

Abstract
Aims
Chronic noise exposure associates with increased cardiovascular disease (CVD) risk; however, the role of confounders and the underlying mechanism remain incompletely defined. The amygdala, a limbic centre involved in stress perception, participates in the response to noise. Higher amygdalar metabolic activity (AmygA) associates with increased CVD risk through a mechanism involving heightened arterial inflammation (ArtI). Accordingly, in this retrospective study, we tested whether greater noise exposure associates with higher: (i) AmygA, (ii) ArtI, and (iii) risk for major adverse cardiovascular disease events (MACE).
Methods and results
Adults (N = 498) without CVD or active cancer underwent clinical 18F-fluorodeoxyglucose positron emission tomography/computed tomography imaging. Amygdalar metabolic activity and ArtI were measured, and MACE within 5 years was adjudicated. Average 24-h transportation noise and potential confounders were estimated at each individual’s home address. Over a median 4.06 years, 40 individuals experienced MACE. Higher noise exposure (per 5 dBA increase) predicted MACE [hazard ratio (95% confidence interval, CI) 1.341 (1.147–1.567), P < 0.001] and remained robust to multivariable adjustments. Higher noise exposure associated with increased AmygA [standardized β (95% CI) 0.112 (0.051–0.174), P < 0.001] and ArtI [0.045 (0.001–0.090), P = 0.047]. Mediation analysis suggested that higher noise exposure associates with MACE via a serial mechanism involving heightened AmygA and ArtI that accounts for 12–26% of this relationship.
Conclusion
Our findings suggest that noise exposure associates with MACE via a mechanism that begins with increased stress-associated limbic (amygdalar) activity and includes heightened arterial inflammation. This potential neurobiological mechanism linking noise to CVD merits further evaluation in a prospective population.
Keywords: Amygdalar activity, Arterial inflammation, Cardiovascular disease, Chronic noise exposure, 18F-FDG-PET/CT
See page 783 for the editorial comment on this article (doi: 10.1093/eurheartj/ehz867)
Introduction
Transportation noise exposure, an inevitable consequence of urbanization and globalization, represents a growing threat to human health.1 Long-term noise exposure precipitates a chronic stress reaction and has been repeatedly shown in epidemiologic studies to contribute to adverse non-auditory physical effects including cognitive impairment, sleep disturbances, psychiatric disease, metabolic disease, cardiovascular disease (CVD), and increased mortality.2–6 Several studies have shown that the association between noise and CVD is independent of important confounders, including traditional and non-traditional CVD risk factors (e.g. air pollution, lower socioeconomic status) that are more prevalent in noisier environments.7–9 However, limited data exist evaluating the relationship between noise exposure and CVD after comprehensively adjusting for confounders in a single population. Moreover, the mechanisms mediating this relationship remain incompletely understood.
Animal and human studies provide important insights into the mechanisms by which noise exposure potentiates CVD. Noise exposure, especially at night, promotes autonomic imbalance and release of stress hormones with consequent oxidative stress, inflammation, metabolic abnormalities, altered gene expression, and endothelial dysfunction in healthy individuals and in individuals with CVD.1,10–15 However, it remains uncertain how noise, as an external stimulus, triggers these pathobiological mechanisms. Among candidate conduits, the brain represents a likely bridge between noise exposure and extra-neural diseases. The amygdala, a limbic structure, plays a critical role in the brain’s response to environmental stressors, including noise.16,17 Activity within the amygdala correlates with perceived stress and is increased in stress-related disorders.18,19 Further, heightened amygdalar activity associates with several of the pathological effects of noise exposure, including increased inflammation [measured as C-reactive protein (CRP)], non-calcified coronary plaque burden, and insulin resistance.20,2118F-fluorodeoxyglucose positron emission tomography/computed tomography (18F-FDG-PET/CT), an advanced imaging technique, provides a reproducible and stable measure of amygdalar metabolic activity (AmygA).22 Furthermore, 18F-FDG-PET/CT enables simultaneous measurement of extra-neural tissue activity, including arterial inflammation (ArtI), an important precursor to major adverse cardiovascular disease events (MACE).19 Using this multi-tissue imaging approach, we previously demonstrated that heightened AmygA associates with increased MACE risk, via a pathway of ↑AmygA→↑ArtI→↑MACE.19 Given the role of the amygdala in responding to noise, we hypothesized the existence of a biological pathway linking higher noise exposure to CVD, whereby noise exposure drives the front end of the aforementioned pathway by increasing AmygA.
Accordingly, we studied a retrospective cohort of individuals with 18F-FDG-PET/CT imaging to investigate the associations between chronic noise exposure and AmygA as well as ArtI. Further, we assessed the association between noise exposure and MACE after accounting for important confounders, including air pollution, socioeconomic factors, healthcare access, and CVD risk factors. Finally, we tested whether the association between noise and MACE is serially mediated by heightened AmygA and ArtI.
Methods
Study cohort selection
The retrospective cohort (N = 498) was identified from a database of 6088 individuals who underwent clinical 18F-FDG-PET/CT imaging, largely for cancer surveillance or screening, at Massachusetts General Hospital (MGH, Boston, MA, USA) from 2005 to 2008 (Figure 1). The cohort was selected from the 1777 patients with imaging without active cancer (no prior malignancy or remission for ≥ 1 year before imaging and throughout follow-up) or CVD (based on medical records). The pre-defined inclusion criteria are provided in Supplementary material online. A subset of individuals (N = 280) provided adequate 18F-FDG-PET/CT brain images for measurement of AmygA. This study was completed in accordance with the Declaration of Helsinki. The Partners Human Research Committee approved the protocol’s ethics.
Figure 1.

Study cohort.
18F-fluorodeoxyglucose positron emission tomography/computed tomography imaging protocol
Whole-body 18F-FDG-PET/CT imaging was performed with a standard scanner (e.g. Biograph 64 Siemens Healthcare, Erlangen, Germany). The Supplementary material online contains further details.
