

Abstract
Cre recombinase-mediated DNA recombination is an established method for conditional control of gene expression in animal models. Regulation of its activity has been accomplished to impart spatial and/or temporal control over recombination of the target gene. In this chapter, optical control of Cre recombinase in developing zebrafish embryos through genetic code expansion is discussed. This method takes advantage of an evolved aminoacyl tRNA synthetase and tRNA pair that can incorporate an unnatural amino acid (UAA) into proteins in response to an amber stop codon (TAG). Genetic code expansion is used to replace a lysine residue critical to Cre recombinase function with a photocaged analogue of lysine, successfully blocking recombination activity until irradiation with 405 nm light. This chapter highlights use of optically controlled Cre recombinase for cell-lineage tracing experiments in zebrafish embryos, demonstrating the ability to target small populations of cells at different developmental timepoints for recombination. Optically controlled Cre recombinase showed no background activation and precise activation upon irradiation, making it a useful new tool for studying development and disease in the zebrafish embryo.
Keywords: Genetic Code Expansion, Zebrafish, Light-Activation, Caged Protein, Cre Recombinase, DNA Recombination
1. Introduction
Cre recombinase is a powerful and well established biological tool that has been used to control gene expression in many contexts to further our understanding of biological processes and disease (Meinke, Bohm, Hauber, Pisabarro, & Buchholz, 2016). Site-specific gene recombination is achieved by targeting loxP recognition sites that flank the gene of interest. Upon recognition, Cre recombinase cleaves the double-stranded DNA backbone and depending on the orientation of the loxP sites in relation to each other, will either invert or excise the flanked gene (Sauer, 1998). In transgenic animal models, this allows for genes of interest to be activated or suppressed with spatial and or temporal control. The most common method of spatial control using this system is to place expression of Cre recombinase under a tissue specific promotor, thus allowing recombination to only take place in that tissue (Pan, Wan, Chia, Tong, & Gong, 2005). This method is limited if targeting of specific regions of an organ like different brain regions are desired, or if the tissue of interest does not have a specific promotor such as in undifferentiated embryonic cells. An established method for temporal control is fusion of Cre recombinase to estrogen receptor (ER), thus preventing nuclear translocation and subsequent recombination until addition of tamoxifen (Hans, Kaslin, Freudenreich, & Brand, 2009). But, while the ER constructs have been mutated to decrease recognition of endogenous hormones, some background recombination can be seen with this method (Feil, Wagner, Metzger, & Chambon, 1997). Both spatial and temporal control of Cre recombination was accomplished using UV light with a photocaged tamoxifen derivative (4-OHT)(Link, Shi, & Koh, 2005) and Cre-ER fusion in zebrafish embryos (Sinha et al., 2010; Zhang et al., 2018). Optical control has also been accomplished through fusion of light-responsive proteins to a split Cre recombinase that dimerizes and activates recombination with blue light, a method that requires continued exposure to light for efficient recombination (Kawano, Okazaki, Yazawa, & Sato, 2016; Kennedy et al., 2010; Schindler et al., 2015). Zebrafish are a versatile animal model for studies of development and disease due to their rapid ex utero development, ease of maintenance, and transparency during embryogenesis, making them ideal for optical manipulation. Light is an ideal inducer for genetic recombination owing to its high spatial and temporal resolution, allowing for targeting of single cells at specific timepoints. Thus, an optically controlled Cre recombinase would be a useful tool for spatiotemporal control of gene expression in zebrafish.
Unnatural amino acid (UAA) incorporation through genetic code expansion is a way to achieve optical control of proteins, requiring replacement of only one critical amino acid with minimal perturbation of overall protein structure and function (Ankenbruck, Courtney, Naro, & Deiters, 2017; Courtney & Deiters, 2018). This methodology uses an engineered aminoacyl tRNA synthetase that recognizes the UAA and will acylate a tRNA that subsequently delivers the UAA in response to an amber stop codon, catalyzed by the ribosome (Figure 1a). Commonly employed amino acyl tRNA synthetases and cognate tRNAs are derived from archaea, such as Methanosarcina mazei or Methanosarcina barkeri, which naturally encode the non-canonical amino acid pyrrolysine (Wan, Tharp, & Liu, 2014). The pyrrolysyl tRNA synthetase (PylRS)/tRNA (PylT) pair has been successfully employed for genetic code expansion in zebrafish (Liu, Hemphill, Samanta, Tsang, & Deiters, 2017) and other animal models (Brown, Liu, & Deiters, 2018). A photocaged analogue of lysine (hydroxycoumarin lysine, HCK, Figure 1b) (Luo et al., 2014) was used to replace K201 and establish an optically controlled Cre recombinase, as K201 is crucial to cleavage of the DNA backbone (Gibb et al., 2010) (Figure 2a). Using a transgenic fish line reporter for Cre recombinase activity (Figure 2b), optically controlled DNA recombination can be used to trace cell fate for targeted cell populations. The high spatiotemporal resolution of the activation of this system would allow for study of tumor cell heterogeneity or stem cell differentiation, which would be difficult or impossible using tissue promotors to control Cre recombinase expression. In the following sections, we will detail the procedures for generating an optically controlled Cre recombinase using UAA incorporation in zebrafish. Next, we describe applying this tool for fate-mapping cells during zebrafish development (Figure 2c).
Fig. 1.
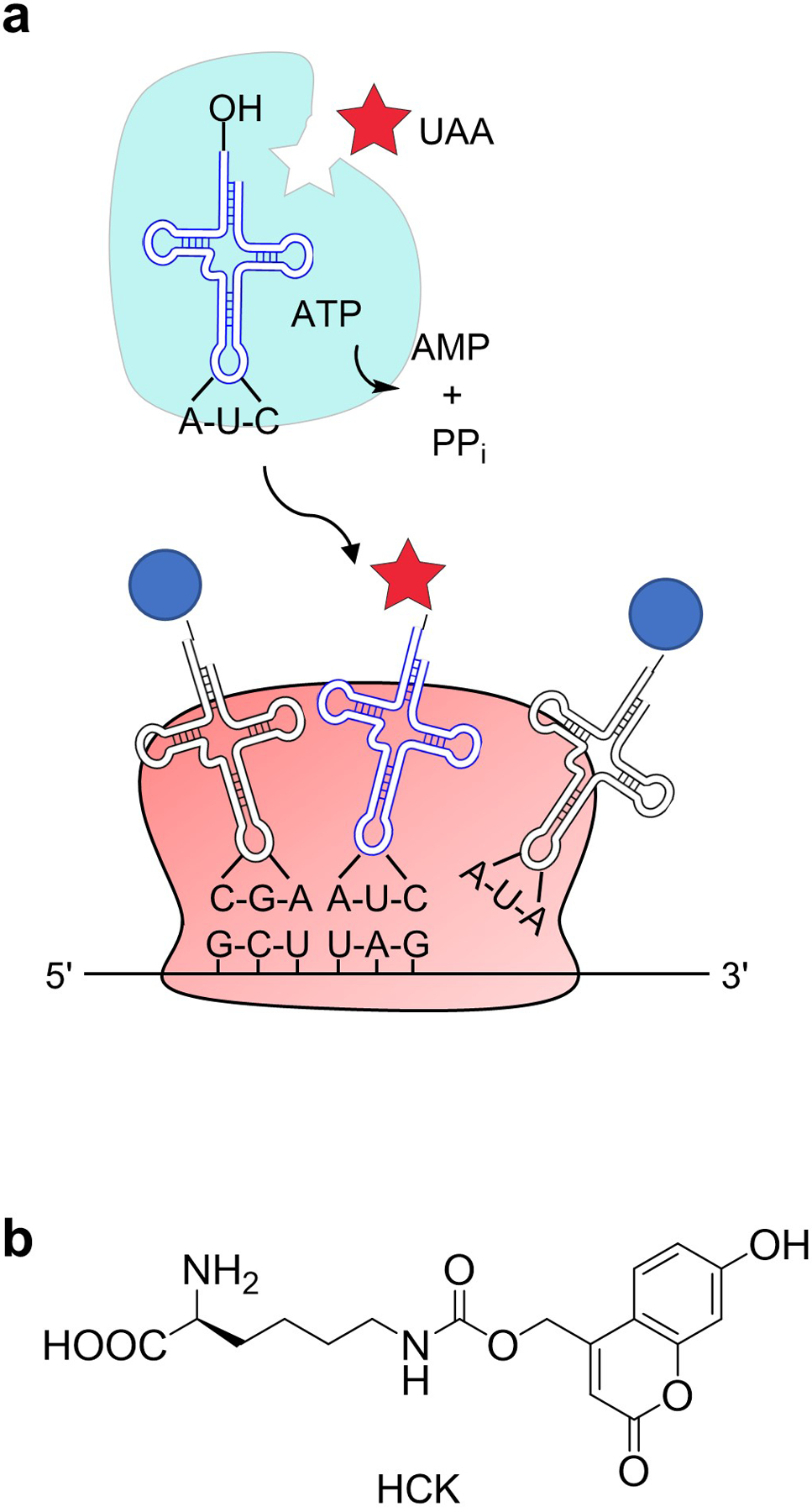
(a) Mechanism behind genetic code expansion. The aminoacyl tRNA synthetase (cyan) was engineered to recognize the unnatural amino acid (UAA, red star) and aminoacylate its cognate tRNA. The tRNA recognizes TAG stop codons and the ribosome then site-specifically incorporates the UAA into the synthesized protein. Adapted with permission from ACS Chem Biol, 13, 2375–2386. Copyright 2018 American Chemical Society. (b) The structure of hydroxycoumarin lysine (HCK), a photocaged lysine UAA used in this method.
Fig. 2.
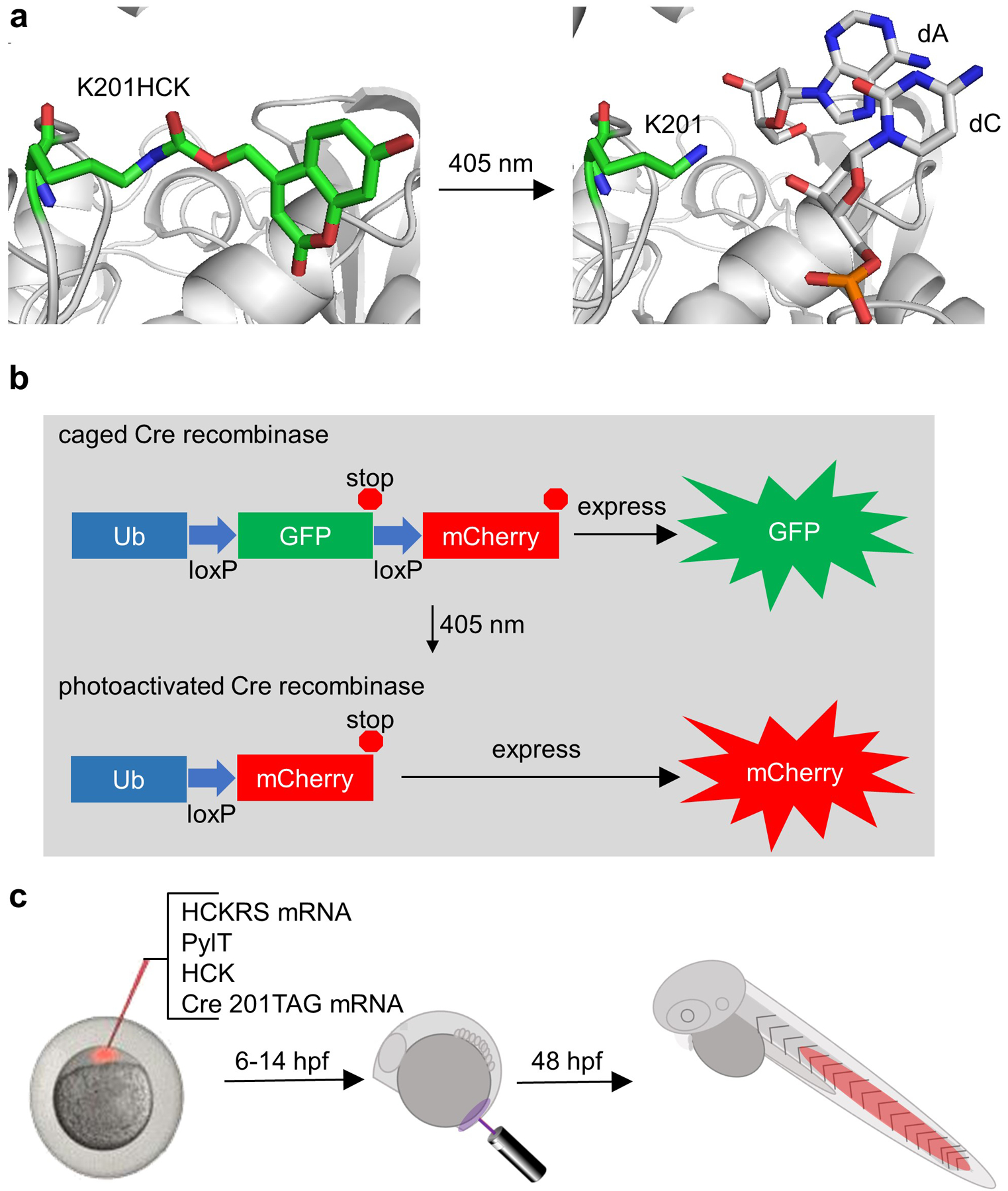
(a) Caging of Cre recombinase activity. The critical lysine K201 is replaced with HCK, blocking function. Upon 405 nm light exposure, the hydroxycoumarin group undergoes photolysis and reveals lysine, restoring activity. (b) In the transgenic fish line Cre recombinase reporter, GFP is expressed until Cre recombinase is activated and expression is switched to mCherry. (c) Embryos at the one-cell stage are injected with mRNA encoding the evolved amino-acyl tRNA synthetase (HCKRS) acylating its cognate tRNA (PylT) with HCK and mRNA encoding Cre recombinase with an amber stop codon at site 201. After 6 or 14 hours, specific embryonic cell populations are irradiated with 405 nm light, and at 48 hours post-fertilization (hfp) embryos are imaged to reveal recombination in the progeny of the originally irradiated cells. Adapted with permission from ChemBioChem, 19, 1244–1249. Copyright 2018 John Wiley and Sons.
