

Abstract
It was demonstrated that styrylquinolizinium derivatives may be applied as photoswitchable DNA ligands. At lower ligand:DNA ratios (≤1.5), these compounds bind to duplex DNA by intercalation, with binding constants ranging from Kb = 4.1 × 104 M to 2.6 × 105 M (four examples), as shown by photometric and fluorimetric titrations as well as by CD and LD spectroscopic analyses. Upon irradiation at 450 nm, the methoxy-substituted styrylquinolizinium derivatives form the corresponding syn head-to-tail cyclobutanes in a selective [2 + 2] photocycloaddition, as revealed by X-ray diffraction analysis of the reaction products. These photodimers bind to DNA only weakly by outside-edge association, but they release the intercalating monomers upon irradiation at 315 nm in the presence of DNA. As a result, it is possible to switch between these two ligands and likewise between two different binding modes by irradiation with different excitation wavelengths.
Keywords: azoniahetarenes, DNA ligands, photodimerization, photoswitches
Introduction
The association of DNA-targeting drugs with nucleic acids [1–8] is considered one of the essential properties that determine their biological activity [9]. Specifically, a ligand may occupy particular binding sites of DNA or induce significant structural changes of the nucleic acid. In turn, both of these processes interfere with biologically relevant recognition processes between DNA and enzymes, e.g., topoisomerase [10]. Therefore, many potential lead structures of chemotherapeutic anticancer drugs exhibit DNA-binding properties [1–10]. Nevertheless, most DNA-binding ligands have an insufficient selectivity towards the targeted nucleic acid, and they also accumulate in healthy tissue, so that the chemotherapeutic treatment of tumors with DNA-binding drugs still suffers from severe side effects because of the intrinsic toxicity of the employed drugs [11–13]. As a result, there is an urgent need for DNA-targeting chemotherapeutic reagents that can be activated with an external stimulus only at the desired point of action. In this context, light offers several distinct advantages to switch on the activity of an otherwise inactive substrate (prodrug) because light is noninvasive, traceless, and easy to apply, and it enables local and temporal control [14]. To this end, photochromic systems appear to be highly attractive as a basis for photocontrollable substrates because they allow to switch the biological activity on and off due to the reversibility of the photoreaction [15]. Indeed, the application of light to induce and control bioactivity of pharmaceuticals or bio(macro)molecules has been convincingly demonstrated in the emerging field of photopharmacology [16–18]. Consequently, several attempts have also been made to develop photochromic DNA binders. Thus, it has been shown with spiropyran [19–21], stilbene [22–23], azobenzene [24–28], dithienylethene [29–32], chromene [33], and spirooxazine [34] derivatives that specifically modified photochromic ligands bind to DNA only with one of the components of the photochromic equilibrium. Moreover, these ligand–DNA interactions can be photochemically switched between the binding and nonbinding form. Interestingly, the photochromic systems applied in this context are almost exclusively photoinduced electrocyclization or E-to-Z isomerization reactions, whereas the well-established photochromic cycloaddition–cycloreversion equilibrium to establish photoswitchable DNA binders has so far been widely neglected. In fact, there is only one reported example for the use of the reversible photoinduced dimerization of stilbene derivatives as photoswitchable DNA ligand [35], and in this case, the structure of the photoproduct was not fully identified. Also, it has been shown that a DNA-binding azoniatetracene may be generated by photoinduced [4 + 4] cycloreversion. However, this system was not applied for photoinduced switching of binding properties [36]. Apparently, styryl-substituted aromatic derivatives could fill this gap because the [2 + 2] photocyclization reaction of stilbenes and derivatives thereof is a well-established reversible photoreaction [37–46], and styryl dyes, in particular cationic ones, were shown to be efficient DNA binders [47–58]. Nevertheless, the photochromic nature of DNA-binding styryl dyes has not been applied to use them as photoswitchable DNA binders. Although, there is one reported example that demonstrates the deactivation of a stilbene tyrosine kinase inhibitor by a [2 + 2] photocycloaddition [59].
As the quinolizinium ion has been established as a versatile platform for the development of DNA intercalators [60], we identified styryl-substituted quinolizinium derivatives as a promising basis for the search for photoswitchable DNA binders based on the photocycloaddition–photocycloreversion equilibrium. In fact, some selected styrylquinolizinium derivatives have already been shown to bind to DNA [61–67], however, their photocycloaddition reaction and the propensity of the corresponding photodimers to release the DNA-binding ligand have not been reported so far. Herein, we report on the photochemical and DNA-binding properties of the selected styrylquinolizinium derivatives 3a–d and demonstrate their ability to operate as photoswitchable DNA ligands.
Results and Discussion
Synthesis
2-Methylquinolizinium tetrafluoroborate (1) was synthesized according to published procedures [68]. The piperidine-catalyzed reaction of the latter with the benzaldehyde derivatives 2a–d gave the 2-styrylquinolizinium derivatives 3a–d in 63–79% yield (Scheme 1). The known products 3a and 3c were identified by comparison with literature data [69], and the new compounds 3b and 3d were fully characterized by NMR spectroscopy (1H, 13C, COSY, HSQC, and HMBC), elemental analyses, and mass spectrometry. In all cases, E-configuration of the alkene double bonds in 3a–d was indicated by characteristic coupling constants of the alkene protons (3JH–H = 16 Hz) [70].
Scheme 1.

Synthesis of styrylquinolizinium derivatives 3a–d.
Absorption and emission properties
The photophysical properties of the styrylquinolizinium derivatives 3a and 3c have already been reported [69], while the ones of 3b and 3d were determined in this work (Table 1 and Figure 1). In acetonitrile, the derivatives 3b and 3d exhibited long-wavelength absorption maxima at λabs = 404 nm and 368 nm, with emission bands at λfl = 548 nm and 419 nm. Derivative 3d was essentially nonfluorescent (Φfl < 0.01 in MeCN), whereas compound 3b (Φfl = 0.17 in MeCN) had the largest fluorescence quantum yield in comparison to the derivatives 3a (Φfl = 0.02 in MeCN) and 3c (Φfl = 0.04 in MeCN). In aqueous solution, the compounds exhibited long-wavelength absorption maxima at 434 nm (3a), 389 nm (3b), 384 nm (3c), and 371 nm (3d) as well as weak emission bands at 630 nm (3a), 538 nm (3b), and 507 nm (3c). In contrast, the emission intensity of 3d was too low to identify a maximum, as usually observed with nitro-substituted fluorophores. Unfortunately, the emission quantum yields of 3a–c could not be determined in water because of the compounds’ tendency to dimerize even at very low concentrations (see below). Overall, the absorption and emission data revealed a significantly less pronounced donor–acceptor interplay in the methoxy-substituted derivatives 3b and 3c as compared to the strong donor–acceptor system 3a, as clearly indicated by the blue-shifted absorption and emission bands of 3b and 3c. Consequently, the absorption bands of the electron acceptor-substituted derivative 3d were shifted to even shorter wavelengths.
Table 1.
Absorption and emission data for styrylquinolizinium derivatives 3a–d in MeCN and water.
| MeCN | H2O | |||||
| λabs/nma | λfl/nmb | Φflc | λabs/nma | λfl/nmb | ||
| 3ad | 474 | 643 | 0.02 | 434 | 630 | |
| 3b | 404 | 548 | 0.17e | 389 | 538 | |
| 3cd | 392 | 517 | 0.04 | 384 | 507 | |
| 3d | 368 | 419 | <0.01f | 371 | –g | |
aLong-wavelength absorption maximum, c(3b/3d) = 20 µM. bFluorescence maximum, λex = 394 nm (3b) and 370 nm (3d). cEmission quantum yield, determined with Abs = 0.10 at λex, estimated error of Φfl: ±10%. dTaken from [71]. eRelative to coumarin 152 (Φfl = 0.28) [71]. fRelative to coumarin 1 (Φfl = 1.00) [71]. gToo weak to be determined.
Figure 1.

Absorption spectra and normalized emission spectrum (Abs. = 0.10, 3b: λex = 394 nm) of derivatives 3b (red) and 3d (black) in MeCN.
DNA-binding properties
The DNA-binding properties of the 2-styrylquinolizinium derivatives 3a–d were investigated by spectrometric titrations of calf thymus DNA (ct DNA) to 3a–d in a phosphate buffered solution at pH 7.0 (Figure 2). During the photometric titrations, the initial absorption maxima continuously decreased and new, bathochromically shifted absorption maxima arose at 464 nm (3a), 404 nm (3b), 399 nm (3c), and 378 nm (3d), respectively (Figure 2), which clearly indicated the association of these ligands with the nucleic acid [72]. In all cases, isosbestic points developed at the beginning of the titration and eventually became indistinct, which already indicated different binding modes at particular stages of the titration.
Figure 2.

Spectrophotometric titration upon the addition of ct DNA to the styrylquinolizinium derivatives 3a (A), 3b (B), 3c (C), and 3d (D) in BPE [cL = 20 µM, cDNA = 1.45 mM (A–C), cDNA = 2.45 mM (D), cDNA in base pairs]. The insets show the plots of absorption vs DNA concentration. The arrows indicate the changes of absorption upon the addition of ct DNA.
The data from the photometric titrations are presented as binding isotherms, and fitting of the experimental data to an established theoretical model [73] gave the corresponding binding constants Kb (cf. Supporting Information File 1). Thus, the largest binding constant was determined for the dimethylamino-substituted styrylquinolizinium derivative 3a (Kb = 2.6 ± 0.1 × 105 M). The nitro-substituted derivative 3d had a slightly lower affinity with Kb = 8.2 ± 0.2 × 104 M, and the methoxy-substituted derivatives had the lowest binding constants of Kb = 4.8 ± 0.1 × 104 M (3b) and 4.1 ± 0.1 × 104 M (3c). Overall, these binding affinities resembled the ones of known DNA-intercalating benzoquinolizinium derivatives [60].
In addition, the changes of the emission properties upon the addition of ct DNA to 2-styrylquinolizinium derivatives 3a–d were determined in fluorimetric titrations (Figure 3). The intensity of the rather weak emission bands of 3a, 3b, and 3c increased significantly upon the addition of DNA. In the case of derivative 3b, a blue-shift of the emission maximum by 10 nm was also observed. Notably, compound 3a had the weakest emission intensity, i.e., it was essentially nonfluorescent in aqueous solution, but when it was bound to DNA, it showed a strong light-up effect of the emission with a factor of I/I0 = 44. For compounds 3b and 3c, significantly smaller light-up factors of I/I0 = 3.3 and 1.6, respectively, were observed. In contrast, the very low emission intensity of 3d did not change upon the addition of ct DNA. The fluorescence light-up effects of the ligands 3a–c upon association with DNA resembled the ones observed for other styryl-substituted quinolizinium derivatives [62–67]. Accordingly, the emission enhancement most likely resulted from the accommodation of the ligand in the constrained binding site of the DNA, which led to a restricted conformational flexibility. As a result, conformational changes of the styryl substituent in the excited state that lead to radiationless deactivation in solution were significantly suppressed within the binding site so that emission became competitive.
Figure 3.

