

Abstract
Background
pWB980 derived from pUB110 is a promising expression vector in Bacillus for its high copy number and high stability. However, the low transformation rate of recombinant plasmids to the wild cells limited the application of it. On the basis of pWB980, constructing an E. coli–B. subtilis shuttle plasmid could facilitate the transformation rate to Bacillus cells. Because the insertion site for E. coli replication origin sequence (ori) is not unique in pWB980, in order to investigate the best insertion site, eight shuttle plasmids (pUC980-1 ~ pUC980-8) containing all possible insertion sites and directions were constructed.
Results
The results showed that all the selected insertion sites could be used to construct shuttle plasmid but some sites required a specific direction. And different insertion sites led to different properties of the shuttle plasmids. The best shuttle plasmids pUC980-1 and pUC980-2, which showed copies more than 450 per cell and segregational stabilities up to 98%, were selected for heterologous expressions of an alkaline pectate lyase gene pelN, an alkaline protease spro1 and a pullulanase gene pulA11, respectively. The highest extracellular activities of PelN, Spro1 and PulA11 were up to 5200 U/mL, 21,537 U/mL and 504 U/mL correspondingly after 54 h, 60 h and 48 h fermentation in a 10 L fermentor. Notably, PelN and Spro1 showed remarkably higher yields in Bacillus than previous reports.
Conclusion
The optimum ori insertion site was the upstream region of BA3-1 in pWB980 which resulted in shuttle plasmids with higher copy numbers and higher stabilities. The novel shuttle plasmids pUC980-1 and pUC980-2 will be promising expression vectors in B. subtilis. Moreover, the ori insertion mechanism revealed in this work could provide theoretical guidance for further studies of pWB980 and constructions of other shuttle plasmids.
Keywords: Expression vectors, pUC980, Bacillus subtilis, Alkaline pectate lyase, Alkaline protease, Pullulanase
Background
Bacillus subtilis is a promising safe host strain for bio-industrial productions for its lack of pathogenicity and endotoxins [1–3]. Its capacity for secreting proteins directly into the medium facilitates the downstream processing greatly [4]. In the last 30 years, B. subtilis expression systems have been well developed. Many foreign proteins have been produced in B. subtilis cells successfully by using different kinds of expression plasmids [5–7]. However, compared to Escherichia coli expression system, relatively difficult in plasmid transformation, instability of plasmids, and lack of diversity for available plasmids became the main obstacles of the development of B. subtilis expression system [8, 9].
At present, the majority of available B. subtilis expression plasmids are derived from pC194 [10–12], pLS1 [13, 14] and pUB110 [3, 15]. In order to improve transformation efficiency of plasmids, a lot of E. coli–B. subtilis shuttle plasmids such as pHCMC05 [16] and pHT43 [17] have been constructed. However, the available plasmids in recent researches remain difficult to combine high copy number and high stability in the fermentation for producing heterologous proteins. As an example of high copy number plasmids, pWB980 [18] derives from plasmid pUB110 has been doing well in heterologous gene expression in B. subtilis [15, 19, 20]. However, the low transformation rate of the ligation products between pWB980 and target genes became a barrier to the widely use of it. On the purpose of solving this problem, many shuttle plasmids were constructed based on pWB980 [20, 21]. But so far, there is still a lack of systematical studies about the manipulations of pWB980.
In this work, the ori region where plasmid replication is originated in E. coli was inserted into four important sites of pWB980 to produce a series of E. coli–B. subtilis shuttle plasmids. Because the replication gene insertions may usually cause the changes of copy number and stability of the plasmids [22, 23], we focused on estimating the copy number and stability of the recombinant plasmids and found some promising E. coli–B. subtilis shuttle plasmids with high copy numbers and good stabilities. Moreover, an alkaline pectate lyase gene pelN from Paenibacillus sp. 0602 [24], an alkaline protease spro1 from alkaliphilic Bacillus sp. 221 [25] and a pullulanase gene pulA11 from Anoxybacillus sp. LM18-11 [26] were expressed more efficiently in the case of specific plasmids in B. subtilis WB600. Thus, we believe that these plasmids will be a wider range of uses in further research of the B. subtilis expression system.
Results
Construction of E. coli–B. subtilis shuttle plasmids
In general, smaller size plasmids usually are easier to be manipulated. On the purpose of shortening pWB980 before construction, the more commonly used gene kan was kept while the bleoR was deleted from it (Fig. 1). As the selective gene bleoR was proved to be unnecessary in other studies and our previous work [19, 20]. The truncated plasmid pWB980-DB was successfully obtained in this work (Fig. 2). It was found to have the same stability with pWB980 along with a higher copy number (272) than that of pWB980 (134) as shown in the Fig. 3.
Fig. 1.

Plasmids constructions. The bleoR was deleted from pWB980, resulting plasmid pWB980-DB. It was integrated with the replication-originated region ori from pUC19 at four essential sites up- and down-stream the membrane-binding region BA3-1 and the replicase-coding gene rep
Fig. 2.
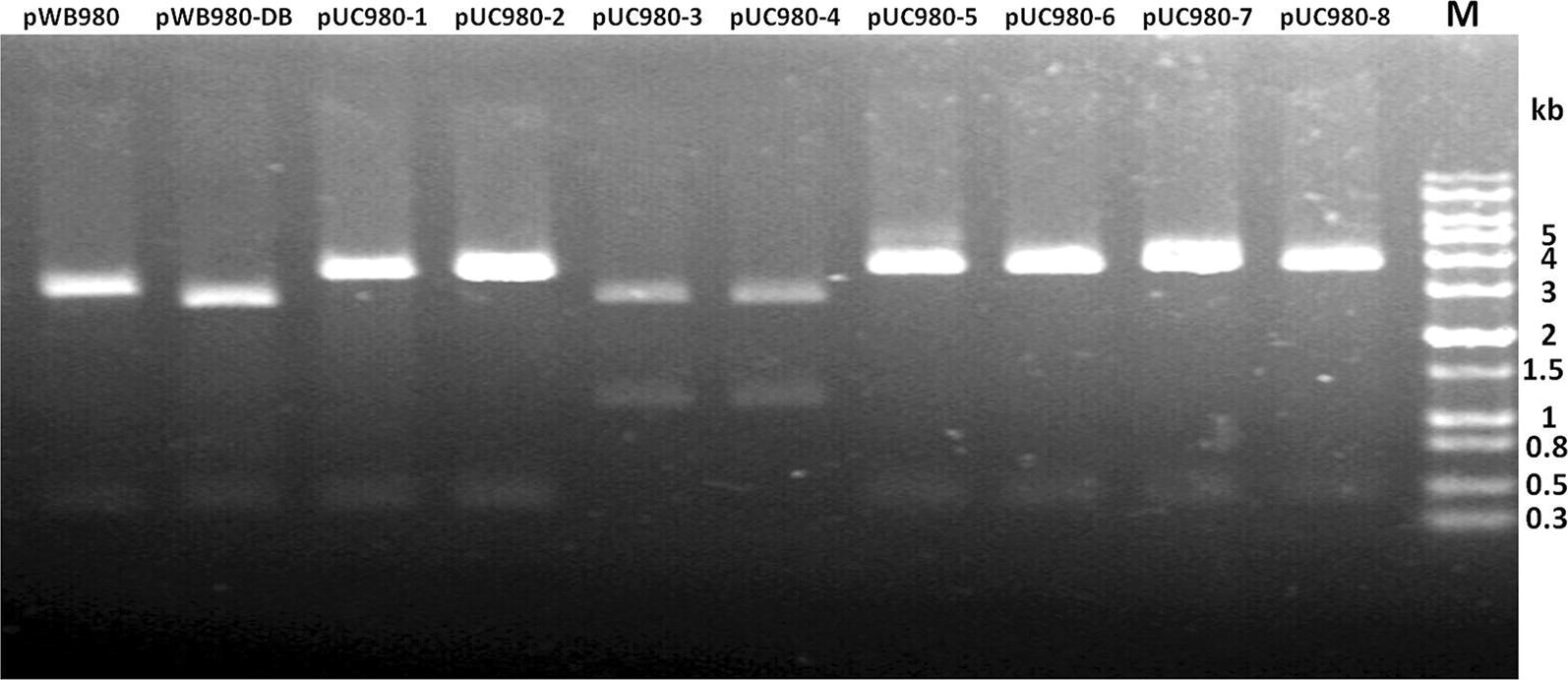
Verifications of constructed plasmids. Constructed plasmids were digested by EcoRI/HindIII restriction enzymes and separation by agarose gel electrophoresis. All plasmids were digested into two parts as shown in lanes. The lanes of pWB980 and pWB980-DB were 3337 bp, 450 bp and 2938 bp, 450 bp respectively. Lanes pUC980-1, pUC980-2, pUC980-5, pUC980-6, pUC980-7 and pUC980-8 were digested into the 3826 bp and 450 bp regions. Lanes pUC980-3 and pUC980-4 resulted into 2938 bp and 1338 bp regions. Lane M was DNA marker
Fig. 3.

Plasmids copy number and segregational stability. a As shown in the graph, all the new plasmids obtained higher copy numbers than pWB980 except pUC980-7 and plasmid pUC980-8 was not counted because it was unreplicapble in B. subtilis cells. b plasmids were classified into three groups from high to low, concerning their copy numbers (C1 to C3) and segregational stabilities (S1 to S3)
Then eight E. coli–B. subtilis shuttle plasmids derived from pWB980-DB were constructed successfully according to the design in Fig. 1. pWB980-DB was in the size of 3388 bp and the eight shuttle plasmids were all in the size of 4276 bp. To verify the plasmids clearly, double enzyme-digestions with EcoRI/HindIII were carried out and the results were shown in Fig. 2. All of them replicated normally both in E. coli and B. subtilis except pUC980-8 which could not replicate in B. subtilis host cells. This might be due to the interruption of reverse ori to the replication of the rep gene.
Copy number of recombinant shuttle plasmids in B. subtilis 168
The copy numbers of all new plasmids were estimated and shown in the Fig. 3. Shuttle plasmids pUC980-1, pUC980-2 and pUC980-6 obtained high copy numbers of 584, 492 and 487 correspondingly. pUC980-1 had the highest copy number, which was over four times than that of pWB980 and twice than that of pWB980-DB. The copy numbers of plasmids pUC980-3 (305), pUC980-4 (295) and pUC980-5 (286) were similar to that of pWB980-DB. Only plasmid pUC980-7 with 107 copies per cell had a lower copy number than that of pWB980. pUC980-8 was not counted due to its un-replicability in B. subtilis. Therefore, the shuttle plasmids were classified into three groups (C1 ~ C3) according to their copy number levels from high to low as shown in Fig. 3b. Plasmids in C1 were determined to be high copy plasmids had copy numbers higher than 400, including pUC980-1, pUC980-2 and pUC980-6. C2 is the middle copy plasmids group included plasmids pUC980-3, pUC980-4, and pUC980-5 with copy numbers from 200 to 400. Plasmid pUC980-7 was ranked into C3 with relatively low copy numbers from 100 to 200.
Stability of recombinant shuttle plasmids in B. subtilis 168
Plasmid stability consists of structural stability and segregation stability. All plasmids have exhibited structural stabilities after 30-days culturing and when digested with enzymes the expected fragments were obtained (Fig. 2). The plasmids were structurally stable and further verified by sequencing (data not shown). pWB980 and pWB980-DB were stable during passages with segregation stabilities of 100% (Fig. 3a). The plasmids from pUC980 series could be classified into three groups based on their segregation stabilities from high to low (Fig. 3b). Plasmids pUC980-1(99%), pUC980-2 (100%) and pUC980-5 (99%) were ranged into group S1 (high stable plasmid group). They acted well with more than 98% transformants retained after 30-days incubation in LB medium showing the similar stability with pWB980 and pWB980-DB. pUC980-4 were included in group S2 (low stable plasmid group) with a segregation stability of 39%. Plasmids in group S3 (unstable plasmids group) contained segregation stabilities lower than 10%, including pUC980-3 (2%), pUC980-6 (4%) and pUC980-7 (0).
Synthetically, all plasmids mentioned above were arranged and stated in Fig. 3b combining their ranks in copy number (C1 to C3) and segregation stability (S1 to S3). pUC980-1 and pUC980-2 were classified both in C1 and S1. Plasmid pUC980-6 in C1 obtained high copy number but low segregation stability in S3. Plasmid pUC980-5 in C2 were listed in S1, having relatively lower copy number than plasmids in C1. pUC980-4 performed moderately both in C2 and S2, while plasmid pUC980-3 in C2 was so unstable that ranked in S3. pUC980-7 was not only the most unstable plasmid but also had the lowest copy number, listed in C3 and S3. Based on the classification, plasmids pUC980-1 and pUC980-2 performing well both in copying and passaging were supposed to be the optimal expression vectors. Plasmids pUC980-5 and pUC980-6 only showed high stability and high copy number respectively. To further test the application potential of these new plasmids in production of industrial enzymes, three different industrial enzymes were selected for heterologous expression using these plasmids in B. subtilis WB600.
Alkaline pectate lyase expression in B. subtilis WB600
The alkaline pectate lyase gene pelN was expressed in the constructed plasmids. All plasmids functioned normally with extracellular secretory expressions after 48 h culturing in shake flasks. The results indicated obvious differences in expression levels as presented in Fig. 4a. The extracellular activities of PelN were more than 1500 U/mL when using pUC980-1, pUC980-2, pUC980-5 and pWB980-DB as expression vectors (Fig. 4a). The highest activity was 2738 U/mL with pUC980-2-pelN, which was nearly 3 times than that of pWB980. It also was the highest expression level in Bacillus [27, 28]. It is worth noting that the plasmids pUC980-3, pUC980-4, pUC980-6 produced activities lower than 200 U/mL. The lowest PelN activity was produced by pUC980-7 (4 U/mL) that was nearly the same to the background of the host cells.
Fig. 4.

Alkaline pectate lyase productions in B. subtilis. a All plasmids were used to express alkaline pectate lyase pelN in B. subtilis WB600. The highest activity was obtained by pUC980-2 with 2738 U/mL. b SDS-PAGE analysis of PelN in culture supernatants. Lane M was protein molecular weight marker. The PelN protein was 48.0 kDa as shown in the frame. Lane 1 was the starting strain WB600, served as negative control. Lanes 2-10 were WB600 with plasmids pWB980, pWB980-DB and pUC980-serial plasmids (pUC980-1 to pUC980-7). Lanes 11-19 represented WB600 with PelN-expressing plasmids pWB980-pelN, pWB980-DB-pelN and pUC980-pelN plasmids (pUC980-1-pelN to pUC980-7-pelN) correspondingly
In addition, comparative analysis of these expressions were carried through SDS-PAGE as shown in Fig. 4b. The SDS-PAGE analysis was performed and the intensity of observed bands correlated well with the PleN activities. The proportions of PelN protein in the supernatants at 48 h culturing with plasmids pWB980, pWB980-DB, pUC980-1 to pUC980-7 were estimated as 48%, 63%, 78%, 80%, 4%, 12%, 65%, 5% and 2% respectively (Fig. 4b).
