

Abstract
Background
Next‐generation sequencing (NGS)‐based panels have gained traction as a strategy for reproductive carrier screening. Their value for screening Ashkenazi Jewish (AJ) individuals, who have benefited greatly from population‐wide targeted testing, as well as Sephardi/Mizrahi Jewish (SMJ) individuals (an underserved population), has not been fully explored.
Methods
The clinical utilization by 6,805 self‐reported Jewish individuals of an expanded NGS panel, along with several ancillary assays, was assessed retrospectively. Data were extracted for a subset of 96 diseases that, during the panel design phase, were classified as being AJ‐, SMJ‐, or pan‐Jewish/pan‐ethnic‐relevant.
Results
64.6% of individuals were identified as carriers of one or more of these 96 diseases. Over 80% of the reported variants would have been missed by following recommended AJ screening guidelines. 10.7% of variants reported for AJs were in “SMJ‐relevant genes,” and 31.2% reported for SMJs were in “AJ‐relevant genes.” Roughly 2.5% of individuals carried a novel, likely pathogenic variant. One in 16 linked cohort couples was identified as a carrier couple for at least one of these 96 diseases.
Conclusion
For maximal carrier identification, this study supports using expanded NGS panels for individuals of all Jewish backgrounds. This approach can better empower at‐risk couples for reproductive decision making.
Keywords: Ashkenazi Jewish, carrier couple, expanded carrier screening, preconception/prenatal genetic testing, Sephardi/Mizrahi Jewish
This retrospective study examines a specific cohort of 6,805 self‐reported Jewish (Ashkenazi/Sephardi/Mizrahi) patients who underwent carrier screening using NGS‐based expanded panels. Results are presented from an extracted set of 96 genes initially classified as “Jewish‐relevant,” albeit that a majority of the patients (79% of cohort) opted for much larger panels that contained this set of 96. The findings highlight the value of large, universal sequence‐based panels, given that (a) self‐reported ethnic information is often unreliable/complex, and (b) many patients want maximal information for their reproductive decision making.
1. INTRODUCTION
Reproductive carrier screening for individuals of Ashkenazi Jewish (AJ; Jews from Central and Eastern Europe; Ostrer & Skorecki, 2013) descent has undergone a remarkable evolution over the past 50 years (Ferreira et al., 2014; Hoffman et al., 2014). The recognition of Tay–Sachs disease (TSD; OMIM: 272800) as one that affects AJ offspring led to enzymatic carrier testing in the 1970s and the near elimination of TSD from this population within a few decades (Kaback, 2000). With the identification of the genetic basis for this and other recessive Mendelian diseases relevant to AJs, genetic testing for population‐specific founder pathogenic variants was developed and quickly gained acceptance. Carrier screening allows at‐risk couples to take advantage of preconception/prenatal options to prevent the birth of affected offspring or, alternatively, to strategize for early intervention and disease management. Ideally this screening should be done before a pregnancy to maximize the options available.
Professional societies such as the American College of Obstetricians and Gynecologists (ACOG) and the American College of Medical Genetics and Genomics (ACMG) have put forth guidelines describing the diseases for which AJ individuals should be offered screening and the criteria for the inclusion of these diseases (ACOG Commitee on Genetics, 2009; Gross, Pletcher, & Monaghan, 2008). The ACMG guidelines from 2008 also opened a window for the addition of diseases over time (Gross et al., 2008), and clinical laboratories began to expand their AJ panels (Hoffman et al., 2014). By adding recessive diseases with AJ carrier frequencies/severities in the range of those of the initial diseases, the cumulative carrier frequency predictably increased, for example, from 1 in 4.3 when testing for the nine ACMG recommended diseases (including cystic fibrosis [CF; OMIM: 219700], which is recommended population‐wide [ACOG Committee on Genetics, 2011; Watson et al., 2004]) to 1 in 2 when testing for a 36‐disease panel (Shi et al., 2017). Acceptance of this screening by the AJ community is widespread, and numerous organizations have arisen to promote awareness and provide education and access to testing (e.g., see https://www.jewishgeneticdiseases.org/resources-for-genetic-screening-2/).
In contrast, there have been limited efforts to address genetic diseases in a population‐wide manner for individuals of Sephardi/Mizrahi Jewish (SMJ; Sephardi = Jews from Southern Europe and North Africa; Mizrahi = Jews from the Middle East/Arab countries) descent (Bloch, 2009; Ostrer & Skorecki, 2013). On the scientific level, this lag can be attributed to the more heterogeneous makeup of this population resulting from specific migrations and bottlenecks and subsequent endogamy. On the communal level, this lag has been associated with a reluctance among its members to participate in research and to access clinical testing due to more limited awareness, inherent superstitions, and fear of stigmatization, which may affect marriage prospects for carriers and their family members (Bloch, 2009). Aside from a few efforts that have been ancestral country‐of‐origin‐based (Kaback et al., 2010; Zlotogora, Grotto, Kaliner, & Gamzu, 2016), carrier screening for SMJ individuals has been invoked primarily in the context of a positive personal/family history of a specific genetic disease.
In this report, we describe results from a cohort of 6,805 self‐reported Jewish individuals who underwent expanded carrier screening using a targeted next‐generation sequencing (NGS)‐based panel over a 2 year time period (June 2015 to June 2017). The panel was designed to not only include an increased set of diseases more prevalent in the AJ population, but also an SMJ disease set, and a set relevant to all Jewish subgroups (as well as other disease genes known to be more relevant to other ethnic groups). As opposed to previous panels that assessed known pathogenic Jewish founder variants by targeted genotyping methods, the NGS panel eliminates biases related to which variants should be ascertained per gene and, when offered to all who report Jewish descent, overcomes being misguided by inaccurate ethnic information (Lazarin & Haque, 2016; Umbarger et al., 2014). The utilization of these types of panels in the reproductive carrier screening realm has been rising (e.g.,see Haque et al. (2016)) due to their increased efficiency and decreases in costs and turnaround times (Gregg, 2018).
Numerous professional societies (ACOG Committee on Genetics, 2017a; Edwards et al., 2015; Grody et al., 2013; Henneman et al., 2016) have outlined the pros and cons of preconception/prenatal expanded carrier screening (ECS) relative to more traditional approaches, and have offered recommendations for its possible laboratory and clinical implementation. In its 2017 committee opinion, ACOG considers ECS “an acceptable strategy,” with the caveat that it “does not replace risk‐based screening” (ACOG Committee on Genetics, 2017a). Findings from our experience with this screening strategy are presented here, with an emphasis on overarching themes relevant for those considering ECS for individuals reporting Jewish descent.