Measurement of arterial inflammation and coronary calcification
A blinded investigator quantified ArtI on 18F-FDG-PET/CT images. The arterial 18F-FDG signal was measured every 3 mm within the ascending aorta from 1 cm above the aortic annulus to the aortic arch. The maximum 18F-FDG uptake intensity of each slice was measured as a standardized uptake value (SUV). The mean value of all slices was corrected for background venous blood activity from the superior vena cava to obtain a target-to-background ratio.19 Using the computed tomography images, coronary artery calcium (CAC) scores, measures of baseline atherosclerosis, were quantified.19 Additional description is provided in Supplementary material online.
Measurement of stress-associated neural metabolic activity
A second blinded investigator measured amygdalar 18F-FDG uptake by placing circular regions of interest (∼15 mm radii) over the right and left amygdalae and measuring the mean tracer accumulation, as an SUV. Amygdalar metabolic activity was defined as the average of the mean SUV of bilateral amygdalar activity corrected for mean temporal lobe activity.19 The Supplementary material online contains further discussion.
Characterization of noise exposure, air pollution, socioeconomic factors, and cardiovascular disease risk factors
Residential addresses and zip codes were obtained from medical records. Using the U.S. Department of Transportation’s Road and Aviation Noise Map, addresses were entered to identify average 24-h equivalent sound levels. These levels approximate average traffic and aircraft noise over 24-h using advanced modelling in A-weighted decibels (dBA) to adjust instrument-measured sound to reflect the relative loudness perceived by the human ear (less sensitive to low-frequency sound). Data for 2014 were provided in 5 dBA increments from <35 to >70 dBA. Noise exposure was analysed: (i) in 5 dBA increments and dichotomized as ≤ or >, (ii) 45 dBA (upper tertile), (iii) 50 dBA (upper quartile), and (iv) 55 dBA [level determined to contribute to CVD by the World Health Organization (WHO)].23 Health insurance status and CVD risk factors were derived from the medical record. Additional information, including descriptions of the derivations of socioeconomic and air pollution data, is provided in Supplementary material online.
Adjudication of major adverse cardiovascular disease events
Two blinded cardiologists adjudicated MACE within 5 years of imaging from clinical records (Supplementary material online). Major adverse cardiovascular disease events were defined as: CVD death, myocardial infarction, unstable angina, cerebrovascular accident, heart failure, and coronary or peripheral artery revascularization.19
Statistical analyses
Statistical analyses were performed using SPSS (Version 25, IBM Corporation, Armonk, NY, USA). Continuous variables are given as mean and standard deviation or, when skewed, as median and interquartile range (IQR). Linear regression was used to test for associations, as β with 95% confidence intervals (CIs), between noise exposure and tissue activities. Cox models and Kaplan–Meier log-rank tests were employed to evaluate hazard ratios (HRs) and event-free survival, respectively. All multivariable analyses incorporated age and sex. Additionally, clinically relevant factors and potential confounders including traditional CVD risk factors, baseline CAC score, healthcare access factors, socioeconomic factors, and air pollution were selected a priori for use in multivariable models. Additional analyses were performed to evaluate noise’s impact in subgroups with lower baseline CVD risk (e.g. no CAC). Major adverse cardiovascular disease event-free survival was also evaluated in subjects stratified by noise exposure and AmygA as well as ArtI. Backwards selection was used where appropriate. Statistical significance was determined as a two-sided P-value <0.05.
Mediation (path) analysis was performed using the SPSS PROCESS macro (IBM Corporation, Armonk, NY, USA).19 We estimated the effect of a hypothesized serial, dual mediator path: ↑noise exposure→↑AmygA→↑ArtI→↑MACE. The Supplementary material online contains additional details.
Results
Study population
Baseline characteristics are summarized in Table 1. The population had a median (IQR) age of 55 (45–66) years and was 42.0% male. Several CVD risk factors predicted MACE. Those with MACE were less likely to have had prior malignancy or malignancy treatment, had lower socioeconomic status, and had greater noise and air pollution exposure.
Table 1.
Baseline characteristics
| Variables | Overall (N = 498) | No MACE (N = 458) | MACE (N = 40) | Cox HR (95% CI) | Cox P-value |
|---|---|---|---|---|---|
| Demographics | |||||
| Age (years), median (IQR) | 55 (45–66) | 54 (44–65) | 67.5 (61–78) | 1.085 (1.056–1.115)a | <0.001 |
| Male sex | 209 (42.0%) | 193 (42.1%) | 16 (40.0%) | 0.940 (0.499–1.769) | 0.847 |
| White race | 451 (90.6%) | 415 (90.6%) | 36 (90.0%) | 0.944 (0.336–2.652) | 0.913 |
| Cardiovascular risk factors | |||||
| Current smoker | 53 (10.7%) | 42 (9.3%) | 11 (27.5%) | 3.141 (1.568–6.293) | 0.001 |
| Hypertension | 174 (35.0%) | 149 (32.6%) | 25 (62.5%) | 3.217 (1.696–6.102) | <0.001 |
| Diabetes mellitus | 44 (8.9%) | 36 (7.9%) | 8 (20.0%) | 2.525 (1.163–5.482) | 0.019 |
| Hyperlipidaemia | 139 (28.0%) | 123 (26.9%) | 16 (40%) | 1.780 (0.946–3.352) | 0.074 |
