

Abstract
Background
VAC14 is a component of a trimolecular complex that tightly regulates the level of phosphatidylinositol 3,5‐bisphosphate [PI (3,5) P2]. VAC14 pathogenic variants cause prominent vacuolation of neurons in basal ganglia of patients with childhood‐onset striatonigral degeneration (SNDC).
Methods
We identified two siblings with SNDC. Whole‐exome sequencing was performed for genetic molecular analysis in these probands.
Results
The patients were compound heterozygotes for two novel variants in the VAC14 gene, p.Ala582Thr and p.Arg681His. The pathogenicity of these variants was indicated by a bioinformatic study and protein three‐dimensional modeling. Eight previously reported SNDC cases and a Yunis–Varón syndrome caused by VAC14 mutations were summarized and compared.
Conclusion
We present novel compound heterozygous variants (c.1744G>A/c.2042G>A) in our proband, and these novel variants were predicted to be likely pathogenic. The affected siblings were clinically severe and lethal; their phenotypes were similar to the majority of previously reported SNDC cases, with the exception of two cases that showed mild clinical manifestations. VAC14 pathogenic variants may be associated with various phenotypes. Herein, we report the Chinese siblings with SNDC, they are the first Asian cases. Our results expanded the spectrum of VAC14 pathogenic variants and the ethnic backgrounds of the affected cases.
Keywords: basal ganglia, striatonigral degeneration, VAC14, variant, whole‐exome sequencing
Pedigree of the patient's family. The proband was compound heterozygotes for two novel variants in the VAC14 gene (NM_018052.4), c.1744G>A (p.Ala582Thr, paternal allele) and c.2042G>A (p.Arg681His, maternal allele). The patient's clinically healthy brother (Ⅰ1) also has a c.2042G>A mutation.

1. INTRODUCTION
The childhood onset of striatonigral degeneration (SNDC, OMIM: 617054) was recently identified to be caused by VAC14 pathogenic variants, and up to now, only eight patients have been reported (Lenk et al., 2016; Lyon et al., 2019; Stutterd et al., 2017; Taghavi et al., 2018). Herein, we present two Chinese siblings with SNDC caused by novel compound heterozygous mutations of VAC14. In addition, we provide a review of all previously published VAC14 mutation‐associated SNDC cases and one Yunis–Varón case (Lines et al., 2017).
2. MATERIALS AND METHODS
2.1. Ethical compliance
Ethical approval for this study was gained through the Institutional Review Board, Children's Hospital of Chongqing Medical University (2018‐64). Informed consent was obtained from the patient's parents.
2.2. Patients and pedigree
Two siblings from a nonconsanguineous Chinese family participated in the present study (Figure 1). Clinical diagnosis was performed in the Department of Neurology, Children's Hospital of Chongqing Medical University, Chongqing, China. Peripheral blood from the family members was collected after signing an informed consent. Then, the proband was followed up in the Department of Pediatrics, Qianjiang Central Hospital of Chongqing.
Figure 1.
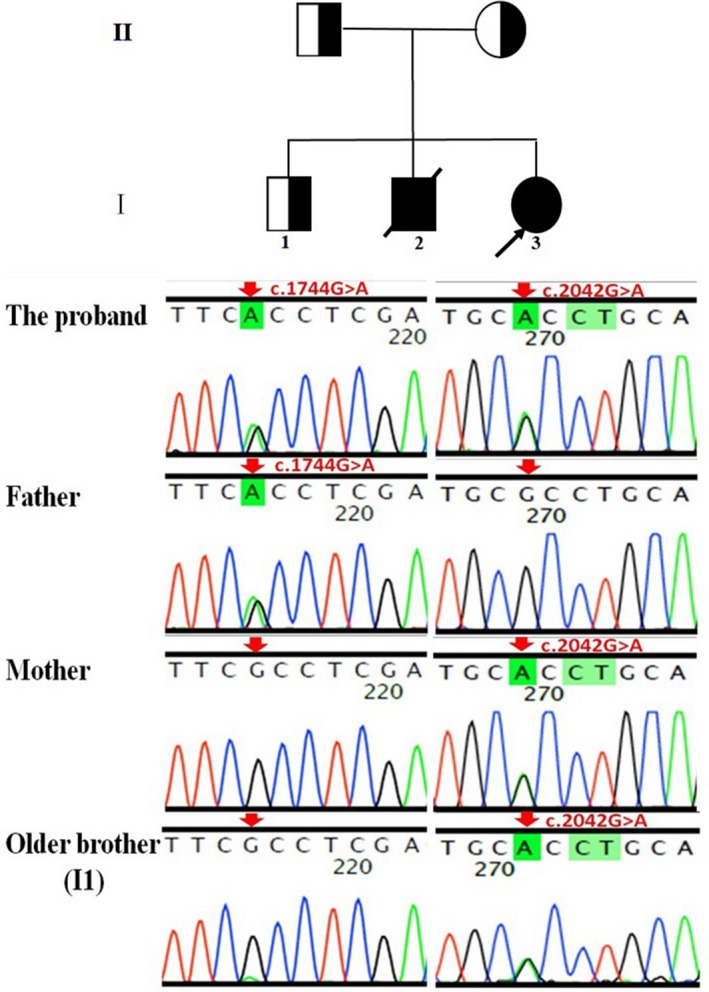
Pedigree of the patient's family. The proband was compound heterozygotes for two novel variants in the VAC14 gene (NM_018052.4), c.1744G>A (p.Ala582Thr, paternal allele) and c.2042G>A (p.Arg681His, maternal allele). The patient's clinically healthy brother (Ⅰ1) also has a c.2042G>A mutation
2.3. WES and Sanger sequencing
Based on human genome reference sequence hg19/GRCh37, trio‐based whole‐exome sequencing (WES) was performed on the proband and parents at B. Braun Precision Medicine Technology Ltd., (Shanghai, China). The Genome Aggregation Database (gnomAD, http://gnomad.broadinstitute.org/) and several other online databases were used as references. To confirm the VAC14 (NM_018052.4) genotyping results from next generation sequencing, two coding mutations (chr16:70,732,632 and chr16:70,726,868) of VAC14 within the family samples were amplified by polymerase chain reaction and confirmed by Sanger DNA sequencing. Primer sequences used are shown in Table S1.
2.4. Bioinformatics analysis
The VAC14‐encoded protein was evolutionarily conserved across representative species [Clustal Omega (https://www.ebi.ac.uk/Tools/msa/clustalo/)] (Figure 2). In silico prediction of the variants was calculated from the output of the programs SIFT, LRT, PROVEAN, Mutation Taster, Mutation Assessor, FATHMM, and FATHMM‐MKL according to the following criteria: 100% consensus = pathogenic/benign, ≥75% consensus = mostly pathogenic/benign, and consensus <75% or no prediction possible = inconsistent. The VAC14 protein structures were modeled using RaptorX (http://raptorx.uchicago.edu).
Figure 2.

The VAC14 missense pathogenic variants (red) of the proband are localized in two highly conserved amino acid sequences (p.Ala582 and p.Arg681) among representative species. The previously reported VAC14 variants are shown (blue)
2.5. Literature review
The databases Wanfang (http://www.wanfangdata.com.cn/index.html) and PubMed (https://www.ncbi.nlm.nih.gov/pubmed/) were searched to retrieve studies using the keywords “VAC14 gene” from inception to June 2019. The publication language was limited to English and Chinese.
3. RESULTS
3.1. Clinical characterization
The proband was a 3‐year 11‐month‐old girl, born to a nonconsanguineous Chinese parent at 36 weeks and 4 days of gestation, G5P3. She developed normally until age 2, and at 2 years 6 months old, she began to walk with a dystonic gait. She first walked on her toes and progressed slowly. One and a half years later, she could only walk several steps. Her verbal ability decreased and was unclear; chewing and swallowing became slow and hard. In addition, she developed increased muscle tone and dystonic movements of the limbs and trunk.
At follow‐up, the patient could not walk by herself at the age of 4 years 3 months old and spoke meaningless vocalization (Video S1). The patient's swallowing has decreased more, she became dependent on a liquid diet and cannot feed herself. During the last follow‐up (4 years 11 months), she can only sit with help, showed dystonic movements, and spoke meaningless words.
The patient's oldest brother is 10 years old and healthy. Her other older brother showed a similar condition of disease onset and progression, and he died at 8 years old. No additional persons with the same condition were found in three generations of relatives in both parent's families. The mother had two spontaneous abortions without identified causes.
Thalamic volume reduction was found in our proband's MRI 6 months after disease onset. Blood/urinary metabolic screening was normal. A CT brain scan was performed of our patient's deceased brother, and no abnormality was observed at the age of 3 years old.
3.2. Molecular genetics
Exome sequencing revealed the compound heterozygous mutations in the VAC14 gene: c.1744G>A; p.Ala582Thr (paternal allele) and c.2042G>A; p.Arg681His (maternal allele) (Figure 1). The actual in silico results are as follows: (c.1744G>A/c.2042G>A), SIFT (0.038/0.0), LRT (0.000/0.000), PROVEAN (−2.72/−4.39), Mutation Taster (1/1), Mutation Assessor (1.655/3.35), FATHMM (−0.21/−0.21), and FATHMM_MKL (0.977/0.972). The mutations were segregated according to a strictly recessive model with full penetrance (Lenk et al., 2016; Lyon et al., 2019; Stutterd et al., 2017; Taghavi et al., 2018). The parents were heterozygous carriers, and the identified mutation (c.2042G>A) was found in the clinically healthy brother (Figure 1). We modeled and predicted the protein structures of the wild‐type and mutated VAC14 proteins (Figure 3a–d). The missense mutations affected the highly conserved amino acid side chain (Figure 2). The variants were considered likely pathogenic according to ACMG Standards (Richards et al., 2015).
Figure 3.
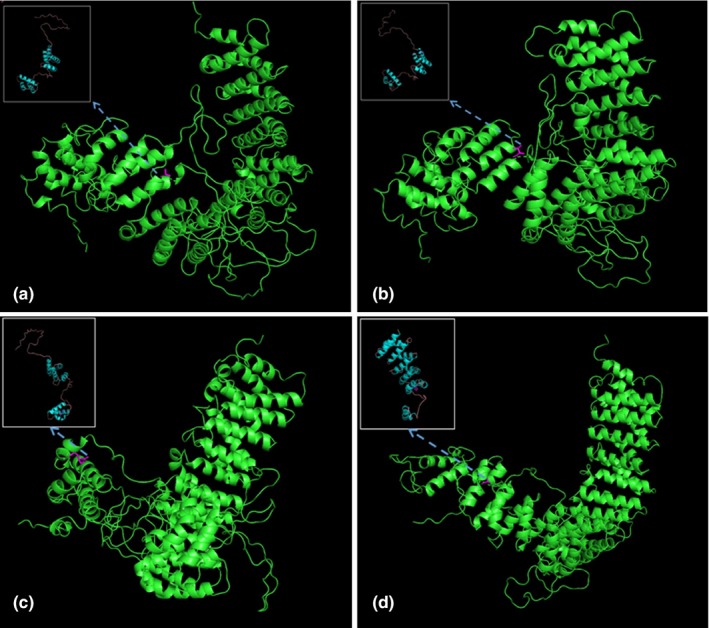
The predicted wild‐type and mutated proteins of VAC14 (NM_018052.4) through in silico analysis. The mutated sites affected the amino acid side chain; the red positions represent the 582nd position of mutations Ala (a; wild type) to Thr (b; mutated). The red positions represent the 681st position of mutations Arg (c; wild type) to His (d; mutated)
3.3. Literature review
All identified articles were read in full and searched to find additional papers not captured by the initial search strategy. A total of eight SNDC cases with VAC14 pathogenic variants were reported (Lenk et al., 2016; Lyon et al., 2019; Stutterd et al., 2017; Taghavi et al., 2018), and the relevant information is summarized in Table 1. A patient of Yunis–Varón syndrome (YVS, OMIM: 216340) with VAC14 mutations is discussed in the following sections (Lines et al., 2017).
Table 1.
VAC14 mutations and phenotypes of affected SNDC cases from the literature and the present findings
| Case 1/2 (Lenk et al., 2016) | Case 3/4 (siblings) (Stutterd et al., 2017) | Case 5/6 (siblings) (Taghavi et al., 2018) | Case 7/8 (siblings) (Lyon et al., 2019) | Our case/her affected brother | ||||
|---|---|---|---|---|---|---|---|---|
| Pregnancy/perinatal period | First/normal; cesarean section, nonconsanguineous parents, no family history | Normal, nonconsanguineous parents, no family history |
NA/normal, nonconsanguineous parents |
NA | First/consanguineous parents, polyhydramnios, respiratory distress, neonatal sepsis, lung atelectasis | 6th/consanguineous parents, | G5P3/G5P2, | |
| Gender | male | Male | Male | Male | Male | Male | Female | Female/male |
| Age of onset | Abrupt 3 years | Abrupt 18 months | Abrupt 3 years 6 months | Abrupt 2 years | From 8 to 13 years | Abrupt 2 years | 18 months | Abrupt 2 years 6 months (both) |
| Life span | Alive | Alive | 5 years | 4 years | Alive | Alive | Alive | alive/8 years |
| First symptoms | Abnormal gait, loss of walking, dystonic leg movements | Abnormal gait, loss of walking | Clumsy walking, | Abnormal movement of leg | Parkinsonism | Unclear speech and an unstable gait | Vision problems | Abnormal gait |
| Main symptoms | Dystonia, episodes of status dystonicus, increased tone in trunk and extremities, dystonic movements of jaw, neck, back, and extremities | Dystonia, weakness of trunk muscles, increased tone in lower extremities | Clumsy walking, frequent falls, intention tremor. dysarthria, impaired truncal balance, muscle spasms, joint contractures, urinary incontinence, weight loss | Abnormal movement of leg, slowed walking and speech, muscle spasms, dystonia, motor function loss | Dystonic gait, dystonia (upper limbs and trunk), dystonic action tremors, profound hypokinesia and bradykinesia, dysarthria | Developmental delay, unsteady gait, drooling, delayed speech, loss of ability to walk, hypotonia, short fingers, delayed speech and language, dysphagia, retinitis pigmentosa | Hypotonia, developmental disabilities and retinitis pigmentosa, drooling | Dystonic gait, frequent falls, muscle spasms, dystonia, motor function loss bradykinesia, dysarthria speech and swallow problem |
| Intellectual capacity | Relative preservation | Relative preservation | Relative preservation | Relative preservation | Normal. | IQ borderline low. | NA | Relative preservation |
| MRI | Striatal abnormalities | Striatal abnormalities | Normal | Normal | Normal in case 5, NA for case 6. | Hypointensity in the globus pallidus and substantia nigra. | NA | Normal |
| Treatments | Baclofen, l‐dopa/carbidopa, steroids, benzodiazepines, biperiden hydrochloride, immunoglobulin, clonidine | l‐dopa/sinemet | Simultaneously with trihexyphenidyl and a cervical cord stimulator with some improvement. | Vigabatrin | No clinical response to levodopa | NA | NA | NA |
| VAC14 mutations (NM_018052) |
c.1271G>T, p.Trp424Leu, c.1528 + 1G>A, |
c.1744G>T, p.Ala582Ser c.1748C>T, p.Ser583Leu, | c.1271 G>T, p.Trp424Leu; c.1096 + 1G>C | c.1685C>T, p. Ala562Val homozygous | c.2005G>T, p. Val669Leu homozygous |
c.1744G>A; p.Ala582Thr c.2042G>A; p.Arg681His |
||
4. DISCUSSION
VAC14 is a component of a trimolecular complex that tightly regulates the level of phosphatidylinositol 3,5‐bisphosphate (PI (3, 5)P2). The complex's content changes dynamically with fission and fusion events that generate or absorb intracellular transport vesicles. PI (3, 5)P2 is critical for the survival of neural cells (Alghamdi et al., 2013; Chow et al., 2007; Sbrissa et al., 2007; Takasuga & Sasaki, 2013). Vac14 lacking mutant mice exhibit massive neurodegeneration, particularly in the midbrain (Zhang et al., 2007). The missense mutation p.Leu156Arg in mouse Vac14 also results in a lethal neurodegenerative disorder (Jin et al., 2008). Furthermore, similar prominent vacuolation of neurons in basal ganglia is observed in VAC14‐related SNDC patients (Stutterd et al., 2017).
VAC14‐related childhood‐onset neurodegeneration was first reported in 2016 and later was named as SNDC with autosomal recessive inheritance in OMIM (https://omim.org/); only, eight cases have been reported up to now (Lenk et al., 2016; Lyon et al., 2019; Stutterd et al., 2017; Taghavi et al., 2018). Herein, we report another patient with novel VAC14 compound heterozygous pathogenic variants; her parents and eldest brother are heterozygous carriers, and they are clinically healthy. Her second elder brother developed similar symptoms with onset at the same age and died at 8 years old. Therefore, we can presume that this brother had SNDC. The inheritance pattern of this pedigree is in accordance with autosomal recessive inheritance with complete penetrance.
All previously reported patients (n = eight) showed normal motor and psychomotor development prior to onset of disease and had no dysmorphic features or seizures (Lenk et al., 2016; Lyon et al., 2019; Stutterd et al., 2017; Taghavi et al., 2018). A total of 6/8 cases developed abrupt onset of disease in early childhood (from 18 months to 3 years), and the two remaining cases disease presentation occurred at an older age (from 8 to 13 years). Symptom onset was marked by dystonia and spasticity except for the vision problems displayed in case 8 (Lyon et al., 2019); however, all cases have relative preservation of cognitive function. The disease course progressed rapidly, and all patients responded poorly to treatments. Premature death was observed as early as two years after onset in the two patients with early onset of disease (Stutterd et al., 2017). Four cases progressively exacerbated in several years and two of them became bedridden five years after disease presentation (Lenk et al., 2016; Taghavi et al., 2018). Two siblings could walk alone, but they both developed retinitis pigmentosa early in life (Lyon et al., 2019). In the present study, the case was affected by progressive dystonic disorder, and the symptoms progressively deteriorated over the following two years. Our two siblings’ disease onset and progression were similar to the four previously reported cases of early onset (Lenk et al., 2016; Stutterd et al., 2017). Our proband was not given any treatment, because her parents knew her disease and refused treatment options.
Marked abnormality in the basal ganglia was reported in two respective patients with VAC14 mutations (Lenk et al., 2016). Hypointensity in the globus pallidus and substantia nigra was observed in one case (Lyon et al., 2019). The remaining four previously reported cases showed normal brain MRIs with no obvious abnormality (Stutterd et al., 2017; Taghavi et al., 2018). However, no abnormal sign was found in the striatum besides thalamic volume reduction in our proband's MRI scan. Accumulation of cases will be helpful in the identification of MRI‐specific signs of SNDC.
The mutation c.1744G>A (p.Ala582Thr) has not been previously reported. However, at the same site, another variant (c.1744G>T, p.Ala582Ser) was reported to be pathogenic in SNDC (Lenk et al., 2016), in which nonpolar hydrophobic amino acids Ala was converted into polar hydrophilic amino acid Ser. In our patient, the nonpolar hydrophobic amino acid Ala was converted into amino acid Thr, which is polar hydrophilic. Through seven in silico prediction tools, our case's variant (c.1744G>A) was predicted to be inconsistent, which means 2/7 of these tools predicted that this variant was benign or uncertain significance, and the other 5 tools predicted that this variant was damaging, deleterious, or disease‐causing. The C‐terminal domain (residues 523–782) of VAC14 has been previously verified to mediate homomeric interactions and confirmed necessary for the formation and maintenance of the PI (3, 5) P2 regulatory complex (Sbrissa, Ikonomov, Fenner, & Shisheva, 2008). Several variants have been identified, and no benign variation has been observed in exon 15 of VAC14. In addition, this variant leads to an amino acid change in an evolutionarily highly conserved position (UCSC Genome Browser) in the C‐terminal Fig4‐binding domain (InterPro, Q08AM6). Therefore, according to the ACMG classification criteria (Richards et al., 2015), c.1744G>A is likely pathogenic (PM1 + PM2 + PM5 + PP3). The c.2042G>A has not been previously reported, which is localized in the highly conserved evolutionary site (UCSC Genome Browser) in the C‐terminal Fig4 binding domain (Q08AM6). The mutation was not found in Clinvar (https://www.ncbi.nlm.nih.gov/clinvar/) or gnomAD. The in silico prediction tools rate the variant as damaging, deleterious, or disease‐causing. So, the c.2042G>A is unknown significance (PM2 and PP3). Therefore, combined with our case's clinical phenotype and family history, we can predict that these VAC14 compound heterozygous variants were likely pathogenic.
In addition, VAC14 pathogenic variants were recently observed in a female neonate of YVS (Lines et al., 2017). YVS is a severe autosomal recessive disorder characterized by skeletal defects, including cleidocranial dysplasia and digital anomalies, and severe neurologic involvement with neuronal loss (Basel‐Vanagaite, Kornreich, Schiller, Yacobovich, & Merlob, 2008; Lines et al., 2017). Enlarged cytoplasmic vacuoles are found in neurons, muscle, and cartilage. YVS is usually lethal in infancy. Most of the pathogenic variants reported in YVS affected FIG4, a lipid phosphatase involved in PI (3, 5)P2 metabolism (Campeau et al., 2013), except for one patient that was recently reported to exhibit biallelic VAC14 mutations, c.1895C>T, p.Thr632Met (paternal allele) and c.923T>A, p.Leu308* (maternal allele) (Lines et al., 2017). Numerous intracytoplasmic vacuoles were found in the YVS patient's fibroblasts. Vacuolization of fibroblasts was also observed in the SNDC patients and may be rescued by transfection of VAC14 cDNA (Lenk et al., 2016). Histology of a SNDC patient's brain showed very prominent vacuolation in the neuropil in the caudate nucleus, putamen, and globus pallidus that was associated with degenerating neurons (Stutterd et al., 2017). Therefore, we conclude that VAC14 pathogenic variants may be associated with different clinical features including SNDC and YVS. More phenotypes are expected to be discovered. The VAC14 variants have relevantly consistent neuropathology of neuron vacuolation, which has been demonstrated in two different VAC14‐mutated mouse models (Zhang et al., 2007; Jin et al., 2008).
In summary, we presented novel compound heterozygous variants (c.1744G>A/c.2042G>A), giving rise to SNDC in two patients. Population data, bioinformatic, and segregation analysis supported the pathogenicity of these novel variants. In addition, we summarized previously reported clinical manifestations of SNDC and described the first siblings of Asian descent. The present results expanded the spectrum of VAC14 pathogenic variants and the ethnic backgrounds of the affected cases.
Supporting information
Liao S, Chen T, Dai Y, Wang Y, Wu F, Zhong M. Novel VAC14 variants identified in two Chinese siblings with childhood‐onset striatonigral degeneration. Mol Genet Genomic Med. 2020;8:e1101 10.1002/mgg3.1101
Shuang Liao and Tingting Chen are contributed equally to this work.
REFERENCES
- Alghamdi, T. A. , Ho, C. Y. , Mrakovic, A. , Taylor, D. , Mao, D. , & Botelho, R. J. (2013). Vac14 protein multimerization is a prerequisite step for Fab1 protein complex assembly and function. The Journal of Biological Chemistry, 288(13), 9363–9372. 10.1074/jbc.M113.453712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basel‐Vanagaite, L. , Kornreich, L. , Schiller, O. , Yacobovich, J. , & Merlob, P. (2008). Yunis‐Varon syndrome: Further delineation of the phenotype. American Journal of Medical Genetics Part A, 146A, 532–537. 10.1002/ajmg.a.32135 [DOI] [PubMed] [Google Scholar]
- Campeau, P. M. , Lenk, G. M. , Lu, J. T. , Bae, Y. , Burrage, L. , Turnpenny, P. , … Lee, B. H. (2013). Yunis‐Varón syndrome is caused by mutations in FIG4, encoding a phosphoinositide phosphatase. American Journal of Human Genetics, 92(5), 781–791. 10.1016/j.ajhg.2013.03.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow, C. Y. , Zhang, Y. , Dowling, J. J. , Jin, N. , Adamska, M. , Shiga, K. , … Meisler, M. H. (2007). Mutation of FIG4 causes neurodegeneration in the pale tremor mouse and patients with CMT4J. Nature, 448, 68–72. 10.1038/emboj.2008.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin, N. , Chow, C. Y. , Liu, L. I. , Zolov, S. N. , Bronson, R. , Davisson, M. , … Weisman, L. S. (2008). VAC14 nucleates a protein complex essential for the acute interconversion of PI3P and PI(3,5)P(2) in yeast and mouse. The EMBO Journal, 27(24), 3221–3234. 10.1038/emboj.2008.248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenk, G. M. , Szymanska, K. , Debska‐Vielhaber, G. , Rydzanicz, M. , Walczak, A. , Bekiesinska‐Figatowska, M. , … Ploski, R. (2016). Biallelic mutations of VAC14 in pediatric‐onset neurological disease. American Journal of Human Genetics, 99(1), 188–194. 10.1016/j.ajhg.2016.05.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lines, M. A. , Ito, Y. , Kernohan, K. D. , Mears, W. , Hurteau‐Miller, J. , Venkateswaran, S. , … Dyment, D. A. (2017). Yunis‐Varón syndrome caused by biallelic VAC14 mutations. European Journal of Human Genetics, 25(9), 1049–1054. 10.1038/ejhg.2017.99 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lyon, G. J. , Marchi, E. , Ekstein, J. , Meiner, V. , Hirsch, Y. , Scher, S. , … Velinov, M. (2019). VAC14 syndrome in two siblings with retinitis pigmentosa and neurodegeneration with brain iron accumulation. Cold Spring Harbor Molecular Case, 5(6), a003715 10.1101/mcs.a003715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. … ACMG Laboratory Quality Assurance Committee . (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–423. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbrissa, D. , Ikonomov, O. C. , Fenner, H. , & Shisheva, A. (2008). ArPIKfyve homomeric and heteromeric interactions scaffold PIKfyve and Sac3 in a complex to promote PIKfyve activity and functionality. Journal of Molecular Biology, 384(4), 766–779. 10.1016/j.jmb.2008.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sbrissa, D. , Ikonomov, O. C. , Fu, Z. , Ijuin, T. , Gruenberg, J. , Takenawa, T. , & Shisheva, A. (2007). Core protein machinery for mammalian phosphatidylinositol 3,5‐bisphosphate synthesis and turnover that regulates the progression of endosomal transport. Novel Sac phosphatase joins the ArPIKfyve‐PIKfyve complex. The Journal of Biological Chemistry, 282(33), 23878–23891. 10.1074/jbc.M611678200 [DOI] [PubMed] [Google Scholar]
- Stutterd, C. , Diakumis, P. , Bahlo, M. , Fanjul Fernandez, M. , Leventer, R. J. , Delatycki, M. , … Lockhart, P. J. (2017). Neuropathology of childhood‐onset basal ganglia degeneration caused by mutation of VAC14. Annals of Clinical and Translational Neurology, 4(12), 859–864. 10.1002/acn3.487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taghavi, S. , Chaouni, R. , Tafakhori, A. , Azcona, L. J. , Firouzabadi, S. G. , Omrani, M. D. , … Paisán‐Ruiz, C. (2018). A clinical and molecular genetic study of 50 families with autosomal recessive parkinsonism revealed known and novel gene mutations. Molecular Neurobiology, 55(4), 3477–3489. 10.1007/s12035-017-0535-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takasuga, S. , & Sasaki, T. (2013). Phosphatidylinositol‐3,5‐bisphosphate: Metabolism and physiological functions. The Journal of Biochemistry, 154(3), 211–218. 10.1093/jb/mvt064 [DOI] [PubMed] [Google Scholar]
- Zhang, Y. , Zolov, S. N. , Chow, C. Y. , Slutsky, S. G. , Richardson, S. C. , Piper, R. C. , … Weisman, L. S. (2007). Loss of Vac14, a regulator of the signaling lipid phosphatidylinositol 3,5‐bisphosphate, results in neurodegeneration in mice. Proceedings of the National Academy of Sciences of United States of America, 104(44), 17518–17523. 10.1073/pnas.0702275104 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials