

Abstract
Methionine synthases are essential enzymes for amino acid and methyl group metabolism in all domains of life. Here, we describe a putatively anciently derived type of methionine synthase yet unknown in bacteria, here referred to as core-MetE. The enzyme appears to represent a minimal MetE form and transfers methyl groups from methylcobalamin instead of methyl-tetrahydrofolate to homocysteine. Accordingly, it does not possess the tetrahydrofolate binding domain described for canonical bacterial MetE proteins. In Dehalococcoides mccartyi strain CBDB1, an obligate anaerobic, mesophilic, slowly growing organohalide-respiring bacterium, it is encoded by the locus cbdbA481. In line with the observation to not accept methyl groups from methyl-tetrahydrofolate, all known genomes of bacteria of the class Dehalococcoidia lack metF encoding for methylene-tetrahydrofolate reductase synthesizing methyl-tetrahydrofolate, but all contain a core-metE gene. We heterologously expressed core-MetECBDB in E. coli and purified the 38 kDa protein. Core-MetECBDB exhibited Michaelis-Menten kinetics with respect to methylcob(III)alamin (KM ≈ 240 µM) and L-homocysteine (KM ≈ 50 µM). Only methylcob(III)alamin was found to be active as methyl donor with a kcat ≈ 60 s−1. Core-MetECBDB did not functionally complement metE-deficient E. coli strain DH5α (ΔmetE::kan) suggesting that core-MetECBDB and the canonical MetE enzyme from E. coli have different enzymatic specificities also in vivo. Core-MetE appears to be similar to a MetE-ancestor evolved before LUCA (last universal common ancestor) using methylated cobalamins as methyl donor whereas the canonical MetE consists of a tandem repeat and might have evolved by duplication of the core-MetE and diversification of the N-terminal part to a tetrahydrofolate-binding domain.
Subject terms: Enzymes, Transferases, Methylases, Molecular evolution
Introduction
Methionine plays an essential role as proteinogenic amino acid in all domains of life, as an initiation amino acid in protein translation1 and as a precursor in the formation of cysteine, carnitine, taurine and lecithin2,3. Moreover, methionine can be converted to S-adenosyl-L-methionine (SAM)4, which represents an activated methyl group donor for many fundamental cellular processes5,6. The final step in methionine de novo synthesis, the methylation of homocysteine to methionine, is catalyzed by different types of methionine synthases including cobalamin-dependent (MetH) and cobalamin-independent methionine synthase (MetE). Some bacteria, e.g. Escherichia coli, possess genes for both enzymes7 and repress the expression of metE in the presence of vitamin B128. Homocysteine methylation in mammals is catalyzed by mammalian methionine synthases (mMS) similar to bacterial MetH9, betaine-L-homocysteine-S-methyltransferase (BHMT) or S-methyl-L-methionine-L-homocysteine-S-methyltransferase (also known as BHMT-2)10. Fungi and plants encode exclusively MetE11 or BHMT-212. All known methionine synthase types contain a zinc ion in the active site that is essential for homocysteine binding and methyl group transfer13.
MetH (EC 2.1.1.13) catalyzes the methyl transfer from 5-methyl-tetrahydrofolate-monoglutamate (5-methyl-THF-Glu) to homocysteine. MetH from E. coli is a large monomeric protein of 1,227 amino acids (136 kDa) and is composed of four functional domains14. In the catalytic cycle the methyl group of methylcob(III)alamin is transferred to homocysteine forming cob(I)alamin and methionine. Subsequently, cob(I)alamin is remethylated using 5-methyl-THF-Glu as the methyl group donor regenerating methylcob(III)alamin. For reactivation of cob(II)alamin to methylcob(III)alamin, which is generated in a side-reaction approximately once in 2,000 turnovers15, SAM is required16.
Canonical MetE proteins (EC 2.1.1.14) are described as a family of zinc-containing metalloenzymes sharing no sequence similarity with MetH1,13. They catalyze the methylation of homocysteine using 5-methyl-THF-Glun (n ≥ 3) as methyl donor without the involvement of cobalamin. MetE in E. coli is a protein of 753 amino acid residues (85 kDa) that is composed of two homologous parts connected by a linker region (Fig. 1b), suggesting that the domains have evolved by gene duplication of a sequence encoding a smaller protein of approximately 340 amino acid residues17–19. We here refer to the canonical E. coli-type MetE as ‘tandem-repeat MetE’ (tr-MetE). The active site of tr-MetE is located within the C-terminal part, where the zinc ion is coordinated by one histidine, two cysteine and one glutamate residue. The binding site for methyl-THF is in the cleft between the two domains of tr-MetE19.
Figure 1.

Bioinformatic analysis of the core-MetECBDB from Dehalococcoides mccartyi strain CBDB1. (a) Maximum-Likelihood phylogenetic tree of MetE representatives was generated with MEGA767. Multiple amino acid sequence alignments of full length with deletion of gaps (MUSCLE algorithm) were used to generate the tree. The analysis involved 39 amino acid sequences including the C-terminus of tandem-repeat methionine synthases (tr-MetE) from bacteria (brown colors) and yeast (green) as well as core-MetEs from archaea (blue colors), Chloroflexi (red) and Clostridiales (brown). The gene loci are in brackets. (b) The crystal structure of core-MetECBDB was calculated with the I-TASSER server65. Overlay of the crystal structure of tr-MetE from Neurospora crassa (PDB No. 4ZTX, grey) with the structural model obtained for core-MetECBDB from D. mccartyi strain CBDB1 (green) was obtained with PyMOL66. Core-MetECBDB matches the C-terminal part of tr-MetENcra (C-score = −0.27) but lacks the N-terminal part and the linker region. (c) Amino acid sequence alignment of selected MetE proteins. The Zn2+-binding site HXCXnC (red) is conserved in annotated tr-MetEs and also in core-MetE homologs. Core-MetECBDB: core-MetE from D. mccartyi strain CBDB1, coreMetEDehly: core-MetE from Dehalogenimonas lykanthroporepellens, core-MetEMMKA: core-MetE from Methanococcus maripaludis, tr-MetEECDH: tandem-repeat MetE from Escherichia coli DH10B, tr-MetESau: tandem-repeat MetE from Staphylococcus aureus.
Dehalococcoides mccartyi strain CBDB1 is an obligately anaerobic, mesophilic bacterium belonging to the phylum Chloroflexi, class Dehalococcoidia20,21. Dehalococcoides species are well known for their ability to use a wide range of persistent and toxic halogenated organic compounds as terminal electron acceptor in anaerobic respiration (“organohalide respiration”) with hydrogen as electron donor22. Strain CBDB1 encodes one of the largest numbers of B12-dependent proteins in known prokaryotes23. The most prominent representatives of B12-dependent proteins in strain CBDB1 are reductive dehalogenases that are responsible for the reduction of halogenated pollutants as a terminal electron acceptor24. Vitamin B12 in the medium is essential for the growth of Dehalococcoides strains25 because Dehalococcoides do not contain many of the genes for de novo biosynthesis of cobalamin26,27. However, Dehalococcoides strains encode corrinoid-specific ABC-transporter and enzymes for the late B12 biosynthesis pathway enabling them to incorporate corrinoid precursors from the environment and to modify them to cobalamin28,29. Although none of the known D. mccartyi genomes contains full gene homologs of metE, metH, bhmt or bhmt-226, D. mccartyi strains synthesize methionine de novo24,30,31. Zhuang et al. demonstrated that acetyl-CoA donates the C2-methyl group for an unconventional methionine biosynthesis pathway independent from methylene-tetrahydrofolate reductase (MTHFR). All D. mccartyi species sequenced so far, lack metF encoding for MTHFR that reduces 5,10-methylene-THF-Glu to 5-methyl-THF-Glu. Furthermore, D. mccartyi strain 195 was not able to incorporate 5-methyl-THF from the environment32.
Here, we found that locus cbdbA481 (NCBI accession number CAI82680) of D. mccartyi strain CBDB1 encodes a 343 amino acid protein that is homologous to the C-terminus of canonical tr-MetE and similar to methylcobalamin:homocysteine methyltransferase (core-MetEMTH) of the methanogenic archaeum Methanobacterium thermoautotrophicum33. In our study we provide biochemical and genetic evidence that the locus cbdbA481 encodes a novel type of bacterial methionine synthase that appears to be an anciently derived MetE-related methionine synthase obtaining its methyl group from an external corrinoid rather than from folate. Together with archaeal methylcobalamin:homocysteine methyltransferases and bacterial homologs the gene product of locus cbdbA481 forms a new group of basal methionine synthases, referred to as core-MetE in the following.
Results
Bioinformatic analysis of locus cbdbA481 in the genome of D. mccartyi strain CBDB1
D. mccartyi strains are able to synthesize methionine de novo, although D. mccartyi genomes do not contain gene homologs of metE, metH, bhmt or bhmt-226. In the KEGG (Kyoto Encyclopedia of Genes and Genomes) database several enzymes of the Dehalococcoides methionine metabolism are annotated34–36. The loci cbdbA476 and cbdbA477 in the genome of strain CBDB1 are annotated as SAM synthetase (EC 2.5.1.6) and S-adenosyl-L-homocysteinase (EC 3.3.1.1), respectively. Since methionine biosynthesis and SAM metabolism are biochemically closely linked, we started our search for a methionine synthase gene in the genome of strain CBDB1 by inspecting the direct neighborhood of the loci cbdbA476 and cbdbA477. Locus cbdbA481 located in the same operon encodes a 343 amino acid polypeptide (here referred to as core-MetECBDB) with a calculated molecular mass of 38 kDa that has sequence similarity with the C-terminal half of canonical tandem-repeat MetE (tr-MetE) proteins. Accordingly, the calculated mass of core-MetECBDB is only about half the size of tr-MetE proteins (e.g. tr-MetEEco of E. coli). Phylogenetic analysis of core-MetECBDB together with 38 other protein sequences including annotated tr-MetE representatives, archaeal methylcobalamin:homocysteine methyltransferase homologs (core-MetEArchaea) and Chloroflexi sequences with high sequence similarity to core-MetECBDB indicates that core-MetECBDB is the prototype of a new bacterial cluster of short MetE sequences (Fig. 1a). This new cluster is phylogenetically well separated from the C-termini of tr-MetE proteins and core-MetEArchaea (Fig. 1a, blue colors)33. Core-MetE proteins are widely distributed in microorganisms with strongly conserved ancient traits (archaea, Clostridiales, Dehalococcoidia and Chloroflexia classes). Within the archaea, only Haloquadratum spp. encode the tr-MetE, whereas many archaea encode a core-MetE homolog (Fig. 1a, blue colors). Bacterial and archaeal core-MetE form a paraphyletic group (excluding the tr-MetE sequences), probably evolved before LUCA (last universal common ancestor) and branched into two groups. Tr-MetEs appear to have evolved from archaeal core-MetE (e.g. core-MetE in Clostridium kluyveri and C. oryzae) (Fig. 1a).
The computational structural model of core-MetECBDB (Fig. 1b, green) resembles the C-terminal domain of annotated tr-MetE proteins, bearing the highest similarity to methionine synthase from Neurospora crassa (C-score = −0.27, identity = 21% and RMSD = 3.52) (PDB No. 4ZTX)37. Compared to tr-MetE proteins, core-MetECBDB lacks the N-terminal domain described to be responsible for 5-methyl-THF binding and the linker region between the C- and N-terminal parts (Fig. 1b, grey). An amino acid alignment of core-MetECBDB with core-MetEDehly from Dehalogenimonas lykanthroporepellens, core-MetEMMKA from Methanococcus maripaludis KA1 and the C-terminal halves of the tr-MetEECDH from E. coli DH10B and tr-MetESau from Staphylococcus aureus shows that the zinc binding motif HXCXnC, essential for L-homocysteine binding and activation13, is conserved in core-MetECBDB (Fig. 1c). The computational model indicates that in strain CBDB1, zinc is coordinated in a tetrahedral fashion by His215, Cys217 and Cys312 (Fig. 1c) which are conserved among all MetEs and by Asp236. In contrast, zinc of tr-MetE from N. crassa is bound by histidine, two cysteine and one glutamate residue.
Heterologous production and purification of core-MetECBDB
To study the function of core-MetECBDB in detail, the recombinant protein was heterologously produced in E. coli and purified. First, production and purification attempts were conducted for a C-terminally Streptavidin-tagged core-MetECBDB using affinity chromatography for purification. However, native polyacrylamide gel electrophoresis (PAGE) indicated misfolding of the protein (Supplementary Figure 1a,b). Therefore, core-MetECBDB was produced without a tag in E. coli and purified using anion exchange chromatography. The purified protein exhibited a molecular mass of approximately 38 kDa, as determined by SDS-PAGE (Supplementary Figure 1c). The identity of core-MetECBDB was verified by liquid chromatography-tandem mass spectrometry (LC-MS/MS) (47 validated unique peptides, 87% coverage). Purified core-MetECBDB was in its monomeric form as indicated by native PAGE (Supplementary Figure 1d) and analytical size exclusion chromatography (data not shown).
Coordination environment of methylcob(III)alamin bound to core-MetECBDB
The binding of methylcob(III)alamin to core-MetECBDB was analyzed spectrophotometrically after mixing methylcob(III)alamin and core-MetECBDB in a 1:1 stoichiometry at a concentration of 10 µM each. Free methylcob(III)alamin exhibited α/β- and γ-absorption bands characteristic for the “base-on” mode (Fig. 2, violet line)38. On binding to core-MetECBDB, the UV/Vis spectrum of methylcob(III)alamin slightly changed: the absorption intensities of β- and γ-bands increased and the absorption maximum of the β-band blue-shifted slightly (Δλ = −5 nm) (Fig. 2, red line).
Figure 2.
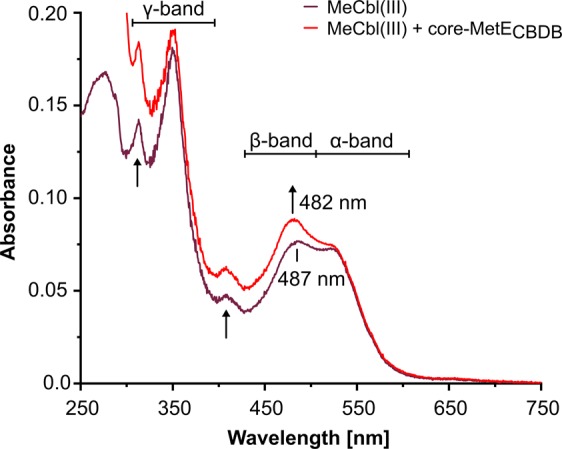
Coordination mode of methylcob(III)alamin (MeCbl(III)) bound to core-MetECBDB from Dehalococcoides mccartyi strain CBDB1. Free MeCbl(III) in the “base-on” mode is characterized by broad α/β-absorbance bands and characteristic maxima at ~ 487 and 524 nm (violet line). The UV/Vis spectrum of MeCbl(III) in the presence of core-MetECBDB at 1:1 stoichiometry was slightly changed. The absorbance intensities of β- and γ-bands increased and the maximum of the β-band shifted (Δλ = −5 nm) (red line), indicating the “base-off/His-on” binding mode of MeCbl(III).
Core-MetECBDB catalyzes methionine formation with methylcob(III)alamin as methyl donor
The enzymatic activity of purified core-MetECBDB was tested using methylcob(III)alamin as methyl donor and homocysteine as methyl acceptor. In the presence of core-MetECBDB, the UV/Vis absorption spectrum of methylcob(III)alamin, exhibiting a characteristic maximum at 524 nm, successively changed over time due to the consumption of methylcob(III)alamin and formation of cob(I)alamin and cob(II)alamin, as indicated by the emergence of absorption features at 681 nm and 474 nm, respectively (Fig. 3a). In the absence of core-MetECBDB or homocysteine, the UV/Vis spectrum of methylcob(III)alamin remained unchanged (Fig. 3b,c). In order to exclude any methyltransferase activity due to impurities of the protein preparation, E. coli cell-free extract was also tested and did not show any activity (Fig. 3d). Finally, in addition to the photometric measurements, the formation of methionine ([M + H]+ = 150.0583 m/z) during the enzymatic reaction was verified via liquid chromatography-mass spectrometry (LC-MS) (Supplementary Figure 2b). In the following, core-MetECBDB enzyme activity was monitored by measuring the increase of absorption at 681 nm (Fig. 3e,f) or the decrease of absorption at 524 nm (Supplementary Figure 2a). Kinetic parameters for core-MetECBDB were determined using an enzyme concentration of 0.1 µM. At a constant D,L-homocysteine concentration of 2 mM and varying methylcob(III)alamin concentrations, methionine was formed with a Vmax = 1664 ± 50 nkat mg−1 and a KM = 236 ± 3 µM for methylcob(III)alamin (Fig. 3e). When different D,L-homocysteine concentrations were used at a fixed methylcob(III)alamin concentration of 0.5 mM, a Vmax = 1582 ± 11 nkat mg−1 and a KM = 98 ± 0 µM for D,L-homocysteine were estimated (Fig. 3f). Since methionine synthase is specific for L-homocysteine, the apparent KM for L-homocysteine might be half of that for D,L-homocysteine33. The maximum turnover number (kcat) was calculated to be about 60 s−1. The substrate specificity of core-MetECBDB was investigated by replacing homocysteine with 2 mM cysteine, 2 mM glutathione or 2 mM dithiothreitol. Core-MetECBDB did not show any activity towards these thiol analogs (data not shown).
Figure 3.

Demethylation of methylcob(III)alamin (MeCbl(III)) catalyzed by purified core-MetECBDB from D. mccartyi strain CBDB1 in the presence of D,L-homocysteine. (a) In the presence of 0.1 µM core-MetECBDB, 0.5 mM methylcob(III)alamin and 2 mM D,L-homocysteine, continuous changes in the UV/Vis spectrum were observed indicating the consumption of methylcob(III)alamin (524 nm) and the formation of cob(I)alamin (681 nm) and cob(II)alamin (474 nm) as highlighted by arrows. (b) In the absence of core-MetECBDB, the UV/Vis spectrum remained unchanged over an incubation time of 30 min. (c) In the absence of D,L-homocysteine, the UV/Vis spectrum remained unchanged. (d) In the presence of 0.5 mg mL−1 E. coli crude extract, instead of core-MetECBDB as catalyst, MeCbl(III) was not transformed. (e) Dependence of core-MetECBDB methyltransferase activity on methylcob(III)alamin concentration. (f) Dependence of core-MetECBDB methyltransferase activity on D,L-homocysteine concentration. The activities in panels (e,f) were determined by following the change of the absorption at 681 nm. Vmax and KM were calculated according to a Hill-Fit plot with R2 = 0.998 for panel (e) and R2 = 0.999 for panel (f), respectively.
Additionally, 5-methyl-THF-Glu3 was tested as a methyl group donor for core-MetECBDB instead of methylcobalamin (Fig. 4b). In the negative control and also in the presence of core-MetECBDB, slow demethylation of 5 methyl-THF-Glu3 occurred abiotically39–41. The demethylation of 5-methyl-THF-Glu3 in the negative control and in the presence of core-MetECBDB was not linked to L methionine formation (Fig. 4b(I)), while in the presence of tr-MetEEco, methionine was formed exhibiting a signal at [M + H]+ = 150.0583 m/z (Fig. 4b(II)).
Figure 4.

Core-MetECBDB from D. mccartyi strain CBDB1 does not catalyze the formation of L-methionine with 5-methyl-THF-Glu3 as the methyl donor. (a) Reaction described for tr-MetE from E. coli (tr-MetEEco) catalyzing the methylation of L-homocysteine with 5-methyl-THF-Glu3 to form L-methionine and THF-Glu380. (b) Representative HPLC chromatograms of a chemical standard of 5-methyl-THF-Glu3 (blue), of reaction products after an enzyme activity assay containing 5-methyl-THF-Glu3, D,L-homocysteine and either core-MetECBDB (red) or tr-MetEEco (green) or no enzyme (black). The peak at RT = 21 min represents dithiothreitol added to all reactions. In the presence of tr-MetEEco, 5-methyl-THF-Glu3 (RT = 18 min) reacts to form THF-Glu3 (RT = 17.4 min). Slow demethylation of 5-methyl-THF-Glu3 to THF-Glu3 did occur in the negative control and also in the presence of core-MetECBDB. To evaluate if this demethylation was linked to L-methionine formation, the products of the activity assays were analyzed by mass spectrometry. I) No L-methionine was formed in the presence of core-MetECBDB; II) The product of tr-MetEEco was identified as L-methionine ([M + H]+ = 150.0583 m/z).
pH-optimum and thermal stability of core-MetECBDB
Methionine synthase activity of core-MetECBDB was observed between pH 5.0 and 9.0, with an optimum between pH 6 and 6.5 (Table 1). The thermal stability of purified tr-MetEEco and core-MetECBDB were assessed by recording protein melting curves using nano differential scanning fluorimetry (nanoDSF). For tr-MetEEco, a melting temperature Tm = 55.8 ± 0.2 °C was determined. In contrast, the Tm of core-MetECBDB was at 68.8 ± 0.0 °C (Supplementary Figure 3).
Table 1.
Demethylation of methylcob(III)alamin (MeCbl(III)) catalyzed by core-MetECBDB from D. mccartyi strain CBDB1 in the presence of D,L-homocysteine at different pH values.
| pH | MeCbl(III) consumption [µM min−1] | relative activity [%] |
|---|---|---|
| 5.0 | 20.53 ± 4.46 | 82.0 |
| 5.5 | 19.12 ± 3.23 | 76.4 |
| 6.0 | 25.05 ± 0.37 | 100 |
| 6.5 | 23.87 ± 1.41 | 95.3 |
| 7.0 | 18.96 ± 0.07 | 75.6 |
| 7.5 | 21.61 ± 0.41 | 86.3 |
| 8.0 | 18.08 ± 1.02 | 72.3 |
| 8.5 | 17.70 ± 0.88 | 70.6 |
| 9.0 | 14.23 ± 0.61 | 56.9 |
Core-MetECBDB does not complement tr-metE-deficient E. coli in vivo
The examination of enzymatic activities of core-MetECBDB in vitro has limitations. However, we were not able to conduct in vivo mutagenesis studies with strain CBDB1 because Dehalococcoides species are not yet genetically accessible. In order to obtain insights into the physiological role of the cbdbA481 gene product in vivo, we tested whether a tr-metE-deficient E. coli strain could be complemented by core-MetECBDB. Therefore, we generated a tr-metE-deficient knockout strain of E. coli DH5α (ΔmetE::kan) that still contained the metH gene for the cobalamin-dependent MetH. This strain was not able to grow in medium without added cyanocobalamin (Supplementary Figure 4, red solid line), but grew when cyanocobalamin was supplemented (Supplementary Figure 4, red dotted line). Next, the growth behavior of the mutant strain carrying different complementation plasmids was investigated. Either the original tr-metEEco gene or the core-MetECBDB nucleotide sequence, both under the control of an arabinose promotor, were provided. Growth experiments with these complementation strains showed that neither of the two strains grew without inducing gene expression by arabinose. After induction with arabinose, tr-metEEco was able to complement the ΔmetE strain as expected (Supplementary Figure 4, blue dotted line), while core-MetECBDB was not (Supplementary Figure 4, green dotted line). These results suggested that core-MetECBDB does not have the same physiological function as the canonical tr-MetEEco.
Discussion
Methionine and SAM have been suggested to belong to the most ancient molecules on earth and might have emerged within or even before the “RNA world”42–44. Although methionine appears to have a continued central metabolic role for more than three billion years, different routes for its biosynthesis have evolved. The biochemically conserved methionine pathway appears to be the product of an evolutionary patchwork involving diverse methionine synthases5. In our study, we identified a novel bacterial MetE-like methionine synthase in D. mccartyi strain CBDB1 that uses methylcobalamin as methyl donor instead of methylated tetrahydrofolate. Our results suggest that this enzyme is the basal form of canonical tandem-repeat MetE (tr-MetE) proteins with roughly half its size and without the domain duplication of canonical MetE proteins evolved to enable tetrahydrofolate binding on the N-terminal domain19. Homologs of this short methionine synthase are encoded in the genomes of several deeply-rooting obligate anaerobic microorganisms from both prokaryotic domains, including all Dehalococcoidia and many Clostridia (e.g. Desulfitobacterium metallireducens, C. kluyveri, C. oryzae) as well as almost all archaea sequenced so far (Fig. 1a). We refer to this short monomeric MetE form as “core-MetE”, because several lines of evidence hint at its basal descendence including the lack of duplication, the exclusive presence in deeply rooting phylogenetic taxa, and the dependence on corrinoids, which are thought to be ancient cofactors45,46, as they participate in fundamental processes such as ribonucleotide reduction47, the Wood-Ljungdahl-pathway48 and methane formation49.