2. MATERIALS AND METHODS
2.1. Editorial policies and ethical considerations
All human subject retrospective research was carried out on anonymized samples in accordance with the policies and procedures of the Mount Sinai Health System and its Institutional Review Board (IRB). For the pilot study (HSM #13‐00743; GCO 13–1611), informed consent was obtained from ~900 self‐reported SMJ individuals who also completed short demographic questionnaires. For the NGS panels and ancillary tests, clinical testing was ordered by healthcare providers, and patients’ written informed consent was obtained.
2.2. Pilot SMJ Study and NGS panel development
For the pilot study, DNA isolated from saliva samples of the SMJ individuals was analyzed for 107 selected variants from 40 disease genes (Table S1) using Agena Bioscience™ iPLEX® pro chemistry on a MassARRAY® platform and Agena Bioscience™ Typer 4.0 genotyping software. Candidate disease selection for this targeted genotyping panel, as well as for the NGS‐based panel, was guided by literature searches, by data from internal laboratory testing databases, and occasionally by family or physician advocacy (see Fedick et al. (2015) for example). Diseases included on the NGS panel met internal criteria of: carrier frequency more common than 1 in 500 in the general population or in a subpopulation (e.g., Jewish), and disease characteristics of (a) early‐onset and severe, or (b) childhood/early adult onset and progressive, or (c) amenable to treatment or intervention. Preclinical validation of the panel was performed using control samples with known pathogenic variants (single nucleotide variants (SNVs) and small indels (insertions/deletions)) previously characterized by standard molecular techniques, according to CLIA (Clinical Laboratory Improvement Amendments), CAP (College of American Pathologists) and NYS‐DOH (New York State‐Department of Health) guidelines.
2.3. NGS clinical testing
Targeted regions (exons, intron‐exon boundaries, and reported pathogenic variants in other regions) were enriched from patients’ genomic DNA with Agilent SureSelectTM QXT® technology on a Bravo NGS platform (Agilent). The barcoded libraries were pooled and sequenced using the Illumina HiSeq2500 system with 100 base pair paired‐end reads. FASTQ sequence files served as inputs to a custom variant of the Broad “GATK best practices” pipeline to obtain variant calls, similar to that described in (Linderman et al., 2014). Genome alignment was to the Broad b37 reference genome. A mean coverage of 300X‐500X was expected over all targeted regions with a minimum average coverage of 200X required for each sample. In addition, a minimum coverage of 20X was required for “promised exons,” which included exons harboring well‐known pathogenic variants as well as exons with multiple variants reported in HGMD (Human Gene Mutation Database; http://www.hgmd.cf.ac.uk/ac/index.php).
Variant curation was performed in real time using a combination of automated (including Cartagenia Bench Lab software (Agilent)) and manual approaches. All novel variants classified as likely pathogenic had to fulfill a minimum of ACMG criteria PVS1 + PM2 (Richards et al., 2015) at the time of curation. Pathogenic and likely pathogenic variants were confirmed by Sanger sequencing and/or MassARRAY according to the NYSDOH guidelines. All novel likely pathogenic variants reported here were submitted to ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/).
Practitioners received single integrated laboratory reports with only pathogenic and likely pathogenic variants reported (Edwards et al., 2015; Richards et al., 2015). Variants of uncertain significance (VUSes) were on occasion unmasked upon provider request. VUSes and likely pathogenic variants are re‐curated by the laboratory on a periodic basis for possible upgrades or downgrades in classification.
Ancillary assays are described in the Supporting Information.
3. RESULTS
3.1. Pilot Study, and targeted NGS panel development
An initial set of candidate pathogenic variants/diseases reported in SMJ individuals was compiled and then culled using inclusion criteria that considered severity, frequency, penetrance, and value for reproductive decision‐making or management. First, a high‐throughput genotyping assay was developed covering 107 reported variants from 39 autosomal recessive genes and one X‐linked disease gene (Table S1). Outreach to SMJ communities spurred support from community leaders who then provided venues for educational talks and sample collection, allowing for the screening of over 900 SMJ individuals from numerous ancestral countries of origin. Carriers were identified for over half of the panel conditions, and several homozygous individuals were identified as well (Table S2; homozygotes identified for familial Mediterranean fever (FMF; OMIM: 249100) and inclusion body myopathy 2 (IBM2; OMIM: 605820)). Four diseases were determined to have a high overall SMJ carrier frequency (>1%) (FMF (~1 in 12), IBM2 (~1 in 11), phenylketonuria (PKU; OMIM: 261600; ~1 in 30) and Wolman disease (LALdef; OMIM: 278000; ~1 in 38)). Carrier frequency generally increased when determined in the context of a relevant subethnic group (e.g., IBM2 carrier frequency in Iranian Jews was 1 in 7 [n = 494]) (data not shown). Finally, ~1,500 anonymized AJ DNAs also were screened using this targeted genotyping panel, and high carrier frequencies (>1%) were found for three diseases (FMF [~1 in 13], TSD [~1 in 25], and glycogen storage disease type II [GSD2; OMIM: 232,300; ~1 in 57]), with some overlapping variants seen between the AJ and SMJ populations for these diseases and others (data not shown).
The above 40 genes formed the core of the SMJ section of the targeted NGS test, a set which was supplemented by another four SMJ disorders (Table S3; SMJ section). Six disorders from the initial 40 SMJ could be classified as pan‐ethnic/pan‐Jewish, together with another four disorders for a total of ten in that category (Table S3; pan‐ethnic/Jewish section). The set of ten includes not only CF as per guidelines (ACOG Committee on Genetics, 2011; Watson et al., 2004), but also spinal muscular atrophy (SMA; OMIM: 253300) which is recommended universally by ACOG and ACMG (ACOG Committee on Genetics, 2017b; Prior, & Professional Practice and Guidelines Committee, 2008), and Fragile X syndrome (FXS; OMIM: 300624), whose appropriateness for universal testing has been debated at length (ACOG Committee on Genetics, 2010; Grody, 2011). With respect to the set of panel genes more relevant for AJ individuals, most came from a 36‐gene panel that already was offered by our laboratory for this subpopulation (Shi et al., 2017). Another 16 relevant disorders met inclusion criteria and were added (Table S3; AJ section). In addition to these 96 genes, the largest ECS panel offered contained 185 additional disease genes that have been reported to be relevant for other ethnic groups.
3.2. Overall experience with offering the expanded panel
Over a 2‐year period, 6,805 individuals of self‐reported Jewish ancestry received carrier screening through our clinical laboratory using the targeted NGS‐based panels. A majority of these patients self‐reported as AJ, but there was a combined SMJ subcohort of 299 individuals as well as an “other Jewish” subcohort that included those of mixed parental or of less certain Jewish backgrounds. Most patients (79%) opted for large panels of 252–281 genes, whereas some preferred to limit their testing to smaller sized panels (Figure 1). Results presented here focus on NGS findings from a set of 96 genes, shown in Table S3, that during the panel design phase were classified as more relevant for AJ and SMJ patients.
Figure 1.
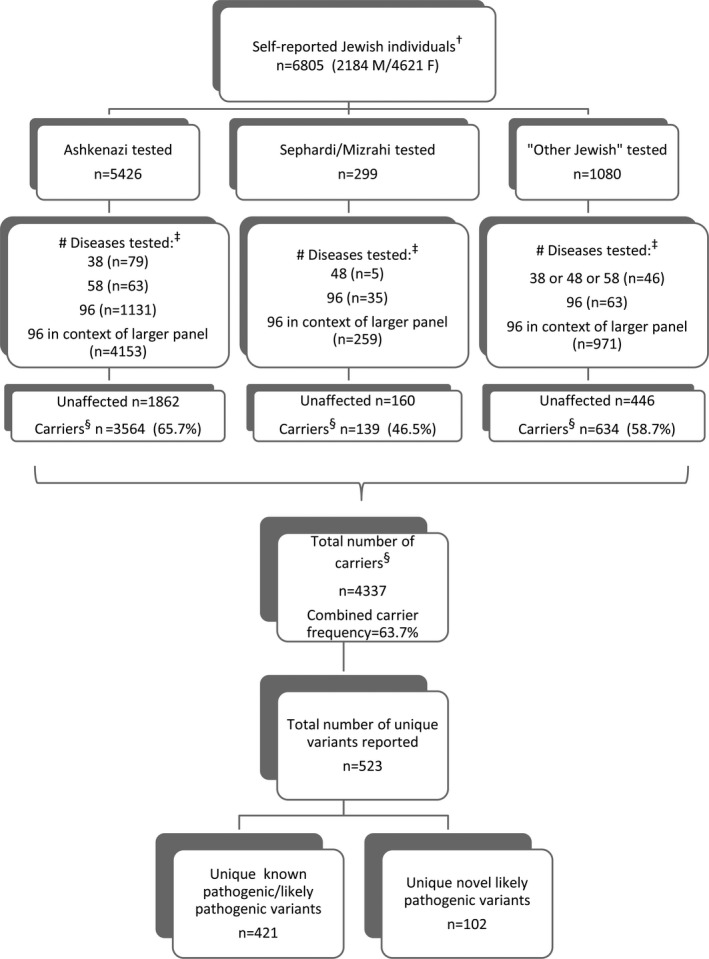
Overview of testing performed for self‐reported Jewish individuals. †Those with an indication other than routine carrier screening have been excluded, albeit that some individuals may have had (more limited) carrier screening previously. ‡The genotyping panel of 38 is that described in (Shi et al., 2017) with the addition of SMA/FXS testing. The panel of 96 is described in the text and shown in Table S3; subpanels of that included 48 SMJ + pan‐ethnic/pan‐Jewish diseases or 58 AJ + pan‐ethnic/pan‐Jewish diseases. Larger panels (of 252–281 diseases) included the full 96‐disease set. §Carrier is defined as someone carrying one or more reported pathogenic/likely pathogenic variants in any of the 96 genes as detected by NGS (or genotyping analysis for certain variants [see methods in Supporting Information]). Abbreviations: M, males; F, females
The overall chance of being a carrier for one or more of these 96 disease genes tested by NGS was 1 in 1.57 (63.7% of individuals) (Figure 1; carriers for FXS and SMA as determined by other methods are not included). Approximately 27% of individuals were carriers for multiple diseases, 1253 were carriers of two diseases, 425 of three diseases, and 134 of four or more diseases (data not shown). Additionally, several homozygous individuals and individuals carrying two distinct variants in the same gene also were identified (data not shown). Finally, many carriers, and some putative Jewish founder variants, also were identified when individuals from this cohort were tested for the additional genes on the panel beyond this set of 96 (manuscript in preparation).
Observed carrier frequencies for the set of 96 diseases are shown in Table S4 for the total cohort as well as the AJ subcohort. AJ carrier frequencies were largely consistent with those reported in the literature, with the six highest frequencies observed being for Gaucher disease (GD; OMIM: 230800, 230900, 231000), FMF, CF, factor 11 deficiency (F11def; OMIM: 612416), nonsyndromic hearing loss (DFNB1A; OMIM: 220290), and PKU. For the limited SMJ subcohort, only results from 31 diseases where two or more carriers were found are shown (Table S5; for another 25 of the 96 diseases, one SMJ carrier for each was observed). Notably, there were only six diseases for which no carriers were identified in any of the subcohorts (Table S4).
For the AJ cohort, 5,779 variants in total were reported for the 96 diseases, and, as shown in Figure 2 (top), differing proportions of these variants would have been detected by previous targeted genotyping panels (9 or 36) or using only the AJ + pan‐ethnic/pan‐Jewish subportions of the NGS panel (58 genes). This latter subpanel would have missed 10.7% of variants that were found in the “SMJ‐relevant genes.” Similarly, if SMJ individuals were tested only for the SMJ + pan‐ethnic/pan‐Jewish subportions of the NGS panel (48 genes), 31.2% of the 176 total variants reported for this cohort would have been missed (Figure 2, bottom), since they were found in “AJ‐relevant genes.” Of a total of 523 unique variants reported for the 96 diseases, there were 102 novel (i.e., not reported in the literature) variants that were classified as likely pathogenic (Richards et al., 2015; Figure 1). Roughly 2.5% of individuals in the cohort carried a novel likely pathogenic variant, and a subset of recurring variants is presented in Table S6. Several of these variants have been found in the gnomAD (Genome Aggregation Database; http://gnomad.broadinstitute.org/) and ExAC (Exome Aggregation Consortium) browser (http://exac.broadinstitute.org/) databases to be at increased frequency in AJs, and may potentially represent novel AJ (or pan‐Jewish) founder mutations (similar to what has been described in Zlotogora, Patrinos, & Meiner (2018)).
Figure 2.
