

Abstract

We investigate the impact of lipidation on the self-assembly of two peptide fragments from the gastrointestinal peptide hormone PYY3–36. The lipopeptides C16IKPEAP and C16IKPEAPGE contain the first 6 and 8 amino acid residues, respectively, from the PYY3–36 peptide sequence, with a palmitoyl C16 tail attached at the N-terminus. These lipopeptides form spherical micelles in aqueous solution, above a critical micelle concentration (cmc), which is pH-dependent. Modeling of small-angle X-ray scattering data along with molecular dynamics simulations shows the formation of micelles with a hydrophobic interior and a well-hydrated exterior. The lipopeptides have a disordered conformation over the pH and temperature ranges studied. The cmc is found to be independent of temperature, pointing to athermal micellization. In contrast to the presence of hydrated micelles in solution, β-sheet amyloid fibrils form in dried samples. Thus, the nanostructure of lipidated PYY3–36 fragment peptides can be tuned by control of pH or concentration, for future applications.
Introduction
Peptide amphiphiles (PAs) are self-assembling peptide-based molecules that contain a bioactive headgroup conjugated to a hydrophobic tail of variable length, and in most cases this is an alkyl chain as in the case of a lipopeptide.1 The hydrophobic tail increases the amphiphilicity of the molecule which drives self-assembly in aqueous solution, to form well-defined and controllable peptide functionalized nanostructures.2 The driving force behind PA self-assembly is the need for the hydrophobic tail to be screened from the aqueous environment, with the hydrophilic peptide exposed on the surface of the aggregated structures.3−6 Many single tail PAs self-assemble into fibers with the hydrophobic tails packing in the core away from the solvent, and the hydrophilic head facing the surface.4,7 Hydrophobic interactions between alkyl tails and β-sheet formation of peptide units favor fiber formation, although other aggregated structures such as micelles or vesicles are also possible.2,6 The self-assembly process may depend on pH, temperature, concentration, and ionic strength.
Lipidated peptides are attractive as therapeutic agents because the level of lipophilicity can be modulated, which also influences absorption, distribution, metabolism, excretion, and bioavailability. Lipidation increases stability and half-life in vivo by facilitating binding to carrier proteins, in particular, serum albumin, which delays renal clearance by the kidneys and thus prolongs biological activity.8,9 As a result, peptide lipidation is an attractive method to convert peptides into drug leads. The palmitoyl chain that is typically attached to the peptide is able to bind to serum albumin, causing steric hindrance, helping to delay proteolytic attack and renal clearance.5,10−12
We investigate the self-assembly of lipidated peptides comprising palmitoylated fragments of the gastrointestinal peptide hormone PYY3–36. PYY belongs to the pancreatic peptide (PP) family, and there are two main endogenous forms, PYY1–36 and PYY3–36. PYY3–36 is of great interest due to its high selectivity for the Y2 receptor, associated with food intake.13 Previous studies on various truncated versions of PYY examined the Y2 receptor affinity and biological function, specifically at the C-terminus.14 The self-assembly in aqueous solution of PYY3–36 lipidated with hexadecyl (palmitoyl) lipids15 or octyl lipid chains16 within the core α-helical peptide domain has been examined. Lipidation leads to the formation of micellar structures at sufficiently low pH (fibrillar structures were observed at high pH for the palmitoylated derivatives15). However, the self-assembly of truncated PYY peptides has not been studied. Here, we investigate the effect of pH, temperature, concentration, and lipidation on the self-assembly of two N-terminal truncated fragments of PYY3–36. The full sequence of PYY3–36 is NH2-IKPEAPGEDASPEELNRYYASLRHYLNLVTRQRY-NH2. The truncated fragments studied here consist of the first six and eight amino acid residues of the whole peptide sequence, with a palmitoyl alkyl chain covalently attached at the N-terminus (IKPEAP and IKPEAPGE) (Figure S1). Studies have shown that PYY3–36 has a partially α-helical secondary structure.17 The short peptide fragments studied here are not within the α-helical part of the whole sequence, and they also contain proline residues which are structure breaking residues that disfavor α-helix conformations.18 This occurs because the amide bond lacks the proton necessary for hydrogen bond stabilization. Lipidation using palmitoyl (C16, hexadecyl) chains is favorable, as it allows the molecule to fuse with the cell membrane and potentially act as a transducing molecule in vivo.19
Results and Discussion
We first determined whether lipidation had an effect on the aggregation behavior of IKPEAP and IKPEAPGE, by measuring critical micelle concentration (cmc) values using ANS (8-anilino-1-naphthalene sulfonic acid) and pyrene as fluorescent probes. ANS interacts with hydrophobic binding sites to cause an increase in fluorescence and a blue shift of the wavelength of maximum fluorescence, λmax.20 Pyrene is also sensitive to the hydrophobic environment, and in the presence of micelles and other macromolecular systems, it becomes encapsulated in the interior hydrophobic regions of the aggregates.21Figure 1a shows results of ANS fluorescence measurements, and calculated cmc values were 0.0056 and 0.0029 wt % for C16IKPEAP and C16IKPEAPGE, respectively, at their native pH values in water (the original fluorescence spectra are shown in Figure S2). Results for the nonlipidated IKPEAP and IKPEAPGE did not show a distinct break in the intensity of fluorescence, and it appears these peptides do not aggregate. Fluorescence experiments using pyrene were also carried out as a function of pH to investigate the effect of pH on aggregation concentration. Results indicate an increase in cmc with increased pH for both peptides (Figures S3 and S4).
Figure 1.
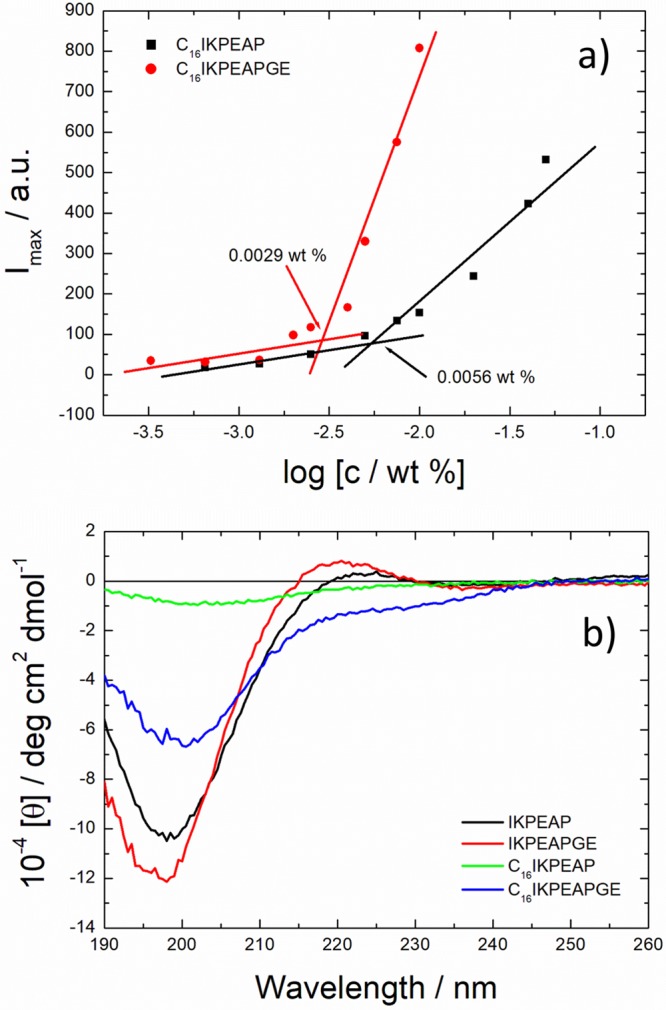
(a) Concentration dependence of ANS Imax fluorescence to show the critical micelle concentration (cmc) of the lipidated peptides at their native pH in water. The intersection of the lines indicates the cmc. (b) CD spectra comparing lipidated and unlipidated peptides at native pH at 20 °C.
The cmc was measured (from pyrene fluorescence experiments) at different temperatures, to determine the enthalpy of micellization using the Gibbs–Helmholtz equation along with the equation for the Gibbs energy of micellization.22 The results indicate that micellization is not affected by temperature since the plot of cmc does not depend significantly on temperature (Figures S5 and S6), i.e., ΔHmic = 0. Values of ΔSmic and ΔGmic at 20 °C were calculated to be 80.45 J K–1 mol–1 and −23.57 kJ mol–1 for C16IKPEAP, and 87.5 J K–1 mol–1 and −25.64 kJ mol–1 for C16IKPEAPGE, respectively. This shows that micellization of these PAs is entropically driven.
Circular dichroism (CD) spectroscopy was used to determine secondary structure at concentrations above the cmc for the lipidated peptides. The same concentrations of nonlipidated peptides were also used for comparison. Spectra of lipidated and unlipidated peptides were compared to determine what effect, if any, the lipidation had on the secondary structure at the peptides. Spectra at the peptides’ native pH showed a disordered conformation for both C16IKPEAP, and C16IKPEAPGE, and a polyproline II helix for IKPEAP and IKPEAPGE, with positive molar ellipticity values near 220 nm7,23 (Figure 1b). CD spectra at other pH values studied (pH 4–12) showed a disordered structure with a minimum at around 195 nm at 20 °C (Figure S7).
Temperature ramp CD experiments to study thermal stability from 20 to 70 °C display similar results, shown in the SI (Figures S8 and S9). Lipidation enhances thermal stability at pH 4 since the CD spectra for the lipidated peptides are much more thermally reversible when compared to those of the peptides themselves. Lipopeptide C16IKPEAP at native pH (pH 6.28) and pH 8 appears to have very little secondary structure. This could be attributed to the measured pH values being close to the calculated isoelectric point which was found to be pH 6.86 for IKPEAP, whereas for IKPEAPGE it was pH 4.15, using Innovagen software.24
Cryogenic transmission electron microscopy (cryo-TEM) images of C16IKPEAP and C16IKPEAPGE showed the presence of micelles with a radius less than 5 nm (Figure 2a). In contrast, cryo-TEM images of IKPEAP and IKPEAPGE showed no self-assembled structures at the same concentration. This is in agreement with the fluorescence probe measurements which revealed no cmc.
Figure 2.

(a) Cryo-TEM images of (i) C16IKPEAP and (ii) C16IKPEAPGE at native pH showing micelles. (b) SAXS intensity profiles and form factor fits in the pH range 2–12. (i) 0.134 wt % C16IKPEAP. (ii) 0.162 wt % C16IKPEAPGE. The data was fitted to a spherical shell form factor.
Dynamic light scattering (DLS) was used to provide an independent measure of the micelle size. The number weighted radius distributions at θ = 90° of the lipidated peptides are shown in Figure S10. The number weighted hydrodynamic radius values are 1.90 and 3.07 nm for C16IKPEAP and C16IKPEAPGE, respectively, which agrees with the cryo-TEM images that show the micelles to be less than 5 nm in radius.
Small-angle X-ray scattering (SAXS) was used to further investigate the shape and size of the self-assembled peptide nanostructures in solution. The intensity profiles with model form factor fits of the lipidated peptide fragments are shown in Figure 2b, and the fitted parameters are shown in Tables S1–S4. The profiles of the lipidated fragments are consistent with the cryo-TEM images that showed spherical micelle structures, and the data can be fitted to a spherical shell form factor. The unlipidated fragments showed completely differently shaped intensity profiles, which were consistent with cryo-TEM images (not shown) where there was no observable self-assembled structure. The SAXS data was fitted to a generalized Gaussian coil form factor to represent monomers in solution (Figure S11).
Ths SAXS data of the lipidated peptide fragments reveals differences in the outer radius as a function of pH. Increasing the pH from 2 to 8 significantly decreases the (inner and outer) radius for both peptide systems. This could be related to the loss of charge on the lysine residue at pH 8, causing an increase in net negative charge in the micelle, thus reducing the size. When comparing the sizes of lipidated peptides to each other at a given pH, there are no obvious differences, suggesting that the length of the peptide does not affect the self-assembly behavior, and that pH is the main contributing factor.
We unexpectedly noticed that drying samples for various measurements led to a profound difference in peptide conformation and self-assembled nanostructure. CD spectra obtained from dried films show a β-sheet secondary structure for both peptides (Figure 3a). TEM images revealed the presence of short fibers with lengths 400–500 nm (Figure 3b), showing that drying leads to disruption of the micelle structures present in solution, as revealed by cryo-TEM (Figure 2).
Figure 3.

(a) CD spectra obtained from dried films. (b) TEM images of dried (i) C16IKPEAP and (ii) C16IKPEAPGE at native pH showing fibers and micelles. (c) Fiber X-ray diffraction intensity profile of C16IKPEAP and C16IKPEAPGE at native pH. (d) SAXS intensity profiles of dried C16IKPEAP and C16IKPEAPGE at native pH. The arrows highlight observed Bragg reflections.
Fiber X-ray diffraction (XRD) was performed to directly examine secondary structure in dried samples. Results for the unlipidated fragments show a lack of secondary structure with d-spacing of 4.52 Å, representative of lipid chain packing (Figure S12). The lipidated peptides have a β-sheet secondary structure as shown by XRD (Figure 3c). The d-spacing of 4.68 Å corresponds to the β-strand spacing, and the peaks at 9.36 Å (C16IKPEAP) or 9.64 Å (IKPEAPGE) correspond to the β-sheet spacing. The peak at 4.11 Å is assigned to chain packing within a fraction of disordered material. SAXS measurements on dried samples (Figure 3d) show Bragg peaks, indicating hexagonal ordering with a d10 spacing = 46.8 Å.
The SAXS model profiles shown in Figure 2b clearly fit the data very well, just using a simple core–shell (two electron density) form factor model. The determined micelle radii (taken as the outer radius, R1, SI Tables S1 and S2) allow determination of micelle volumes, which along with estimated molecular volumes enables the association number p to be calculated. Molecular volumes were calculated using the software Gaussian25 and were found to be 960 Å3 for C16IKPEAP (slightly larger, 1048 Å3 for a stretched conformation) and 1161 Å3 for C16IKPEAPGE. Using micelle radii determined from the SAXS fits at pH 8 (SI Tables S1 and S2) leads to estimated association numbers p = 83 and p = 72 approximately, for C16IKPEAP and C16IKPEAPGE, respectively. The uncertainties on these values are estimated to be ±20% approximately, considering the errors on both the outer radius values and the estimates of molecular volume. These values are notably larger than association numbers in the range p = 5–10 for lipidated versions of the full PYY3–36 peptide,15,16 which is due to the much smaller size of the peptide headgroup which greatly facilitates packing in larger micelles. There is also a distinction in that the PYY3–36 peptide was not lipidated N-terminally, but rather via lysine derivatives within the core of the peptide sequence.
On the basis of these association numbers, we performed MD simulations on model micelles of C16IKPEAP containing p = 80, 101, 143, 225 molecules as detailed in the Materials and Methods section. Values of the inner and outer radius of gyration were computed, with the values being listed in Table S5 and plotted in Figure S13. This shows good agreement with the experimental data for the systems with p = 80 and p = 101. The larger assemblies produce values of the radii of gyration that are too large and are therefore not considered further. Figure S14 shows the equilibration of the MD system along the MD trajectory as a root-mean-square distance from the first snapshot (RMSD values). Micelle structures were generated from frames in the last 10 ns of the production run. This was then used to generate pdb files of micelle structures. The form factors were then computed using the Debye formula:
Here, N is the number of atoms in the micelle (i.e., p × Nat, where Nat = 145 for C16IKPEAP), and dij denotes the distance between atom i and atom j. The atomic scattering factors fi(q) are computed allowing both for displaced solvent (excluded volume effect) and the presence of a hydration layer around the micelle surface. The SAXS profile was calculated using the software FoXS,26 which allows for both of these terms (the hydration layer is computed via the solvent-accessible surface, computed from the rolling sphere method27). The software CRYSOL developed earlier28 can be used to analyze solution SAXS data in a similar manner to FoXS. The use of software designed to model protein solution structures to compute form factors for micellar systems is unprecedented to our knowledge and has great potential to provide detailed information on hydration effects. Interestingly, the model fits indicate that the C16IKPEAP micelles are highly hydrated (the FoXS hydration parameter c2(26) was in the range c2 = −1.4 to 2.58 indicating a denser hydration layer than the bulk solvent) and have minimally enlarged excluded volume26 (c1 = 0.99–1.05); i.e., each atom has between a 1% decrease and a 5% increase in radius due to the excluded volume effect of the water. An average form factor curve computed from 10 different configurations of micelles considering polydispersity in association number (micelles with p = 80 and 101) is shown in Figure 4a and provides a good representation of the data even when only considering a small number of micelle configurations. Micelles with larger association numbers p = 143 or p = 225 produced SAXS profiles in poor agreement with the experimental data (data not shown). A representative image of a micelle containing 80 molecules is shown in Figure 4b.
Figure 4.

(a) SAXS data at native pH (open circles) for C16IKEAP fitted to an atomistic model based on three configurations of micelles containing p = 80 and 101 molecules (red line). (b) Representative image of a model C16IKPEAP micelle containing 80 molecules.
Conclusions
In summary, adding a lipid tail to N-terminal fragments of the PYY3–36 peptide drives self-assembly into micelles, above a critical micelle concentration. Fluorescence spectroscopy using ANS as the fluorescent probe enabled determination of cmc values. CD results showed the two lipopeptides have disordered conformations. The insights into micelle structure available from an atomistic model to analyze the SAXS data, applying for the first time methods used to analyze solution protein solution structures, provide unique information on the presence of a substantial hydration layer at the surface of the C16IKPEAP micelles at native pH. Our modeling of the SAXS data from micelles using a similar method to that employed for globular proteins in solution points to an interesting analogy between the self-assembled micelles and compact protein structures. Of course, the latter are covalently fixed in contrast to the dynamically self-assembling lipopeptide molecules which constitute the micelles. Our findings show that lipidation of short fragments of PYY3–36 peptide leads to the formation of micelles with an association number (p ≈ 80) much more similar to that of conventional surfactants than the small “micelles” formed by (laterally) lipidated versions of the long PYY3–36 peptide which in fact are more like oligomeric clusters with only p = 5–10 molecules.15,16 Temperature-dependent measurements of the cmc show, unexpectedly, that micellization is essentially athermal showing that the process is driven by the favorable entropy change associated with the hydrophobic effect as in protein folding.
Remarkably, the two lipopeptides studied form β-sheet fibrils on drying, pointing to the formation of “amyloid-like” structures under these “denaturation” conditions. Possibly for lipopeptides as is proposed for proteins, amyloid may represent a common stable native state.29 Our findings may be useful in the development of proto-globular protein model systems and in the creation of therapeutic agents based on PYY3–36 fragment molecules. In a different direction we are presently investigating the utility of the proline coating at the surface of C16IKPEAP-based micelles in the creation of biocatalytic nanoparticles.
Materials and Methods
Materials
IKPEAP (TFA salt) was synthesized by Biomatik, Canada. The molecular weight by mass spectrometry was 653.8 g mol–1 (expected: 653.37 g mol–1), and the purity by HPLC was 99.8%.
IKPEAPGE (TFA salt) was synthesized by Biomatik, Canada. The molecular weight by mass spectrometry was 839.98 g mol–1 (expected: 839.44 g mol–1), and the purity by HPLC was 99.7%.
C16IKPEAP (ammonium acetate salt) was synthesized by Peptide Synthetics, UK. The molecular weight by mass spectrometry was 892.18 g mol–1 (expected: 891.60 g mol–1), and the purity by HPLC was >95.0%.
C16IKPEAPGE (ammonium acetate salt) was synthesized by Peptide Synthetics, UK. The molecular weight by mass spectrometry was 1078.346 g mol–1 (expected: 1077.67 g mol–1), and the purity by HPLC was >95.0%.
Fluorescence Spectroscopy
Fluorescence spectra were recorded with a Varian Cary Eclipse fluorescence spectrometer (Varian IEEE-488, Australia) with samples in 4 mm inner quartz cuvettes. The ANS assays were performed using 2.5 × 10–4 to 0.13 wt % peptide, in 2.1 × 10–3 wt % ANS solution. The samples were excited at λex = 356 nm, and the fluorescence emission was measured for λ = 400–670 nm. Pyrene assays were performed using 2.5 × 10–4 to 0.13 wt % peptide, in 2.167 × 10–5 wt % pyrene solution. The samples were excited at λex = 339 nm, and the fluorescence emission was measured for λ = 360–500 nm.
Temperature-dependent fluorescence assays of cmc were carried out using the same method, but a temperature controlled water bath was also used and measurements were collected in the temperature range 20–70 °C.
Circular Dichroism (CD)
CD spectra were recorded using a Chirascan spectropolarimeter (Applied Photophysics, UK). Spectra are presented with absorbance A < 2 at any measured point with a 0.5 nm step, 1 nm bandwidth, and a 1 s collection time per step. The CD signal from the background was subtracted from the CD signal of the sample solution. Ellipticity is reported as the mean residue ellipticity ([θ], in deg cm2 dmol–1) and calculated as
where [θ]obs is the ellipticity measured in millidegrees, MRW is the mean residue molecular weight of the peptide (molecular weight divided by the number of amino acid residues), c is the concentration of the sample in mg/mL, and l is the optical path length of the cell in centimeters.
CD spectra were measured in the temperature range 20–70 °C, using a 10 °C temperature step. Samples were equilibrated at each temperature for 2 min before measurements were recorded. Quartz plaques (0.1 and 0.01 mm thick) were used for the experiments, and a pH range of 2–12 was measured. 0.1–1 wt % samples were used. Experiments were carried out in D2O. CD spectra were also measured on dried peptides using quartz plates (0.01 mm).
Small-Angle X-ray Scattering (SAXS)
Solution and dried SAXS experiments were performed on the bioSAXS beamline BM29 at the ESRF, Grenoble, France, and the bioSAXS beamline B21 at Diamond light source, U.K. Solutions containing 0.162 wt % C16IKPEAPGE and IKPEPAGE, and 0.134 wt % of C16IKPEAP and IKPEAP, were loaded in PCR tubes in an automated sample changer. SAXS data were collected using a Pilatus 1 M detector. The sample–detector distance was 2.84 m. The X-ray wavelength was 0.99 Å. The wavenumber q = 4π sin θ/λ scale was calibrated using silver behenate, where λ is the X-ray wavelength and 2θ is the scattering angle. Dried SAXS measurements were performed by drying the peptides onto kapton tape and inserting them into a gel cell. Data was collected using a Pilatus 2M detector at a fixed camera length of 3.9 m with a wavelength λ = 1 Å.
Fiber X-ray Diffraction (XRD)
XRD was performed on peptide stalks prepared by drawing a fiber of peptide solution between the ends of wax-coated capillaries. 3 wt % peptide solutions were used for IKPEAP and IKPEAPGE, and 5 wt % peptide solutions for C16IKPEAP and C16IKPEAPGE. After drying, the capillaries were separated and a stalk was left on the end of one of the capillaries. Stalks were vertically mounted onto the goniometer of an Oxford Diffraction Gemini Ultra instrument, equipped with a Sapphire CCD detector. The sample-to-detector distance was 44 mm. The X-ray wavelength was λ = 1.54 Å. The wavenumber scale (q = 4π sin θ/λ where 2θ is the scattering angle) was geometrically calculated. The software CLEARER30 was used to reduce the 2D data to a one-dimensional intensity profile.
Dynamic Light Scattering (DLS)
Experiments were performed using an ALV CGS-3 system with a 5003 multidigital correlator. The light source was a 20 mW HeNe laser, linearly polarized, with λ = 633 nm. Samples were filtered through 0.20 μm Anotop organic membrane filters from Whatman into standard 0.5 cm diameter cylindrical glass cells.
Cryogenic Transmission Electron Microscopy (Cryo-TEM)
Imaging was carried out using a field emission cryo-electron microscope (JEOL JEM-3200FSC) operating at 200 kV. Images were taken using bright-field mode and zero loss energy filtering (Ω type) with a slit width of 20 eV. Micrographs were recorded using a CCD camera (Gatan Ultrascan 4000, USA). The specimen temperature was maintained at −187 °C during the imaging. Vitrified specimens were prepared using an automated FEI Vitrobot device using Quantifoil 3.5/1 holey carbon copper grids, with 3.5 μm hole sizes. Grids were cleaned using a Gatan Solarus 9500 plasma cleaner just prior to use and then transferred into the environmental chamber of a FEI Vitrobot at room temperature and 100% humidity. Following this, 3 μL of sample solution at 1 wt % concentration was applied on the grid, blotted once for 1 s, and then vitrified in a 1/1 mixture of liquid ethane and propane at −180 °C. Grids with vitrified sample solutions were maintained in a liquid nitrogen atmosphere and then cryo-transferred into the microscope.
Transmission Electron Microscopy (TEM)
Imaging was performed using a JEOL AMT XR-401 TEM instrument. A thin film of peptide was added (0.134 wt % of C16IKPEAP and 0.162 wt % C16IKPEAPGE) to the surface of a carbon film coated TEM grid and stained with 1 wt % uranyl acetate solution for 1 min, followed by washing with distilled water by applying enough water to cover the grid and leaving it for 1 min. The grids were then taken and placed in the TEM instrument, and images were taken, using a 4.0 megapixel CMOS camera, at various magnifications.
Molecular Dynamics Simulations (MD Simulations)
Lipopeptide C16IKPEAP was simulated at pH 7. Under these conditions, Lys is protonated, and Glu and the C terminus are deprotonated, leading to a total charge (−1). Four micelle systems made of 80, 101, 143, and 225 lipopeptides (corresponding to a broad range around the experimentally determined association number) were built with an initial spherical distribution of lipopeptides (LP). Simulations were performed in explicit water, with each lipopeptide micelle being neutralized with Na+ ions and solvated in a orthorhombic simulation box with enough water molecules to allow an approximately 12 Å buffer region around the solute. Specifically, the number of solvent molecules contained in the simulation box were 41 735, 42 247, 69 345, and 53 955 water molecules, respectively.
All MD trajectories were performed using the AMBER 16 computer program.31 The energy of each system was computed using the AMBER ff14SB force field32 and the TRIP3P33 water model. Initially, the four systems were minimized over 2500 steps to relax prebuilt spherical micelle structures, heated up to 298 K, and, finally, equilibrated using a NPT ensemble for 0.5 ns at 1 atm and 298 K (2 fs time steps). A restraint over lipopeptide and sodium atoms of 10 kcal mol–1 Å–2 was applied in both thermalization and equilibration steps. The SHAKE algorithm34 was used to keep the bond lengths involving hydrogen atoms at their equilibrium distance. van der Waals interactions were computed by applying 10 Å atom pair distance cutoffs. Electrostatic interactions were computed using the nontruncated electrostatic potential by means of Ewald Summations.35 The production run consisted of a 250 ns trajectory using a NPT ensemble, and similar conditions of previous equilibration steps without any restriction on the system atoms. Snapshots structures were obtained for statistics every 20 ps.
Acknowledgments
This work was supported by a studentship awarded to J.A.H. cofunded by Medimmune and the University of Reading. We are grateful for their support. The work of I.W.H. was also supported by EPSRC Platform grant EP/L020599/1. We acknowledge the use of facilities in the Chemical Analysis Facility at the University of Reading. C.A. and J.T. acknowledge MINECO/FEDER (MAT2015-69367-R) and the Agència de Gestió d’Ajuts Universitaris i de Recerca (2017SGR359) for financial support. Support for the research of C.A. was received through the prize “ICREA Academia” for excellence in research funded by the Generalitat de Catalunya. We thank Diamond (beamtime ref. SM19477-1) and ESRF (MX-1869) for the award of SAXS beamtime.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jpcb.8b11097.
Tables of SAXS fitting parameters and radii of gyration from MD simulations, CD and fluorescence spectra, temperature-dependent cmc plots, DLS data, SAXS fits and XRD data for unlipidated peptides, and plots of RMSD from MD trajectory (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Hamley I. W.; Dehsorkhi A.; Castelletto V.; Walter M. N. M.; Connon C. J.; Reza M.; Ruokolainen J. Self-Assembly and Collagen-Stimulating Activity of a Peptide Amphiphile Incorporating a Peptide Sequence from Lumican. Langmuir 2015, 31, 4490–4495. 10.1021/acs.langmuir.5b00057. [DOI] [PubMed] [Google Scholar]
- Trent A.; Marullo R.; Lin B.; Black M.; Tirrell M. Structural properties of soluble peptide amphiphile micelles. Soft Matter 2011, 7, 9572–9582. 10.1039/c1sm05862b. [DOI] [Google Scholar]
- Cui H. G.; Webber M. J.; Stupp S. I. Self-Assembly of Peptide Amphiphiles: From Molecules to Nanostructures to Biomaterials. Biopolymers 2010, 94, 1–18. 10.1002/bip.21328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehsorkhi A.; Castelletto V.; Hamley I. W. Self-assembling amphiphilic peptides. J. Pept. Sci. 2014, 20 (7), 453–67. 10.1002/psc.2633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchinson J. A.; Burholt S.; Hamley I. W. Peptide hormones and lipopeptides: from self-assembly to therapeutic applications. J. Pept. Sci. 2017, 23, 82–94. 10.1002/psc.2954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamley I. W. Lipopeptides: from self-assembly to bioactivity. Chem. Commun. 2015, 51, 8574–83. 10.1039/C5CC01535A. [DOI] [PubMed] [Google Scholar]
- Paramonov S. E.; Jun H. W.; Hartgerink J. D. Self-assembly of peptide-amphiphile nanofibers: The roles of hydrogen bonding and amphiphilic packing. J. Am. Chem. Soc. 2006, 128, 7291–7298. 10.1021/ja060573x. [DOI] [PubMed] [Google Scholar]
- Irwin N.; Green B. D.; Gault V. A.; Greer B.; Harriott P.; Bailey C. J.; Flatt P. R.; O’Harte F. P. M. Degradation, insulin secretion, and antihyperglycemic actions of two palmitate-derivitized N-terminal pyroglutamyl analogues of glucose-dependent insulinotropic polypeptide. J. Med. Chem. 2005, 48, 1244–1250. 10.1021/jm049262s. [DOI] [PubMed] [Google Scholar]
- Ward B. P.; Ottaway N. L.; Perez-Tilve D.; Ma D. J.; Gelfanov V. M.; Tschop M. H.; DiMarchi R. D. Peptide lipidation stabilizes structure to enhance biological function. Mol. Metab. 2013, 2, 468–479. 10.1016/j.molmet.2013.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.; Shao M. X.; Zheng X. M.; Kong W. L.; Zhang J. N.; Gong M. Self-Assembling Peptides Improve the Stability of Glucagon-like Peptide-1 by Forming a Stable and Sustained Complex. Mol. Pharmaceutics 2013, 10, 3356–3365. 10.1021/mp4001734. [DOI] [PubMed] [Google Scholar]
- Gao Z. H.; Bai G.; Chen J. Q.; Zhang Q.; Pan P. W.; Bai F.; Geng P. Development, Characterization, and Evaluation of a Fusion Protein of a Novel Glucagon-Like Peptide-1 (GLP-1) Analog and Human Serum Albumin in Pichia pastoris. Biosci., Biotechnol., Biochem. 2009, 73, 688–694. 10.1271/bbb.80742. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Lomakin A.; Kanai S.; Alex R.; Benedek G. B. Transformation of Oligomers of Lipidated Peptide Induced by Change in pH. Mol. Pharmaceutics 2015, 12, 411–419. 10.1021/mp500519s. [DOI] [PubMed] [Google Scholar]
- Dumont Y.; Fournier A.; Stpierre S.; Quirion R. Characterization of Neuropeptide-Y Binding-Sites in Rat-Brain Membrane Preparations Using [125I] [Leu31,Pro34]Peptide YY and [125I] Peptide YY3–36 as Selective Y1 and Y2 Radioligands. J. Pharmacol. Exp. Ther. 1995, 272, 673–680. [PubMed] [Google Scholar]
- Ehrlich G. K.; Michel H.; Truitt T.; Riboulet W.; Pop-Damkov P.; Goelzer P.; Hainzl D.; Qureshi F.; Lueckel B.; Danho W.; Conde-Knape K.; Konkar A. Preparation and Characterization of Albumin Conjugates of a Truncated Peptide YY Analogue for Half-Life Extension. Bioconjugate Chem. 2013, 24, 2015–2024. 10.1021/bc400340z. [DOI] [PubMed] [Google Scholar]
- Hutchinson J. A.; Burholt S.; Hamley I. W.; Lundback A.-K.; Uddin S.; Gomes dos Santos A.; Reza M.; Seitsonen J.; Ruokolainen J. The Effect of Lipidation on the Self-Assembly of the Gut Derived Peptide Hormone PYY3–36. Bioconjugate Chem. 2018, 29, 2296–2308. 10.1021/acs.bioconjchem.8b00286. [DOI] [PubMed] [Google Scholar]
- Castelletto V.; Hamley I. W.; Seitsonen J.; Ruokolainen J.; Harris G.; Bellmann-Sickert K.; Beck-Sickinger A. G. Conformation and Aggregation of Selectively PEGylated and Lipidated Gastric Peptide Hormone Human PYY3–36. Biomacromolecules 2018, 19, 4320–4332. 10.1021/acs.biomac.8b01209. [DOI] [PubMed] [Google Scholar]
- Lerch M.; Mayrhofer M.; Zerbe O. Structural similarities of micelle-bound peptide YY (PYY) and neuropeptide Y (NPY) are related to their affinity profiles at the Y receptors. J. Mol. Biol. 2004, 339, 1153–1168. 10.1016/j.jmb.2004.04.032. [DOI] [PubMed] [Google Scholar]
- Creighton T. E.Proteins: Structures and Molecular Properties; W.H.Freeman: New York, 1993. [Google Scholar]
- Brunsveld L.; Waldmann H.; Huster D. Membrane binding of lipidated Ras peptides and proteins - The structural point of view. Biochim. Biophys. Acta, Biomembr. 2009, 1788, 273–288. 10.1016/j.bbamem.2008.08.006. [DOI] [PubMed] [Google Scholar]
- Hawe A.; Sutter M.; Jiskoot W. Extrinsic Fluorescent Dyes as Tools for Protein Characterization. Pharm. Res. 2008, 25, 1487–99. 10.1007/s11095-007-9516-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyanasundaram K.; Thomas J. K. Environmental Effects on Vibronic Band Intensities in Pyrene Monomer Fluorescence and their Application in Studies of Micellar Systems. J. Am. Chem. Soc. 1977, 99, 2039–2044. 10.1021/ja00449a004. [DOI] [Google Scholar]
- Hamley I. W.Introduction to Soft Matter, revised ed.; Wiley: Chichester, 2007. [Google Scholar]
- Castelletto V.; Hamley I. W.; Cenker C.; Olsson U.; Adamcik J.; Mezzenga R.; Miravet J. F.; Escuder B.; Rodriguez-Llansola F. Influence of End-Capping on the Self-Assembly of Model Amyloid Peptide Fragments. J. Phys. Chem. B 2011, 115, 2107–2116. 10.1021/jp111168s. [DOI] [PubMed] [Google Scholar]
- Innovagen Peptide property calculator. http://pepcalc.com/. Accessed in 2018.
- Frisch M. J. T.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Montgomery J. A. Jr.; Vreven T.; Kudin K. N.; Burant J. C.; et al. Gaussian 03; Gaussian, Inc: Wallingford, CT, 2004.
- Schneidman-Duhovny D.; Hammel M.; Tainer J. A.; Sali A. Accurate SAXS Profile Computation and its Assessment by Contrast Variation Experiments. Biophys. J. 2013, 105, 962–974. 10.1016/j.bpj.2013.07.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards F. M. Areas, Volumes, Packing, and Protein-Structure. Annu. Rev. Biophys. Bioeng. 1977, 6, 151–176. 10.1146/annurev.bb.06.060177.001055. [DOI] [PubMed] [Google Scholar]
- Svergun D.; Barberato C.; Koch M. H. J. CRYSOL - A program to evaluate x-ray solution scattering of biological macromolecules from atomic coordinates. J. Appl. Crystallogr. 1995, 28, 768–773. 10.1107/S0021889895007047. [DOI] [Google Scholar]
- Baldwin A. J.; Knowles T. P. J.; Tartaglia G. G.; Fitzpatrick A. W.; Devlin G. L.; Shammas S. L.; Waudby C. A.; Mossuto M. F.; Meehan S.; Gras S. L.; et al. Metastability of Native Proteins and the Phenomenon of Amyloid Formation. J. Am. Chem. Soc. 2011, 133, 14160–14163. 10.1021/ja2017703. [DOI] [PubMed] [Google Scholar]
- Makin O. S.; Sikorski P.; Serpell L. C. CLEARER: a new tool for the analysis of X-ray fibre diffraction patterns and diffraction simulation from atomic structural models. J. Appl. Crystallogr. 2007, 40, 966–972. 10.1107/S0021889807034681. [DOI] [Google Scholar]
- Case D. A.; Betz R. M.; Cerutti D. S.; Cheatham T. E. I.; Darden T. A.; Duke R. E.; Giese T. J.; Gohlke H.; Goetz A. W.; Homeyer N.; et al. AMBER 2016; University of California: San Francisco, 2016.
- Maier J. A.; Martinez C.; Kasavajhala K.; Wickstrom L.; Hauser K. E.; Simmerling C. ff14SB: Improving the Accuracy of Protein Side Chain and Backbone Parameters from ff99SB. J. Chem. Theory Comput. 2015, 11, 3696–3713. 10.1021/acs.jctc.5b00255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L.; Chandrasekhar J.; Madura J. D.; Impey R. W.; Klein M. L. Comparison of Simple Potential Functions for Simulating Liquid Water. J. Chem. Phys. 1983, 79, 926–935. 10.1063/1.445869. [DOI] [Google Scholar]
- Ryckaert J. P.; Ciccotti G.; Berendsen H. J. C. Numerical-Integration of Cartesian Equations of Motion of a System with Constraints - Molecular-Dynamics of N-Alkanes. J. Comput. Phys. 1977, 23, 327–341. 10.1016/0021-9991(77)90098-5. [DOI] [Google Scholar]
- Toukmaji A. Y.; Board J. A. Ewald summation techniques in perspective: A survey. Comput. Phys. Commun. 1996, 95, 73–92. 10.1016/0010-4655(96)00016-1. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.