Compared with tr-MetEEco, core-MetECBDB is more stable towards pH changes18 and thermal denaturation. The turnover number kcat ≈ 60 s−1 of core-MetECBDB is very high in comparison to other methionine synthases such as tr-MetEEco with 0.4 s−1 50 or E. coli MetH with 26 s−1 51. The activity of MetH is based on domain movements, which could contribute to the lower catalytic rate in comparison to a small monomeric core-MetE. The relatively slow conversion rate of tr-MetE proteins can be due to the poor methylation power of 5-methyl-THF-Glun (Fig. 4a) and the weak nucleophilicity of homocysteine at physiological pH52. In tr-MetE and MetH, 5-methyl-THF must be activated for the nucleophilic attack by protonation at N5 15. In both MetE and MetH, the nucleophilicity of homocysteine is enhanced by coordination with Zn2+ that serves as Lewis acid13. While in the “base-on” form the dimethylbenzimidazole (Dmbz) base of methylcob(III)alamin is coordinated to the cobalt center of the corrin ring, in the “base-off” mode Dmbz is dissociated from the cobalt. Stabilization of the transition state of methylcob(III)alamin in the “base-off” or “base-off/His-on” binding mode enable nucleophilic attack of homocysteine by weakening the Co-C bond and by reducing the thermodynamic barrier53–55. Thus, only binding modes “base-off” or “base-off/His-on” enable methyl transfer from methylcob(III)alamin. However, the “base-off” mode of methylcob(III)alamin which is characterized by strong spectral changes namely, a significant blue shift in the UV/Vis spectrum and reduced intensity of the γ-band38,56,57, was not observed in our study (Fig. 2). It is difficult to precisely distinguish between the “base-on” and “base-off/His-on” form because, the UV/Vis spectra of them are very similar56. The formation of methionine and cob(I)alamin (Fig. 3a) can only take place if methylcob(III)alamin and homocysteine are bound to core-MetECBDB in a stable and catalytically favorable configuration. Due to the minor spectral changes, we propose that methylcob(III)alamin is utilized by core-MetECBDB in the “base-off/His-on” binding mode. The “base-off/His-on” binding mode is found in many B12-dependent proteins with a consensus motif DxHxxG, where His represents the lower axial ligand replacing the Dmbz moiety58. In our computational model of core-MetECBDB, His122 points towards the active site of the protein and probably belongs to a truncated B12-binding motif with the sequence HxxG, conserved among all Dehalococcoidia (Fig. 5).
Figure 5.
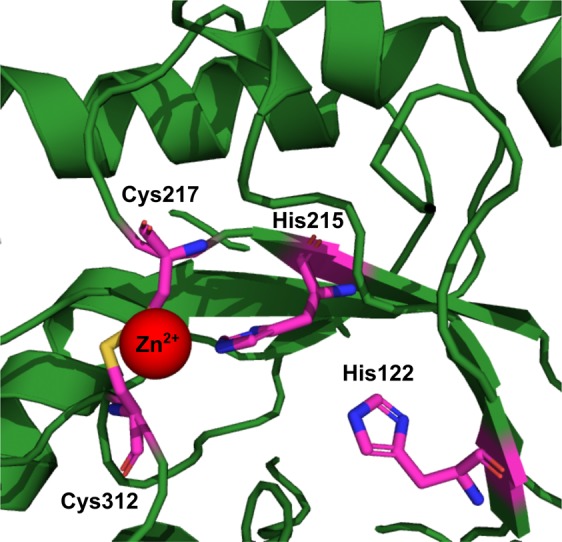
Computational model of the active site of core-MetECBDB from Dehalococcoides mccartyi strain CBDB1. His122 is tuned towards the active site of the protein where a zinc atom is coordinated by His215, Cys217 and Cys312. His122 might replace the dimethylbenzimidazole moiety of cobalamin in the “bae-off/His-on” mode. The structure was calculated with the I-TASSER server65 and visualized with PyMOL66.
The determined KM-value of core-MetECBDB for methylcob(III)alamin of ~ 240 µM is likely much higher than the intracellular concentration of free methylcobalamin29,59. Therefore, the physiological methyl donor might not be soluble methyl(III)cobalamin. We hypothesize that the physiological methyl donor is a corrinoid protein that directly interacts with core-MetECBDB. Needless to say, that inference of physiological characteristics from the determination of enzyme activity in vitro is limited. Examining the role of core-MetE in vivo could shed more light on the essentiality and functionality of the enzyme. However, genetic modification of Dehalococcoides strains is not possible yet.
The methyl group transferred by Dehalococcoides methionine synthase origins from exogenously supplied acetate, as has been shown by Zhuang et al.32. Acetate is activated in Dehalococcoides by acetyl-CoA synthetase (ACS) to acetyl-CoA31. Acetyl-CoA is then cleaved to free coenzyme A, carbon monoxide (which leaves the cell) and a methyl group originating from the C2-atom of acetate. This reaction is catalyzed by acetyl-CoA decarbonylase/synthase (AcsB), an enzyme known mostly for its activity in the opposite direction for carbon fixation via the Wood-Ljungdahl pathway60. In D. mccartyi strain CBDB1, AcsB represents the acetyl-CoA decarbonylase and AcsCD a dimeric corrinoid iron-sulfur protein (CoFeSP) to which the methyl group from acetyl-CoA is transferred61. Zhuang et al. hypothesized that the methyl group is then transferred from AcsCD to tetrahydrofolate and from there to homocysteine, but Dehalococcoides neither encode the methyltransferases acsE, responsible for the methyl transfer from CoFeSP to tetrahydrofolate nor the classical metE/metH, responsible for methyl transfer from methyl-tetrahydrofolate to homocysteine (Fig. 6a,b)32. Our results can now explain these two gaps by hypothesizing that the methyl group from AcsCD/CoFeSP is directly transferred to homocysteine by core-MetECBDB instead of taking the diversion via tetrahydrofolate (Fig. 6c). This hypothesis would also explain the absence of carbon monoxide dehydrogenase in Dehalococcoides which would be needed if the Wood-Ljungdahl pathway was employed for CO2 fixation. With the direct transfer of methyl groups from CoFeSP to homocysteine, the cells would be independent from methyl-tetrahydrofolate and indeed metF encoding methylene-tetrahydrofolate reductase (MTHFR) is missing in all Dehalococcoides genomes27. This might be an unusual pathway in extant microbiology but in our view could represent a very early evolutionary stage in which methyl metabolism could have been independent from folates. This view is supported by the fact that methionine, SAM, corrinoids and coenzyme A are conserved between archaea and bacteria but tetrahydrofolate/tetrahydromethanopterin are not45,46.
Figure 6.

Pathways of L-glycine and L-homocysteine methylation in Dehalococcoides species and hypothesized involvement of THF and corrinoid proteins. (a) Genes annotated to be involved in the incomplete Wood-Ljungdahl pathway encoded in D. mccartyi strain 195 and the respective homologous genes in other Dehalococcoidia. (b) L-serine formation via glycine hydroxymethyltransferase (GlyA, grey) as proposed by Zhuang et al. is shown32. The methyl group is derived most probably from formate with the aid of formyl-tetrahydrofolate synthase (Fhs, yellow) and methylene-tetrahydrofolate dehydrogenase/cyclohydrolase (FolD, blue). (c) Methylation of L-homocysteine is conducted by core-MetECBDB encoded by the locus cbdbA481 (purple) in D. mccartyi strain CBDB1. The original source of the methyl group is acetate, which is activated to acetyl-CoA and then cleaved by acetyl-CoA decarbonylase (AcsB, green) into HSCoA, carbon monoxide (CO) and a methyl group. The standard activity of AcsB is to transfer the methyl group to a corrinoid iron-sulfur protein complex (CoFeSP) AcsCD (red). We speculate that the methyl group is directly transferred from the CoFeSP to the core-MetECBDB (A481) for L-homocysteine methylation (dashed arrow) but this transfer could also be indirect via a yet unidentified participant.
Enzymes similar to the core-MetE identified in Dehalococcoides were also found in the majority of archaea (Fig. 1a, blue colors). M. thermoautotrophicum and other methanogens are described to encode methylcobalamin:homocysteine methyltransferase (core-MetEArchaea)33, a protein of 308 amino acids that is also homologous to the C-terminal part of tr-MetE proteins. In vitro experiments showed, that core-MetEArchaea utilizes methylated corrinoids for the methylation of homocysteine, similar to what we now found for the core-MetECBDB. Schröder and Thauer concluded that soluble methylcobalamin is unlikely the physiological methyl group donor and hypothesized that a corrinoid protein with yet unknown function could play this role. The gene products of MTH124 or MTH1156 were proposed as possible candidates. MTH1156 encodes MtrH, a protein with sequence similarity to the 5-methyl-THF-Glu-binding domain of MetH (26% identity)33. MtrH is part of the methyl-tetrahydromethanopterin-coenzyme M methyltransferase complex and catalyzes the methylation of cob(I)alamin to methylcob(III)alamin using methyl-tetrahydromethanopterin as methyl group donor62. In contrast to methyl-THF biosynthesis in Dehalococcoides strains, methanopterin biosynthesis, a functional equivalent to THF in archaea, is fully encoded in all methanogenic archaea49,63. The methyl group of methionine in methanogenic archaea is derived from methyl-tetrahydromethanopterin49,64, which might be the primary methyl donor of archaeal methylcobalamin:homocysteine methyltransferases.
In conclusion, our findings show that bacterial core-MetECBDB homologs together with archaeal core-MetE representatives form a basal group of methionine synthases using methylcobalamin in vitro as co-substrate. Due to the fact that organisms encoding core-MetE enzymes are slowly growing strict anaerobes with strongly conserved ancient traits, we speculate that the core-MetE homologs are similar to an ancient methionine synthase encoded already in the genome of a predecessor of LUCA and therefore basal to both archaea and bacteria. We speculate that such basal methionine synthases were active in the metabolism of ancient microorganisms using methylcobalamin-containing proteins as methyl donor. Core-MetECBDB is the first biochemically described bacterial representative of these core-MetE proteins resembling the methylcobalamin:homocysteine methyltransferase from M. thermoautotrophicum33. Tr-MetE proteins appear to have evolved by duplications of core-MetE and subsequently acquired the capacity to bind folate at the N-terminal part. In our phylogenetic analysis tr-MetE clusters with archaeal core-MetE genes (Fig. 1a).
Materials and Methods
General
All chemicals were purchased from Sigma-Aldrich (Munich, Germany) or Carl Roth (Karlsruhe, Germany). Whenever methionine or homocysteine are mentioned in the text, the L-form is meant. Chemicals used for mass spectrometry were obtained in LC-MS grade from Carl Roth. Pteroyltri-γ-L-glutamic acid (PteGlu3) was acquired from Schircks Laboratories (Jona, Switzerland). Restriction enzymes, DNA polymerase, DNA and protein standards were obtained from New England BioLabs (Frankfurt/Main, Germany). Oligonucleotides and sequencing services were provided by Seqlab (Göttingen, Germany). All oligonucleotide primers, plasmids and strains used in this study are listed in Supplementary Tables 1 and 2. Anaerobic experiments were performed in a COY glovebox (Grass Lake, USA).
Bioinformatics
The structural model of CbdbA481 (core-MetECBDB) was calculated using the I-TASSER server65. Broadly defined, the server aligns the template protein with proteins of similar folds or with super-secondary structures from the PDB library by LOMETS. The overlay of core-MetECBDB and tr-MetE from N. crassa was generated with PyMOL66. The amino acid sequences of tr-MetEs were trimmed approximately at the position 370. For the multiple sequence alignment and construction of the phylogenetic tree, only the C-termini of truncated tr-MetEs from bacteria, yeast and complete amino acid sequences of core-MetEs from archaea, Chloroflexi and Clostridiales were used. MEGA767 was used to calculate multiple amino acid sequence alignments using the implemented MUSCLE algorithm with default settings68. The evolutionary relationship between different methionine synthase amino acid sequences was inferred by using the Maximum Likelihood method based on the JTT matrix model69. Evolutionary distances were computed using Poisson correction and are expressed as the number of amino acid substitutions per site70.
Construction of expression and complementation plasmids
Based on pBAD30, expression and complementation plasmids were generated as described in supplementary information. The resulting plasmids pBAD_MetE and pBAD_CbdbA481 were used for the complementation experiments as well as for the heterologous production and purification of tandem-repeat MetE (tr-MetEEco) from E. coli and core-MetECBDB from D. mccartyi strain CBDB1.
Production and purification of recombinant tr-MetEEco and core-MetECBDB
A preculture of E. coli DH10B containing pBAD30_MetE or pBAD30_CbdbA481 was set up in Luria-Bertani (LB) medium containing 100 µg mL−1 ampicillin and grown overnight at 37 °C and 140 rpm. On the following day, 1% (v/v) of the overnight culture was used to inoculate fresh LB medium containing the appropriate antibiotic. The cultures were grown at 37 °C under agitation at 140 rpm until the OD600 reached 0.4–0.5. Then, the production of either tr-MetEEco or core-MetECBDB was induced by the addition of 0.05% (w/v) L-arabinose. Additionally, the medium was supplemented with 1 mM ZnSO4. MetE and CbdbA481 were produced for 5 h at 37 °C and 140 rpm. Then, the cells were harvested by centrifugation and washed with 50 mM Tris/HCl, pH 7.5. Purification of tr-MetEEco and core-MetECBDB was performed under anoxic conditions in an anaerobic chamber. Both enzymes were purified by anion exchange chromatography using a MonoQ 5/50 GL column connected to an ÄKTA purifier FPLC system (GE Healthcare Life Sciences) as described in detail in the supplementary information.
SDS-PAGE and native PAGE
The purity of core-MetECBDB and tr-MetEEco protein preparations was evaluated by 10% SDS-PAGE. In addition, the oligomeric state of both proteins was investigated via 10% discontinuous native PAGE71.
Protein identification from SDS-PAGE by LC-MS/MS
Qualitative identification of the purified core-MetECBDB and tr-MetEEco proteins was conducted mass spectrometrically. Therefore, protein bands at the height of 38 kDa and 80 kDa were excised from 10% SDS-PAGE gels. Acetonitrile, 10 mM DTT and 100 mM iodoacetamide were used to destain, to reduce and to alkylate the proteins within the gel slices. Subsequently, the proteins were digested with 0.1 µg trypsin (Promega) at 37 °C for 18 h. The resulting peptides were extracted from the gel matrix with 50% (v/v) acetonitrile and 5% (v/v) formic acid and dried. The peptides were again dissolved in 10 µL 0.1% formic acid and subsequently desalted using C18 ZipTip Pipette Tips (Merck Millipore) and dried in a vacuum centrifuge. Prior analysis, the peptides were resuspended in 20 µL 0.1% formic acid. Samples were analyzed on an LC-MS/MS system composed of a nano-UPLC system (UltiMate 3000 RSLCnano System, Thermo Fisher Scientific) equipped with an Acclaim PepMap 100 75 µm × 25 cm C18 column and connected to an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) via an electrospray ion source (TriVersa NanoMate, Advion). Sample volumes of 5 µL were injected onto the column and separated applying a flow rate of 0.3 µL min−1 with the aid of a 60 min gradient from 3.2% to 44% acetonitrile in water containing 0.1% formic acid. The mass spectrometer was operated in positive-ionization mode. The spray voltage was set at 2.2 kV and an electron spray ionization source temperature at 220 °C. Full MS1 scans were obtained over a mass range of 300–2000 m/z and the resolution in the Orbitrap was set to 240,000. The most intense ions (threshold ion count above 5.0 × 104) were selected for fragmentation with the quadrupole, setting the isolation window to 1.6 m/z. Ions were fragmented by ETciD (ETD reaction time 100 ms, CID collision energy 35%). The resulting fragment ion spectra were obtained achieved in the Orbitrap at a resolution of 60,000 and a maximum injection time of 120 ms.
Protein and peptide identification
The raw mass spectrometric data were converted to mgf-files using ProteoWizard MSConvert v3.072. The software SearchGUI (v3.3.5)73 and the OMSSA search algorithm were used for peptide identification. Mass spectrometric data were searched against the E. coli proteome database obtained from UniProt (Taxon identifier 316385). A precursor ion mass tolerance of 10 ppm was used at the MS1 level and up to two missed cleavages were allowed. The fragment ion mass tolerance was set to 0.2 Da for the Orbitrap MS2 detection. The oxidation of methionine was considered as variable modification and carbamidomethylation on cysteines as fixed modification. The false discovery rate (FDR) in peptide identification was limited to a maximum of 0.01 by using a decoy database. The analyzed data were visualized with the PeptideShaker software (v1.16.27) (CompOmics, Ghent University)74.
Photometric analysis of methylcob(III)alamin binding to core-MetECBDB
The binding of methylcob(III)alamin to core-MetE was followed spectrophotometrically in the range between 250 and 800 nm (0.5 nm steps) in 50 mM Tris/HCl (pH 6.5), 150 mM NaCl and 10% glycerol. First, the UV/Vis spectrum of 10 µM free methylcob(III)alamin was recorded. To assess the binding mode of methylcob(III)alamin to core-MetECBDB, the protein solution was mixed with methylcob(III)alamin in a 1:1 stoichiometry (10 µM each). The UV/Vis spectra of free and bound methylcob(III)alamin were compared.
Enzyme activity assay with methylcob(III)alamin as methyl group donor
Enzyme activity assays were set up at dim light under strictly anoxic conditions. The standard enzyme assay mix contained 50 mM Tris/HCl (pH 6.5), 150 mM NaCl, 10% glycerol, 0.5 mM methylcob(III)alamin and 0.1 µM enzyme. After 5 min preincubation at room temperature, the reaction was started by the addition of 2 mM D,L-homocysteine. The reaction was photometrically monitored either at 524 nm indicating the consumption of methylcob(III)alamin (ε524 = 6,200 M−1 cm−1) or at 681 nm indicating the formation of cob(I)alamin (ε681 = 1,200 M−1 cm−1) (Fig. 2a and Supplementary Figure 2)75. Enzyme kinetics of core-MetECBDB were performed at concentrations of either 0.5 mM methylcob(III)alamin or 2 mM D,L-homocysteine while the concentration of the second substrate was varied.