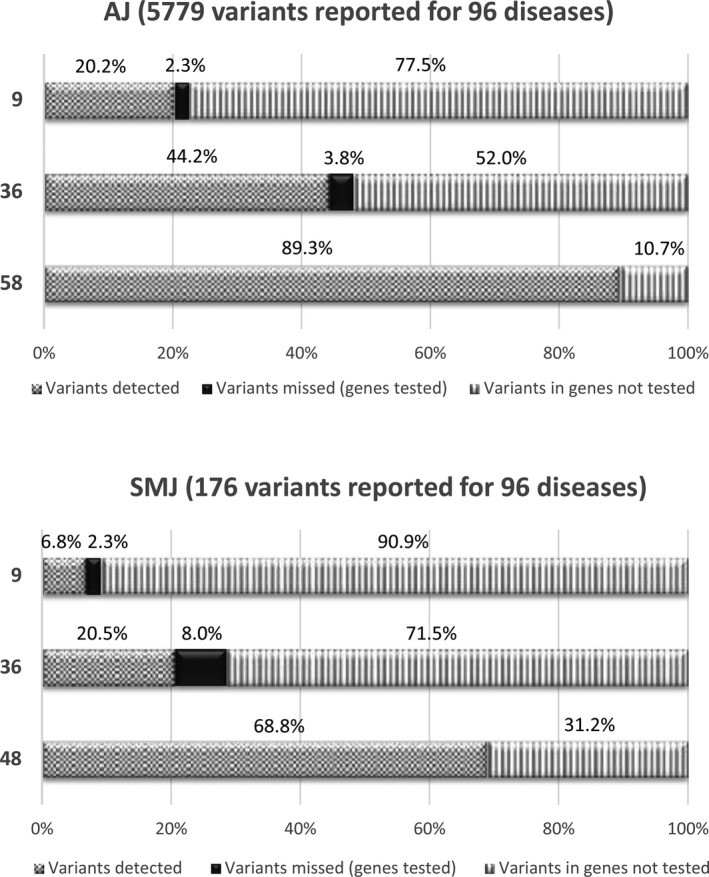
Increased detection rates with panel expansion. Each bar represents the breakdown of the three categories (labeled on x‐axis) for a given testing panel (y‐axis) relative to the total number of reported “variants detected” when the 96 disease gene set of the NGS panel is used for AJs (top) or SMJs (bottom), each of which is taken to be 100%. The genotyping panel of 36 diseases is described in Shi et al. (2017), and the genotyping panel of nine diseases includes CF plus the eight ACMG diseases (Gross et al., 2008). NGS panels of 48 and 58 are described in the text; both exclude SMA and FXS carriers that are detected by other means. Abbreviations: AJ, Ashkenazi Jewish; SMJ, Sephardi/Mizrahi Jewish
3.3. Special Interest Diseases
Carrier status for several of the diseases was analyzed by ancillary or alternative methods (see Supporting Information). For TSD, 226 HEXA carriers via DNA analysis were identified; this number includes one individual carrying a novel likely pathogenic variant (c.1A > C), but excludes 12 pseudodeficiency allele carriers (Figure 3 and Table S7). Seventy‐six percent of those HEXA carriers via DNA analysis also were tested for Hexosaminidase A (HexA) enzyme activity, and 92% were HEXA carriers via enzyme analysis, 6% were inconclusive, and one (a c.1274_1277dupTATC carrier) was unexpectedly a noncarrier by enzyme analysis. Additionally, the carrier of a B1 allele (c.533G>A) was classified as a noncarrier by enzyme analysis and represents a false negative (see (Mahuran, 1999) for review). Of the 167 patients that were determined to be HEXA carriers via enzyme analysis, 96.4% were also HEXA carriers via DNA analysis. Six individuals determined to be HEXA carriers via enzyme analysis not carry reportable HEXA variants, albeit that three of the six did carry a VUS (data not shown). Thirty‐two putative Sandhoff disease (OMIM: 268800) carriers also were identified based on high total HexA levels (Mahuran, 1999), and these individuals had follow‐up HEXB analysis (Figure 3b and data not shown).
Figure 3.
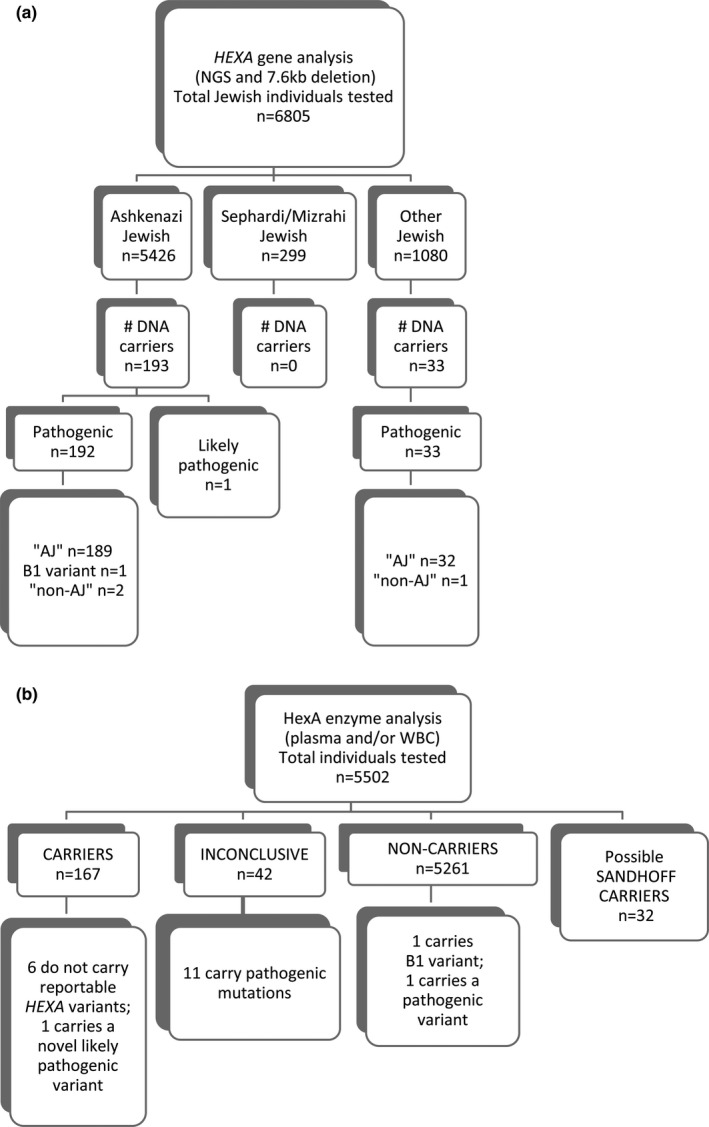
Overview of results from TSD screening. DNA (a) and enzyme (b) testing results are shown. DNA carriers do not include those with pseudodeficiency alleles. Abbreviations: AJ, Ashkenazi Jewish; NGS, next‐generation sequencing
For SMA, gene dosage analysis was performed to assess loss of SMN1 exon 7, and deletion carriers were identified at a frequency of 1 in 76 in the AJ cohort (Table 1), similar to what has been reported in comparably sized cohorts (Sugarman et al., 2012). Previously we described an SMN1 g.27134T > G intron 7 variant in individuals of AJ (and other) descent that is present in ~ 50% of all AJ SMN1 duplication events (Luo et al., 2014). By extension, the presence of this founder variant in the context of two copies of SMN1 can identify a subset of likely SMA silent (2 + 0) carriers (Feng et al., 2017; Luo et al., 2014). Genotyping analysis for this variant identified 17 of these likely silent carriers (of 4,842 tested individuals, 0.35%) (Table 1). Notably, the AJ residual risk after a negative result from copy number analysis (i.e., two copies detected) decreases from 1 in 672 to 1 in 978 when also negative for this variant, and increases to 1 in 10 when positive for the variant.