Spectrofluorimetric titration upon the addition of ct DNA to the styrylquinolizinium derivatives 3a (A), 3b (B) and 3c (C) in BPE buffer [cL = 5 µM (A, C), cL = 1 µM (B), cDNA = 1.45 mM, cDNA in base pairs]. The insets show the plots of relative emission intensity vs DNA concentration. The arrows indicate the changes of emission intensity upon the addition of ct DNA.
The DNA-binding properties of the ligands 3a–d were further investigated by circular dichroism (CD) and flow linear dichroism (LD) spectroscopy [74–75] in phosphate buffer at different ligand-to-DNA ratios (LDR). The mixtures of compounds 3a–d with ct DNA showed clear induced circular dichroism (ICD) and LD bands in the absorption region of the ligands that further confirmed the binding of the ligands (Figure 4). In all cases, a positive ICD signal developed, and with increasing LDR, the characteristic CD bands of duplex DNA at 254 nm and 277 nm [76] increased slightly. Ligand 3a exhibited a strong positive and a weak negative ICD signal at 473 nm and 583 nm, respectively, in the presence of DNA, along with a weaker positive signal at 346 nm (Figure 4A1). For LD spectroscopic analysis, the DNA molecules were oriented in a hydrodynamic field of a rotating couette (flow linear dichroism). The corresponding LD spectra were the result of the differential absorption of linearly polarized light, which was polarized parallel and perpendicular to a reference axis, respectively, thus indicating the orientation of the transition moment of the chromophores relative to the electric field vector of the light [75]. The LD spectrum of DNA-bound 3a displayed a negative band in the absorption range of the ligand at small LDR (≤1.0) at 506 nm, whereas at higher values, a positive band developed, which led to a distorted bisignate band. In the case of ligands 3b and 3c, a similar development of LD bands was observed with increasing LDR, however, the effect was more pronounced with a strong positive LD signal at 397 nm (3b) and 382 nm (3c) at LDR = 0.5 (Figure 4B2 and Figure 4C2). Interestingly, the CD spectra of 3b and 3c did not resemble the ones of 3a. Both ligands showed a clear positive ICD band at 400–407 nm (3b) and 382 nm (3c), but only in the case of 3b, a weak blue-shifted ICD band also appeared at lower LDR (Figure 4B1 and Figure 4C1). Ligand 3d exhibited positive ICD and negative LD signals at 382 nm upon binding to DNA (Figure 4D). Altogether, the CD and LD spectra of ligands 3a–c at low LDR as well as the ones of 3d in general showed the characteristic signatures of DNA intercalators. Namely, the negative LD bands of the bound ligands unambiguously revealed an intercalative mode [75–76], whereas the positive ICD bands indicated an essentially perpendicular alignment of the transition dipole moments of the ligands relative to the ones of the DNA base pairs [75–76]. Considering a dipole moment of the donor–acceptor systems 3a–c along the long molecular axis, a binding mode in which the ligand is accommodated in the intercalation site with its long molecular axis perpendicular to the long axis of the binding site could be deduced. With increasing LDR, however, another binding mode became predominant for the ligands 3a–c, as particularly indicated by the development of a positive LD band in the absorption range of the ligand that denoted groove binding [74–76]. It is proposed that with increasing ligand concentration, i.e., at larger LDR, the ligands tended to form aggregates, as commonly observed for donor–acceptor dyes, that stacked along the grooves of DNA.
Figure 4.

CD and LD spectra of the styryl derivatives 3a (A), 3b (B), 3c (C), and 3d (D) with ct DNA in BPE buffer [cct DNA = 20 µM (A1, B1), cct DNA = 50 µM (C1, D1), and cct DNA = 500 µM (A2–D2), with LDR = 0 (black), 0.5 (red), 1.0 (blue), 1.5 (green), and 2.0 (magenta), cct DNA in base pairs).
Photocycloaddition reactions
The photochemical properties of the derivatives 3a–d were investigated. Firstly, the substrates were irradiated in acetonitrile solution at 520–535 nm (3a), 420–470 nm (3b and 3c), and >395 nm (3d), and the photoreaction was monitored photometrically. Notably, the amino-substituted derivative 3a did not react under these conditions, as indicated by only marginal changes of the absorption spectrum (Figure 5A). Presumably, the strong donor–acceptor system in 3a led to an intramolecular charge-transfer (ICT) state that did not lead to a subsequent photoreaction [77]. In contrast, the absorption bands of the substrates 3b–d decreased relatively fast upon irradiation, but the maxima did not disappear completely (Figure 5B–D). Even after 4 h, compound 3b exhibited a weak band at λabs = 404 nm, whereas the newly formed band at λabs = 332 nm did not increase further (Figure 5B). In this case, additional 1H NMR spectroscopic analysis showed that the derivatives 3c and 3d were initially converted to the Z-isomer by irradiation at λ = 450 nm or λ = 360 nm in acetonitrile, as indicated by the upfield shift of the signals of the alkene double bonds and the characteristic coupling constants of Z-configured protons (3JH–H = 12 Hz). Notably, the derivative 3c did not react any further under these conditions (cf. Supporting Information File 1). However, it was observed that further irradiation of the nitro-substituted derivative 3d furnished the dimer in acetonitrile, as shown by the development of the characteristic cyclobutane protons at 4.85–4.95 ppm. In contrast, the NMR-spectroscopic analysis in D2O showed that the derivative 3b gave the corresponding cycloaddition product much faster, i.e., within a few minutes under these conditions, and the formation of the corresponding Z-isomer proceeded only to a marginal extent.
Figure 5.

Spectrophotometric monitoring of the irradiation of styrylquinolizinium derivatives 3a (A), 3b (B), 3c (C), and 3d (D) in acetonitrile [cL = 10 µM (A), cL = 20 µM (B, C, D)]. The arrows indicate the changes of absorption upon irradiation.
In aqueous solution, the substrates 3a–d showed essentially the same photochemical behavior, however, with different reaction times and conversions. Thus, the photoreaction of derivative 3b was complete after 90 min (Figure 6A), whereas the reaction of derivative 3c took more than 5 h. The early stages of the photoreaction of substrate 3b in water were monitored in short time intervals (1 s) to identify possible primary photoprocesses (Figure 7A). The initial maximum of the monomer 3b decreased substantially by approximately half within a second, whereas further reaction was indicated by the appearance of the absorption maximum of 4b at 317–331 nm. Notably, no additional intermediate absorption band appeared, and three isosbestic points developed at 239 nm, 310 nm, and 337 nm after the initial steps. These observations provided evidence that the phototransformation of the styrylquinolizinium species 3b to its photodimer 4b was a two-step process.
Figure 6.

Absorption of the monomers (c = 20 µM, red) 3b (A) and 3c (B) and their dimers (black) 4b and 4c in H2O after 1.5 h and 4 h, respectively, at ca. 450 nm.
Figure 7.

Photometric monitoring of the photoreaction of 3b (c = 20 µM) to the dimer 4b by irradiation at ca. 450 nm in H2O (A) and of the photoinduced cycloreversion of 4b (c = 20 µM) to the monomer 3b at 315 nm in H2O (B).
Preparative-scale photoreactions were performed with the methoxy-substituted derivatives 3b and 3c because the photometric studies (see above) indicated reasonable reaction times. Unfortunately, it turned out that due to the low solubility of these derivatives in water, the concentrations required for a bimolecular reaction could not be accomplished. However, it is well known that [2 + 2] photodimerizations can also be performed in the solid state or with a thoroughly stirred suspension [37,43,78]. Therefore, suspensions of 3b und 3c in water were irradiated with an LED lamp at 450–470 nm to give the 2,2'-(2,4-diphenyl-1,3-cyclobutanediyl)bisquinolizinium 4b and 4c as photoproducts in quantitative yield. The products 4b and 4c were fully characterized by NMR spectroscopy (1H, 13C, COSY, HSQC, HMBC, and ROESY) and mass spectrometry, which revealed a cyclobutane structure, specifically by the appearance of the characteristic NMR signals of the cyclobutane at 4.89–5.00 ppm [42–46]. Unfortunately, detailed 2D NMR and spectroscopic analyses did not allow a conclusive assignment of the configuration of the products. Even in the ROESY NMR spectra, only unspecific correlations were detected. However, as both products could be obtained as single crystals after slow evaporation, their structure was determined by single crystal X-ray diffraction (XRD) analysis (Figure 8, cf. Supporting Information File 1). The cyclobutane 4b crystallized from water in the monoclinic space group P21/n, and the derivative 4c crystallized from water in the triclinic space group 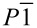 . Both XRD analyses clearly showed that both cyclobutane products were formed as rctt configured dimers 4b and 4c (Figure 8).
. Both XRD analyses clearly showed that both cyclobutane products were formed as rctt configured dimers 4b and 4c (Figure 8).
Figure 8.