Synthesis of (6R,S)-5-methyl-5,6,7,8-tetrahydropteroyltri-γ-L-glutamic acid (5-methyl-THF-Glu3)
The synthesis of 5-methyl-THF-Glu3 was accomplished from commercially available PteGlu3 under anoxic conditions following the modified protocol of Yeo and Wagner76 and as described in detail in the supplementary information. 5-methyl-H4PteGlu3 was stored at −20 °C.
Enzyme activity assay with 5-methyl-THF-Glu3 as methyl group donor
Enzyme assays were performed under strictly anoxic conditions. The standard assay was set up in 25 mM Tris/HCl (pH 7.2) or 50 mM KH2PO4/K2HPO4 (pH 7.2), 100 µM MgSO4, 100 µM ZnSO4, 10 mM dithiothreitol (DTT), 2 mM D,L-homocysteine and 150 µM 5-methyl-THF-Glu3. The reaction was started by the addition of 0.25 µM tr-MetEEco or core-MetECBDB. After an incubation time of 60 min at 37 °C, the reactions were stopped by heat denaturation at 80 °C for 10 min, then centrifuged at 15,000 rpm for 5 min (Eppendorf Centrifuge 5424 R) and analyzed by HPLC. A negative control under same conditions without protein was run to evaluate abiotic transformation of 5-methyl-THF-Glu3. 5-methyl-THF-Glu3, other folate derivatives and PteGlu3 were analyzed with a JASCO HPLC 2000 series system equipped with an Equisil BDS C18 column (250 × 4.6 mm, 5 μm; Dr. Maisch HPLC GmbH, Ammerbuch-Entringen/Germany) following the modified protocol of Patring77. The identities of PteGlu3, 5-methyl-THF-Glu3 and L-methionine were confirmed via liquid chromatography-mass spectrometry in direct injection mode.
Generation of E. coli knockout strain
The E. coli DH5α (ΔmetE::kan) knockout strain was generated using the Quick & Easy E. coli Gene Deletion Kit (GeneBridges GmbH, Heidelberg, Germany) according to the manufacturer’s protocol78. Hereby, metE gene in E. coli DH5α was replaced by a linear kanamycin cassette. The introduction of the kanamycin cassette allowed to screen for the knockout strain on 20 µg mL−1 kanamycin agar plates.
In vivo complementation of E. coli DH5α (ΔmetE::kan) and cultivation procedure
The metE-deficient E. coli DH5α (E. coli DH5α (ΔmetE::kan)) strain was transformed with pBAD30 (negative control), pBAD30_MetE (positive control) or pBAD30_CbdbA481. The first subculture was grown in 5 mL LB medium with 100 µg mL−1 ampicillin and 20 µg mL−1 kanamycin at 37 °C and 140 rpm overnight. An inoculum of 1% (v/v) of the first subculture was then used to inoculate the second subculture of 5 mL M9 minimal medium79 supplemented with 1 mM MgSO4, 0.1 mM CaCl2, 10 µM FeCl3/EDTA, 1.2 mM thiamine, 0.3 mM L-leucine, 0.4% (v/v) glycerol and 0.4 µM cyanocobalamin, that was grown at 37 °C and 140 rpm overnight. Then, several 10 mL-tubes of fresh M9 medium containing all supplements except cyanocobalamin were inoculated with 1% (v/v) of the second subculture. The following main cultures were set up:
E. coli DH5α wild type with or without 0.4 µM cyanocobalamin,
E. coli DH5α (ΔmetE::kan) with or without 0.4 µM cyanocobalamin,
E. coli DH5α (ΔmetE::kan) + pBAD30_MetE with or without 0.4 µM cyanocobalamin and with or without 0.05% (w/v) L-arabinose,
E. coli DH5α (ΔmetE::kan) + pBAD_CbdbA481 with or without 0.4 µM cyanocobalamin and with or without 0.05% (w/v) L-arabinose.
The main cultures were then incubated at 37 °C and 140 rpm and growth was monitored by measuring the OD600. E. coli DH5α still encodes the arabinose operon. However, arabinose at a concentration of 0.05% (w/v) sufficed to induce the production of core-MetECBDB and tr-MetEEco. In our experiments, reciprocal metabolism leads to preferential use of glycerol instead of arabinose.
Supplementary information
Acknowledgements
We thank Benjamin Scheer for assistance with mass spectrometric experiments. Mass spectrometry was done at the Centre for Chemical Microscopy (ProVIS) at the Helmholtz Centre for Environmental Research – UFZ, which is supported by European regional development funds (EFRE-Europe Funds Saxony) and the Helmholtz Association. We thank Dr. Jochen Müller from Helmholtz Centre for Environmental Research (Environmental Biotechnology) for providing us the Quick & Easy E. coli Gene Deletion Kit.
Author contributions
L.A. and D.D. conceived the study. D.D., R.H., G.L. and S.S. designed the experiments in coordination with L.A. R.H., D.D. and S.S. conducted the lab experiments. R.H. and D.D. analyzed the data. D.D. and L.A. wrote the manuscript, G.L. revised it.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-020-58873-z.
References
- 1.Kozak M. Initiation of translation in prokaryotes and eukaryotes. Gene. 1999;234:187–208. doi: 10.1016/S0378-1119(99)00210-3. [DOI] [PubMed] [Google Scholar]
- 2.Stipanuk MH. Metabolism of sulfur-containing amino acids. Ann. Rev. Nutr. 1986;6:179–209. doi: 10.1146/annurev.nu.06.070186.001143. [DOI] [PubMed] [Google Scholar]
- 3.Stipanuk MH, Ueki I. Dealing with methionine/homocysteine sulfur: cysteine metabolism to taurine and inorganic sulfur. J. Inherit. Metab. Dis. 2011;34:17–32. doi: 10.1007/s10545-009-9006-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantoni GL. S-adenosylmethionine; a new intermediate formed enzymatically from L-methionine and adenosinetriphosphate. J. Biol. Chem. 1953;204:403–416. [PubMed] [Google Scholar]
- 5.Gophna U, Bapteste E, Doolittle WF, Biran D, Ron EZ. Evolutionary plasticity of methionine biosynthesis. Gene. 2005;355:48–57. doi: 10.1016/j.gene.2005.05.028. [DOI] [PubMed] [Google Scholar]
- 6.Frey PA, Ballinger MD, Reed GH. S-adenosylmethionine. A ‘poor man’s coenzyme B12’ in the reaction of lysine 2,3-aminomutase. Biochm Soc. Trans. 1998;26:304–310. doi: 10.1042/bst0260304. [DOI] [PubMed] [Google Scholar]
- 7.Foster MA, Tejerina G, Guest JR, Woods DD. Two enzymic mechanisms for the methylation of homocysteine by extracts of Escherichia coli. Biochem. 1964;92:476–488. doi: 10.1042/bj0920476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Weissbach H, Brot N. Regulation of methionine synthesis in Escherichia coli. Mol. Microbiol. 1991;5:1593–1597. doi: 10.1111/j.1365-2958.1991.tb01905.x. [DOI] [PubMed] [Google Scholar]
- 9.Wolthers KR, et al. Crystal structure and solution characterization of the activation domain of human methionine synthase. FEBS J. 2007;274:738–750. doi: 10.1111/j.1742-4658.2006.05618.x. [DOI] [PubMed] [Google Scholar]
- 10.Szegedi SS, Castro CC, Koutmos M, Garrow TA. Betaine-homocysteine-S-methyltransferase-2 is an S-methylmethionine-homocysteine methyltransferase. J. Biol. Chem. 2008;283:8939–8945. doi: 10.1074/jbc.M710449200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kacprzak MM, Lewandowska I, Matthews RG, Paszewski A. Transcriptional regulation of methionine synthase by homocysteine and choline in Aspergillus nidulans. Biochem. J. 2003;376:517–524. doi: 10.1042/bj20030747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bourgis F, et al. S-methylmethionine plays a major role in phloem sulfur transport and is synthesized by a novel type of methyltransferase. Plant. Cell. 1999;11:1485–1497. doi: 10.1105/tpc.11.8.1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Matthews RG, Goulding CW. Enzyme-catalyzed methyl transfers to thiols: the role of zinc. Curr. Opin. Chem. Biol. 1997;1:332–339. doi: 10.1016/S1367-5931(97)80070-1. [DOI] [PubMed] [Google Scholar]
- 14.Datta S, Koutmos M, Pattridge KA, Ludwig ML, Matthews RG. A disulfide-stabilized conformer of methionine synthase reveals an unexpected role for the histidine ligand of the cobalamin cofactor. Proc. Natl Acad. Sci. USA. 2008;105:4115–4120. doi: 10.1073/pnas.0800329105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drummond JT, Huang S, Blumenthal RM, Matthews RG. Assignment of enzymic function to specific protein regions of cobalamin-dependent methionine synthase from Escherichia coli. Biochem. 1993;32:9290–9295. doi: 10.1021/bi00087a005. [DOI] [PubMed] [Google Scholar]
- 16.Goulding CW, Postigo D, Matthews RG. Cobalamin-dependent methionine synthase is a modular protein with distinct regions for binding homocysteine, methyltetrahydrofolate, cobalamin, and adenosylmethionine. Biochem. 1997;36:8082–8091. doi: 10.1021/bi9705164. [DOI] [PubMed] [Google Scholar]
- 17.Ferrer J-L, Ravanel S, Robert M, Dumas R. Crystal structures of cobalamin-independent methionine synthase complexed with zinc, homocysteine, and methyltetrahydrofolate. J. Biol. Chem. 2004;279:44235–44238. doi: 10.1074/jbc.C400325200. [DOI] [PubMed] [Google Scholar]
- 18.Fu T-M, et al. Crystal structures of cobalamin-independent methionine synthase (MetE) from Streptococcus mutans. A dynamic zinc-inversion model. J. Mol. Biol. 2011;412:688–697. doi: 10.1016/j.jmb.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 19.Pejchal R, Ludwig ML. Cobalamin-independent methionine synthase (MetE): a face-to-face double barrel that evolved by gene duplication. PLoS Biol. 2005;3:254–265. doi: 10.1371/journal.pbio.0030254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Adrian L, Szewzyk U, Wecke J, Görisch H. Bacterial dehalorespiration with chlorinated benzenes. Nat. 2000;408:580–583. doi: 10.1038/35046063. [DOI] [PubMed] [Google Scholar]
- 21.Hölscher T, Gorisch H, Adrian L. Reductive dehalogenation of chlorobenzene congeners in cell extracts of Dehalococcoides sp. strain CBDB1. Appl. Env. Microbiol. 2003;69:2999–3001. doi: 10.1128/AEM.69.5.2999-3001.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Löffler FE, et al. Dehalococcoides mccartyi gen. nov., sp. nov., obligately organohalide-respiring anaerobic bacteria relevant to halogen cycling and bioremediation, belong to a novel bacterial class, Dehalococcoidia classis nov., order Dehalococcoidales ord. nov. and family Dehalococcoidaceae fam. nov., within the phylum Chloroflexi. Int. J. Syst. Evol. Microbiol. 2013;63:625–635. doi: 10.1099/ijs.0.034926-0. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y, Rodionov DA, Gelfand MS, Gladyshev VN. Comparative genomic analyses of nickel, cobalt and vitamin B12 utilization. BMC genomics. 2009;10:78. doi: 10.1186/1471-2164-10-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Adrian L, Rahnenführer J, Gobom J, Hölscher T. Identification of a chlorobenzene reductive dehalogenase in Dehalococcoides sp. strain CBDB1. Appl. Env. Microbiol. 2007;73:7717–7724. doi: 10.1128/AEM.01649-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schipp CJ, Marco-Urrea E, Kublik A, Seifert J, Adrian L. Organic cofactors in the metabolism of Dehalococcoides mccartyi strains. Philos. Trans. R. Soc. Lond. 2013;368:20120321. doi: 10.1098/rstb.2012.0321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kube M, et al. Genome sequence of the chlorinated compound-respiring bacterium Dehalococcoides species strain CBDB1. Nat. Biotechnol. 2005;23:1269–1273. doi: 10.1038/nbt1131. [DOI] [PubMed] [Google Scholar]
- 27.Seshadri R, et al. Genome sequence of the PCE-dechlorinating bacterium Dehalococcoides ethenogenes. Sci. 2005;307:105–108. doi: 10.1126/science.1102226. [DOI] [PubMed] [Google Scholar]
- 28.Yi S, et al. Versatility in corrinoid salvaging and remodeling pathways supports corrinoid-dependent metabolism in Dehalococcoides mccartyi. Appl. Env. Microbiol. 2012;78:7745–7752. doi: 10.1128/AEM.02150-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yan J, Ritalahti KM, Wagner DD, Löffler FE. Unexpected specificity of interspecies cobamide transfer from Geobacter spp. to organohalide-respiring Dehalococcoides mccartyi strains. Appl. Env. Microbiol. 2012;78:6630–6636. doi: 10.1128/AEM.01535-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tang YJ, et al. Investigation of carbon metabolism in “Dehalococcoides ethenogenes” strain 195 by use of isotopomer and transcriptomic analyses. J. Bacteriol. 2009;191:5224–5231. doi: 10.1128/JB.00085-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Marco-Urrea E, Seifert J, Bergen Mvon, Adrian L. Stable isotope peptide mass spectrometry to decipher amino acid metabolism in Dehalococcoides strain CBDB1. J. Bacteriol. 2012;194:4169–4177. doi: 10.1128/JB.00049-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhuang W-Q, et al. Incomplete Wood-Ljungdahl pathway facilitates one-carbon metabolism in organohalide-respiring Dehalococcoides mccartyi. Proc. Natl Acad. Sci. USA. 2014;111:6419–6424. doi: 10.1073/pnas.1321542111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Schröder I, Thauer RK. Methylcobalamin:homocysteine methyltransferase from Methanobacterium thermoautotrophicum. Eur. J. Biochem. 1999;263:789–796. doi: 10.1046/j.1432-1327.1999.00559.x. [DOI] [PubMed] [Google Scholar]
- 34.Kanehisa M, Goto S. KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000;28:27–30. doi: 10.1093/nar/28.1.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M. KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 2016;44:457–462. doi: 10.1093/nar/gkv1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017;45:353–361. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wheatley RW, Ng KKS, Kapoor M. Fungal cobalamin-independent methionine synthase: Insights from the model organism, Neurospora crassa. Arch. Biochem. Biophys. 2016;590:125–137. doi: 10.1016/j.abb.2015.11.037. [DOI] [PubMed] [Google Scholar]
- 38.Bandarian V, Matthews RG. Measurement of energetics of conformational change in cobalamin-dependent methionine synthase. Methods Enzymol. 2004;380:152–169. doi: 10.1016/S0076-6879(04)80007-7. [DOI] [PubMed] [Google Scholar]
- 39.Steindal AH, Juzeniene A, Johnsson A, Moan J. Photodegradation of 5-methyltetrahydrofolate: biophysical aspects. Photochem. Photobiol. 2006;82:1651–1655. doi: 10.1111/j.1751-1097.2006.tb09826.x. [DOI] [PubMed] [Google Scholar]
- 40.Verlinde PHCJ, et al. Influence of reducing carbohydrates on (6S)-5-methyltetrahydrofolic acid degradation during thermal treatments. J. Agric. Food Chem. 2010;58:6190–6199. doi: 10.1021/jf9041134. [DOI] [PubMed] [Google Scholar]
- 41.Liu Y, et al. Thermal oxidation studies on reduced folate, L-5-methyltetrahydrofolic acid (L-5-MTHF) and strategies for stabilization using food matrices. J. Food Sci. 2012;77:C236–43. doi: 10.1111/j.1750-3841.2011.02561.x. [DOI] [PubMed] [Google Scholar]
- 42.Parker ET, et al. Primordial synthesis of amines and amino acids in a 1958 Miller H2S-rich spark discharge experiment. Proc. Natl Acad. Sci. USA. 2011;108:5526–5531. doi: 10.1073/pnas.1019191108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Parker ET, et al. Prebiotic synthesis of methionine and other sulfur-containing organic compounds on the primitive Earth: a contemporary reassessment based on an unpublished 1958 Stanley Miller experiment. Orig. Life Evol. Biosph. 2011;41:201–212. doi: 10.1007/s11084-010-9228-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Laurino P, Tawfik DS. Spontaneous emergence of S-adenosylmethionine and the evolution of methylation. Angew. Chem. Int. Ed. 2017;56:343–345. doi: 10.1002/anie.201609615. [DOI] [PubMed] [Google Scholar]
- 45.Decker K, Jungermann K, Thauer RK. Energy production in anaerobic organisms. Angew. Chem. Int. Ed. 1970;9:138–158. doi: 10.1002/anie.197001381. [DOI] [PubMed] [Google Scholar]
- 46.Martin WF, Sousa FL. Early microbial evolution: the age of anaerobes. Cold Spring Harb. Perspect. Biol. 2015;8:a018127. doi: 10.1101/cshperspect.a018127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dickman SR. Ribonucleotide reduction and the possible role of cobalamin in evolution. J. Mol. Evol. 1977;10:251–260. doi: 10.1007/BF01764600. [DOI] [PubMed] [Google Scholar]
- 48.Menon S, Ragsdale SW. The role of an iron-sulfur cluster in an enzymatic methylation reaction. Methylation of CO dehydrogenase/acetyl-CoA synthase by the methylated corrinoid iron-sulfur protein. J. Biol. Chem. 1999;274:11513–11518. doi: 10.1074/jbc.274.17.11513. [DOI] [PubMed] [Google Scholar]
- 49.Thauer RK. Biochemistry of methanogenesis: a tribute to Marjory Stephenson. Microbiol. 1998;144:2377–2406. doi: 10.1099/00221287-144-9-2377. [DOI] [PubMed] [Google Scholar]
- 50.González JC, Peariso K, Penner-Hahn JE, Matthews RG. Cobalamin-independent methionine synthase from Escherichia coli: a zinc metalloenzyme. Biochem. 1996;35:12228–12234. doi: 10.1021/bi9615452. [DOI] [PubMed] [Google Scholar]
- 51.Frasca V, Banerjee RV, Dunham WR, Sands RH, Matthews RG. Cobalamin-dependent methionine synthase from Escherichia coli B: electron paramagnetic resonance spectra of the inactive form and the active methylated form of the enzyme. Biochem. 1988;27:8458–8465. doi: 10.1021/bi00422a025. [DOI] [PubMed] [Google Scholar]
- 52.Matthews RG, et al. Cobalamin-dependent and cobalamin-independent methionine synthases: are there two solutions to the same chemical problem? Helvetica Chim. Acta. 2003;86:3939–3954. doi: 10.1002/hlca.200390329. [DOI] [Google Scholar]
- 53.Martin BD, Finke RG. Cobalt-carbon homolysis and bond dissociation energy studies of biological alkylcobalamins: methylcobalamin, including a >1015 Co-CH3 homolysis rate enhancement at 25 °C following one-electron reduction. J. Am. Chem. Soc. 1990;112:2419–2420. doi: 10.1021/ja00162a053. [DOI] [PubMed] [Google Scholar]
- 54.Martin BD, Finke RG. Methylcobalamin’s full- vs. half-strength cobalt-carbon σ bonds and bond dissociation enthalpies: A > 1015 Co-CH3 homolysis rate enhancement following one-antibonding-electron reduction of methlycobalamin. J. Am. Chem. Soc. 1992;114:585–592. doi: 10.1021/ja00028a027. [DOI] [PubMed] [Google Scholar]
- 55.Jensen KP, Ryde U. Conversion of homocysteine to methionine by methionine synthase. A density functional study. J. Am. Chem. Soc. 2003;125:13970–13971. doi: 10.1021/ja034697a. [DOI] [PubMed] [Google Scholar]
- 56.Männel-Croisé C, Zelder F. Immobilised vitamin B12 as a biomimetic model for base-off/histidine-on coordination. Chem. Commun. 2011;47:11249–11251. doi: 10.1039/c1cc15093f. [DOI] [PubMed] [Google Scholar]
- 57.Ragsdale SW, Lindahl PA, Münck E. Mössbauer, EPR, and optical studies of the corrinoid/iron-sulfur protein involved in the synthesis of acetyl-CoA by Clostridium thermoaceticum. J. Biol. Chem. 1987;262:14289–14297. [PubMed] [Google Scholar]
- 58.Drennan C, Huang S, Drummond J, Matthews R, Lidwig M. How a protein binds B12. A 3.0 A X-ray structure of B12-binding domains of methionine synthase. Science. 1994;266:1669–1674. doi: 10.1126/science.7992050. [DOI] [PubMed] [Google Scholar]
- 59.Yan J, et al. The corrinoid cofactor of reductive dehalogenases affects dechlorination rates and extents in organohalide-respiring Dehalococcoides mccartyi. ISME J. 2016;10:1092–1101. doi: 10.1038/ismej.2015.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ljungdahl LG, Wood HG. Total synthesis of acetate from CO2 by heterotrophic bacteria. Annu. Rev. microbiology. 1969;23:515–538. doi: 10.1146/annurev.mi.23.100169.002503. [DOI] [PubMed] [Google Scholar]
- 61.Matthews RG, Koutmos M, Datta S. Cobalamin-dependent and cobamide-dependent methyltransferases. Curr. Opin. Struct. Biol. 2008;18:658–666. doi: 10.1016/j.sbi.2008.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Hippler, B. & Thauer, R. K. The energy conserving methyltetrahydromethanopterin:coenzyme M methyltransferase complex from methanogenic archaea: function of the subunit MtrH. FEBS Lett. 449, 165–168 (1999). [DOI] [PubMed]
- 63.Ferry JG. Fundamentals of methanogenic pathways that are key to the biomethanation of complex biomass. Curr. Opin. Biotechnol. 2011;22:351–357. doi: 10.1016/j.copbio.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Länge S, Fuchs G. Autotrophic synthesis of activated acetic acid from CO2 in Methanobacterium thermoautotrophicum. Eur. J. Biochem. 1987;163:147–154. doi: 10.1111/j.1432-1033.1987.tb10748.x. [DOI] [PubMed] [Google Scholar]
- 65.Zhang Y. I-TASSER server for protein 3D structure prediction. BMC Bioinfomatics. 2008;9:1–8. doi: 10.1186/1471-2105-9-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.DeLano, W. L. PyMol: an open-source molecular graphics tool. CCP4 Newsletter on Protein Crystallography.40, 82–92 (2002).
- 67.Kumar S, Stecher G, Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016;33:1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Edgar RC. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004;32:1792–1797. doi: 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jones DT, Taylor WR, Thornton JM. The rapid generation of mutation data matrices from protein sequences. Comput. Appl. Biosci. 1992;8:275–282. doi: 10.1093/bioinformatics/8.3.275. [DOI] [PubMed] [Google Scholar]
- 70.Zuckerkandl, E. & Pauling, L. In Evolving genes and proteins (Elsevier), pp. 97–166 (1965).
- 71.Ornstein L, Davis BJ. Disc electrophoresis-I: background and theory. Ann. NY. Acad. Sci. 1964;121:321–349. doi: 10.1111/j.1749-6632.1964.tb14207.x. [DOI] [PubMed] [Google Scholar]
- 72.Holman JD, Tabb DL, Mallick P. Employing ProteoWizard to convert raw mass spectrometry data. Curr. Protoc. Bioinforma. 2014;46:13.24.1–9. doi: 10.1002/0471250953.bi1324s46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Vaudel M, Barsnes H, Berven FS, Sickmann A, Martens L. SearchGUI: an open-source graphical user interface for simultaneous OMSSA and X!Tandem searches. Proteom. 2011;11:996–999. doi: 10.1002/pmic.201000595. [DOI] [PubMed] [Google Scholar]
- 74.Vaudel M, et al. PeptideShaker enables reanalysis of MS-derived proteomics data sets. Nat. Biotechnol. 2015;33:22–24. doi: 10.1038/nbt.3109. [DOI] [PubMed] [Google Scholar]
- 75.Sauer K, Thauer RK. Methanol:coenzyme M methyltransferase from Methanosarcina barkeri - substitution of the corrinoid harbouring subunit MtaC by free cob(I)alamin. Eur. J. Biochem. 1999;261:674–681. doi: 10.1046/j.1432-1327.1999.00355.x. [DOI] [PubMed] [Google Scholar]
- 76.Yeo EJ, Wagner C. Purification and properties of pancreatic glycine N-methyltransferase. J. Biol. Chem. 1992;267:24669–24674. [PubMed] [Google Scholar]
- 77.Patring JDM, Jastrebova JA, Hjortmo SB, Andlid TA, Jägerstad IM. Development of a simplified method for the determination of folates in baker’s yeast by HPLC with ultraviolet and fluorescence detection. J. Agric. Food Chem. 2005;53:2406–2411. doi: 10.1021/jf048083g. [DOI] [PubMed] [Google Scholar]
- 78.Baba T, et al. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the Keio collection. Mol. Sys Biol. 2006;2:1–11. doi: 10.1038/msb4100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Harwood, C. R. & Cutting, S. M. In Molecular biological methods in Bacillus, edited by C. R. Harwood & S. M. Cutting (Wiley, Chichester), Vol. 1, p. 548 (1990).
- 80.Eichel J, González JC, Hotze M, Matthews RG, Schröder J. Vitamin-B12-Independent Methionine Synthase from a Higher Plant (Catharanthus Roseus) Eur. J. Biochem. 1995;230:1053–1058. doi: 10.1111/j.1432-1033.1995.tb20655.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.