Table 1.
SMA deletion and likely silent AJ carriers identified. Screening results from SMN1 copy number and g.27134T > G genotyping analyses are shown for the AJ (Ashkenazi Jewish) subcohort. Carrier frequencies for the likely silent carriers may be underestimates, since some are missed even with g.27134T > G genotyping (Luo et al., 2014). Residual risk estimates are noted in the text and were calculated as per (Luo et al., 2014)
| Category | Copy number and genotyping results | n | 1 in |
|---|---|---|---|
| 1 (deletion carriers) | SMN1 copies = 1, g.27134T>G: Negative | 64 | 76 |
| 2 | SMN1 copies = 2, g.27134T>G: Negative | 4,094 | |
| 3 (likely silent carriers) | SMN1 copies = 2, g.27134T>G: Positive | 17 | 285 |
| 4 | SMN1 copies≥=3, g.27134T>G: Negative | 365 | |
| 5 | SMN1 copies≥=3, g.27134T>G: Positive | 302 | |
| Total tested | 4,842 |
For FXS, CGG repeat expansion sizes were determined for females only, since these alleles do not typically expand in the male germline (ACOG Committee on Genetics, 2017b). Carrier frequencies (premutation + full mutation) of 1 in 128 in the total cohort and 1 in 118 in the AJ subcohort were observed (Table S8), consistent with published frequencies (Hantash et al., 2011). Only one full mutation carrier (262 repeats), and only two premutation carriers with ≥90 repeats and an associated highly significant chance of expansion (80% chance; ACOG Committee on Genetics, 2017b) were identified. Although rare FMR1 intragenic pathogenic variants have been reported for carriers (Lugenbeel, Peier, Carson, Chudley, & Nelson, 1995), these were not seen in the 6,805 individuals tested by NGS.
3.4. Clinical outcomes and decision making after receiving ECS results
There were 831 linked couples identified in which both members of the couple were part of this cohort and were tested either in tandem or sequentially. Of these, 53 couples were shown to be carrier couples for the same genetic disorder among the set of 96 genes, and most of these couples were AJ‐AJ pairs (Figure 4a). There were 19 diseases for which carrier couples were detected (Figure 4b): ten diseases were classified on the panel as AJ, five as pan‐ethnic/pan‐Jewish, and four as SMJ. Of note, several AJ‐AJ couples were carrier couples for the diseases more relevant to SMJs (data not shown). When assessing the 53 couples by the nature of the specific variants identified, 26 of the allele combinations were expected to be fully penetrant, and 30 of reduced penetrance (three couples were double carrier couples; Figure 4b). Finally, at the time of testing, 30% of the 53 couples were pregnant, and 56% of the pregnant couples already knew their carrier couple status (mostly for variants in genes/disorders on the ACMG eight‐gene panel [Gross et al., 2008] or in CFTR) (Figure 4c and data not shown).
Figure 4.
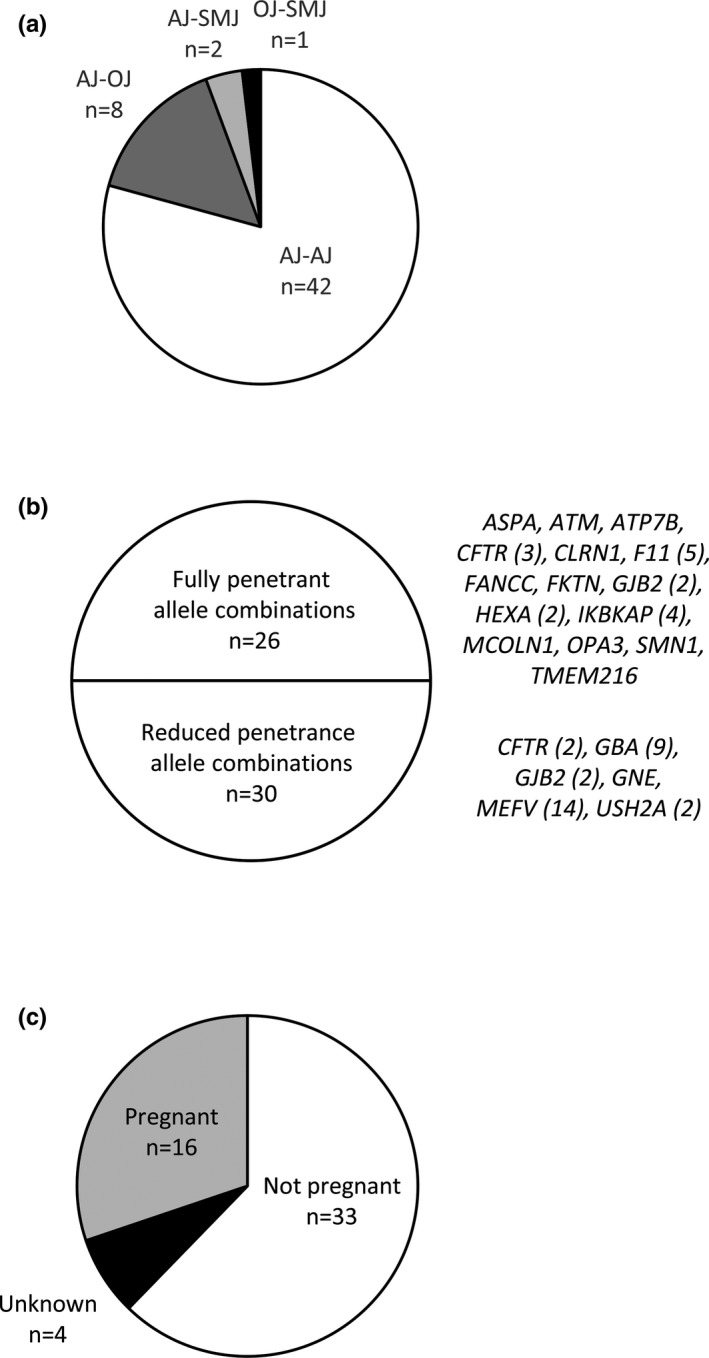
Characteristics of carrier couples for autosomal recessive diseases. (a) Distribution of Jewish subethnicities of identified couples wherein both members were carriers for the same genetic disorder among the set of 96 genes. (b) Distribution of these carrier couples by penetrance of their allelic combinations. The number of couples who were carriers for a given gene is shown in parentheses. CFTR and GJB2 fall into both groups, depending on the specific variants detected. (c) Pregnancy status of the identified carrier couples at the time of testing. Abbreviations: AJ, Ashkenazi Jewish; OJ, Other Jewish; SMJ, Sephardi/Mizrahi Jewish
4. DISCUSSION
This report describes findings from NGS‐based reproductive carrier screening in individuals of different Jewish backgrounds (Ashkenazi, Sephardi and/or Mizrahi), with a particular focus on a 96 disease gene subset of a larger expanded panel. The recovery of an extensive set of pathogenic/likely pathogenic variants from this type of universal testing yielded a high cumulative carrier frequency for these 96 diseases. In the total cohort, 64.6% of individuals were identified as carriers of one or more diseases (when NGS carriers, SMA deletion + silent carriers, and FXS FM + PM carriers all are included). While the substantial representation of AJs in the total pool of carriers is expected based on this ethnicity being “high risk,” almost 50% of the SMJ individuals were found to be carriers (Figure 1). We recognize that our SMJ carrier frequencies were determined in a comparatively smaller‐sized subcohort, which could relate in part to the smaller number of SMJs in the United States relative to AJs (Bloch, 2009). It has been estimated that only approximately 5% of Jews living in the United States are not AJ. Additionally, because SMJs have been an underserved population in terms of carrier screening, there also is likely a continuing need to educate SMJ individuals (and physicians) about the benefits of carrier screening and the normalcy of being a carrier (Bloch, 2009).