ORTEP drawings of cyclobutane derivatives 4b (A) and 4c (B) in the solid state (thermal ellipsoids indicate 50% probability). The tetrafluoroborate counterions were omitted for clarity.
The products 4b and 4c may have formed by a syn head-to-tail dimerization of the E-configured substrate 3a and 3b or by an anti head-to-tail photodimerization of the initially formed Z-isomers Z-3a and Z-3b, with both processes generally being possible starting from 3b and 3c (Scheme 2). On the one hand, the photometric monitoring as well as the 1H NMR spectroscopic studies of the photoreaction of 3b indicated a preceding E-to-Z isomerization (cf. Supporting Information File 1) that may have been followed by a [2 + 2] photodimerization (Scheme 2). However, it is difficult to explain why the photocycloaddition of the Z-isomers Z-3b and Z-3c led exclusively to the dimer, because such a selectivity has not been reported so far for (Z)-stilbenes. On the other hand, the reaction was performed using suspensions, so that the reaction may also have taken place with undissolved solid in which the E-to-Z isomerization was most likely suppressed due to the restricted space in the confined medium. Thus, the selective formation of the dimers 4b and 4c is reminiscent of the high stereoselectivity observed for [2 + 2] photodimerizations in organized media or in the solid state [37–41].
Scheme 2.

Possible pathways for the selective photodimerization of styrylquinolizinium derivatives 3b and 3c.
Considering the pronounced donor–acceptor interplay in 3b and 3c and the resulting strong dipole moment, it may be proposed that these compounds form dimeric aggregates in the solid state and even in solution through dipole–dipole interactions and directional π stacking (Scheme 2), as observed, for example, with donor-substituted benzoquinolizinium derivatives [79] or donor-substituted styrylpyridinium derivatives [80–82]. Hence, an ideal overlap of the π systems and antiparallel alignment of dipole moments is realized in a syn head-to-tail complex where irradiation would lead directly to the photodimers 4b and 4c in a topochemical reaction (Scheme 2).
Notably, the cyclobutane derivatives 4b and 4c were not persistent in solution for extended periods of time. As already shown for several cyclobutane derivatives, these compounds tend to isomerize to the corresponding rttt isomers [83–86].
With derivative 4b as a representative example, it was demonstrated that the photodimers can be transformed back to the monomers. Thus, upon irradiation of cyclobutane 4b at 315 nm in H2O, the monomer 3b formed, as indicated by the development of its characteristic absorption band (Figure 7B). After 30 min, the reaction was almost complete, however, dimer 4b still remained in solution in the photostationary state.
Interactions of the photodimer 4b with DNA
The interactions of dimer 4b with DNA were investigated by photometric titrations as well as by CD and LD spectroscopy (Figure 9). Upon the addition of ct DNA to compound 4b in buffered solution, the absorption maximum decreased slightly, but apart from a broadening of the band at the long-wavelength tail, the overall shape of the spectrum did not change (Figure 9). Furthermore, only a small positive ICD band in the absorption region of ligand 4b appeared at 300–350 nm that developed into a significantly broader band with increasing LDR. At the same time, the signal of the DNA did not change in the presence of the ligand. Additionally, the LD experiment showed a small positive signal at 300–350 nm, and the negative band of the ct DNA at 254 nm decreased. These spectroscopic data indicated a very weak interaction of the substrate 4b with DNA, and the band broadening in the absorption region already confirmed an aggregation of the molecules along the DNA backbone at very high ligand concentrations. Nevertheless, the CD and LD spectroscopic data revealed at least some specific binding interactions of 4b with DNA that caused a distinct orientation of the aromatic units relative to the host DNA. In particular, the weak positive LD band indicated an alignment of the aromatic units along the DNA grooves. In addition, the close vicinity of the quinolizinium substituents to the DNA helix was further confirmed by the CD band in the absorption region of the quinolizinium moiety, as it resulted from the coupling of its transition dipole moment with the ones of the DNA bases. Overall, these data revealed a loose binding of the cyclobutane derivative 4b to DNA through outside-edge binding of the ligand that enabled the association of one or two aromatic units in the DNA grooves.
Figure 9.