| Mean total cholesterol (SD) (mg/dL) | 190.96 (42.41) | 191.79 (42.90) | 186.11 (39.58) | 0.997 (0.989–1.005)a | 0.467 |
| LDL (mg/dL), mean (SD) | 110.20 (36.96) | 110.58 (37.49) | 108.03 (34.193) | 0.999 (0.990–1.008)a | 0.786 |
| Statin therapy | 99 (19.9%) | 83 (18.2%) | 16 (40.0%) | 2.799 (1.487–5.270) | 0.001 |
| Framingham risk score, median (IQR) | 3.00 (1.00–8.00) | 2.00 (1.00–6.00) | 8.00 (3.00–13.50) | 1.095 (1.051–1.142)a | <0.001 |
| Body mass index (kg/m2), median (IQR) | 26.34 (23.41–30.91) | 26.30 (23.29–30.88) | 27.02 (24.13–31.35) | 1.021 (0.959–1.087) | 0.514 |
| Coronary artery calcium score | |||||
| 0–10 | 319 (73.5%) | 306 (75.7%) | 13 (43.3%) | 1.990 (1.332–2.972) | 0.001 |
| 11–99 | 59 (13.6%) | 50 (12.4%) | 9 (30.0%) | ||
| ≥100 | 56 (12.9%) | 48 (11.9%) | 8 (26.7%) | ||
| Malignancy history | |||||
| Previous cancer | 420 (84.3%) | 396 (86.5%) | 24 (60.0%) | 0.279 (0.148–0.526) | <0.001 |
| Previous chemotherapy/radiation | 378 (75.9%) | 360 (78.6%) | 18 (45%) | 0.255 (0.137–0.476) | <0.001 |
| Psychiatric history | |||||
| History of depression/anxiety | 29 (10.5%) | 27 (10.3%) | 2 (12.5%) | 1.192 (0.271–5.246) | 0.816 |
| Antidepressant therapy | 27 (9.7%) | 25 (9.6%) | 2 (12.5%) | 1.237 (0.281–5.445) | 0.778 |
| Socioeconomic and environmental factors | |||||
| Average noise exposure (dBA), median (IQR) | 35–40 (<35–55) | 35–40 (<35–55) | 52.5–57.5 (35–65) | 1.396 (1.203–1.620)a | <0.001 |
| Noise exposure >45 dBA (upper tertile) | 170 (34.1%) | 145 (31.7%) | 25 (62.5%) | 3.368 (1.776–6.389) | <0.001 |
| Noise exposure >50 dBA (upper quartile) | 118 (23.7%) | 98 (21.4%) | 20 (50%) | 3.499 (1.881–6.506) | <0.001 |
| Noise exposure >55 dBA (WHO cut-off) | 63 (12.7%) | 49 (10.7%) | 14 (35%) | 4.175 (2.178–8.004) | <0.001 |
| Mean air pollution, particulate matter ≤2.5 μm, μg/m3, median (IQR) | 5.80 (4.60–7.60) | 5.30 (4.60–7.60) | 6.70 (5.30–8.80) | 1.346 (1.129–1.605)a | 0.001 |
| Income ($), median (IQR) | 78 420 (61 619–100 162) | 79 540 (61 828–100 301) | 76 527 (50 923–87 381) | 0.985 (0.973–0.998)a,b | 0.022 |
| High school graduation rate (%), median (IQR) | 94.05 (90.00–96.50) | 94.20 (90.10–96.60) | 91.50 (82.20–94.625) | 0.949 (0.920–0.979)a | 0.001 |
| Total annual crimes, median (IQR) | 718 (303–1907) | 712.5 (281–1907) | 1327 (369–1990) | 1.028 (0.990–1.067)a,b | 0.070 |
| Health insurance | 435 (87.3%) | 409 (89.3%) | 26 (65.0%) | 0.174 (0.090–0.336) | <0.001 |
| In-state residence | 437 (87.8%) | 399 (87.1%) | 38 (95.0%) | 2.694 (0.650–11.169) | 0.172 |
Data are presented as N (%) unless specified. Bold faced values indicate P-value < 0.05.
Continuous predictor.
HR per 1000 units.
We evaluated associations between noise exposure (per 5 dBA) and CVD risk factors as well as socioeconomic and environmental factors (Supplementary material online, Table S1). Higher noise exposure associated with a higher incidence of hypertension, lower socioeconomic status, and greater air pollution.
Noise exposure associates with amygdalar activity and arterial inflammation
In univariable and almost all multivariable models, including those evaluating quadratic age effects, higher noise exposure associated with heightened AmygA (Table 2, Supplementary material online, Tables S2 and S3). The distribution of AmygA across noise exposure quartiles is shown (Figure 2A, linear trend P = 0.001). Furthermore, we observed a positive association between increasing noise exposure (per 5 dBA) and ArtI (P = 0.047) in a model adjusted for age and sex. However, the relationship between noise and ArtI was attenuated in fully adjusted models.
Table 2.
Associations between noise exposure and tissue activities
| Noise exposure | Tissue activity | Unadjusted standardized β (95% CI) | P-value | Adjusted standardized βa (95% CI) | P-value |
|---|---|---|---|---|---|
| Per 5 dBA | Amygdalar activity | 0.122 (0.060 to 0.184) | <0.001 | 0.112 (0.051 to 0.174) | <0.001 |
| Arterial inflammation | 0.039 (−0.007 to 0.085) | 0.097 | 0.045 (0.001 to 0.090) | 0.047 | |
| Venous blood (control) | 0.007 (−0.039 to 0.053) | 0.774 | 0 (−0.045 to 0.046) | 0.989 | |
| >45 dBA (upper tertile) vs. others | Amygdalar activity | 0.572 (0.325 to 0.820) | <0.001 | 0.535 (0.289 to 0.781) | <0.001 |
| Arterial inflammation | 0.145 (−0.042 to 0.332) | 0.127 | 0.164 (−0.017 to 0.344) | 0.075 | |
| Venous blood (control) | 0.002 (−0.184 to 0.188) | 0.983 | −0.027 (−0.212 to 0.158) | 0.777 | |
| >50 dBA (upper quartile) vs. others | Amygdalar activity | 0.475 (0.197 to 0.754) | 0.001 | 0.418 (0.140 to 0.697) | 0.003 |
| Arterial inflammation | 0.124 (−0.084 to 0.333) | 0.242 | 0.140 (−0.061 to 0.341) | 0.173 | |
| Venous blood (control) | 0.044 (−0.163 to 0.252) | 0.675 | 0.015 (−0.192 to 0.221) | 0.889 | |
| >55 dBA (WHO cut-off) vs. others | Amygdalar activity | 0.467 (0.096 to 0.838) | 0.014 | 0.411 (0.042 to 0.779) | 0.029 |
| Arterial inflammation | 0.174 (−0.092 to 0.440) | 0.200 | 0.230 (−0.027 to 0.486) | 0.079 | |
| Venous blood (control) | 0.113 (−0.152 to 0.378) | 0.401 | 0.072 (−0.191 to 0.335) | 0.540 |
Bold faced values indicate P-value < 0.05.
Amygdalar activity: range 0.42–1.74, SD 0.124.
Arterial inflammation: range 1.0–3.3, SD 0.285.
Venous background: range 0.41–2.96 MBq/mL, SD 0.225 MBq/mL.
Adjusted for age and sex.
Figure 2.
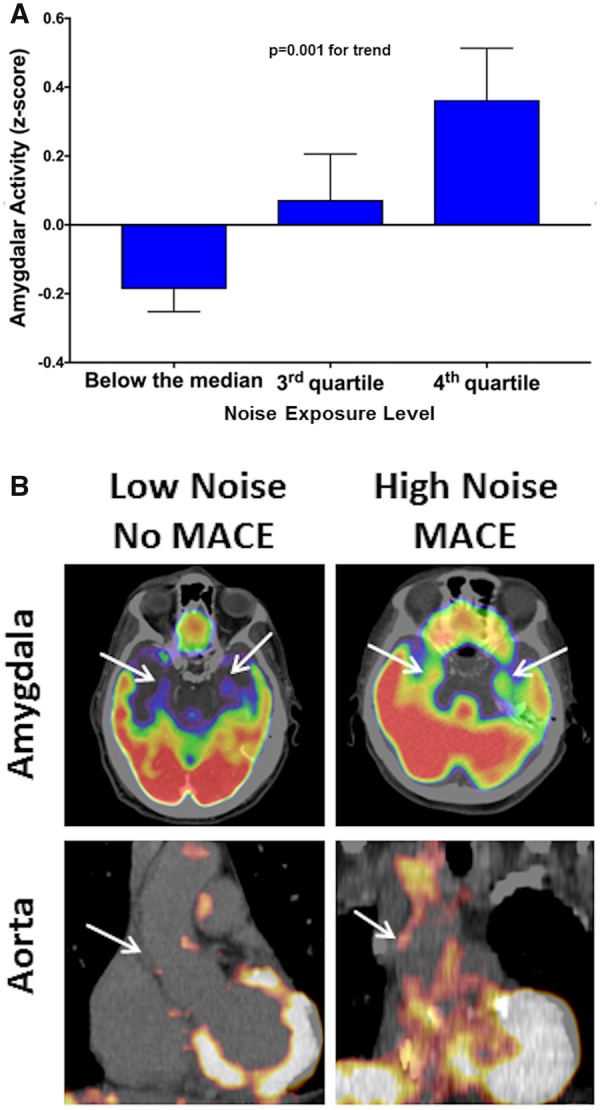
Noise exposure and tissue 18F-fluorodeoxyglucose uptake. (A) Amygdalar metabolic activity by noise exposure (quartiles), adjusted for age and sex. Error bars represent standard error of the mean. (B) Amygdalar and arterial 18F-fluorodeoxyglucose uptake (arrows) from subjects with and without heightened noise exposure (>45 dBA vs. less) and subsequent major adverse cardiovascular disease events. Amygdalar (upper) and aortic (lower) 18F-fluorodeoxyglucose uptake were increased in a patient with increased noise exposure with subsequent major adverse cardiovascular disease event (right) vs. an individual with lower noise exposure without major adverse cardiovascular disease event (left).
Higher noise exposure predicts major adverse cardiovascular disease events and reduced major adverse cardiovascular disease event-free survival
Over a median (IQR) follow-up of 4.06 (2.96-5.00) years, 40 (8.0%) individuals developed MACE. There were 18 myocardial infarctions, 10 unstable angina presentations, 8 cerebrovascular accidents, and 4 peripheral artery disease revascularizations.
An unadjusted histogram of MACE by noise exposure is shown in Supplementary material online, Figure S1. Individuals exposed to higher noise had greater risk of MACE [HR per 5 dBA increase (95% CI) 1.396 (1.203–1.620), P < 0.001]. A similarly heightened risk was seen with the pre-specified noise thresholds (Tables 1 and 3). In separate analyses, MACE-free survival was lower among individuals with greater noise exposure by all thresholds (log-rank P < 0.001 for all, Figures 2B and 3A–C). Moreover, noise exposure remained associated with MACE after accounting for confounders, including CVD risk factors, air pollution, socioeconomic factors, and healthcare access (Table 3).
Table 3.
Cox-proportional hazard ratios for noise vs. MACE risk
| Covariables | per 5 dBA |
>45 dBA (upper tertile) vs. others |
>50 dBA (upper quartile) vs. others |
>55 dBA (WHO cut-off) vs. others |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| HR (95% CI) | P-value | C-index | HR (95% CI) | P-value | C-index | HR (95% CI) | P-value | C-index | HR (95% CI) | P-value | C-index | |
| Age and sex | 1.341 (1.147–1.567) | <0.001 | 0.804 | 2.556 (1.336–4.890) | 0.005 | 0.795 | 2.872 (1.540–5.356) | 0.001 | 0.791 | 3.477 (1.794–6.741) | <0.001 | 0.796 |
| Framingham risk scorea (N = 252) | 1.269 (1.074–1.498) | 0.005 | 0.794 | 2.285 (1.131–4.616) | 0.021 | 0.791 | 2.366 (1.231–4.548) | 0.010 | 0.783 | 3.151 (1.593–6.231) | 0.001 | 0.790 |
| CVD risk factorsa,b (N = 494) | 1.289 (1.095–1.519) | 0.002 | 0.835 | 2.149 (1.115–4.143) | 0.022 | 0.823 | 2.493 (1.295–4.797) | 0.006 | 0.821 | 3.130 (1.544–6.347) | 0.002 | 0.831 |
| Coronary artery calcium scorea (N = 434) | 1.290 (1.077–1.545) | 0.006 | 0.800 | 2.383 (1.138–4.990) | 0.021 | 0.793 | 2.412 (1.170–4.974) | 0.017 | 0.786 | 3.636 (1.678–7.881) | 0.001 | 0.799 |
| Previous cancer and chemotherapy/radiationa (N = 473) | 1.297 (1.106–1.522) | 0.001 | 0.824 | 2.170 (1.122–4.197) | 0.021 | 0.818 | 2.549 (1.357–4.788) | 0.004 | 0.813 | 3.231 (1.634–6.389) | 0.001 | 0.820 |
| Air pollutiona (N = 473) | 1.313 (1.101–1.565) | 0.002 | 0.821 | 2.104 (1.011–4.377) | 0.047 | 0.815 | 2.476 (1.276–4.802) | 0.007 | 0.811 | 3.065 (1.532–6.133) | 0.002 | 0.818 |
| Socioeconomic factorsa,c (N = 370) | 1.283 (1.069–1.540) | 0.008 | 0.802 | 2.154 (0.994–4.670) | 0.052 | 0.796 | 2.588 (1.235–5.421) | 0.012 | 0.795 | 3.036 (1.423–6.477) | 0.004 | 0.801 |
| Statin therapya (N = 497) | 1.330 (1.135–1.557) | <0.001 | 0.811 | 2.437 (1.271–4.673) | 0.007 | 0.803 | 2.724 (1.452–5.111) | 0.002 | 0.798 | 3.325 (1.708–6.473) | <0.001 | 0.802 |
| Healthcare access factorsa,d (N = 498) | 1.292 (1.106–1.509) | 0.001 | 0.837 | 2.464 (1.280–4.743) | 0.007 | 0.824 | 2.547 (1.346–4.821) | 0.004 | 0.821 | 2.684 (1.343–5.364) | 0.005 | 0.824 |
| Fully adjusted modela,e (N = 470) | 1.326 (1.129–1.558) | 0.001 | 0.862 | 2.278 (1.158–4.482) | 0.017 | 0.778 | 2.776 (1.451–5.313) | 0.002 | 0.855 | 2.639 (1.298–5.365) | 0.007 | 0.863 |
Bold faced values indicate P-value < 0.05.
Adjusted for age and sex.
CVD risk factors = diabetes, hypertension, hyperlipidaemia, and current smoking.
Socioeconomic factors = median income, percent high school graduates, and town total crimes.
Healthcare access factors = Massachusetts state address and health insurance status.
Fully adjusted = CVD risk factors, statin use, neighbourhood pollution and income, previous cancer and chemotherapy/radiation, and healthcare access factors.
Figure 3.
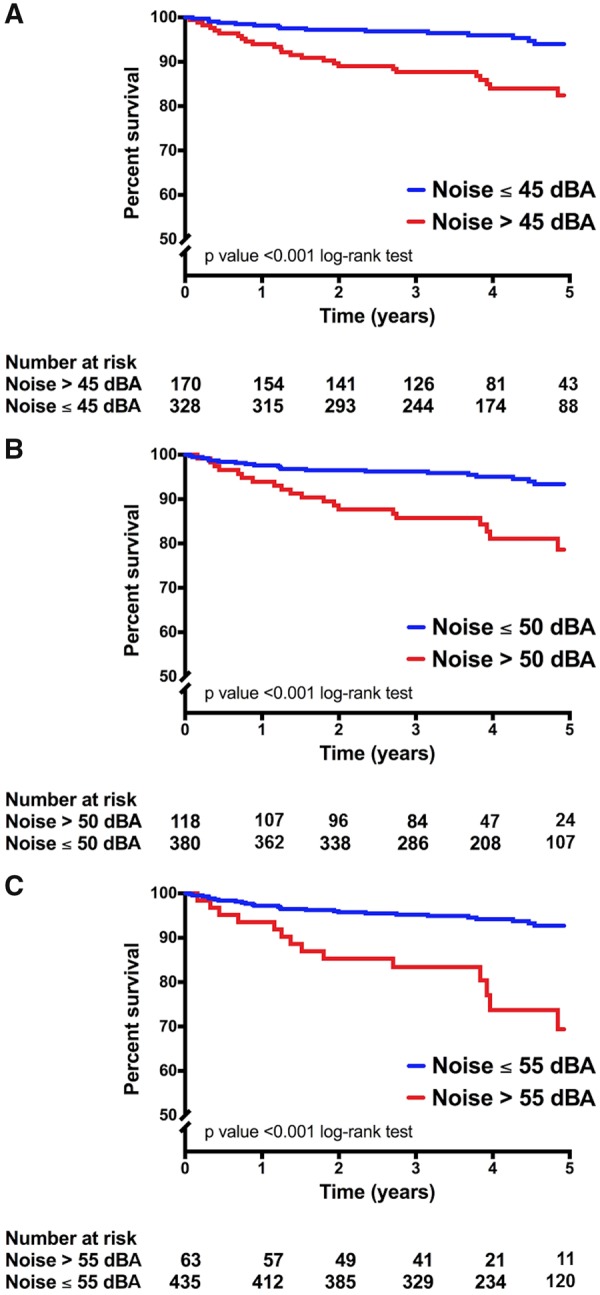
Major adverse cardiovascular disease event-free survival by noise exposure. Major adverse cardiovascular disease event-free survival for individuals with noise exposure ≤ (blue) vs. > (red): (A) 45 dBA (upper tertile), (B) 50 dBA (upper quartile), and (C) 55 dBA (WHO cut-off). Log-rank P-values are shown.
There were no significant two-way interactions between noise exposure and covariables often found in noisier locations with MACE (Supplementary material online, Table S4). In the event that an interaction was not detected due to limited power or a threshold effect, we conducted subgroup analyses to more thoroughly evaluate these relationships after excluding individuals with socioeconomic and environmental variables often found in noisier locations that could indirectly account for the described associations. In these analyses (Supplementary material online, Table S5), noise exposure remained associated with MACE among individuals with: greater healthcare access (i.e. those with insurance or in-state address), higher socioeconomic status (i.e. those in the upper two tertiles of neighbourhood income), and less neighbourhood air pollution (i.e. those in the lower two tertiles of pollution). Furthermore, since CVD risk factors may be more prevalent in noisier neighbourhoods, we assessed the relationship between noise exposure and MACE among those without baseline subclinical atherosclerosis (i.e. CAC). In that analysis that also adjusted for CVD risk factors, noise exposure remained associated with MACE.
Tissue activities predict major adverse cardiovascular disease events among individuals exposed to higher noise
Both AmygA and ArtI independently associate with MACE.17 Because higher noise exposure associates with increased AmygA and ArtI, we conducted stratified subgroup analyses to evaluate whether AmygA and ArtI associate with MACE among individuals exposed to higher noise (>50 dBA). Among this subgroup, AmygA remained predictive of MACE [standardized HR (95% CI) 1.599 (1.206–2.120), Cox P = 0.001, log-rank P = 0.044, Supplementary material online, Figure S2]. Similarly, ArtI remained predictive of MACE [1.556 (1.051–2.303), Cox P = 0.027, log-rank P < 0.001, Supplementary material online, Figure S3].
The relationship between noise exposure and major adverse cardiovascular disease events may be mediated by amygdalar activity and arterial inflammation
We performed mediation analysis to estimate the effect of the hypothesized path of: ↑noise exposure→↑AmygA→↑ArtI→↑MACE. In a model adjusted for CVD risk factors, the pre-specified path was significant [standardized log odds ratio (95% CI) 0.11 (0.02–0.30), Figure 4, >45 dBA]. It generally remained significant for all pre-defined measures of noise exposure and with multivariable adjustments (Supplementary material online, Table S6) with the indirect path accounting for 12–26% of the relationship between noise exposure and MACE.
Figure 4.
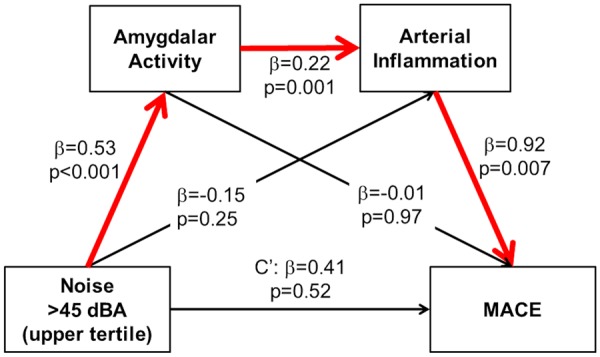
Mediation analysis for the hypothesized pathway from noise exposure to major adverse cardiovascular disease events. Mediation analysis estimated the effect of a serial two-mediator path: ↑noise exposure (>45 dBA)→↑AmygA→↑ArtI→↑MACE. The proposed indirect path (red arrows) was significant [standardized log odds ratio (95% confidence interval) 0.11 (0.02–0.30)], suggesting that amygdalar metabolic activity and arterial inflammation serially mediate the association between increased noise exposure and major adverse cardiovascular disease events (accounting for ∼26% of the total effect) after adjustment for age, sex, and cardiovascular disease risk factors. c’, residual direct effect of noise on major adverse cardiovascular disease events (independent of mediated effects).
Discussion
Chronic exposure to heightened transportation noise associates with an increased incidence of CVD. Until now, basic gaps remained in our understanding of the underlying mechanisms, including how noise initially triggers a physiologic response and results in physical disease. The current study implemented 18F-FDG-PET/CT imaging of the brain and arteries to provide new insights. We observed that higher noise exposure associates with heightened amygdalar metabolic activity and increased atherosclerotic inflammation. Further, we observed that higher noise exposure associates with an increased risk of MACE after adjustments for important confounders including CVD risk factors, baseline subclinical atherosclerosis, socioeconomic status, air pollution, and healthcare access. Moreover, we observed that the link between noise and MACE may be mediated in part by a multi-organ pathway that begins with up-regulated amygdalar activity and involves heightened arterial inflammation.
The independent relationship between noise exposure and cardiovascular disease
According to the WHO, 45 000 life-years are lost annually to noise-related CVD in Western Europe, and the Global Burden of Disease Study acknowledges that occupational noise exposure increases the risk for developing disease.24,25 Several studies have shown that the risk of MACE is dose-dependent above 50–60 dB.5,25 Other studies have shown that the association between noise and CVD is independent of CVD risk factors, socioeconomic factors, behavioural factors, and air pollution.7–9 The current study demonstrates a greater risk for CVD consequent to noise than previously reported and adds to prior observations by accounting for important confounders to confirm an independent relationship between noise exposure and MACE. Moreover, subgroup analyses showed that increased noise exposure was a significant risk factor for MACE even in individuals with seemingly lower clinical CVD risk (e.g. those without baseline CAC). In other words, chronic noise exposure is an independent and clinically under-recognized CVD risk factor and may have an important effect even in individuals otherwise presumed to have low CVD risk.
Mechanistic insights
Although much of the underlying biology linking noise to CVD is well described, our findings may shed further light on a multiorgan mechanism linking noise exposure to CVD. This mechanism likely begins in the limbic system of the brain and involves the amygdala. The amygdala is intimately linked to the response to bothersome auditory stimuli,16,17 and noise exposure increases amygdalar blood flow.16 Noise disrupts the circadian rhythm and leads to oxidative stress and inflammation within neural tissues with subsequent activation of the hypothalamic–pituitary–adrenal (HPA) axis and sympathetic nervous system (SNS).12–14,17 These changes activate haematopoietic tissues and trigger altered gene expression and increased oxidative stress, endothelial dysfunction, and inflammation that lead to increased ArtI, non-calcified coronary plaque burden, and CRP.14,19,21 Hence, prior studies provide a framework that supports the amygdala as a potential conduit linking noise exposure to CVD.
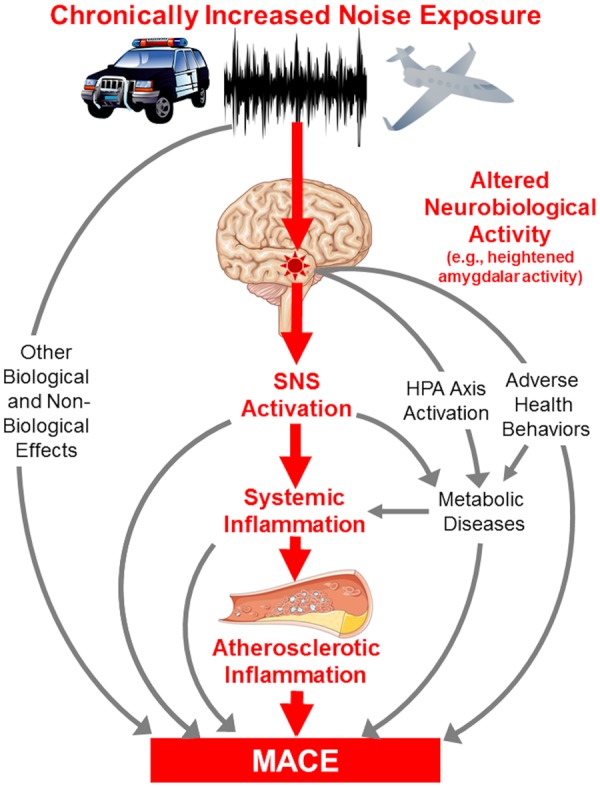
Herein, we provide evidence of such a pathway in humans. We show for the first time that increased noise exposure associates with higher AmygA, ArtI, and MACE risk. Moreover, mediation analysis suggests that the association between noise and CVD involves a pathway of: ↑AmygA →↑ArtI→↑MACE. Additional mechanisms undoubtedly contribute to noise-associated CVD. Among them, altered health behaviours, metabolic impairment, and sleep disruption may also be promoted by altered neural activity involving the amygdala.12,20,21 Furthermore, noise exposure associates with other pathological mechanisms that could also be impacted by altered neural activity and activation of the SNS, HPA axis, and renin–angiotensin–aldosterone system, including endothelial dysfunction, oxidative stress, and inflammation.1,15,21 Accordingly, the current findings suggest an important role for the brain, and particularly the amygdala, in the link between noise and CVD (Take home figure).
Take home figure.
Hypothesized mechanism linking noise exposure to major adverse cardiovascular disease events through up-regulated amygdalar metabolic activity and arterial inflammation.
Future directions
Our findings identify several targets that could potentially be modulated to interrupt the pathway linking noise to CVD. New technologies and regulations to reduce transportation noise may provide benefits. Better urban planning and sound insulation also merit consideration. Additionally, the impact of individualized noise reduction techniques (e.g. earplugs) could be evaluated. Beyond these modifications, this study’s mechanistic insights into the noise-induced activation of the neural–arterial axis suggest other potential therapeutic targets. Because we observed that those with lower AmygA or ArtI in the presence of increased noise exposure were relatively protected from MACE, both AmygA and ArtI may also represent targets to mitigate noise-associated CVD.
Limitations
The current study was a single-centre retrospective cohort study of select individuals without baseline CVD or active cancer referred for clinical 18F-FDG-PET/CT. Most (84.3%) had a history of prior malignancy. Further, we excluded patients with systemic inflammatory diseases. The retrospective nature and inclusion criteria may limit our findings’ generalizability to a subset of individuals exposed to transportation noise. However, a similar relationship between AmygA and atherosclerosis was shown in patients with psoriasis, suggesting that these results may also be relevant in chronic inflammatory conditions.21 Health behaviour data, direct measures of SNS and HPA axis activity, and classical measures of inflammation and oxidative stress were unavailable. Noise exposure was quantified as a 24-h average without nocturnal weighting at each individual’s home; therefore, the impact of exposure in other locations was not measured. Additionally, noise data were available in 5 dBA increments (starting at 35 dBA) from 2014, potentially providing sources of misclassification. Socioeconomic factors (e.g. neighbourhood income) were evaluated at the zip code rather than individual level, follow-up was variable and uncontrolled, and we could not account for the potential effect of relocation, all of which could have influenced our findings. Due to the limitations of clinical whole-body 18F-FDG-PET/CT imaging, we could not evaluate all brain regions involved in stress perception. Finally, this retrospective study cannot establish a causal relationship between noise and AmygA, ArtI, and CVD risk. It is possible that persons with higher AmygA, ArtI, and/or CVD risk, or genetic predispositions to these tend to move into noisier neighbourhoods. However, this interpretation is unlikely given the persistence of the findings after adjustment for socioeconomic factors. Furthermore, animal work, in which experimentally varied noise was associated with deleterious effects, supports a causal inference.11,12 Interventional studies are needed to demonstrate a causal role for the hypothesized mediators. Nonetheless, these limitations are outweighed by several innovations that provide novel insights into the biological mechanisms linking noise to CVD.
Conclusion
Transportation noise exposure associates with increased risk for CVD events after comprehensively accounting for confounders, including CVD risk factors, baseline subclinical atherosclerosis, socioeconomic factors, healthcare access, and air pollution. Moreover, the link between noise exposure and CVD may be partially mediated by a mechanism that begins with up-regulated stress-associated neurobiological activity and involves heightened arterial inflammation. Although a prospective study is needed to confirm the generalizability of these results, the findings point to a potentially important biologic pathway that could be targeted to reduce CVD associated with noise exposure.
Funding
The authors were partially supported by the U.S. National Institutes of Health (T32HL076136 and KL2TR002542 to M.T.O. and P01HL131478 to Z.A.F. and A.T.) and American Heart Association (18CDA34110366 to M.T.O.).
Conflict of interest: A.T. received institutional grants from Genentech and Actelion and personal fees from Actelion for unrelated research. All the remaining authors have no conflict of interest.
Supplementary Material
References
- 1. Munzel T, Schmidt FP, Steven S, Herzog J, Daiber A, Sorensen M.. Environmental noise and the cardiovascular system. J Am Coll Cardiol 2018;71:688–697. [DOI] [PubMed] [Google Scholar]
- 2. Clark C, Paunovic K.. WHO environmental noise guidelines for the European Region: a systematic review on environmental noise and quality of life, wellbeing and mental health. Int J Environ Res Public Health 2018;15:2400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Kempen EV, Casas M, Pershagen G, Foraster M.. WHO environmental noise guidelines for the European Region: a systematic review on environmental noise and cardiovascular and metabolic effects: a summary. Int J Environ Res Public Health 2018;15:379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Basner M, Babisch W, Davis A, Brink M, Clark C, Janssen S, Stansfeld S.. Auditory and non-auditory effects of noise on health. Lancet 2014;383:1325–1332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Halonen JI, Hansell AL, Gulliver J, Morley D, Blangiardo M, Fecht D, Toledano MB, Beevers SD, Anderson HR, Kelly FJ, Tonne C.. Road traffic noise is associated with increased cardiovascular morbidity and mortality and all-cause mortality in London. Eur Heart J 2015;36:2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Stansfeld SA, Haines MM, Burr M, Berry B, Lercher P.. A review of environmental noise and mental health. Noise Health 2000;2:1–8. [PubMed] [Google Scholar]
- 7. Roswall N, Raaschou-Nielsen O, Ketzel M, Gammelmark A, Overvad K, Olsen A, Sørensen M.. Long-term residential road traffic noise and NO2 exposure in relation to risk of incident myocardial infarction—a Danish cohort study. Environ Res 2017;156:80–86. [DOI] [PubMed] [Google Scholar]
- 8. Selander J, Nilsson ME, Bluhm G, Rosenlund M, Lindqvist M, Nise G, Pershagen G.. Long-term exposure to road traffic noise and myocardial infarction. Epidemiology 2009;20:272–279. [DOI] [PubMed] [Google Scholar]
- 9. Hansell AL, Blangiardo M, Fortunato L, Floud S, de Hoogh K, Fecht D, Ghosh RE, Laszlo HE, Pearson C, Beale L, Beevers S, Gulliver J, Best N, Richardson S, Elliott P.. Aircraft noise and cardiovascular disease near Heathrow airport in London: small area study. BMJ 2013;347:f5432.. [DOI] [PubMed] [Google Scholar]
- 10. Schmidt F, Kolle K, Kreuder K, Schnorbus B, Wild P, Hechtner M, Binder H, Gori T, Munzel T.. Nighttime aircraft noise impairs endothelial function and increases blood pressure in patients with or at high risk for coronary artery disease. Clin Res Cardiol 2015;104:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Munzel T, Daiber A, Steven S, Tran LP, Ullmann E, Kossmann S, Schmidt FP, Oelze M, Xia N, Li H, Pinto A, Wild P, Pies K, Schmidt ER, Rapp S, Kroller-Schon S.. Effects of noise on vascular function, oxidative stress, and inflammation: mechanistic insight from studies in mice. Eur Heart J 2017;38:2838–2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kroller-Schon S, Daiber A, Steven S, Oelze M, Frenis K, Kalinovic S, Heimann A, Schmidt FP, Pinto A, Kvandova M, Vujacic-Mirski K, Filippou K, Dudek M, Bosmann M, Klein M, Bopp T, Hahad O, Wild PS, Frauenknecht K, Methner A, Schmidt ER, Rapp S, Mollnau H, Munzel T.. Crucial role for Nox2 and sleep deprivation in aircraft noise-induced vascular and cerebral oxidative stress, inflammation, and gene regulation. Eur Heart J 2018;39:3528–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Schmidt FP, Basner M, Kroger G, Weck S, Schnorbus B, Muttray A, Sariyar M, Binder H, Gori T, Warnholtz A, Munzel T.. Effect of nighttime aircraft noise exposure on endothelial function and stress hormone release in healthy adults. Eur Heart J 2013;34:3508–3514a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Daiber A, Kröller‐Schön S, Frenis K, Oelze M, Kalinovic S, Vujacic‐Mirski K, Kuntic M, Bayo Jimenez MT, Helmstädter J, Steven S, Korac B, Münzel T.. Environmental noise induces the release of stress hormones and inflammatory signaling molecules leading to oxidative stress and vascular dysfunction-Signatures of the internal exposome. Biofactors 2019;45:495–506. [DOI] [PubMed] [Google Scholar]
- 15. Babisch W. Updated exposure-response relationship between road traffic noise and coronary heart diseases: a meta-analysis. Noise Health 2014;16:1–9. [DOI] [PubMed] [Google Scholar]
- 16. Zald DH, Pardo JV.. The neural correlates of aversive auditory stimulation. Neuroimage 2002;16:746–753. [DOI] [PubMed] [Google Scholar]
- 17. Spreng M. Central nervous system activation by noise. Noise Health 2000;2:49–58. [PubMed] [Google Scholar]
- 18. Zhu Y, Du R, Zhu Y, Shen Y, Zhang K, Chen Y, Song F, Wu S, Zhang H, Tian M.. PET mapping of neurofunctional changes in a posttraumatic stress disorder model. J Nucl Med 2016;57:1474–1477. [DOI] [PubMed] [Google Scholar]
- 19. Tawakol A, Ishai A, Takx RA, Figueroa AL, Ali A, Kaiser Y, Truong QA, Solomon CJ, Calcagno C, Mani V, Tang CY, Mulder WJ, Murrough JW, Hoffmann U, Nahrendorf M, Shin LM, Fayad ZA, Pitman RK.. Relation between resting amygdalar activity and cardiovascular events: a longitudinal and cohort study. Lancet 2017;389:834–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Osborne MT, Ishai A, Hammad B, Tung B, Wang Y, Baruch A, Fayad ZA, Giles JT, Lo J, Shin LM, Grinspoon SK, Koenen KC, Pitman RK, Tawakol A.. Amygdalar activity predicts future incident diabetes independently of adiposity. Psychoneuroendocrinology 2019;100:32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goyal A, Dey AK, Chaturvedi A, Elnabawi YA, Aberra TM, Chung JH, Belur AD, Groenendyk JW, Lerman JB, Rivers JP, Rodante JA, Harrington CL, Varghese NJ, Sanda GE, Baumer Y, Sorokin AV, Teague HL, Genovese LD, Natarajan B, Joshi AA, Playford MP, Bluemke DA, Chen MY, Alavi A, Pitman RK, Powell-Wiley TM, Tawakol A, Gelfand JM, Mehta NN.. Chronic stress-related neural activity associates with subclinical cardiovascular disease in psoriasis: a prospective cohort study. JACC Cardiovasc Imaging 2018;doi:10.1016/j.jcmg.2018.08.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Schaefer SM, Abercrombie HC, Lindgren KA, Larson CL, Ward RT, Oakes TR, Holden JE, Perlman SB, Turski PA, Davidson RJ.. Six-month test-retest reliability of MRI-defined PET measures of regional cerebral glucose metabolic rate in selected subcortical structures. Hum Brain Mapp 2000;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.European Environment Agency. Noise in Europe 2014. EEA Report No. 10/2014, pp. 1–68.
- 24.Global Risk Factors Collaborators. Global, regional, and national comparative risk assessment of 84 behavioural, environmental and occupational, and metabolic risks or clusters of risks, 1990-2016: a systematic analysis for the Global Burden of Disease Study 2016. Lancet 2017;390:1345–1422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Fritschi L, Brown AL, Kim R, Schwela DH, Kephalopoulos SE.. Burden of Disease from Environmental Noise: Quantification of Healthy Life Years Lost in Europe. Bonn: World Health Organization, Regional Office for Europe and European Commission Joint Research Centre; 2011. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.