While breaking down the panel into AJ, SMJ, and pan‐ethnic/pan‐Jewish subsections was helpful for panel design purposes, this study does not support using sub‐Jewish‐oriented panels for testing (as some patients did; see Figure 1), since AJ carriers of “SMJ diseases” and SMJ carriers of classically “AJ diseases” were identified (Figure 2, Tables S4 and S5). Specific pathogenic variants also are shared between these groups (data not shown), and these variations may have predated the formation of Jewish Diaspora populations (Ostrer & Skorecki, 2013). Moreover, if the end‐goal is maximal carrier identification, then this study also does not support using only the 96 gene subpanel on self‐reported Jewish patients (as some patients did; see Figure 1), since many carriers, and some putative Jewish founder variants, also were identified when testing was done for additional genes beyond the set of 96 (manuscript in preparation). This is consistent with the trending of the field towards offering a universal panel for all individuals screened by ECS (Lazarin & Haque, 2016), especially since ethnic identification is often unreliable/complex, but also because this would be a more equitable approach.
Increased panel uptake by individuals of any Jewish descent will allow for more accurate determination of carrier frequencies and residual risk estimates, an objective that also will be aided by increasing detection rates through technological improvements such as the ability to assess copy number variation (see also Beauchamp et al. (2017)). Over time, shared findings from functional studies and clinical cases also will lead to enhanced abilities to assign definitive classifications to novel/rare variants and to resolve a proportion of the variants classified as VUS. This becomes particularly poignant for couples in which one member is a carrier of a known pathogenic variant and the other carries a variant whose pathogenicity is indeterminate.
To improve detection rates, the current study supports the utilization of ancillary assays for certain diseases, albeit that these are labor‐ and time‐intensive to perform and antithetical to the trend toward multiplexing. The continued used of HexA enzyme analysis (also see Hoffman et al. (2013)) is supported by our identification of one carrier of a novel likely pathogenic variant (HEXA c.1A>C start loss variant) who was definitively a TSD carrier by enzyme analysis. Additionally, several TSD carriers by enzyme analysis were identified who did not carry reportable DNA variants (Figure 3 and Table S7); these individuals are being been counseled as TSD carriers until proven otherwise. While the enzyme assay is sensitive to storage and transport conditions and biological factors including hormonal medications and pregnancy, DNA testing is limited by the ability to classify a variant as pathogenic due to available reports in the literature. The two assays performed in conjunction have a greater ability to identify carriers of the disease than either one alone.
Ancillary testing also proved valuable for the identification of some AJ likely silent (2 + 0) SMA carriers who harbored the SMN1 g.27134T>G intron 7 variant (Luo et al., 2014). Approximately 50% of silent (2 + 0) SMA carriers are expected to be identified using this method. Of note, one of the likely silent carriers belonged to a couple whose other member was a deletion carrier, and therefore they were counseled as an SMA carrier couple (Figure 4b). As has been done with this SMN1 variant, there may be other diseases associated with complex alleles for which supplemental testing could be helpful for detecting additional carriers and improving residual risk estimates after a negative result.
In addition to the SMA carrier couple, there were 52 other carrier couples found among the 831 linked couples, representing a rate of 1 in 16 (or 1 in 32 if only fully penetrant allelic combinations are considered). In some respects, this high rate of carrier couples may appear to be higher than expected, as these patients have been tested for many more diseases than previously reported in other Jewish cohorts. Additionally, some of these couples were likely pursuing testing because the first member of the couple was found to be a carrier, thus increasing the carrier couple rate. However, this high rate of carrier couples in this cohort is expected to be an underestimate, since (a) carrier couples were not included if only one of the two members was part of this cohort, and (b) carrier couples for diseases beyond the 96 gene subset also were not included. Of the identified 53 couples, 42% already knew their carrier couple status from previous (more limited) testing, and it is likely that some had more readily pursued expanded testing as it became available. Future studies will monitor decision making in these and other carrier couples, as well as assess whether the nature of the allelic combinations affects these choices (see also (Ghiossi, Goldberg, Haque, Lazarin, & Wong, 2017)). Given the complex nature of ECS results, posttest genetic counseling of carrier couples must include a careful and user‐friendly review of the nature of the diseases and the specific variants identified. This is especially important given that pretest genetic counseling education is likely overwhelming for patients due to the quantity and diversity of the panel genes and the complexity of possible NGS results.
The clinical utility of ECS lies in empowering at‐risk couples with maximal, useful knowledge to assist them in making reproductive decisions that also will be informed by the couples’ personal values and beliefs. We recognize that there are opponents of using ECS as a reproductive screening strategy, and also that the inclusion of certain genes/diseases on targeted ECS panels may be criticized based on their being too rare, associated with mild/variable phenotypes, having later onset, among other concerns (ACOG Committee on Genetics, 2017a; Gregg, 2018; Grody, 2016). However, since this testing is available and has high analytical sensitivity and specificity, one can argue that the decisions of whether to pursue this, and whether/how to act on the returned results, should be left up to the patient/couple. Patients also need to be aware that, at least in a cohort such as this one, the chance of being a carrier or part of a carrier couple is high, and the testing process itself does not affect that chance.
CONFLICT OF INTEREST
All authors except GA, NSA, MD, and BW are employees of Sema4, a clinical laboratory that performs carrier screening and receives monetary reimbursement for testing services. Sema4 employees are compensated by Sema4, and NSA and BW receive compensation as consultants for Sema4.
AUTHORS CONTRIBUTION
LE, GA, AHB, NSA, EES, and RK were involved in conceptualization of the project. GA, AHB, XC, GC, LS, CY, GMV, ASM, and BDW were involved in data curation. AHB, NSA, JZ, and GA were involved in formal analysis. EES and LE were involved in funding acquisition. GA, AHB, LE, MWD, and BDW were involved in investigation. XC, GC, LS, and CY were involved in methodology. LE and RK were involved in project administration. LE and EES were involved in resourcing. ASM, AHB, and GC were involved in software. LE, RK, and FS were involved in supervision. LE, RK, AHB, XC, GC, LS, GMV, and CY were involved in validation. NSA, GA, and AHB were involved in visualization. NSA, GA, and AHB were involved in writing the first draft. All authors were involved in review and editing of the second draft.
Supporting information
ACKNOWLEDGMENTS
We acknowledge the contributions of the Sema4 testing technologists, variant curators, and bioinformaticians. We thank those individuals who participated in the SMJ pilot study and the organization SHORE (https://www.shoreforlife.org/) for its involvement therein.
Akler G, Birch AH, Schreiber‐Agus N, et al. Lessons learned from expanded reproductive carrier screening in self‐reported Ashkenazi, Sephardi, and Mizrahi Jewish patients. Mol Genet Genomic Med. 2020;8:e1053 10.1002/mgg3.1053
Akler and Birch should be considered joint first authors.
Contributor Information
Ruth Kornreich, Email: ruth.kornreich@sema4.com.
Lisa Edelmann, Email: lisa.edelmann@sema4.com.
REFERENCES
- ACOG Committee on Genetics (2009). ACOG Committee opinion no. 442: Preconception and prenatal carrier screening for genetic diseases in individuals of Eastern European Jewish descent. Obstetrics and Gynecology, 114(4), 950–953. 10.1097/AOG.0b013e3181bd12f4 [DOI] [PubMed] [Google Scholar]
- ACOG Committee on Genetics (2010). ACOG Committee opinion no. 469: Carrier screening for fragile X syndrome. Obstetrics and Gynecology, 116(4), 1008–1010. 10.1097/AOG.0b013e3181fae884 [DOI] [PubMed] [Google Scholar]
- ACOG Committee on Genetics (2017a). Committee opinion no. 690: Carrier screening in the age of genomic medicine. Obstetrics and Gynecology, 129(3), e35–e40. 10.1097/AOG.0000000000001951 [DOI] [PubMed] [Google Scholar]
- ACOG Committee on Genetics (2017b). Committee opinion no. 691: Carrier screening for genetic conditions. Obstetrics and Gynecology, 129(3), e41–e55. 10.1097/AOG.0000000000001952. [DOI] [PubMed] [Google Scholar]
- Beauchamp, K. A. , Muzzey, D. , Wong, K. K. , Hogan, G. J. , Karimi, K. , Candille, S. I. , … Haque, I. S. (2017). Systematic design and comparison of expanded carrier screening panels. Genetics in Medicine, 20(1), 55–63 10.1038/gim.2017.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bloch, T. (2009). Sephardi Jews lack screening programs for their genetic diseases. The Forward. Retrieved from https://www.haaretz.com/1.5092392 [Google Scholar]
- ACOG Committee on Genetics . (2011). ACOG Committee Opinion No. 486: Update on carrier screening for cystic fibrosis. Obstetrics and Gynecology, 117(4), 1028–1031. 10.1097/AOG.0b013e31821922c2 [DOI] [PubMed] [Google Scholar]
- Edwards, J. G. , Feldman, G. , Goldberg, J. , Gregg, A. R. , Norton, M. E. , Rose, N. C. , … Watson, M. S. (2015). Expanded carrier screening in reproductive medicine‐points to consider: A joint statement of the American College of Medical Genetics and Genomics, American College of Obstetricians and Gynecologists, National Society of Genetic Counselors, Perinatal Quality Foundation, and Society for Maternal‐Fetal Medicine. Obstetrics and Gynecology, 125(3), 653–662. 10.1097/AOG.0000000000000666 [DOI] [PubMed] [Google Scholar]
- Fedick, A. M. , Shi, L. , Jalas, C. , Treff, N. R. , Ekstein, J. , Kornreich, R. , … Savage, S. A. (2015). Carrier screening of RTEL1 mutations in the Ashkenazi Jewish population. Clinical Genetics, 88(2), 177–181. 10.1111/cge.12459 [DOI] [PubMed] [Google Scholar]
- Feng, Y. , Ge, X. , Meng, L. , Scull, J. , Li, J. , Tian, X. , … Zhang, J. (2017). The next generation of population‐based spinal muscular atrophy carrier screening: Comprehensive pan‐ethnic SMN1 copy‐number and sequence variant analysis by massively parallel sequencing. Genetics in Medicine, 19(8), 936–944. 10.1038/gim.2016.215 [DOI] [PubMed] [Google Scholar]
- Ferreira, J. C. , Schreiber‐Agus, N. , Carter, S. M. , Klugman, S. , Gregg, A. R. , & Gross, S. J. (2014). Carrier testing for Ashkenazi Jewish disorders in the prenatal setting: Navigating the genetic maze. American Journal of Obstetrics and Gynecology, 211(3), 197–204. 10.1016/j.ajog.2014.02.001 [DOI] [PubMed] [Google Scholar]
- Ghiossi, C. E. , Goldberg, J. D. , Haque, I. S. , Lazarin, G. A. , & Wong, K. K. (2017). Clinical utility of expanded carrier screening: reproductive behaviors of at‐risk couples. Journal of Genetic Counseling, 27(3), 616–625. 10.1007/s10897-017-0160-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregg, A. R. (2018). Expanded carrier screening. Obstetrics and Gynecology Clinics of North America, 45(1), 103–112. 10.1016/j.ogc.2017.10.005 [DOI] [PubMed] [Google Scholar]
- Grody, W. W. (2011). Expanded carrier screening and the law of unintended consequences: From cystic fibrosis to fragile X. Genetics in Medicine, 13(12), 996–997. 10.1097/GIM.0b013e31823c49a2 [DOI] [PubMed] [Google Scholar]
- Grody, W. W. (2016). Where to draw the boundaries for prenatal carrier screening. JAMA, 316(7), 717–719. 10.1001/jama.2016.10888 [DOI] [PubMed] [Google Scholar]
- Grody, W. W. , Thompson, B. H. , Gregg, A. R. , Bean, L. H. , Monaghan, K. G. , Schneider, A. , & Lebo, R. V. (2013). ACMG position statement on prenatal/preconception expanded carrier screening. Genetics in Medicine, 15(6), 482–483. 10.1038/gim.2013.47 [DOI] [PubMed] [Google Scholar]
- Gross, S. J. , Pletcher, B. A. , & Monaghan, K. G. . (2008). Carrier screening in individuals of Ashkenazi Jewish descent. Genetics in Medicine, 10(1), 54–56. 10.1097/GIM.0b013e31815f247c [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hantash, F. M. , Goos, D. M. , Crossley, B. , Anderson, B. , Zhang, K. , Sun, W. , & Strom, C. M. (2011). FMR1 premutation carrier frequency in patients undergoing routine population‐based carrier screening: Insights into the prevalence of fragile X syndrome, fragile X‐associated tremor/ataxia syndrome, and fragile X‐associated primary ovarian insufficiency in the United States. Genetics in Medicine, 13(1), 39–45. 10.1097/GIM.0b013e3181fa9fad [DOI] [PubMed] [Google Scholar]
- Haque, I. S. , Lazarin, G. A. , Kang, H. P. , Evans, E. A. , Goldberg, J. D. , & Wapner, R. J. (2016). Modeled fetal risk of genetic diseases identified by expanded carrier screening. JAMA, 316(7), 734–742. 10.1001/jama.2016.11139 [DOI] [PubMed] [Google Scholar]
- Henneman, L. , Borry, P. , Chokoshvili, D. , Cornel, M. C. , van El, C. G. , Forzano, F. , … Peterlin, B. (2016). Responsible implementation of expanded carrier screening. European Journal of Human Genetics, 24(6), e1–e12. 10.1038/ejhg.2015.271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, J. D. , Greger, V. , Strovel, E. T. , Blitzer, M. G. , Umbarger, M. A. , Kennedy, C. , … Towne, C. (2013). Next‐generation DNA sequencing of HEXA: A step in the right direction for carrier screening. Molecular Genetics and Genomic Medicine, 1(4), 260–268. 10.1002/mgg3.37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffman, J. D. , Park, J. J. , Schreiber‐Agus, N. , Kornreich, R. , Tanner, A. K. , Keiles, S. , … Heim, R. A. (2014). The Ashkenazi Jewish carrier screening panel: Evolution, status quo, and disparities. Prenatal Diagnosis, 34(12), 1161–1167. 10.1002/pd.4446 [DOI] [PubMed] [Google Scholar]
- Kaback, M. (2000). Population‐based genetic screening for reproductive counseling: The Tay‐Sachs disease model. European Journal of Pediatrics, 159(Suppl 3), S192–195. 10.1007/PL00014401 [DOI] [PubMed] [Google Scholar]
- Kaback, M. , Lopatequi, J. , Portuges, A. R. , Quindipan, C. , Pariani, M. , Salimpour‐Davidov, N. , & Rimoin, D. L. (2010). Genetic screening in the Persian Jewish community: A pilot study. Genetics in Medicine, 12(10), 628–633. 10.1097/GIM.0b013e3181edef5b [DOI] [PubMed] [Google Scholar]
- Lazarin, G. A. , & Haque, I. S. (2016). Expanded carrier screening: A review of early implementation and literature. Seminars in Perinatology, 40(1), 29–34. 10.1053/j.semperi.2015.11.005 [DOI] [PubMed] [Google Scholar]
- Linderman, M. D. , Brandt, T. , Edelmann, L. , Jabado, O. , Kasai, Y. , Kornreich, R. , … Schadt, E. E. (2014). Analytical validation of whole exome and whole genome sequencing for clinical applications. BMC Medical Genomics, 7, 20 10.1186/1755-8794-7-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugenbeel, K. A. , Peier, A. M. , Carson, N. L. , Chudley, A. E. , & Nelson, D. L. (1995). Intragenic loss of function mutations demonstrate the primary role of FMR1 in fragile X syndrome. Nature Genetics, 10(4), 483–485. 10.1038/ng0895-483 [DOI] [PubMed] [Google Scholar]
- Luo, M. , Liu, L. , Peter, I. , Zhu, J. , Scott, S. A. , Zhao, G. , … Edelmann, L. (2014). An Ashkenazi Jewish SMN1 haplotype specific to duplication alleles improves pan‐ethnic carrier screening for spinal muscular atrophy. Genetics in Medicine, 16(2), 149–156. 10.1038/gim.2013.84 [DOI] [PubMed] [Google Scholar]
- Mahuran, D. J. (1999). Biochemical consequences of mutations causing the GM2 gangliosidoses. Biochimica Et Biophysica Acta, 1455(2–3), 105–138. 10.1016/S0925-4439(99)00074-5 [DOI] [PubMed] [Google Scholar]
- Ostrer, H. , & Skorecki, K. (2013). The population genetics of the Jewish people. Human Genetics, 132(2), 119–127. 10.1007/s00439-012-1235-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prior, T. W. , & Professional Practice and Guidelines Committee (2008). Carrier screening for spinal muscular atrophy. Genetics in Medicine, 10(11), 840–842. 10.1097/GIM.0b013e318188d069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards, S. , Aziz, N. , Bale, S. , Bick, D. , Das, S. , Gastier‐Foster, J. , … Rehm, H. L. (2015). Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in Medicine, 17(5), 405–424. 10.1038/gim.2015.30 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, L. , Webb, B. D. , Birch, A. H. , Elkhoury, L. , McCarthy, J. , Cai, X. , … Kornreich, R. (2017). Comprehensive population screening in the Ashkenazi Jewish population for recurrent disease‐causing variants. Clinical Genetics, 91(4), 599–604. 10.1111/cge.12834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugarman, E. A. , Nagan, N. , Zhu, H. , Akmaev, V. R. , Zhou, Z. , Rohlfs, E. M. , … Allitto, B. A. (2012). Pan‐ethnic carrier screening and prenatal diagnosis for spinal muscular atrophy: Clinical laboratory analysis of >72,400 specimens. European Journal of Human Genetics, 20(1), 27–32. 10.1038/ejhg.2011.134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Umbarger, M. A. , Kennedy, C. J. , Saunders, P. , Breton, B. , Chennagiri, N. , Emhoff, J. , … Porreca, G. J. (2014). Next‐generation carrier screening. Genetics in Medicine, 16(2), 132–140. 10.1038/gim.2013.83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, M. S. , Cutting, G. R. , Desnick, R. J. , Driscoll, D. A. , Klinger, K. , Mennuti, M. , … Grody, W. W. (2004). Cystic fibrosis population carrier screening: 2004 revision of American College of Medical Genetics mutation panel. Genetics in Medicine, 6(5), 387–391. https://doi.org/10.109701.GIM.0000139506.11694.7C [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotogora, J. , Grotto, I. , Kaliner, E. , & Gamzu, R. (2016). The Israeli national population program of genetic carrier screening for reproductive purposes. Genetics in Medicine, 18(2), 203–206. 10.1038/gim.2015.55 [DOI] [PubMed] [Google Scholar]
- Zlotogora, J. , Patrinos, G. P. , & Meiner, V. (2018). Ashkenazi Jewish genomic variants: Integrating data from the Israeli National Genetic Database and gnomAD. Genetics in Medicine, 20(8), 867–871. 10.1038/gim.2017.193 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials