , Baars, H. F. , & Asselbergs, F. W. (2017). Truncating titin (TTN) variants in chemotherapy‐induced cardiomyopathy. J Card Fail, 23(6), 476–479. 10.1016/j.cardfail.2017.03.003 [DOI] [PubMed] [Google Scholar]
- Meder, B. , Rühle, F. , Weis, T. , Homuth, G. , Keller, A. , Franke, J. , … Katus, H. A. (2014). A genome‐wide association study identifies 6p21 as novel risk locus for dilated cardiomyopathy. European Heart Journal, 35(16), 1069–1077. 10.1093/eurheartj/eht251 [DOI] [PubMed] [Google Scholar]
- Niederlander, N. , Fayein, N. A. , Auffray, C. , & Pomies, P. (2004). Characterization of a new human isoform of the enigma homolog family specifically expressed in skeletal muscle. Biochemical and Biophysical Research Communications, 325(4), 1304–1311. 10.1016/j.bbrc.2004.10.178 [DOI] [PubMed] [Google Scholar]
- Pashmforoush, M. , Pomiès, P. , Peterson, K. L. , Kubalak, S. , Ross, J. , Hefti, A. , … Chien, K. R. (2001). Adult mice deficient in actinin‐associated LIM‐domain protein reveal a developmental pathway for right ventricular cardiomyopathy. Nature Medicine, 7(5), 591–597. 10.1038/87920 [DOI] [PubMed] [Google Scholar]
- Pinto, Y. M. , Elliott, P. M. , Arbustini, E. , Adler, Y. , Anastasakis, A. , Böhm, M. , … Charron, P. (2016). Proposal for a revised definition of dilated cardiomyopathy, hypokinetic non‐dilated cardiomyopathy, and its implications for clinical practice: A position statement of the ESC working group on myocardial and pericardial diseases. European Heart Journal, 37(23), 1850–1858. 10.1093/eurheartj/ehv727 [DOI] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roncarati, R. , Viviani Anselmi, C. , Krawitz, P. , Lattanzi, G. , von Kodolitsch, Y. , Perrot, A. , … Robinson, P. N. (2013). Doubly heterozygous LMNA and TTN mutations revealed by exome sequencing in a severe form of dilated cardiomyopathy. European Journal of Human Genetics, 21(10), 1105–1111. 10.1038/ejhg.2013.16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schafer, S. , de Marvao, A. , Adami, E. , Fiedler, L. R. , Ng, B. , Khin, E. , … Cook, S. A. (2017). Titin‐truncating variants affect heart function in disease cohorts and the general population. Nature Genetics, 49(1), 46–53. 10.1038/ng.3719 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sequeira, V. , Nijenkamp, L. L. , Regan, J. A. , & van der Velden, J. (2014). The physiological role of cardiac cytoskeleton and its alterations in heart failure. Biochimica Et Biophysica Acta, 1838(2), 700–722. 10.1016/j.bbamem.2013.07.011 [DOI] [PubMed] [Google Scholar]
- Sobreira, N. , Schiettecatte, F. , Valle, D. , & Hamosh, A. (2015). GeneMatcher: A matching tool for connecting investigators with an interest in the same gene. Human Mutation, 36(10), 928–930. 10.1002/humu.22844 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szklarczyk, R. , Megchelenbrink, W. , Cizek, P. , Ledent, M. , Velemans, G. , Szklarczyk, D. , & Huynen, M. A. (2016). WeGET: Predicting new genes for molecular systems by weighted co‐expression. Nucleic Acids Research, 44(D1), D567–D573. 10.1093/nar/gkv1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vatta, M. , Mohapatra, B. , Jimenez, S. , Sanchez, X. , Faulkner, G. , Perles, Z. , … Towbin, J. A. (2003). Mutations in Cypher/ZASP in patients with dilated cardiomyopathy and left ventricular non‐compaction. Journal of the American College of Cardiology, 42(11), 2014–2027. 10.1016/j.jacc.2003.10.021 [DOI] [PubMed] [Google Scholar]
- Verdonschot, J. A. J. , Hazebroek, M. R. , Derks, K. W. J. , Barandiarán Aizpurua, A. , Merken, J. J. , Wang, P. , … Heymans, S. R. B. (2018). Titin cardiomyopathy leads to altered mitochondrial energetics, increased fibrosis and long‐term life‐threatening arrhythmias. European Heart Journal, 39(10), 864–873. 10.1093/eurheartj/ehx808 [DOI] [PubMed] [Google Scholar]
- Verdonschot Job, A. J. , Hazebroek Mark, R. , Ware James, S. , Prasad Sanjay, K. , & Heymans Stephane, R. B. (2019). Role of targeted therapy in dilated cardiomyopathy: The challenging road toward a personalized approach. Journal of the American Heart Association, 8(11), e012514 10.1161/JAHA.119.012514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villard, E. , Perret, C. , Gary, F. , Proust, C. , Dilanian, G. , Hengstenberg, C. , … Cambien, F. (2011). A genome‐wide association study identifies two loci associated with heart failure due to dilated cardiomyopathy. European Heart Journal, 32(9), 1065–1076. 10.1093/eurheartj/ehr105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, D. , Fang, J. , Lv, J. , Pan, Z. , Yin, X. , Cheng, H. , & Guo, X. (2019). Novel polymorphisms in PDLIM3 and PDLIM5 gene encoding Z‐line proteins increase risk of idiopathic dilated cardiomyopathy. Journal of Cellular and Molecular Medicine, 23(10), 7054–7062. 10.1111/jcmm.14607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, J. S. , Amor‐Salamanca, A. , Tayal, U. , Govind, R. , Serrano, I. , Salazar‐Mendiguchía, J. , … Garcia‐Pavia, P. (2018). Genetic etiology for alcohol‐induced cardiac toxicity. Journal of the American College of Cardiology, 71(20), 2293–2302. 10.1016/j.jacc.2018.03.462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ware, J. S. , & Cook, S. A. (2018). Role of titin in cardiomyopathy: From DNA variants to patient stratification. Nature Reviews Cardiology, 15(4), 241–252. 10.1038/nrcardio.2017.190 [DOI] [PubMed] [Google Scholar]
- Ware, J. S. , Li, J. , Mazaika, E. , Yasso, C. M. , DeSouza, T. , Cappola, T. P. , … Arany, Z. (2016). Shared genetic predisposition in peripartum and dilated cardiomyopathies. New England Journal of Medicine, 374(3), 233–241. 10.1056/NEJMoa1505517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamazaki, T. , Wälchli, S. , Fujita, T. , Ryser, S. , Hoshijima, M. , Schlegel, W. , … Maturana, A. D. (2010). Splice variants of enigma homolog, differentially expressed during heart development, promote or prevent hypertrophy. Cardiovascular Research, 86(3), 374–382. 10.1093/cvr/cvq023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Cheng, H. , Banerjee, I. , & Chen, J. (2010). ALP/Enigma PDZ‐LIM domain proteins in the heart. Journal of Molecular Cell Biology, 2(2), 96–102. 10.1093/jmcb/mjp038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, M. , Cheng, H. , Li, X. , Zhang, J. , Cui, L. , Ouyang, K. , … Chen, J. (2009). Cardiac‐specific ablation of Cypher leads to a severe form of dilated cardiomyopathy with premature death. Human Molecular Genetics, 18(4), 701–713. 10.1093/hmg/ddn400 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials