

Abstract
Background
Rett syndrome (RTT) is a neurodevelopmental disorder that predominantly affects girls. Its causative gene is the X‐linked MECP2 encoding the methyl‐CpG‐binding protein 2 (MeCP2). The gene comprises four exons and generates two isoforms, namely MECP2_e1 and MECP2_e2. However, it remains unclear whether both MeCP2 isoforms have similar function in the brain.
Methods
We report a case of a boy with typical RTT. Male cases with MECP2 variants have been considered inviable, but somatic mosaicism of the variants can cause RTT in males. Whole‐exome sequencing was performed to search for the genetic background.
Results
A novel nonsense and mosaic variant was identified in exon 1 of MECP2, and the variant allele fraction (VAF) was 28%. Our patient had the same level of VAF as that in reported male cases with mosaic variants in MECP2 exon 3 or 4, but manifested RTT symptoms that were milder in severity compared to those in these patients.
Conclusion
This is probably because the variants in MECP2 exon 3 or 4 disrupt both isoforms of MeCP2, whereas the variant in exon 1, as presented in this study, disrupts only MeCP2_e1 but not MeCP2_e2. Therefore, our findings indicate that MeCP2_e2 may partially compensate for a deficiency in MeCP2_e1.
Keywords: male with Rett syndrome, MeCP2 isoform, MeCP2_e1, MeCP2_e2, somatic mosaicism
A male case with a novel nonsense and mosaic variant in exon 1 of methyl‐CpG‐binding protein 2 (MECP2) manifested Rett syndrome symptoms that were milder in severity compared to those in other male cases with mosaic variants in MECP2 exon 3 or 4. This is probably because the variants in MECP2 exon 3 or 4 disrupt both isoforms of MeCP2, whereas the variant in exon 1 disrupts only MeCP2_e1 but not MeCP2_e2. Therefore, our findings indicate that MeCP2_e2 may partially compensates for a deficiency in MeCP2_e1.
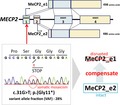
1. INTRODUCTION
Rett syndrome (RTT) is a neurodevelopmental disorder primarily occurring in girls. It is caused by a loss‐of‐function variant in one copy of the X‐linked gene MECP2 (OMIM #300005) that encodes methyl‐CpG‐binding protein 2 (MeCP2). In females with typical RTT due to random X‐chromosome inactivation (XCI), approximately 50% of the cells express the variant MECP2 and the other half express the wild‐type MECP2. Males manifesting the symptoms of typical RTT also have an MECP2 variant that is found in females with typical RTT. These males have either an extra X‐chromosome (Klinefelter syndrome) or somatic mosaicism of the variant (Kleefstra et al., 2004; Schonewolf‐Greulich et al., 2019; Schwartzman, Bernardino, Nishimura, Gomes, & Zatz, 2001; Topcu et al., 2002; Villard, 2007; Zhang et al., 2019). Evidence can be obtained from male RTT patients with somatic mosaicism of the MECP2 variant to better understand the relationship between variant allele fractions (VAFs) and the clinical severity of RTT, because the effect of XCI on the phenotype does not need to be considered in male cases.
MECP2 comprises four exons and generates two isoforms: MECP2_e1 and MECP2_e2 as a result of the alternative splicing of exon 2. MeCP2_e1 is translated from a start site in exon 1, and exon 2 is skipped through alternative splicing, whereas MeCP2_e2 is translated from a start site in exon 2 (Mnatzakanian et al., 2004). In this study, we sought to investigate the case of a male RTT patient mosaic for nonsense variant in exon 1 of MECP2 that disrupts only MeCP2_e1 but not MeCP2_e2. To examine whether MeCP2_e2 can ameliorate some neurological symptoms due to the affected MeCP2_e1 functions, we compared the clinical severity of the present case with those of other reported male cases carrying the mosaic variants that affect both MeCP2_e1 and MeCP2_e2.
2. MATERIALS AND METHODS
2.1. Patient background and informed consent
The patient was a young boy with typical RTT phenotype who fulfilled the diagnostic criteria for the disorder (Neul et al., 2010). He and his parents gave informed consent to participate in this study. The experimental protocols were approved by the Committee for Ethical issues at Asahikawa Medical University.
2.2. Mutation analysis of the MECP2
For Sanger sequencing, their DNA was used as the template for polymerase chain reaction (PCR). Appropriate primers were used to yield DNA fragments spanning the entire MECP2 coding region and the intron–exon boundaries (Takahashi et al., 2008). The PCR fragments were analyzed using automated sequencing. Whole‐exome sequencing of the DNA was performed on a HiSeq2000 sequencer (Illumina) with 101 bp paired end reads and 6 bp index reads. Exome data processing, variant calling, and variant annotation were performed as previously described (Itoh et al., 2018). To confirm the variant identified in the Sanger and whole‐exome sequencing, the DNA fragment encompassing the variation site was amplified by PCR using the primers 5′‐CATCACAGCCAATGACGGGC‐3′ (forward) and 5′‐CATCCGCCAGCCGTGTCGTC‐3′ (reverse), and it was subsequently digested with restriction endonuclease Dde I. The reaction products were then visualized through ethidium bromide staining after electrophoresis on a 2% agarose gel.
2.3. RNA isolation and RT‐PCR
To examine the expression levels of MECP2_e1 and MECP2_e2 isoforms, total RNA was extracted from the peripheral blood cells using the PAXgene Blood RNA Kit (QIAGEN GmbH). Reverse transcription (RT) was performed using the SuperScript First‐Strand Synthesis System (Invitrogen Corporation) for generation of cDNA using 1 μg of total RNA in a 20 μl reaction. Primers were designed for simultaneous amplification of both isoforms: a forward primer in exon 1 (exon1F, 5′‐GAGAGGGCTGTGGTAAAAGC‐3′) and a reverse primer in exon 3 (exon3R, 5′‐GATGGAGCGCCGCTGTTTGG‐3′), which generated a 328‐bp product for MECP2_e1 and a 452‐bp product for MECP2_e2. As an internal control, glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as described (Itoh et al., 2012). The PCR products were visualized by ethidium bromide staining, following electrophoresis on 2% agarose gels. The optical densities of the bands were quantified using an image analysis system and ImageJ software (National Institutes of Health; Bethesda, MD). The obtained PCR products were purified from an agarose gel and directly sequenced on an ABI 3130 Genetic Analyzer (Applied Biosystems).
3. RESULTS
3.1. Case report
The 9‐year‐old male patient was born after 39 weeks of uneventful pregnancy without asphyxia. His birth weight and head circumference were 2,865 g (−0.7 SD) and 33.0 cm (−0.4 SD), respectively. He acquired head control at 3 months of age, walked alone and spoke meaningful words at 9 months, and had no apparent developmental delay until he was about 2 years old. Thereafter, his development stagnated, and it was followed by a period of regression. He became less interested in his toys and hardly had any speech. At 3 years and 6 months, he was diagnosed with autism spectrum disorder and intellectual disability. At 4 years, his purposeful hand skills began to regress, and stereotypic movements such as hand wringing appeared. At 6 years, he lost his minimal spoken language, his gait became unsteady and wide based, and his head circumference was only 47.8 cm (−2.2 SD), making his postnatal microcephaly more evident. He developed epilepsy at 7 years, but his seizures were eventually controlled following treatment with carbamazepine and topiramate. The chromosomal analysis revealed normal 46,XY karyotype. No abnormal findings were observed in brain MRI and various tests related to congenital metabolic disorders.
3.2. Molecular studies
We performed the Sanger sequencing of MECP2 for genetic diagnosis, but the initial analysis failed to identify the pathogenic variant of this gene. Subsequently, we performed whole‐exome sequencing and identified a novel nonsense and mosaic variant in exon 1 of MECP2, NM_001110792.1: c.31G>T; p.(Gly11*). Deep sequencing confirmed the presence of the T allele uniquely in the patient sample at a ratio of 25 mutant T allele reads to 63 G allele reads, a VAF of 28%. The reexamination of the MECP2 variant using Sanger sequencing confirmed the wild‐type sequence, with only a small amount of the variant allele, suggesting somatic mosaicism (Figure 1a). This nonsense variant created a new Dde I restriction site. Consequently, PCR‐restriction digestion analysis of the DNA obtained from the patient and his parents revealed novel fragments in addition to the estimated wild‐type fragment only in the patient sample, further confirming that the mosaic variant occurred de novo (Figure 1b). RT‐PCR results revealed that the variant did not affect the expression levels of both MECP2_e1 and MECP2_e2 (Figure 2a). Analysis of cDNA showed the presence of an abnormal transcript with the nonsense variant of exon 1 in a mosaic state (Figure 2b).
Figure 1.
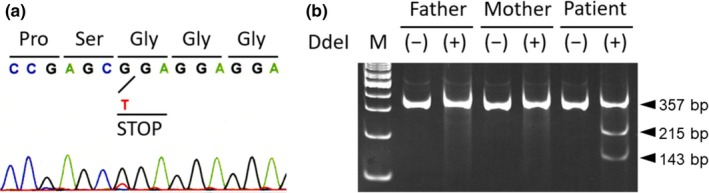
A novel mosaic variant of MECP2 in a male patient with typical Rett syndrome. Electropherogram shows the nonsense variant in exon 1 of MECP2 [NM_001110792.1: c.31G>T: p.(Gly11*)] in mosaicism (a). Dde I digestion of the PCR product encompassing the variation site shows additional fragments (143 and 215 bp), which resulted from the G‐to‐T transition creating a new Dde I restriction site in the patient but not in his parents (b). These additional fragments are observed together with the 358 bp wild‐type fragment, confirming the mosaic variant in the patient
Figure 2.
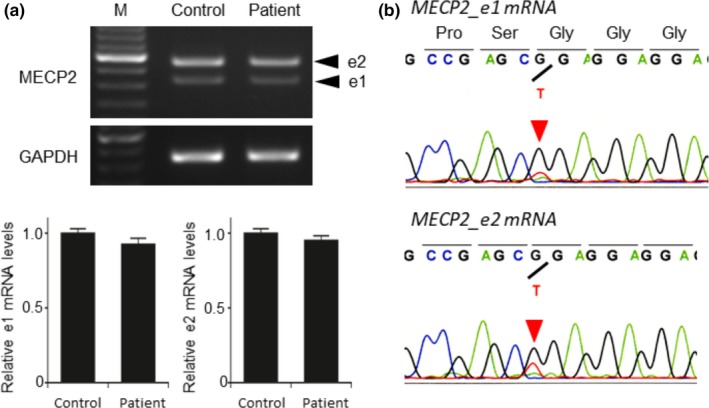
Equal amounts of MECP2_e1 and MECP2_e2 mRNA and abnormal transcript with the nonsense variant in a mosaic state. RT‐PCR results reveal that both MECP2_e1 and MECP2_e2 mRNA amounts are unaffected in the patient. Glyceraldehyde‐3‐phosphate dehydrogenase (GAPDH) was used as an internal control (a). Sequencing analysis of the obtained PCR products shows the presence of abnormal transcript with the nonsense variant in a mosaic state (b)
3.3. Compensatory role of MeCP2_e2 for lack of MeCP2_e1
The nonsense variant identified in the present case affected the coding sequence of MECP2_e1 but not of MECP2_e2. Based on this finding, we further examined whether MeCP2_e2 is able to ameliorate the affected MeCP2_e1 function by comparing the clinical severity of the present case with those of six previously reported RTT males carrying MECP2 mosaic variants that affect both MeCP2_e1 and MeCP2_e2 (Table 1). This comparison revealed that the median VAFs and the age at onset of regression of the reported male patients were 25% (range 9–36) and 13 months (range 8–18), respectively, whereas in the present case with 28% VAF, the developmental problems were not noticed until the patient was 2 years old. At 9 years, he was already able to walk independently, although his balance was poor. Among the six previously reported cases, only three were able to walk with or without support.
Table 1.
Relationship between variant allele fractions and the clinical severity in males with typical Rett syndrome associated with mosaic MECP2 variants
| Age at evaluation (Authors) | Variants | VAFsa | Onset of regression | Hand skills | Language | Gait | Stereotypy | |
|---|---|---|---|---|---|---|---|---|
| Nucleotide change | Predicted effect on protein sequence | |||||||
| 9 years (Present case) | c.31G>T | p.(Gly11*) | 28% | 24 months | Poor | Lost | Poor | Present |
| 12 years (Topcu et al., 2002) | c.808C>T | p.(Arg270*) | 36% | 11 months | Lost | Never | Never | Present |
| 11 years (Kleefstra et al., 2004) | c.473G>T | p.(Thr158Met) | 25% | 13 months | Lost | Lost | Lost | Present |
| 2 years (Zhang et al., 2019) | c.316C>T | p.(Arg106Trp) | 26% | 18 months | Never | Never | Never | Present |
| 2 years (Zhang et al., 2019) | c.353G>A | p.(Gly118Val) | 20% | 13 months | Poor | Lost | Poor | Present |
| 8 years (Schonewolf‐Greulich et al., 2019) | c.1308dupT | p.(Gln437Serfs*50) | 9% (15%) | 18 months | Poor | Lost | Poor | Present |
| 9 years (Schonewolf‐Greulich et al., 2019) | c.808C>T | p.(Arg270*) | 36% (45%) | 8 months | Lost | Never | Poor | Present |
VAFs, variant allele fractions in blood lymphocytes (in fibroblasts).
4. DISCUSSION
We present clinical and molecular findings in a male RTT mosaic for a novel nonsense variant of MECP2. Deep sequencing with a next‐generation sequencing method revealed the small amount of the variant allele c.31G>T; p.(Gly11*) with 28% VAF, which the initial Sanger sequencing failed to identify. The PCR‐restriction digestion analysis further confirmed the somatic mosaicism of the variant. RT‐PCR results revealed that both MECP2_e1 and MECP2_e2 mRNA amounts were unaffected in the patient, suggesting that the nonsense variant in exon 1 of MECP2 likely escapes nonsense‐mediated mRNA decay (NMD) and does not affect transcription of MECP2_e2. The level of sensitivity of a premature‐termination codon (PTC) ‐ containing mRNA to NMD is multifactorial. It has been shown that mRNAs carrying PTCs in close proximity to the translation initiation AUG codon escape NMD, called the “AUG‐proximity effect” (Silva, Ribeiro, Inacio, Liebhaber, & Romao, 2008). Consequently, this variant may lead to translational reinitiation at a downstream AUG codon producing an N‐terminally truncated protein functionally distinct from wild‐type MeCP2_e1, but does not affect the translation of MECP2_e2.
The majority of the RTT‐associated MECP2 variants are located in exons 3 and 4, which simultaneously disrupt both the MeCP2_e1 and MeCP2_e2 isoforms. RTT associated with exon 1 variants of MECP2 is rare, and its detection rate is 8.1% in patients with typical or atypical RTT (Saunders, Minassian, Chow, Zhao, & Vincent, 2009). However, exon 2 variants that exclusively affect MeCP2_e2 have never been identified in RTT, suggesting that MeCP2_e2 does not have an essential function in the brain. Supporting this notion, a mouse model with a deletion in MECP2 exon 2 failed to recapitulate the neurologic symptoms characteristic of RTT (Itoh et al., 2012). In this study, we presented the nonsense variant in exon 1 of MECP2 that disrupted MeCP2_e1 but not MeCP2_e2. Notably, however, we examined the variant using DNA extracted from peripheral blood leukocytes, so that it is uncertain whether the VAF in brain will be equivalent to that observed in the present study. Nonetheless, this study demonstrated that a male carrying an MECP2 exon 1 mosaic variant and expressing 28% less MeCP2_e1 in blood than normal individuals exhibited the typical RTT phenotype, indicating that even a mild reduction of MeCP2_e1 is sufficient to cause RTT.
In a previous study using the isoform‐specific knockout mice in which Mecp2_e1 was lacking while the expression of Mecp2_e2 was preserved, the neurologic deficits of RTT were recapitulated (Yasui et al., 2014). The study implied that an RTT phenotype may occur even in the presence of MeCP2_e2, which is unable to compensate for the lack of MeCP2_e1. Nevertheless, recent animal studies have revealed the partial rescue of Rett‐like symptoms in Mecp2‐null mice through the reexpression of Mecp2_e2 (Jugloff et al., 2008; Kerr et al., 2012). However, there had been no clinical evidence whether MeCP2_e2 is able to ameliorate some neurological symptoms due to the affected MeCP2_e1 functions. Genotype–phenotype correlations are difficult to make in female RTT patients because of the differences in XCI. Examination of male RTT patients with somatic mosaicism of the MECP2 variant allowed us to assess the relationship between VAFs and clinical severity. Comparison of the clinical severity of the present case involving the MECP2_e1‐specific variant with those of other reported male cases with mosaic variants that affect both MeCP2_e1 and MeCP2_e2 revealed that the present case had a milder phenotype even though the VAFs were almost the same (Table 1). In conclusion, this study is the first to present clinical and molecular evidence that MeCP2_e2 may partially compensate for the deficiency of MeCP2_e1, although further functional studies are needed.
CONFLICT OF INTEREST
None of the authors declare any conflict of interest related to this study.
AUTHOR CONTRIBUTIONS
RT and ST conceived and planned the study, and drafted the manuscript. MI performed whole‐exome sequencing. RT, ST, MK, RT, and NS performed genetic analysis. YT, YI, and NS acquired clinical phenotype data. All authors provided important feedback on the analysis and manuscript, and approved the final version.
ACKNOWLEDGMENTS
We sincerely thank the patient and his parents, whose help and participation made this work possible.
Takeguchi R, Takahashi S, Kuroda M, et al. MeCP2_e2 partially compensates for lack of MeCP2_e1: A male case of Rett syndrome. Mol Genet Genomic Med. 2020;8:e1088 10.1002/mgg3.1088
REFERENCES
- Itoh, M. , Ide, S. , Iwasaki, Y. , Saito, T. , Narita, K. , Dai, H. , … Arima, M. (2018). Arima syndrome caused by CEP290 specific variant and accompanied with pathological cilium; clinical comparison with Joubert syndrome and its related diseases. Brain and Development, 40(4), 259–267. 10.1016/j.braindev.2017.11.002 [DOI] [PubMed] [Google Scholar]
- Itoh, M. , Tahimic, C. G. T. , Ide, S. , Otsuki, A. , Sasaoka, T. , Noguchi, S. , … Kurimasa, A. (2012). Methyl CpG‐binding protein isoform MeCP2_e2 is dispensable for Rett syndrome phenotypes but essential for embryo viability and placenta development. Journal of Biological Chemistry, 287(17), 13859–13867. 10.1074/jbc.M111.309864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jugloff, D. G. , Vandamme, K. , Logan, R. , Visanji, N. P. , Brotchie, J. M. , & Eubanks, J. H. (2008). Targeted delivery of an Mecp2 transgene to forebrain neurons improves the behavior of female Mecp2‐deficient mice. Human Molecular Genetics, 17(10), 1386–1396. 10.1093/hmg/ddn026 [DOI] [PubMed] [Google Scholar]
- Kerr, B. , Soto, C. J. , Saez, M. , Abrams, A. , Walz, K. , & Young, J. I. (2012). Transgenic complementation of MeCP2 deficiency: Phenotypic rescue of Mecp2‐null mice by isoform‐specific transgenes. European Journal of Human Genetics, 20(1), 69–76. 10.1038/ejhg.2011.145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleefstra, T. , Yntema, H. G. , Nillesen, W. M. , Oudakker, A. R. , Mullaart, R. A. , Geerdink, N. , … Hamel, B. C. J. (2004). MECP2 analysis in mentally retarded patients: Implications for routine DNA diagnostics. European Journal of Human Genetics, 12(1), 24–28. 10.1038/sj.ejhg.5201080 [DOI] [PubMed] [Google Scholar]
- Mnatzakanian, G. N. , Lohi, H. , Munteanu, I. , Alfred, S. E. , Yamada, T. , MacLeod, P. J. M. , … Minassian, B. A. (2004). A previously unidentified MECP2 open reading frame defines a new protein isoform relevant to Rett syndrome. Nature Genetics, 36(4), 339–341. 10.1038/ng1327 [DOI] [PubMed] [Google Scholar]
- Neul, J. L. , Kaufmann, W. E. , Glaze, D. G. , Christodoulou, J. , Clarke, A. J. , Bahi‐Buisson, N. , … Percy, A. K. (2010). Rett syndrome: Revised diagnostic criteria and nomenclature. Annals of Neurology, 68(6), 944–950. 10.1002/ana.22124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, C. J. , Minassian, B. E. , Chow, E. W. , Zhao, W. , & Vincent, J. B. (2009). Novel exon 1 mutations in MECP2 implicate isoform MeCP2_e1 in classical Rett syndrome. American Journal of Medical Genetics Part A, 149A(5), 1019–1023. 10.1002/ajmg.a.32776 [DOI] [PubMed] [Google Scholar]
- Schonewolf‐Greulich, B. , Bisgaard, A. M. , Duno, M. , Jespersgaard, C. , Rokkjaer, M. , Hansen, L. K. , … Tumer, Z. (2019). Mosaic MECP2 variants in males with classical Rett syndrome features, including stereotypical hand movements. Clinical Genetics, 95(3), 403–408. 10.1111/cge.13473 [DOI] [PubMed] [Google Scholar]
- Schwartzman, J. S. , Bernardino, A. , Nishimura, A. , Gomes, R. R. , & Zatz, M. (2001). Rett syndrome in a boy with a 47,XXY karyotype confirmed by a rare mutation in the MECP2 gene. Neuropediatrics, 32(3), 162–164. 10.1055/s-2001-16620 [DOI] [PubMed] [Google Scholar]
- Silva, A. L. , Ribeiro, P. , Inacio, A. , Liebhaber, S. A. , & Romao, L. (2008). Proximity of the poly(A)‐binding protein to a premature termination codon inhibits mammalian nonsense‐mediated mRNA decay. RNA, 14(3), 563–576. 10.1261/rna.815108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi, S. , Ohinata, J. , Makita, Y. , Suzuki, N. , Araki, A. , Sasaki, A. , … Fujieda, K. (2008). Skewed X chromosome inactivation failed to explain the normal phenotype of a carrier female with MECP2 mutation resulting in Rett syndrome. Clinical Genetics, 73(3), 257–261. 10.1111/j.1399-0004.2007.00944.x [DOI] [PubMed] [Google Scholar]
- Topcu, M. , Akyerli, C. , Sayi, A. , Toruner, G. A. , Kocoglu, S. R. , Cimbis, M. , & Ozcelik, T. (2002). Somatic mosaicism for a MECP2 mutation associated with classic Rett syndrome in a boy. European Journal of Human Genetics, 10(1), 77–81. 10.1038/sj.ejhg.5200745 [DOI] [PubMed] [Google Scholar]
- Villard, L. (2007). MECP2 mutations in males. Journal of Medical Genetics, 44(7), 417–423. 10.1136/jmg.2007.049452 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasui, D. H. , Gonzales, M. L. , Aflatooni, J. O. , Crary, F. K. , Hu, D. J. , Gavino, B. J. , … Lasalle, J. M. (2014). Mice with an isoform‐ablating Mecp2 exon 1 mutation recapitulate the neurologic deficits of Rett syndrome. Human Molecular Genetics, 23(9), 2447–2458. 10.1093/hmg/ddt640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Q. , Yang, X. , Wang, J. , Li, J. , Wu, Q. , Wen, Y. , … Bao, X. (2019). Genomic mosaicism in the pathogenesis and inheritance of a Rett syndrome cohort. Genetics in Medicine, 21(6), 1330–1338. 10.1038/s41436-018-0348-2 [DOI] [PMC free article] [PubMed] [Google Scholar]