Alkaline protease expression in B. subtilis WB600
As shown above, the pUC980-1 and pUC980-2 were proved to be useful in over-expressions of heterologous proteins. They were further used to express alkaline protease gene spro1 in B. subtilis WB600, employing pWB980 and pWB980-DB as controls. After culturing in shake-flask for 48 h, the extracellular activities of protease expressed with pUC980-1-spro1 and pUC980-2-spro1 were 4590 U/mL and 4818 U/mL (Fig. 5a), which were 2.3 times and 2.4 times than that with pWB980-spro1 (1991 U/mL), respectively. The proportions of the Spro1 in the supernatants were 72%, 77%, 92% and 95% in which expressed with plasmids pWB980, pWB980-DB, pUC980-1 and pUC980-2, respectively (Fig. 5b).
Fig. 5.

Alkaline protease productions in B. subtilis WB600. a Protease activities in supernatants were determined during Spro1-expressions with different plasmids in B. subtilis WB600. The highest activity was obtained by pUC980-2 with 4818 U/mL. b SDS-PAGE analysis of Spro1 in culture supernatants. Lane M was protein molecular weight marker. The Spro1 protein was 26.7 kDa as shown in the frame
Pullulanase expression in B. subtilis WB600
Meanwhile, plasmids pUC980-1 and pUC980-2 were used to express a pullulanase gene pulA11 in WB600 in shake flask culturing for 48 h. Enzyme activities of plasmids pUC980-1-pulA11 and pUC980-2-pulA11 in the supernatants were 173 U/mL and 187 U/mL respectively, which were 1.9 times and twice than pWB980 (92 U/mL) correspondingly as stated in Fig. 6a. The percentages of PulA11 proteins in the supernatants after 48 h fermentation of the recombinant strains containing plasmids pWB980-pulA11, pWB980-DB-pulA11, pUC980-1-pulA11, and pUC980-2-pulA11 were 6%, 8%, 27% and 30%, respectively (Fig. 6b).
Fig. 6.

Pullulanase productions in B. subtilis WB600. a Activities of pullulanase expressed by different plasmids were determined in supernatants of WB600. The highest activity was obtained by pUC980-2 with 187 U/mL. b SDS-PAGE analyses of pullulanase expressions, M was the protein molecular weight marker. The PulA11 protein was 81.7 kDa as shown in the frame
Scale up fermentation of the optimum recombinant strains in a 10 L fermenter
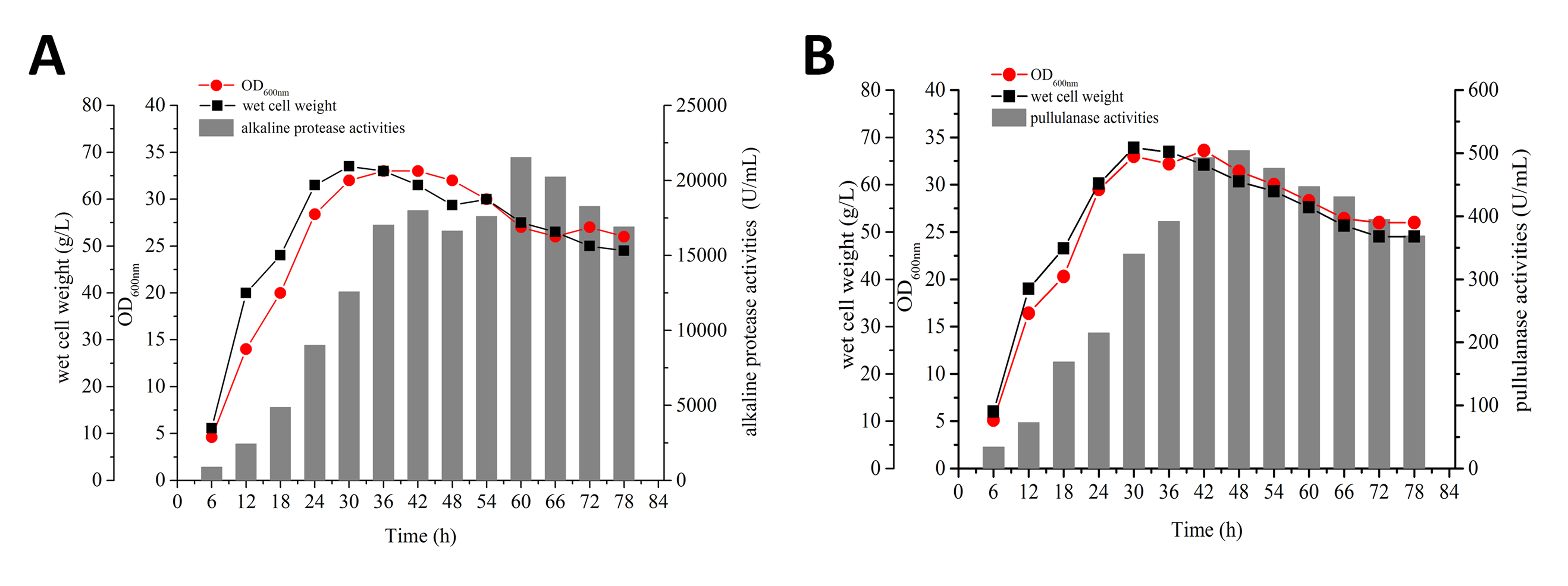
The above results indicated that the recombinant strains containing pUC980-2-pelN, pUC980-2-spro1, and pUC980-2- pulA11 showed the highest expression level in shake flask culturing basing on the remarkable properties of pUC980-2. In order to verify the stable production capacity of these strains in scale up fermentation, we performed batch-feed fermentations in a 10 L fermenter. The highest APL activity reached to 5200 U/mL after 54 h fermentation of the recombinant strain B. subtilis WB600 (pUC980-2-pelN). The curves for cell-growth and wet weight were shown in Fig. 7a. The cells grown into the stationary phase at 30 h with OD600nm in 34.1 and 66.3 g/L of wet cell weight. The fermentation of this strain in a 10 L fermenter were repeated 20 times and the activities fluctuated little as shown in Fig. 7b. Furthermore, the highest production of Spro1 was 21,537 U/mL at 60 h and the highest production of pullulanase was 504 U/mL at 48 h (Additional file 1: Fig S1). So far, the extracellular yields of PelN and Spro1 by the recombinant strains B. subtilis WB600 (pUC980-2-pelN) and B. subtilis WB600 (pUC980-2-spro1) showed the highest level in Bacillus subtilis [29, 30].
Fig. 7.

The 10 L fermentations of WB600 (pUC980-2-pelN). a The cell wet-weights, OD600nm and the pectate lyase activities were determined for every 6 h. Cells went into the stationary phase after 30-hours fermentation and the cell wet-weight was 66.3 g/mL with OD600nm of 34.1. The highest activity were obtained at 54 h with 5200 U/mL. Then the enzyme productions decreased gradually. b The fermentations of WB600 (pUC980-2-pelN) have been repeated for 20 times and the PelN activities kept stable and repeatable
Discussion
The properties of pUB110-derived plasmid pWB980 were regulated by the rep gene and membrane-biding region BA3-1 according to previous studies [31–34]. The rep gene, encoding the replicase protein, was kept unchanged to fully support functioning of the pWB980 plasmid [18, 35–37]. The BA3-1 region was related to plasmid partitioning into the daughter cells [31, 38–40]. It has long been suspected that plasmid replication origin ori might be important for plasmids replications and partitioning [41, 42]. On these basis, the rep gene and the membrane-biding region have been manipulated with ori insertion separately.
Before ori insertions, the plasmid size was reduced for easier manipulation. The occasionally used resistance gene bleoR was deleted from pWB980, resulting plasmid pWB980-DB with a few higher copy numbers than pWB980 (Fig. 3a). The improvement of plasmid replication might be due to the elimination of the bleoR gene near the initial signal in BA3-1 region for lagging strand synthesis. Using pWB980-DB as the original plasmids, ori was inserted into four positions upstream and downstream the rep gene and the BA3-1 region.
As for shuttle plasmids derived from pWB980-DB, ori insertions at certain sites influenced the properties of each shuttle plasmid differently. Firstly, all ori insertions flanking the BA3-1 improved the plasmids copies in host cells concluded from the enhancements of copy numbers of plasmids pUC980-1, pUC980-2, pUC980-3 and pUC980-4. Moreover, insertions downstream BA3-1 affected plasmid segregations much more than the upstream ones and depressed plasmid stabilities sharply. These depressions strongly suggested the importance of this region to the distribution of plasmids in the cell division. In addition, ori insertions upstream the rep gene in plasmids pUC980-5 and pUC980-6 were conducive to plasmid replication. While there was a direction bias in plasmid segregations as the forward insertion was largely more stable than the reverse one. Furthermore, insertions of the ori downstream the rep gene resulted in poor performances in copy number and stability of pUC980-7 and pUC980-8 plasmids.
All plasmids (pUC980-1 ~ pUC980-7) kept structural stabilities in B. subtilis. They were systematically classified based on their copy numbers and segregation stabilities and further used to produce heterogenous proteins in B. subtilis WB600. Plasmids with good segregation stabilities dramatically outperformed the unstable ones, stated by the disparity between S1 plasmids and others. Moreover, the production of heterogenous proteins increased along with the copy numbers when the plasmids had similar stabilities. This conclusion was approved by the gradually improved expressions along the copy numbers of plasmids in group S1. And it was also supported by the plasmids in group S3. As for plasmids with similar copies, improvements in segregation stabilities enhanced the productions of heterogenous proteins notably, corroborated by plasmids in every segregation stability group.
Plasmids pUC980-1 and pUC980-2 performed well in expressing the tested heterogenous proteins, especially plasmid pUC980-2, with which the highest productions of alkaline pectate lyase PelN and alkaline protease Spro1 were obtained in B. subtilis. The high producing levels of foreign proteins have proved the high efficiency and universality of these two plasmids and stated that they were capable of expressing many other proteins. Furthermore, the practicability of pUC980-2 in industrial applications were strongly illustrated by the stable expressions of heterogenous proteins with pUC980-2 in larger scale fermentations.
As for the relatively lower expressions of PulA11 than the other two proteins, the result indicated that the foreign gene itself also make important effect in the final expression level. Meanwhile, the expression levels of protein PulA11 with plasmid pUC980-2 in B. subtilis was still promoted to a certain extent compared with our previous studies in E. coli expression system [43–45]. These improvements further confirmed the availability of pUC980-2 in production of heterogenous proteins.
Many E. coli–B. subtilis shuttle plasmids have been constructed and employed in heterogenous productions mainly based on natural plasmids such as pBS72, pUB110 and pC194 in previous studies [16, 46, 47]. Some of them are inducible, such as pHCMC05 and pHT43 derived from pBS72, which were seldom considerable in economy because of the expensive inducers [48]. There were also some constitutive plasmids originated from pUB110, e.g. pBNS2 and pBSG, which have shown potentials in foreign expressions [49, 50]. However, further developments of these shuttle plasmids were restricted by the lack of systematical analysis about plasmids constructions. In this context, the analyses carried out in this work may have provided some conducts in shuttle plasmids manipulations in the B. subtilis expression system. According to our knowledge, it is the first time to classify manipulations with ori-insertions to pUB110-derived plasmid in this work.
Conclusion
In this work, some novel rules about the constructions of E. coli-B. subtilis shuttle plasmids based on pWB980 were concluded: (1) The deletion of bleoR gene improved the plasmid copying of pWB980; (2) The ori insertion was generally beneficial to plasmid replication in B. subtilis, while its impacts on plasmids segregation stabilities differed with insertion sites; (3) The site upstream the membrane binding region BA3-1 was the optimum position for plasmids manipulations with improvements both in copy number and stability. Based on these construction rules, two plasmids pUC980-1 and pUC980-2 with high copy numbers and high stabilities were selected out and performed well in tested protein productions. The highest production levels in B. subtilis of pectate lyase PelN and alkaline protease Spro1 were achieved by using plasmid pUC980-2 as expressing vector through 10 L fermentations. Plasmid pUC980-2 was proved to be capable and valuable in further industrial productions by these promising results.
Methods
Bacterial strains, plasmids and materials
The bacterial strains and plasmids used in this study are listed in Additional file 2: Table S1. E. coli DH5α was used in plasmids construction and amplification. B. sutilis 168 were used in the determinations of plasmids properties. B. subtilis WB600 was employed in gene expressions. The sequences of the alkaline pectate lyase from Paenibacillus sp. 0602 (pelN, GenBank: KC351190.1), the alkaline protease gene spro1 from alkaliphilic Bacillus sp. 221 (spro1, Sequence ID: D13157.1) and the pullulanase gene pulA11 from Anoxybacillus sp. LM18-11 (GeneBnak ID: HQ844266.1) were deposited in NCBI. The restriction endonucleases and DNA polymerase were commercially supplied by Thermo Fisher Scientific Co., Ltd. All other enzymes, chemicals and reagents were purchased from TaKaRa Biotechnology (Dalian, China) Co., Ltd.
Cultivation conditions
All cells were routinely grown at 37 °C and 200 rpm in Luria–Bertani (LB) medium or fermentation medium. LB medium (1 L) consisted of tryptone 10 g, yeast extract 5 g and NaCl 5 g. 1 L of fermentation medium consisted of tryptone 16 g, soluble starch 37 g, CaCl2 1.5 g, NaCl 2 g, MgSO4·7H2O 0.2 g, KH2PO4 1 g, FeSO4·7H2O 0.05 g and (NH4)2SO4 1.5 g. Different antibiotics (50 μg/mL kanamycin and 25 μg/mL kanamycin) were added in the medium for the relevant recombinant strains.
B. subtilis WB600 strains containing recombinant plasmids were cultured in Shake flask (SHUNIU, GG-17, Sichuan SHUBO Co., LTD, China) or in a 10-L fermenter (5BG, bxbio, China) to produce foreign proteins. The shake-flask culturing was performed as followed. A 5 mL aliquot of LB medium supplemented with 25 μg/mL kanamycin was inoculated with a frozen glycerol stock of recombinant strain (20 μL), and then incubated for up to 14 h at 37 °C and 200 rpm in a rotary shaker (ZQWY-200G, Shanghai Zhichu Instruments Co., Ltd., China). An aliquot of this preculture (0.5 mL) was transferred into a 250 mL shake-flask that was loaded with 50 mL of LB medium supplemented with 25 μg/mL kanamycin, which was then shaken in the rotary shaker (200 rpm) at 37 °C. After culturing for 48 h, samples of each culture were collected and analyzed for enzyme activities. The culture was harvested by centrifugation at 12,000×g for 10 min at 4 °C to obtain the culture supernatant. Enzyme activities in the supernatant were determined with corresponding assays. Then the culture supernatant was analyzed with SDS-PAGE. The percentages of the produced extracellular proteins were calculated by a software Gel-Pro analyzer 4.0 (Media Cybernetics, CA, USA).
Fermentation of the recombinant B. subtilis WB600 strain was performed in a 10-L fementer. The seed culture was prepared as described in the above section and then used to inoculate an initial batch of fermentation medium supplemented with 25 μg/mL kanamycin. The biomass and enzyme activities were analyzed at designated time intervals. The cell density was determined by measuring the OD600nm with a UV-1800/PC spectrophotometer (Epoch 2, BioTek, USA). To determine the wet cell weight (WCW), a 2-mL sample of the culture broth were centrifuged at 12,000×g for 10 min at 4 °C. The resulting pellet was collected and weighed. The enzyme activities in the culture supernatant were further determined with corresponding methods. The whole fermentation process was stopped and finished when continuous reductions of biomass and enzyme activities were observed.