2. Expression of photocaged Cre recombinase in zebrafish embryos
Production of exogenous protein in zebrafish is readily performed by injection of mRNA into the embryo at the one-cell stage (Kardash, 2012). mRNA is generated through in vitro transcription (IVT) reactions using commercially available kits. The first step is to take the gene of interest, in this case Cre recombinase, and clone it into a vector for IVT such as pCS2+ which contains an SP6 RNA polymerase promotor and an SV40 poly-A sequence. Mammalian codon optimized genes are efficiently expressed in zebrafish due to similar codon usage (Horstick et al., 2015). Once in the pCS2 vector, site-directed mutagenesis is conducted to replace the critical lysine K201 with an amber stop codon (TAG). This will be the site where HCK is incorporated during translation (Luo et al., 2016). The evolved PylRS for incorporating HCK (HCKRS) (Luo et al., 2014) will also need to be placed in a pCS2 vector for IVT. Injection of the mRNA encoding the Cre201TAG and HCKRS along with PylT and HCK into embryos at the one-cell stage will result in expression of photocaged Cre recombinase. Activation of Cre recombination can be performed by irradiating a small region (or even single cells) using a confocal imaging laser, or by irradiating the whole embryo with an LED light source. Imaging of the Cre recombinase reporter line at 48 hpf reveals a switch from green to red fluorescence only in the progeny of the cells irradiated, demonstrating tight spatial control. Successful activation of recombination at different stages of development also highlights temporal control over Cre recombinase activity. Quantification of recombination can be performed by fluorescence measurement or by qPCR methods.
3. Protocol
3.1. Materials
mMessage mMachine SP6 Transcription Kit (Ambion)
MEGAscript T7 Transcription Kit (Ambion)
NotI-HF restriction enzyme (NEB)
phenol:chloroform:isoamyl alcohol mix
sodium acetate
ethanol 200 proof
phenol red solution
hydroxy coumarin lysine (100 mm in DMSO)(Luo et al., 2014)
petri dishes (35 mm and 10 cm)
mineral oil
needle puller
microcapillary tubes
razor blade or scalpel
wash bottles with E3 medium
agarose microinjection plate
eyelash embryo manipulator
transfer pipettes
pneumatic microinjector (World Precision Instruments Pneumatic Picopump)
micromanipulator
Cre recombinase reporter transgenic zebrafish adults (Mosimann et al., 2011)
zebrafish breeding tanks
egg catcher
low melting point agarose
confocal microscope with 405 nm laser (Zeiss LSM 700 laser scanning confocal microscope)
fine-tipped tweezers
tricaine
fluorescence stereo microscope (Leica m205 FA microscope)
sodium hydroxide
Genomic DNA Purification Kit (GeneJET Genomic DNA Purification Kit, Thermo Scientific)
qPCR thermocycler (CFX96 Touch™ Real-Time PCR Detection System, Bio-Rad)
SYBR Green qPCR mix
3.2. In vitro transcription of mRNA and PylT
-
3.2.1
Using NotI-HF to linearize the pCS2 plasmid, digest ~10 μg of plasmid DNA.
-
3.2.2
Purify the linearized plasmid by column purification or if working with smaller amounts of template, phenol:chloroform:isoamyl alcohol (PCIA) extraction and ethanol precipitation.
-
3.2.3
Using the SP6 mMessage mMachine Transcription Kit (Ambion), generate 5’ capped mRNA from the linearized pCS2 vector using DNA template (1 μg) in a 20 μl reaction at 37 °C for 5 hours. Add Turbo DNase (1 μl, included in kit) at the end and incubate at 37 °C for 15 additional minutes to remove template DNA.
-
3.2.4
Add ultra-pure water (30 μl) and purify with PCIA extraction (50 μl) and precipitate with ethanol (137.5 μl) overnight at −20 °C. PCIA extraction will remove most free nucleotides and further purification is not necessary. Pellet the mRNA by max centrifugation (≥ 13,000 x g) at 4 °C, wash with 70% ethanol (200 μl), re-pellet, remove ethanol, and air-dry on ice for 5 minutes. Resuspend the pellet in ultrapure water (20 μl). Quantification of concentration is obtained by absorbance at 260 nm (Nanodrop) and a concentration of 2–6 μg/μl can be expected, which is sufficient for injections. The quality of the mRNA is confirmed by native agarose gel electrophoresis for signs of smearing indicating degradation. Cre recombinase mRNA (1038 bp) appears as one band at ~ 800 bp compared to a DNA ladder, which is not unusual as RNA can run differently than DNA on a native gel. The HCKRS mRNA appears as one band at ~ 1000 bp. The mRNA solution is aliquoted to prevent multiple freeze-thaw cycles and is stored at −80 °C.
-
3.2.5
The pyrrolysine tRNA (PylT) is generated through IVT using the MEGAscript T7 Transcription Kit (Ambion). Amplify the template PylT with the following sequence: 5’-taatacgactcactataggaaacctgatcatgtagatcgaacggactctaaatccgttcagccgggttagattcccggggtttccgcca (purchased from IDT) with the U25C mutation and CCA at the 3’ end to increase aminoacylation efficiency (Liu et al., 2017). Use PCR primers (forward: 5’-aatacgactcactatagga; reverse: 5’-tggcggaaaccccgggaatctaa) to install a truncated T7 promotor at the 5’ end (PCR protocol: 95 °C for 3 minutes followed by 34 cycles of 95 °C for 30 seconds, 45 °C for 30 seconds, then 72 °C for 90 seconds, then finished with 72 °C for 5 minutes). Purify the PCR product by PCIA extraction and ethanol precipitation, because the amplicon is too small for column purifications.
-
3.2.6
Use 1 μg of this purified PCR product in a 20 μl IVT reaction incubated at 37 °C for 5 hours. Four identical reactions running in parallel can be set up to generate a more concentrated PylT stock at the end by ethanol precipitating all reactions in one tube and resuspending in ultra-pure water (20 μl). The final concentration should be 15–30 μg/μl. Add TurboDNase (1 μl) at the end of the IVT reaction and incubate for 15 minutes at 37 °C, followed by PCIA extraction (50 μl) and ethanol precipitation (137.5 μl) overnight at −20 °C. Pellet the PylT by max centrifugation (≥ 13000 x g) at 4 °C, wash with 70% ethanol (200 μl), re-pellet, remove the ethanol, and air-dry the pellet on ice for 5 minutes. Resuspend the pellet in ultrapure water (20 μl). Measure the concentration by absorbance at 260 nm, and check the PylT quality on a 1.5% agarose gel, which should appear as one band at ~ 100 bp. Aliquot the PylT solution and store at −80 °C.
3.3. Microinjection of zebrafish embryos
-
3.3.1
Thaw the mRNA solutions for Cre recombinase and HCKRS and the PylT solution on ice. HCK is synthesized according to literature protocol (Luo et al., 2014). Make the following injection solution at a total volume of 1.5 μl with a final concentration of 200 ng/μl of Cre recombinase K201TAG mRNA, 300 ng/μl of HCKRS mRNA, 15–30 μg/μl of PylT, and 10 mM HCK (from a 100 mM stock in DMSO). Add phenol red (0.1 μl), which will turn yellow in the presence of HCK. Protect the solution from light and place a drop onto a 35 mm diameter petri dish and cover it with mineral oil to avoid evaporation. The mixture should be injected immediately, but can be stored on ice if this is not possible.
-
3.3.2
Use a needle puller to make a few needles from microcapillary tubes. Only one will be needed, but bring extras in case they break during preparation or injection. Mark every millimeter along the needle starting at the beveled edge and moving away from the point for 5 mm.
-
3.3.3
The evening before embryo injections, setup the transgenic fish, such as the Tg(ubi:loxP-EGFP-loxP-mCherry) line (Mosimann et al., 2011) used in the cell fate tracing experiments, by placing 5–8 fish (depending on the size of the tank) of each sex in a segregated breeding tank. After preparing injection solutions in the morning, transfer the fish to a fresh tank and remove the segregating barrier to begin mating. Plan experiments so that mating begins within an hour after the lights turn on to increase the yield of fertilized eggs as compared to starting mating later in the morning. Allow for 1–2 weeks of rest between breeding the same sets of fish. Usually, within 30 minutes there will be enough embryos to start injections, but they should be checked frequently in order to inject at the one-cell stage for highest recombination efficiency.
-
3.3.4
Working at a stereoscope equipped with a pneumatic microinjector, remove the very tip of the needle (2–3 mm) using a scalpel or razor blade, insert the needle into the microinjector mounted on a micromanipulator (for ease of needle movement during injections) and suck up the injection mix into the needle. Calibrate the pneumatic microinjector to dispense 2 nl of the injection mixture by measuring the time for ejection of 1 mm as marked on the needle, which is approximately a volume of 30 nl. The injector can then be set to eject for a certain amount of time to achieve consistent injection of 2 nl for each embryo.
-
3.3.5
Collect embryos by transferring the adult fish to a new tank and pouring the water and embryos into an egg catcher. Gently rinse the embryos into a 10 cm plate with E3 media from a wash bottle. With a transfer pipette, line up embryos on an injection tray made of 1.5% agarose with divots to hold each embryo and remove excess water.
-
3.3.6
Inject 2 nl of the injection mix into either the yolk or a single cell under a dissecting microscope. Both yolk- and cell-injection have worked for cell fate tracing experiments (Brown, Liu, Tsang, & Deiters, 2018). Once all embryos have been injected, wash them into a 10 cm petri dish with E3 media and incubate at 28.5 °C, making sure to protect them from light. It should be noted that the minimal light exposure necessary during injections was not enough to cause noticeable decaging of the injected HCK. Incubate the embryos for 6–7 hours until they reach shield stage.
3.4. Optical activation of Cre recombinase in zebrafish
-
3.4.1
By shield stage (6–7 hpf), the Cre recombinase containing HCK at K201 (Cre201HCK) should be expressed. This timepoint is also chosen for cell-lineage tracing because several populations of cells are organized at this stage and can be targeted for recombination. Immobilize the embryos in 1.5% low melting point agarose in a 35 mm petri dish. Let the agarose cool to RT before adding just enough to cover the embryos (~ 500 μl). Orient the embryos before the agarose hardens to allow for visualization of the target region for irradiation by a confocal microscope. Once the agarose has solidified, add E3 media on top.
-
3.4.2
Using a laser scanning confocal microscope with a water immersion objective, identify the target region of cells. With the bleaching option, irradiate a small population of cells with a 405 nm laser at 30% power (5 mW laser, 1.6 μs dwell time) for 20 iterations to ensure complete photolysis and activation of the photocaged Cre recombinase in that region of cells. Representative results are highlighted below. Recombination in cells that will form the telencephalon is accomplished by irradiating the outermost layer of cells at the tip of the animal pole (Figure 3a). Targeted DNA recombination can also be accomplished at later time points in development. At the 10-somite stage (~14 hpf) the embryo tail was irradiated leading to DNA recombination in cells that form the tail of the 48 hpf embryo (Figure 3b).
-
3.4.3
Manually release the embryos from the agarose with fine-tipped tweezers and incubate at 28.5 °C until 48 hpf for fluorescent imaging. Dechorionate the 48 hpf embryos with tweezers and anesthetize with tricaine (16 mg/ml, 6.1 μM) in E3 media. Image with bright field, GFP, and mCherry channels.
-
3.4.4
Activation of whole embryo recombination is best performed by irradiation at the shield stage with an 405 nm LED (Luxeonstar, 350 mW) for 5 minutes. Swirl the embryos periodically during irradiation to help ensure all cells are exposed. Representative results are shown below. Red fluorescence indicating recombination can be seen throughout the whole embryo by 48 hpf, but EGFP is also present, suggesting either incomplete recombination or persistence of EGFP protein produced before illumination (Figure 4). qPCR is used to quantify recombination in globally irradiated fish.
Fig 3.
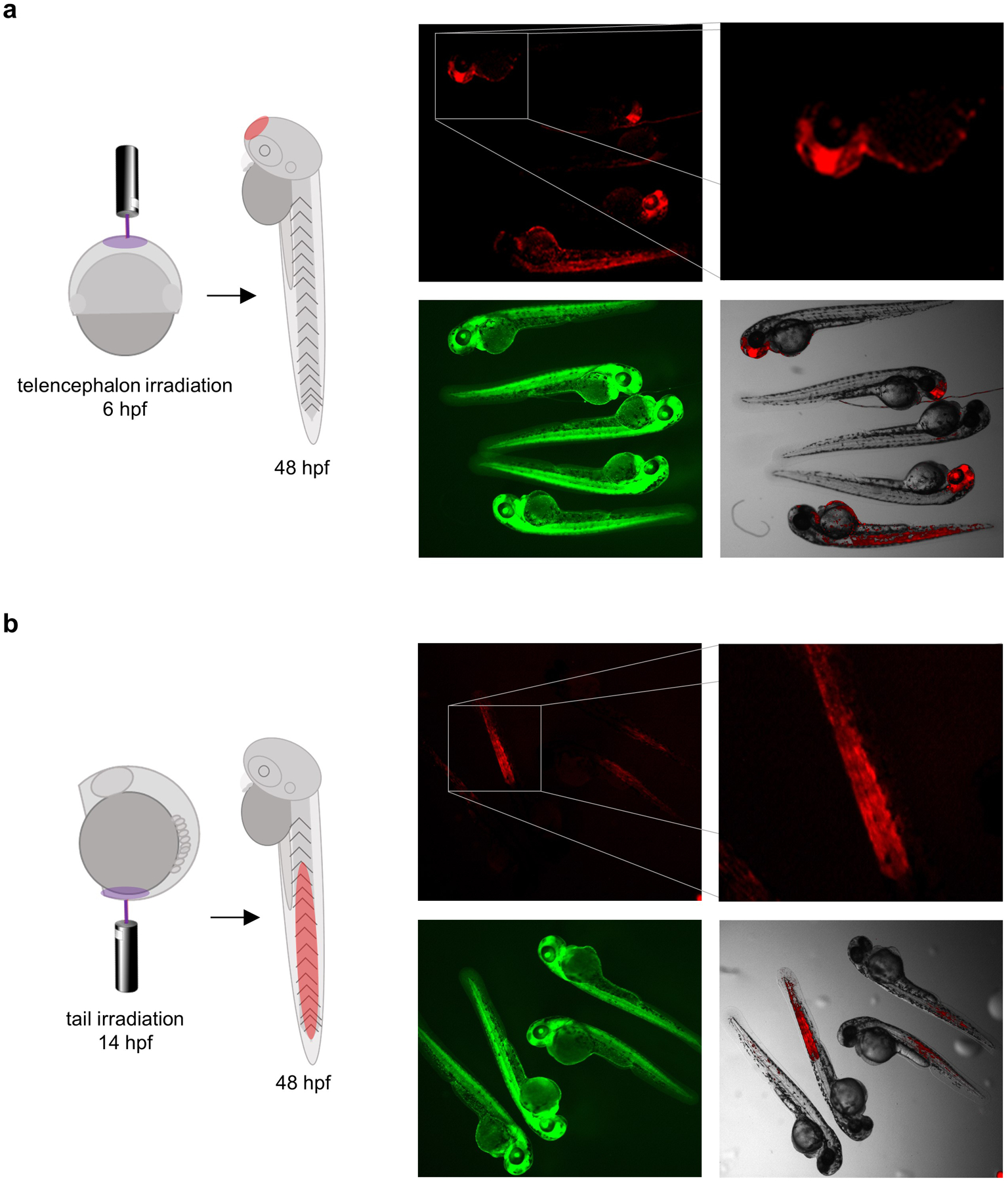
Targeted irradiation of cells that would form the telencephalon (a) and the tail (b) at either shield stage or 10-somite stage in the developing zebrafish embryo. Adapted with permission from ChemBioChem, 19, 1244–1249. Copyright 2018 John Wiley and Sons.
Fig. 4.
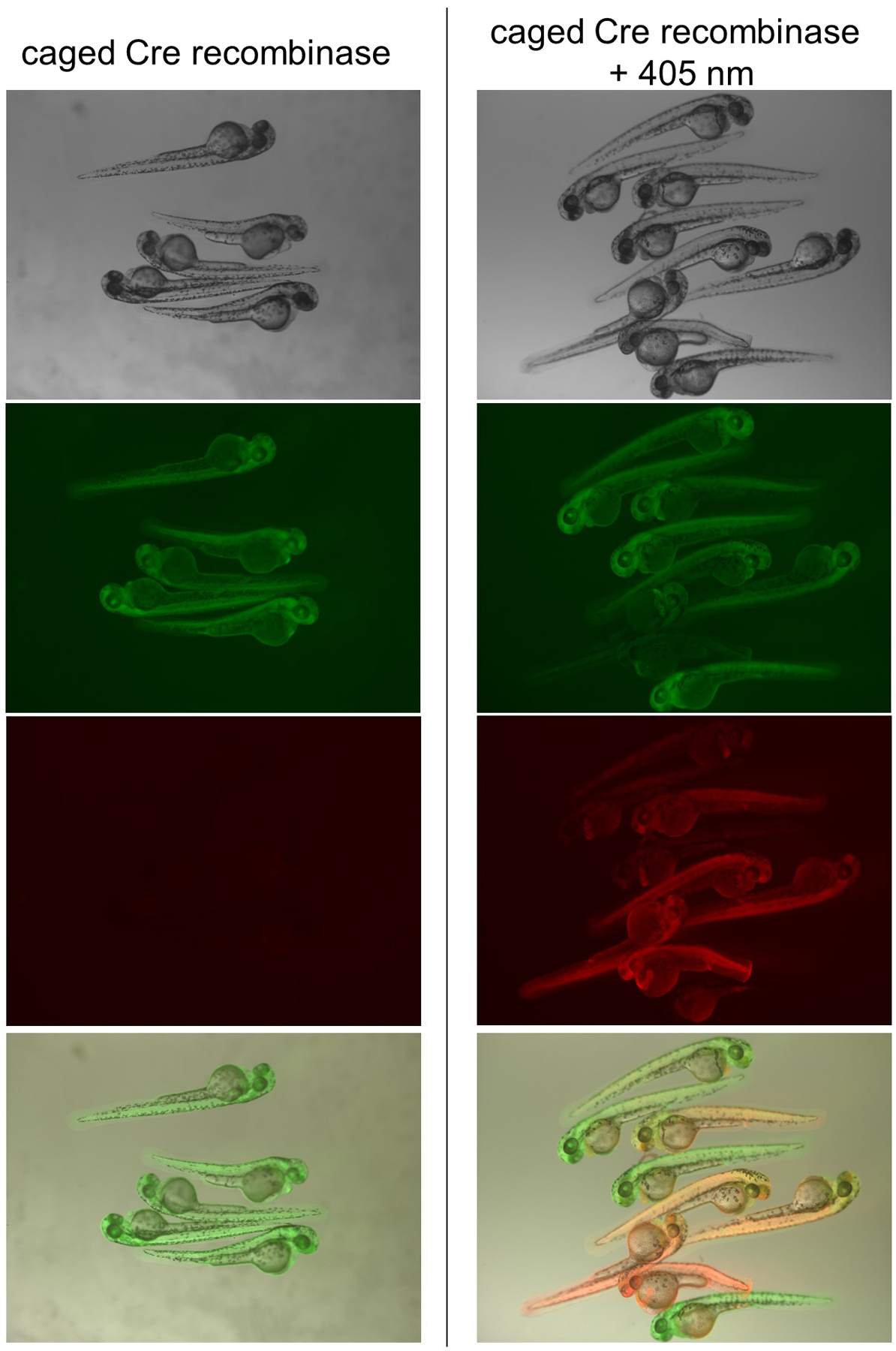
Global irradiation of zebrafish embryos. mCherry expression signals recombination and can be seen throughout the embryo expressing Cre201HCK after irradiation, but not in the absence of irradiation. Adapted with permission from ChemBioChem, 19, 1244–1249. Copyright 2018 John Wiley and Sons.
3.5. Quantification of recombination using fluorescence or qPCR
-
3.5.1
Image globally irradiated embryos and collect at 48 hpf in pools of three in Eppendorf tubes. For reference, embryos injected with 50 pg of wild-type Cre recombinase mRNA are also collected at 48 hpf, as well as non-injected embryos and non-irradiated embryos expressing Cre201HCK.
-
3.5.2
Measure total fluorescence intensity using ImageJ software. This is done by measuring total fluorescence intensity of the embryo (integrated density) and subtracting background intensity. Background intensity is calculated by multiplying the intensity of a background pixel (mean gray value) with the total number of pixels that constitute the embryo in the image (area). First, convert the image to 16-bit grayscale, and to set what types of measurements to collect, go to the measurements tab and select the boxes “integrated density”, “mean gray area”, and “area”. Draw a region around the outline of the embryo and take a measurement. Also measure an area outside the embryo for background. The size of the area drawn for background does not matter as we only need the mean intensity of the background (mean gray value). The difference in measured areas between embryos should not vary by much since all are at the same developmental stage. Repeat this for three embryos and three background measurements. Calculate corrected total embryo fluorescence with the following equation: corrected total embryo fluorescence = embryo integrated density – (area of embryo x mean gray value of background). Representative results are shown in Figure 5a–b.
-
3.5.3
Extract genomic DNA by adding 60 mM NaOH (50 μl) to each tube and incubate at 95 °C for 10 minutes. Neutralize the solution with 1 M Tris-HCl pH 8 (~ 2 μl) and then centrifuge at max speed (≥ 13,000 x g) for 10 minutes (Nathan D. Meeker, 2007). Purify the genomic DNA (GeneJET Genomic DNA Purification Kit, Thermo Scientific).
-
3.5.4
The recombined gene was detected using a SYBR Green assay on a qPCR machine (CFX96 Touch™ Real-Time PCR Detection System, Bio-Rad). The primers (forward: 5’-gggagaagtgcaaaacatacattattggctag; reverse: 5’-cttgatgatggccatgttgtcctc) amplify the 3’ of the Ub promotor to the 5’ of the mCherry gene, an amplicon too long for the qPCR parameters for efficient amplification in the non-recombined gene, but short enough and efficiently amplified in the recombined gene (Weis, Schmidt, Lyko, & Linhart, 2010). Amplification of EF1α is used as a reference using established primers (Tang R, 2007). PCR conditions: 95 °C for 5 minutes, 70 cycles of 95 °C for 5 seconds then 56.6 °C for 60 seconds.
-
3.5.5
Analyze the data with the ΔΔCt method (Livak & Schmittgen, 2001) to compare the levels of recombination to wild-type Cre recombinase injected embryos. Representative results are shown below. The amount of recombination varies with the method of irradiation, with the highest levels of whole embryo recombination at about 50% that of the wild-type Cre recombinase mRNA-injected embryos using a 405 nm LED (350 mW) for 5 minutes. The other conditions use the confocal 405 nm laser to irradiate 10 z-planes of the embryo with a 1.6 μs or 3.1 μs dwell time and demonstrated less recombination overall, but did show a dose response to light (Figure 5c). Importantly, no recombination took place in non-irradiated embryos expressing Cre201HCK.
Fig. 5.
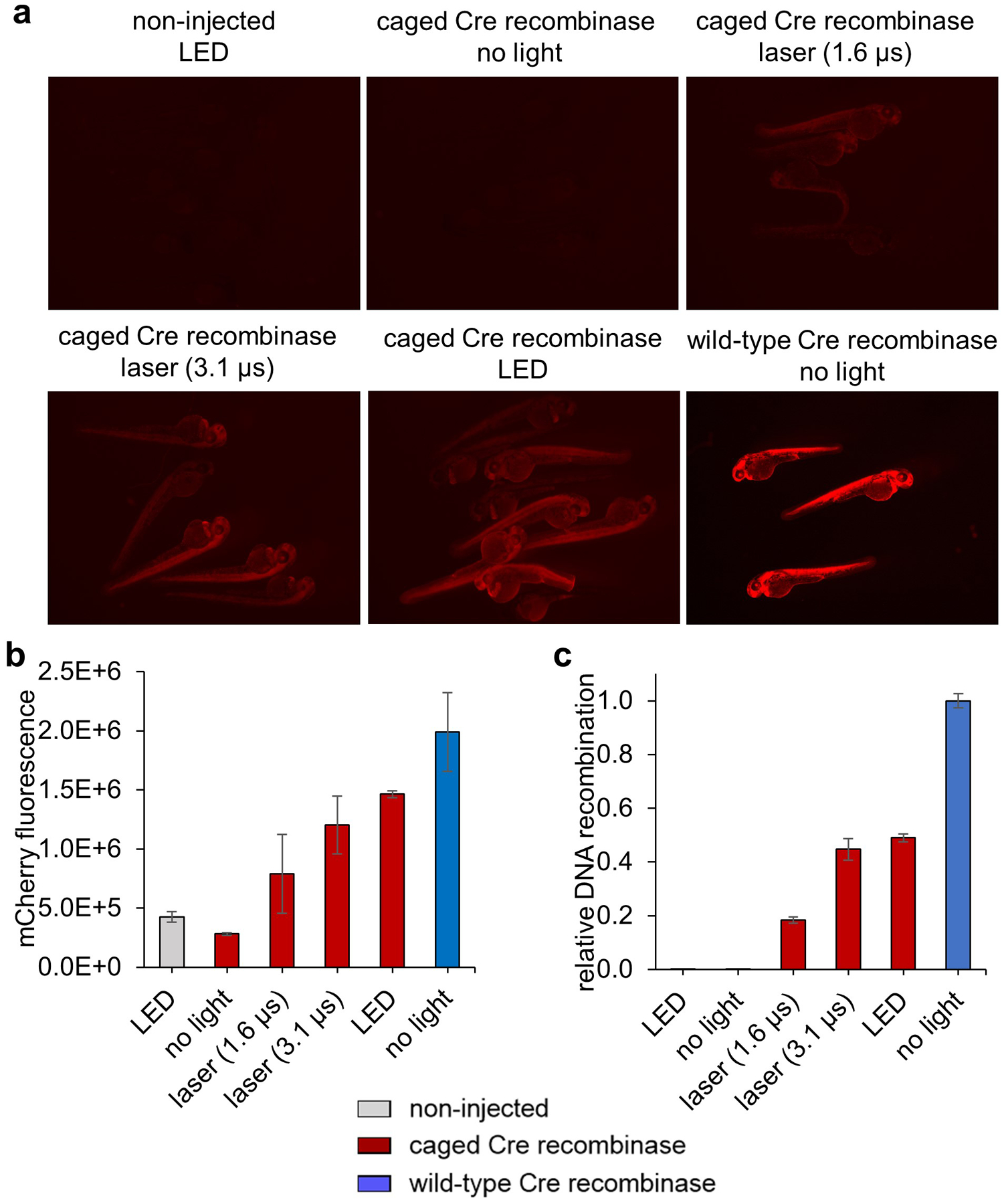
Quantification of recombination after global irradiation. (a) Representative mCherry fluorescence images of 48 hpf embryos after various irradiation conditions. (b) Quantification of mCherry fluorescence in the 3 brightest embryos using ImageJ software. (c) qPCR results after ΔΔCt analysis with comparison of percent recombination to the wild-type Cre recombinase mRNA injected embryos. Adapted with permission from ChemBioChem, 19, 1244–1249. Copyright 2018 John Wiley and Sons.
4. Summary
Optical control of Cre recombinase allows for activation of gene recombination with exquisite spatial and temporal resolution. This chapter describes an approach for generating Cre recombinase activated by 405 nm light through incorporation of a photocaged lysine analogue into the active site of the enzyme in zebrafish embryos(Brown, Liu, Tsang, et al., 2018; Luo et al., 2016). Success of this method was demonstrated in a transgenic fish line with fluorescent reporters for Cre recombinase activity, where early cell populations that would form the brain and tail were irradiated and their progeny traced at a later developmental timepoint. Light controlled recombination can be extended to any transgenic fish line with a gene of interest flanked by loxP sites. Targeted recombination in single cells, in tissues that may not have a specific tissue promotor, or in a subset of cells in an organ or tumor will facilitate study of embryo development, disease, and tumor heterogeneity. Moreover, following the methods described here, other proteins might be optically controlled in zebrafish embryos as well, such as RNA polymerases (Hemphill, Chou, Chin, & Deiters, 2013) (REF), DNA polymerases (Chou, Young, & Deiters, 2009), CRISPR endonucleases (Hemphill, Borchardt, Brown, Asokan, & Deiters, 2015), protease (Luo, Torres-Kolbus, Liu, & Deiters, 2017), or helicases (Luo, Kong, et al., 2017).
5. Acknowledgement
Support is gratefully acknowledged from NIH grants T32GM088119 (WB) and R01GM112728 (AD).
7. References
- Ankenbruck N, Courtney T, Naro Y, & Deiters A (2017). Optochemical Control of Biological Processes in Cells and Animals. Angewandte Chemie International Edition, 57(11), 2768–2798. doi: 10.1002/anie.201700171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W, Liu J, & Deiters A (2018). Genetic Code Expansion in Animals. ACS Chemical Biology, 13(9), 2375–2386. doi: 10.1021/acschembio.8b00520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown W, Liu J, Tsang M, & Deiters A (2018). Cell-Lineage Tracing in Zebrafish Embryos with an Expanded Genetic Code. ChemBioChem, 19(12), 1244–1249. doi: 10.1002/cbic.201800040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou C, Young DD, & Deiters A (2009). A Light-Activated DNA Polymerase. Angewandte Chemie International Edition, 48(32), 5950–5953. doi: 10.1002/anie.200901115 [DOI] [PubMed] [Google Scholar]
- Courtney T, & Deiters A (2018). Recent advances in the optical control of protein function through genetic code expansion. Current Opinion in Chemical Biology, 46, 99–107. doi: 10.1016/j.cbpa.2018.07.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feil R, Wagner J, Metzger D, & Chambon P (1997). Regulation of Cre Recombinase Activity by Mutated Estrogen Receptor Ligand-Binding Domains. Biochemical and Biophysical Research Communications, 237(3), 752–757. doi: 10.1006/bbrc.1997.7124 [DOI] [PubMed] [Google Scholar]
- Gibb B, Gupta K, Ghosh K, Sharp R, Chen J, & Van Duyne GD (2010). Requirements for catalysis in the Cre recombinase active site. Nucleic Acids Research, 38(17), 5817–5832. doi: 10.1093/nar/gkq384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans S, Kaslin J, Freudenreich D, & Brand M (2009). Temporally-Controlled Site-Specific Recombination in Zebrafish. PLOS ONE, 4(2), e4640. doi: 10.1371/journal.pone.0004640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill J, Borchardt EK, Brown K, Asokan A, & Deiters A (2015). Optical Control of CRISPR/Cas9 Gene Editing. Journal of the American Chemical Society, 137(17), 5642–5645. doi: 10.1021/ja512664v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hemphill J, Chou C, Chin JW, & Deiters A (2013). Genetically Encoded Light-Activated Transcription for Spatiotemporal Control of Gene Expression and Gene Silencing in Mammalian Cells. Journal of the American Chemical Society, 135(36), 13433–13439. doi: 10.1021/ja4051026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstick EJ, Jordan DC, Bergeron SA, Tabor KM, Serpe M, Feldman B, & Burgess HA (2015). Increased functional protein expression using nucleotide sequence features enriched in highly expressed genes in zebrafish. Nucleic acids research, 43(7), e48–e48. doi: 10.1093/nar/gkv035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kardash E (2012). Current Methods in Zebrafish Research. Materials and Methods, 2(109). [Google Scholar]
- Kawano F, Okazaki R, Yazawa M, & Sato M (2016). A photoactivatable Cre–loxP recombination system for optogenetic genome engineering. Nature Chemical Biology, 12, 1059. doi: 10.1038/nchembio.2205 [DOI] [PubMed] [Google Scholar]
- Kennedy MJ, Hughes RM, Peteya LA, Schwartz JW, Ehlers MD, & Tucker CL (2010). Rapid blue-light–mediated induction of protein interactions in living cells. Nature Methods, 7, 973. doi: 10.1038/nmeth.1524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Link KH, Shi Y, & Koh JT (2005). Light activated recombination. Journal of the American Chemical Society, 127(38), 13088–13089. doi: 10.1021/ja0531226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Hemphill J, Samanta S, Tsang M, & Deiters A (2017). Genetic Code Expansion in Zebrafish Embryos and Its Application to Optical Control of Cell Signaling. Journal of the American Chemical Society, 139(27), 9100–9103. doi: 10.1021/jacs.7b02145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, & Schmittgen TD (2001). Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods, 25(4), 402–408. doi: 10.1006/meth.2001.1262 [DOI] [PubMed] [Google Scholar]
- Luo J, Arbely E, Zhang J, Chou C, Uprety R, Chin JW, & Deiters A (2016). Genetically encoded optical activation of DNA recombination in human cells. Chemical Communications, 52(55), 8529–8532. doi: 10.1039/C6CC03934K [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Kong M, Liu L, Samanta S, Van Houten B, & Deiters A (2017). Optical Control of DNA Helicase Function through Genetic Code Expansion. ChemBioChem, 18(5), 466–469. doi: 10.1002/cbic.201600624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo J, Torres-Kolbus J, Liu J, & Deiters A (2017). Genetic Encoding of Photocaged Tyrosines with Improved Light-Activation Properties for the Optical Control of Protease Function. ChemBioChem, 18(14), 1442–1447. doi: 10.1002/cbic.201700147 [DOI] [PubMed] [Google Scholar]
- Luo J, Uprety R, Naro Y, Chou C, Nguyen DP, Chin JW, & Deiters A (2014). Genetically encoded optochemical probes for simultaneous fluorescence reporting and light activation of protein function with two-photon excitation. Journal of the American Chemical Society, 136(44), 15551–15558. doi: 10.1021/ja5055862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meinke G, Bohm A, Hauber J, Pisabarro MT, & Buchholz F (2016). Cre Recombinase and Other Tyrosine Recombinases. Chemical Reviews, 116(20), 12785–12820. doi: 10.1021/acs.chemrev.6b00077 [DOI] [PubMed] [Google Scholar]
- Mosimann C, Kaufman CK, Li P, Pugach EK, Tamplin OJ, & Zon LI (2011). Ubiquitous transgene expression and Cre-based recombination driven by the ubiquitin promoter in zebrafish. Development, 138(1), 169–177. doi: 10.1242/dev.059345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meeker Nathan D., H SA, Ho Linh, Trede Nikolaus S. (2007). Method for isolation of PCR-ready genomic DNA from zebrafish tissues. BioTechniques, 43(5), 610–614. [DOI] [PubMed] [Google Scholar]
- Pan X, Wan H, Chia W, Tong Y, & Gong Z (2005). Demonstration of site-directed recombination in transgenic zebrafish using the Cre/loxP system. Transgenic Research, 14(2), 217–223. doi: 10.1007/s11248-004-5790-z [DOI] [PubMed] [Google Scholar]
- Sauer B (1998). Inducible Gene Targeting in Mice Using the Cre/loxSystem. Methods, 14(4), 381–392. doi: 10.1006/meth.1998.0593 [DOI] [PubMed] [Google Scholar]
- Schindler SE, McCall JG, Yan P, Hyrc KL, Li M, Tucker CL, . . . Diamond MI. (2015). Photo-activatable Cre recombinase regulates gene expression in vivo. Scientific Reports, 5, 13627. doi: 10.1038/srep13627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinha DK, Neveu P, Gagey N, Aujard I, Saux TL, Rampon C, . . . Vriz S (2010). Photoactivation of the CreERT2 Recombinase for Conditional Site-Specific Recombination with High Spatiotemporal Resolution. Zebrafish, 7(2), 199–204. [DOI] [PubMed] [Google Scholar]
- Tang R, D. A, Lai D, McNabb WC, Love DR. (2007). Validation of zebrafish (Danio rerio) reference genes for quantitative real-time RT-PCR normalization. Acta Biochimica et Biophysica Sinica, 39(5), 384–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan W, Tharp JM, & Liu WR (2014). Pyrrolysyl-tRNA synthetase: An ordinary enzyme but an outstanding genetic code expansion tool. Biochimica et Biophysica Acta (BBA) - Proteins and Proteomics, 1844(6), 1059–1070. doi: 10.1016/j.bbapap.2014.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weis B, Schmidt J, Lyko F, & Linhart HG (2010). Analysis of conditional gene deletion using probe based Real-Time PCR. BMC Biotechnology, 10, 75–75. doi: 10.1186/1472-6750-10-75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Hamouri F, Feng Z, Aujard I, Ducos B, Ye S, . . . Bensimon D. (2018). Control of Protein Activity and Gene Expression by Cyclofen-OH Uncaging. ChemBioChem. doi: 10.1002/cbic.201700630 [DOI] [PubMed] [Google Scholar]