Shuttle plasmids construction
Construction of shuttle plasmids is schematically presented in Fig. 1. All plasmids were constructed through the method of MEGAWHOP [51]. All of the specific primers used for PCR amplification were synthesized by the Beijing Genomics Institute (BGI) and listed in Table 1 and the isolation and manipulation of recombinant DNA were performed according to the previously published protocol [52]. 1.5 mL of culture was poured into a 2 mL EP tube and centrifuged at 12,000×g for 10 min at 4 °C. The supernatant was discarded and the bacterial pellet was collected. The bacterial samples were treated with solution I (100 mL of solution I consisted of 1 M pH 8.0 Tris–HCl, 2.5 mL; 0.5 M EDTA 2 mL; ddH2O 91 mL; 20% sterile glucose 4.5 mL), solution II (100 mL of solution II consisted of 10% SDS 50 mL; 2 M NaOH 50 mL), solution III (500 mL of solution III consisted of KAc 147 g; HAc 57.5 mL, ddH2O was added to 500 mL), mixture of phenol: chloroform (volume ration 1:1) and 70% ethanol successively according to the protocol. The DNA sample was dissolved with 80 μL ddH2O and stored in − 20 °C for further experiments.
Table 1.
Primers used in this study
| Primers | Sequence (5′-3′) |
|---|---|
| DB-1 | AATCTATTATTAATCTGTTCAGCAATC |
| DB-2 | TCTCACGCATAAAATCCCC |
| P1-S | GGGGATTTTATGCGTGAGACTGTCAGACCAAGTTTACTCAT |
| P1-A | GATTGCTGAACAGATTAATAATAGATTAGCGGTATCAGCTCACTC |
| P2-S | GATTGCTGAACAGATTAATAATAGATTCTGTCAGACCAAGTTTACTCAT |
| P2-A | GGGGATTTTATGCGTGAGAAGCGGTATCAGCTCACTC |
| P3-S | ATTGAACATACGGTTGATTTAATAACTGACTGTCAGACCAAGTTTACTCAT |
| P3-A | GCTTTAGCAAGAGGGTGATGTTTGAGCGGTATCAGCTCACTC |
| P4-S | GCTTTAGCAAGAGGGTGATGTTTGCTGTCAGACCAAGTTTACTCAT |
| P4-A | GAACATACGGTTGATTTAATAACTGAAGCGGTATCAGCTCACTC |
| P5-S | CAAGAAAAACACGATTTAGAACCCTGTCAGACCAAGTTTACTCAT |
| P5-A | GAGTTAGTTCAAATTCGTTCTTTTTAAGCGGTATCAGCTCACTC |
| P6-S | GAGTTAGTTCAAATTCGTTCTTTTTACTGTCAGACCAAGTTTACTCAT |
| P6-A | CAAGAAAAACACGATTTAGAACCAGCGGTATCAGCTCACTC |
| P7-S | TGCTTAGGAAGACGAGTTATTAATACTGTCAGACCAAGTTTACTCAT |
| P7-A | GAATATTTGGAGAGCACCGTTCTTATTCAGCAGCGGTATCAGCTCACTC |
| P8-S | GAATATTTGGAGAGCACCGTTCTTATTCAGCCTGTCAGACCAAGTTTACTCAT |
| P8-A | TGCTTAGGAAGACGAGTTATTAATAAGCGGTATCAGCTCACTC |
| 62-S | CATAAAAAAGGAGACATGAACGATGGCGGGCAATGCAGATTAC |
| 62-A | CCCCGGGTACCGAGCTCGATTAATAGCTCGTCTTCAGCCAGTTGTCC |
| spro-S | GCGCAACTCAAGCTTTTGCCATGAAGCTTAAGAAACCGTTG |
| spro-A | CCCGGGTACCGAGCTCGATTAGCGTGTTGCCGCTTC |
| pulA-S | GAGGCGCAACTCAAGCTTTTGCCCCCCCAAAACAACAGTCGTTTGAAG |
| pulA-A | GATCCCCGGGTACCGAGCTCGATCAACATTGAATTAATACCCACGCAC |
| kanS | CGGATATTGAGATGATGTGTGTCATGTC |
| kanA | GACCATGTGTAAGCGGCCAATC |
| dnaNS | GCACTTGCCGCAGATTGA |
| dnaNA | AATGCAAGACGGTGGCTATC |
According to the map obtained after sequencing, the 169 bp fragment of the membrane-binding region BA3 originating from pUB110 was deleted during construction of pWB980. In order to be mentioned conveniently, the remaining 346 bp fragment of BA3 was named BA3-1 in this work as shown in Fig. 1. pWB980 is consist of five parts: BA3-1, P43 promoter cohered with a signal-peptide sequence, the replicase-coding gene rep in B. subtilis, the kanamycin-resistance gene kanR and the bleomycin gene bleoR gene (Fig. 1). In this work, the bleoR was deleted with primers DB-1/DB-2. The plasmid replication origin ori (888 bp) from pUC19 was inserted at the four sites up- and down-stream BA3-1 region and rep gene in pWB980-DB (3388 bp) separately. With primer pairs P1-S/P1-A and P2-S/P2-A, ori integrated into the site (659 bp) upstream the BA3-1 with forward and reverse directions. The recombinant plasmids were named as pUC980-1 and pUC980-2 respectively. At the site (169 bp) downstream the BA3-1, primers P3-S/P3-A and P4-S/P4-A were used in ori-insertions, resulting plasmids pUC980-3 and pUC980-4 in forward and reverse directions. Upstream the rep gene, the ori was inserted at the 3118 site using primers P5-S/P5-A and P6-S/P6-A. The recombinant plasmids were named as pUC980-5 and pUC980-6 with forward and reverse ori-insertions respectively. Ori was put into the 1722 site downstream the rep by using primers P7-S/P7-A and P8-S/P8-A. The plasmid with forward ori insertion was pUC980-7 and the other one was pUC980-8.
With primer pair 62-S/62-A, the alkaline pectate lyase gene pelN was inserted into the sites after the P43 promoter of plasmids pWB980, pWB980-DB and all pUC980-serial plasmids except pUC980-8. The recombinant plasmids were pWB980-pelN, pWB980-DB-pelN, pUC980-1-pelN, pUC980-2-pelN, pUC980-3-pelN, pUC980-4-pelN, pUC980-5-pelN, pUC980-6-pelN and pUC980-7-pelN correspondently.
Determination of plasmid copy number by quantitative real time PCR (qPCR)
The quantitative real-time PCR (qPCR) was performed using iQ™ SYBR® Green Supermix (Bio-Rad) as described by Turgeon [53]. The dnaN and kanR genes were chosen as a single copy reference gene on the B. subtilis chromosome and the test gene in plasmids correspondingly [54–56]. Specific primer pairs dnaNS/dnaNA and kanS/kanA (Table 1) were designed to generate products of approximately 150 bp. The standard curves were prepared based on the pMD-18-T vector. The PCR mixtures (total volume 20 μL) contained 10 μL 2xTakara SYBR Green Real-Time PCR Master Mix, 1.5 μL forward and reverse primers (10 μM) and 1 μL diluted (10−1 to 10−4) sample DNA. The reaction condition was as followed: 95 °C for 10 min, 45 cycles of 95 °C for 10 s, 62 °C for 20 s, and 72 °C for 30 s, followed by a gradient temperature from 55 °C to 95 °C. The calculated mean of the kanR gene in each compared plasmid was divided by the dnaN gene (single-copy reference), resulting the copy number. All experiments were performed with three independent biological replicates.
Plasmid stability assay
Plasmid segregational stability in B. subtilis 168 was detected according to the method of Bron and Luxen [39] with minor modifications. Single colonies on kanamycin selective (25 μg/mL) LB agar plate were used to inoculate 5 mL kanamycin selective (25 μg/mL) LB medium. After 24 h culturing at 37 °C, the cultures were diluted 1:1000 in fresh LB media without antibiotics and incubated at 37 °C. This iterative subculturing process was repeated every 24 h for 30 consecutive days. Samples taken at appropriate intervals were checked for the fraction of plasmid-containing cells by replica plating on LB agar plates containing 25 μg/mL kanamycin.
N: Total amount of colonies on the selectable plate.
Plasmids were extracted from host cells after 30-days continuously culturing and detected the structural stability by EcoRI/HindIII enzyme-digestion verifications.
Alkaline pectate lyase assay
The pectate lyase activity was measured using the method described previously by Wang et al. [57]. 20 μL of the diluted enzyme solutions were added to 400 μL of 0.2% pectin in 50 mmol/L glycine–NaOH buffer (with 50 μmol/L CaCl2) at pH 9.0. The reaction mixture was incubated at 65 °C for 10 min, and the reaction was terminated by adding 580 μL of 30 mmol/L H3PO4. The product yields were detected using a spectrophotometer at 235 nm (Epoch 2, BioTek, USA). One standard enzyme unit was defined as the yield of 1 μmol unsaturated pectin per minute under the above-mentioned conditions.
Alkaline protease assay
The alkaline protease activity was measured using the modified method described by Tjalsma et al. [28]. One standard enzyme unit was defined as the as the amount of enzyme that hydrolyzes 1 μg casein minute under the situation of pH 9.0, 50 °C. 250 μL of the diluted enzyme solution were mixed with 250 μL solution containing 2% casein in pH 9.0 buffer. The mixture was incubated at 50 °C for 10 min, and the reaction was terminated by adding 500 μL 0.4 M TCA. The productions were detected using a spectrophotometer (Epoch 2, BioTek, USA) at 280 nm. Casein with concentrations from 0 to 500 μg/mL were used as standard.
Pullulanase assay
Pullulanase activity was measured using the modified dinitrosalicylic acid (DNS) method [58]. One standard enzyme unit was defined as the amount of enzyme that releases 1 μmol reducing sugar per minute under the assay conditions. Glucose with concentrations from 0 to 400 mg/mL were used as standard. 50 μL of the diluted enzyme solutions were added into the 450 μL reaction mixture containing 5% pullulan solutions and the pH 6.0 buffer at the volume ratio of 1:8. After incubations at pH 6.0, 60 °C for 30 min, 500 μL DNS were used to stop the reactions. The productions were detected using a spectrophotometer (Epoch 2, BioTek, USA) at 540 nm. The amount of reducing sugar released during incubations were tested as the enzyme-index.
SDS-PAGE
B. subtilis WB600 transformant cells harboring recombinant plasmids were cultured in LB medium containing 25 μg/mL kanamycin at 37 °C for 48 h. The cultures (0.5 mL respectively) were centrifuged at 5000 rpm, 4 °C for 10 min and the supernatant solutions were reserved. The cell pellets were re-suspended in 0.5 mL ddH2O, followed by sonication to release intracellular content. Extracellular proteins were precipitated with 13% (w/v) TCA for 30 min on ice, then the precipitates which were collected by 10 min centrifugation at 4 °C and 13,000×g, were carefully washed with ice-cold acetone and dried under vacuum. The collective proteins were dissolved in 20 μL urea (6 M) and mixed with appropriate volume of 4 × SDS loading buffer. Samples were then heated in boiling water bath for 10 min and centrifuged at 13,000×g for 10 s. 12 μL of the samples were loaded onto 10% SDS-PAGE and run at 20 mA for around 1 h. Gel was stained using Coomassie brilliant blue dye.
Supplementary information
{kind=link}
Additional file 1: Figure S1. 10 L fermentations of Spro1 and PulA11. Alkaline protease Spro1 and pullulanase PulA11 were further produced in 10 L fermentator in B. subtilis WB600 with plasmid pUC980-2. The cell growth curves and activities were tested every 6 h. A. During the fermentation of Spro1, cells reached at the stationary phase at 30 h and the highest activity was 21537 U/mL at 60 h. B. the growth curve of the WB600 (pUC980-2-pulA11) was similar to the PelN and Spro1 productions. The cells went into the stationary phase at 30 h and the highest production was 504 U/mL at 48 h.
Additional file 2: Table S1. Strains and plasmids used in this study.
Acknowledgements
Not applicable.
Abbreviations
- SDS-PAGE
Sodium dodecyl sulfate–polyacrylamide gel electrophoresis
- BGI
The Beijing Genomics Institute
- qPCR
The quantitative real-time PCR
Authors’ contributions
XYZ and JYX designed the experiments together. XYZ led the performance of the experiments, analysis of the data and writing of the paper. MT, JZ, WJS and SBY participated in experiments and analysis. HCZ, HS and YHM participated in editing the paper. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Fund of China (Grant 31701534), the Tianjin outstanding talent training program, the Tianjin Science & Technology Planning Project (Grant 14ZCZDSY00157 and 15PTCYSY00020) and Yantai Marine economy innovation development demonstration project (Grant YHCX-SW-L-201703).
Availability of data and materials
We declared that materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
XingYa Zhao, JianYong Xu and Ming Tan contributed equally to this work
Contributor Information
YanHe Ma, Email: ma_yanhe@tib.cas.cn.
HongChen Zheng, Email: zheng_hc@tib.cas.cn.
Hui Song, Email: song_h@tib.cas.cn.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12934-020-1296-5.
References
- 1.Dong H, Zhang D. Current development in genetic engineering strategies of Bacillus species. Microb Cell Fact. 2014;13:63. doi: 10.1186/1475-2859-13-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang K, Su L, Duan X, Liu L, Wu J. High-level extracellular protein production in Bacillus subtilis using an optimized dual-promoter expression system. Microb Cell Fact. 2017;16:32. doi: 10.1186/s12934-017-0649-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ji S, Li W, Rasheed Baloch A, Wang M, Li H, Cao B, Zhang H. Efficient biosynthesis of a Cecropin A-melittin mutant in Bacillus subtilis WB700. Scientific Reports. 2017;7:40587. doi: 10.1038/srep40587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Li W, Zhou X, Lu P. Bottlenecks in the expression and secretion of heterologous proteins in Bacillus subtilis. Res Microbiol. 2004;155:605–610. doi: 10.1016/j.resmic.2004.05.002. [DOI] [PubMed] [Google Scholar]
- 5.Marcus S, Ajay S, Ward OP. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50:1. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- 6.Hallberg ZF, Su Y, Kitto RZ, Ming CH. Engineering and in vivo applications of riboswitches. Annu Rev Biochem. 2017;86:515–539. doi: 10.1146/annurev-biochem-060815-014628. [DOI] [PubMed] [Google Scholar]
- 7.Willenbacher J, Mohr T, Henkel M, Gebhard S, Mascher T, Syldatk C, Hausmann R. Substitution of the native srfA promoter by constitutive P veg in two B. subtilis strains and evaluation of the effect on Surfactin production. J Biotechnol. 2016;224:14–17. doi: 10.1016/j.jbiotec.2016.03.002. [DOI] [PubMed] [Google Scholar]
- 8.Maeda T, Sanchez-Torres V, Wood TK. Hydrogen production by recombinant Escherichia coli strains. Microb Biotechnol. 2012;5:214–225. doi: 10.1111/j.1751-7915.2011.00282.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lu Z, Yang S, Yuan X, Shi Y, Ouyang L, Jiang S, Yi L, Zhang G. CRISPR-assisted multi-dimensional regulation for fine-tuning gene expression in Bacillus subtilis. Nucleic Acids Res. 2019;47:e40. doi: 10.1093/nar/gkz072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Iannelli F, Santagati M, Santoro F, Oggioni MR, Stefani S, Pozzi G. Nucleotide sequence of conjugative prophage Φ1207.3 (formerly Tn1207.3) carrying the mef(A)/msr(D) genes for efflux resistance to macrolides in Streptococcus pyogenes. Front Microbiol. 2014;5:687. doi: 10.3389/fmicb.2014.00687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu H, Xiao T, Chen CH, Li W, Liu SX. Sequence determinants of improved CRISPR sgRNA design. Genome Res. 2015;25:1147–1157. doi: 10.1101/gr.191452.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gros MF, Te Riele H, Ehrlich SD. Rolling circle replication of single-stranded DNA plasmid pC194. EMBO J. 1987;6:3863–3869. doi: 10.1002/j.1460-2075.1987.tb02724.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pluta R, Espinosa M. Antisense and yet sensitive: copy number control of rolling circle-replicating plasmids by small RNAs. WIREs RNA. 2018;9:e1500. doi: 10.1002/wrna.1500. [DOI] [PubMed] [Google Scholar]
- 14.Liu M, Kwong SM, Jensen S, Brzoska A, Firth N. Biology of the staphylococcal conjugative multiresistance plasmid pSK41. Plasmid. 2013;70:42–51. doi: 10.1016/j.plasmid.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 15.Yihan L, Fuping L, Guanqun C, Snyder CL, Jing S, Yu L, Jianling W, Jing X. High-level expression, purification and characterization of a recombinant medium-temperature alpha-amylase from Bacillus subtilis. Biotech Lett. 2010;32:119–124. doi: 10.1007/s10529-009-0112-4. [DOI] [PubMed] [Google Scholar]
- 16.Nguyen HD, Nguyen QA, Ferreira RC, Ferreira LC, Tran LT, Schumann W. Construction of plasmid-based expression vectors for Bacillus subtilis exhibiting full structural stability. Plasmid. 2005;54:241–248. doi: 10.1016/j.plasmid.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 17.Phan TT, Nguyen HD, Schumann W. Novel plasmid-based expression vectors for intra- and extracellular production of recombinant proteins in Bacillus subtilis. Protein Expr Purif. 2006;46:189–195. doi: 10.1016/j.pep.2005.07.005. [DOI] [PubMed] [Google Scholar]
- 18.Wu SC, Wong SL. Development of improved pUB110-based vectors for expression and secretionstudies in Bacillus subtilis. J Biotechnol. 1999;72:185–195. doi: 10.1016/S0168-1656(99)00101-7. [DOI] [PubMed] [Google Scholar]
- 19.Reinhard B, Sui-Lam W, Dieter J. Production of PHA depolymerase A (PhaZ5) from Paucimonas lemoignei in Bacillus subtilis. FEMS Microbiol Lett. 2002;209:237–241. doi: 10.1111/j.1574-6968.2002.tb11137.x. [DOI] [PubMed] [Google Scholar]
- 20.Lin B, Li Z, Zhang H, Wu J, Luo M. Cloning and expression of the γ-polyglutamic acid synthetase genepgs BCA in Bacillus subtilis WB600. Biomed Res Int. 2016;2016:1–7. doi: 10.1155/2016/3073949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sun TY, Shao JX, Gao YY, Chen LM, Gao ZQ, Lin WP, Fu-Ping LU. Establishment of high-efficiency transformation and expression of the gene encoding alkaline protease PB92 in Bacillus subtilis DB104. Jiangsu J Agric Sci. 2009;25:534–537. [Google Scholar]
- 22.Kramer MG, Espinosa M, Misra TK, Khan SA. Characterization of a single-strand origin, ssoU, required for broad host range replication of rolling-circle plasmids. Mol Microbiol. 2010;33:466–475. doi: 10.1046/j.1365-2958.1999.01471.x. [DOI] [PubMed] [Google Scholar]
- 23.Gryczan TJ, Dubnau D. Construction and properties of chimeric plasmids in Bacillus subtilis. Proc Natl Acad Sci USA. 1978;75:1428–1432. doi: 10.1073/pnas.75.3.1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhou Z, Liu Y, Chang Z, Wang H, Leier A, Marquez-Lago TT, Ma Y, Li J, Song J. Structure-based engineering of a pectate lyase with improved specific activity for ramie degumming. Appl Microbiol Biotechnol. 2016;101:1–11. doi: 10.1007/s00253-016-7994-6. [DOI] [PubMed] [Google Scholar]
- 25.van der Laan JC, Gerritse G, Mulleners LJ, van der Hoek RA, Quax WJ. Cloning, characterization, and multiple chromosomal integration of a Bacillus alkaline protease gene. Appl Environ Microbiol. 1991;57:901–909. doi: 10.1128/AEM.57.4.901-909.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu J, Ren F, Huang CH, Zheng Y, Zhen J, Sun H, Ko TP, He M, Chen CC, Chan HC, et al. Functional and structural studies of pullulanase from Anoxybacillus sp. LM18-11. Proteins. 2014;82:1685–1693. doi: 10.1002/prot.24498. [DOI] [PubMed] [Google Scholar]
- 27.Zhou M, Wu J, Wang T, Gao L, Yin H, Lü X. The purification and characterization of a novel alkali-stable pectate lyase produced by Bacillus subtilis PB1. World J Microbiol Biotechnol. 2017;33:190. doi: 10.1007/s11274-017-2357-8. [DOI] [PubMed] [Google Scholar]
- 28.Tjalsma H, Koetje EJ, Kiewiet R, Kuipers OP, Kolkman M, Laan J, Daskin R, Ferrari E, Bron S. Engineering of quorum-sensing systems for improved production of alkaline protease by Bacillus subtilis. J Appl Microbiol. 2004;96:569–578. doi: 10.1111/j.1365-2672.2004.02179.x. [DOI] [PubMed] [Google Scholar]
- 29.Feto NA. Bacillus spp. and their biotechnological roles in green industry. Cham: Springer; 2016. [Google Scholar]
- 30.Liu L, Liu Y, Shin HD, Chen RR, Wang NS, Li J, Du G, Chen J. Developing Bacillus spp. as a cell factory for production of microbial enzymes and industrially important biochemicals in the context of systems and synthetic biology. Appl Microbiol Biotechnol. 2013;97:6113–6127. doi: 10.1007/s00253-013-4960-4. [DOI] [PubMed] [Google Scholar]
- 31.Bron S, Luxen E, Swart P. Instability of recombinant pUB110 plasmids in Bacillus subtilis: plasmid-encoded stability function and effects of DNA inserts. Plasmid. 1988;19:231–241. doi: 10.1016/0147-619X(88)90041-8. [DOI] [PubMed] [Google Scholar]
- 32.Maciag IE, Viret JF, Alonso JC. Replication and incompatibility properties of plasmid pUB110 in Bacillus subtilis. Mol Gen Genet. 1988;212:232–240. doi: 10.1007/BF00334690. [DOI] [PubMed] [Google Scholar]
- 33.Tanaka T, Sueoka N. Site-specific in vitro binding of plasmid pUB110 to Bacillus subtilis membrane fraction. J Bacteriol. 1983;154:1184–1194. doi: 10.1128/JB.154.3.1184-1194.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Viret JF, Alonso JC. A DNA sequence outside the pUB110 minimal replicon is required for normal replication in Bacillus subtilis. Nucleic Acids Res. 1988;16:4389–4406. doi: 10.1093/nar/16.10.4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thomas CD, Jennings LJ. RepD/D*: a protein-DNA adduct arising during plasmid replication. Biochem Soc Trans. 1995;23:442S. doi: 10.1042/bst023442s. [DOI] [PubMed] [Google Scholar]
- 36.Alonso JC, Viret JF, Tailor RH. Plasmid maintenance in Bacillus subtilis recombination-deficient mutants. Mol Genet Genomics. 1987;208:349–352. doi: 10.1007/BF00330464. [DOI] [PubMed] [Google Scholar]
- 37.Noirot-Gros MF, Ehrlich SD. Change of a catalytic reaction carried out by a DNA replication protein. Science. 1996;274:777–780. doi: 10.1126/science.274.5288.777. [DOI] [PubMed] [Google Scholar]
- 38.Alonso JC, Stiege CA, Tailor RH, Viret JF. Functional analysis of the dna (Ts) mutants of Bacillus subtilis: plasmid pUB110 replication as a model system. Mol Gen Genet. 1988;214:482–489. doi: 10.1007/BF00330484. [DOI] [PubMed] [Google Scholar]
- 39.Bron S, Luxen E. Segregational instability of pUB110-derived recombinant plasmids in Bacillus subtilis. Plasmid. 1985;14:235–244. doi: 10.1016/0147-619X(85)90007-1. [DOI] [PubMed] [Google Scholar]
- 40.Wassenaar TM, Cabal A. The mobile dso-gene-sso element in rolling-circle plasmids of staphylococci reflects the evolutionary history of its resistance gene. Lett Appl Microbiol. 2017;65:192–198. doi: 10.1111/lam.12767. [DOI] [PubMed] [Google Scholar]
- 41.Costa A, Hood IV, Berger JM. Mechanisms for initiating cellular DNA replication. Annu Rev Biochem. 2013;82:25–54. doi: 10.1146/annurev-biochem-052610-094414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bleichert F, Botchan MR. Mechanisms for initiating cellular DNA replication. Science. 2017;355:eaah6317. doi: 10.1126/science.aah6317. [DOI] [PubMed] [Google Scholar]
- 43.Li SF, Xu JY, Bao YJ, Zheng HC, Song H. Structure and sequence analysis-based engineering of pullulanase from Anoxybacillus sp. LM18-11 for improved thermostability. J Biotechnol. 2015;210:8–14. doi: 10.1016/j.jbiotec.2015.06.406. [DOI] [PubMed] [Google Scholar]
- 44.Zeng Y, Zheng H, Shen Y, Xu J, Tan M, Liu F, Song H. Identification and analysis of binding residues in the CBM68 of pullulanase PulA from Anoxybacillus sp. LM18-11. J Biosci Bioeng. 2019;127:8–15. doi: 10.1016/j.jbiosc.2018.06.007. [DOI] [PubMed] [Google Scholar]
- 45.Zeng Y, Xu J, Fu X, Tan M, Liu F, Zheng H, Song H. Effects of different carbohydrate-binding modules on the enzymatic properties of pullulanase. Int J Biol Macromol. 2019;137:973–981. doi: 10.1016/j.ijbiomac.2019.07.054. [DOI] [PubMed] [Google Scholar]
- 46.Phan TTP, Tran LT, Schumann W, Nguyen HD. Development of Pgrac100-based expression vectors allowing high protein production levels in Bacillus subtilis and relatively low basal expression in Escherichia coli. Microb Cell Fact. 2015;14:72. doi: 10.1186/s12934-015-0255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zyprian E, Matzura H. Characterization of signals promoting gene expression on the Staphylococcus aureus plasmid pUB110 and development of a gram-positive expression vector system. DNA. 1986;5:219–225. doi: 10.1089/dna.1986.5.219. [DOI] [PubMed] [Google Scholar]
- 48.Nguyen HD, Phan TT, Schumann W. Expression vectors for the rapid purification of recombinant proteins in Bacillus subtilis. Curr Microbiol. 2007;55:89–93. doi: 10.1007/s00284-006-0419-5. [DOI] [PubMed] [Google Scholar]
- 49.Guo S, Tang JJ, Wei DZ, Wei W. Construction of a shuttle vector for protein secretory expression in Bacillus subtilis and the application of the mannanase functional heterologous expression. J Microbiol Biotechnol. 2014;24:431–439. doi: 10.4014/jmb.1311.11009. [DOI] [PubMed] [Google Scholar]
- 50.Guan C, Cui W, Cheng J, Liu R, Liu Z, Zhou L, Zhou Z. Construction of a highly active secretory expression system via an engineered dual promoter and a highly efficient signal peptide in Bacillus subtilis. New Biotechnol. 2016;33:372–379. doi: 10.1016/j.nbt.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 51.Miyazaki K. MEGAWHOP cloning: a method of creating random mutagenesis libraries via megaprimer PCR of whole plasmids. Methods Enzymol. 2011;498:399–406. doi: 10.1016/B978-0-12-385120-8.00017-6. [DOI] [PubMed] [Google Scholar]
- 52.Green MR, Sambrook J. Molecular cloning: a laboratory manual (fourth edition): three-volume set. Cold Spring Harbor Laboratory Press 2012, 1:part1.
- 53.Turgeon N, Laflamme C, Ho J, Duchaine C. Evaluation of the plasmid copy number in B. cereus spores, during germination, bacterial growth and sporulation using real-time PCR. Plasmid. 2008;60:118–124. doi: 10.1016/j.plasmid.2008.05.001. [DOI] [PubMed] [Google Scholar]
- 54.Friehs K. Plasmid copy number and plasmid stability. Adv Biochem Eng Biotechnol. 2004;86:47. doi: 10.1007/b12440. [DOI] [PubMed] [Google Scholar]
- 55.Maidak BL, Larsen N, Mccaughey MJ, Overbeek R, Olsen GJ, Fogel K, Blandy J, Woese CR. The ribosomal database project. Nucleic Acids Res. 1994;22:3485–3487. doi: 10.1093/nar/22.17.3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Providenti MA, O’Brien JM, Ewing RJ, Paterson ES, Smith ML. The copy-number of plasmids and other genetic elements can be determined by SYBR-Green-based quantitative real-time PCR. J Microbiol Methods. 2006;65:476–487. doi: 10.1016/j.mimet.2005.09.007. [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Li X, Ma Y, Song J. Process optimization of high-level extracellular production of alkaline pectate lyase in recombinant Escherichia coli BL21 (DE3) Biochem Eng J. 2015;93:38–46. doi: 10.1016/j.bej.2014.08.020. [DOI] [Google Scholar]
- 58.He XS, Shyu YT, Nathoo S, Wong SL, Doi RH. Construction and use of a Bacillus subtilis mutant deficient in multiple protease genes for the expression of eukaryotic genes. Ann N Y Acad Sci. 2010;646:69–77. doi: 10.1111/j.1749-6632.1991.tb18565.x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Figure S1. 10 L fermentations of Spro1 and PulA11. Alkaline protease Spro1 and pullulanase PulA11 were further produced in 10 L fermentator in B. subtilis WB600 with plasmid pUC980-2. The cell growth curves and activities were tested every 6 h. A. During the fermentation of Spro1, cells reached at the stationary phase at 30 h and the highest activity was 21537 U/mL at 60 h. B. the growth curve of the WB600 (pUC980-2-pulA11) was similar to the PelN and Spro1 productions. The cells went into the stationary phase at 30 h and the highest production was 504 U/mL at 48 h.
Additional file 2: Table S1. Strains and plasmids used in this study.
Data Availability Statement
We declared that materials described in the manuscript, including all relevant raw data, will be freely available to any scientist wishing to use them for non-commercial purposes, without breaching participant confidentiality.