A) Spectrophotometric titration of ct DNA to dimer 4b in BPE buffer (cL = 20 µM, cct DNA = 1.45 mM, cct DNA in base pairs). The arrow indicates the changes of absorption upon the addition of ct DNA. B) CD spectra of the dimer 4b with ct DNA (50 µM) in BPE buffer with LDR = 0 (black), 0.5 (red), 1.0 (blue), and 2.0 (green). C) LD spectra of dimer 4b with ct DNA (c = 500 µM) in BPE buffer with LDR = 0 (black), 0.04 (red), 0.08 (blue), and 2.0 (green).
Photoswitching of the DNA binding properties
Finally, it was tested whether the DNA-binding quinolizinium derivative 3b could be released photochemically from cyclobutane 4b in the presence of DNA. For that purpose, a mixture of the photodimer and DNA was irradiated at 315 nm using an LED, and the reaction was monitored by absorption and CD spectroscopy (Figure 10). In the course of the photoreaction, the formation of 3b was indicated by the emergence of its characteristic long-wavelength absorption band, whose shape and shift matched its DNA-bound form. The association of the released monomer 3b with DNA was also clearly demonstrated by the ICD band of the DNA-bound ligand. It should be noted, however, that the photoinduced conversion of the dimer was not complete, indicating a photostationary state. Noteworthy, irradiation of the bound ligand at ca. 450 nm using an LED regenerated the cyclobutane dimer, as shown unambiguously by the formation of the characteristic signature of its absorption and CD bands and by the disappearance of the monomer’s signals (Figure 10). Although the sequence of photocycloreversion and photoaddition could be performed four times, a slight but steady photobleaching or photodecomposition was observed. It should be noted that the DNA-bound ligand did not dimerize upon irradiation because within the intercalation site, it could not approach another quinolizinium molecule that was required for the photoreaction. Instead, the photodimerization most likely involved the free or loosely backbone-associated ligands that were in a dynamic equilibrium with the respective intercalator–DNA complexes, as shown for aryl stilbazonium ligands [35]. At the same time, the photoinduced cycloreversion may have taken place both with the free or DNA-bound dimer. Specifically, the dimer is only loosely bound to the DNA backbone so that the cycloreversion reaction does not experience steric constrains that may hinder the photoreaction. Furthermore, it has been demonstrated that the photoinduced cycloreversion of quinolizinium dimers is even enhanced in the presence of DNA [36].
Figure 10.

A) Photometric and B) CD spectroscopic monitoring of the photoinduced switching (4b: λex = 315 nm, 3b: λex = 450 nm) between 4b (c = 20 µM, black) and 3b (red) in the presence of ct DNA (c = 20 µM) in BPE buffer. DNA concentration in base pairs.
Conclusion
In summary, we have shown that appropriately substituted styrylquinolizinium derivatives constitute a new class of photoswitchable DNA ligands. It was shown that these ligands bind to duplex DNA mainly by intercalation and that their syn head-to-tail photodimers, obtained by selective [2 + 2] photocycloaddition, bind to DNA only weakly by outside-edge association. Most notably, it was possible to switch between those two binding modes by irradiation with different excitation wavelengths (Scheme 3). Although the system still has to be improved with respect to photostability, it may be considered as a promising complementary approach toward the development of photoswitchable bioactive compounds.
Scheme 3.

Photoinduced switching of the DNA binding properties of styrylquinolizinium compound 3b.
Supporting Information
Additional spectroscopic data, detailed experimental procedures, 1H NMR spectra, and crystallographic data.
Acknowledgments
We thank Ms. Sandra Uebach, Ms. Jennifer Hermann, and Mr. Aboubakr Hamad for technical assistance.
This article is part of the thematic issue "Molecular switches" and is dedicated to Prof. Dr. Hans-Jörg Deiseroth, University of Siegen, on the occasion of his 75th birthday.
Funding Statement
Financial support by the Deutsche Forschungsgemeinschaft (DFG) is gratefully acknowledged.
References
- 1.Rahman A, O'Sullivan P, Rozas I. Med Chem Commun. 2019;10:26–40. doi: 10.1039/c8md00425k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bhaduri S, Ranjan N, Arya D P. Beilstein J Org Chem. 2018;14:1051–1086. doi: 10.3762/bjoc.14.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang M, Yu Y, Liang C, Lu A, Zhang G. Int J Mol Sci. 2016;17:No. 779. doi: 10.3390/ijms17060779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rescifina A, Zagni C, Varrica M G, Pistarà V, Corsaro A. Eur J Med Chem. 2014;74:95–115. doi: 10.1016/j.ejmech.2013.11.029. [DOI] [PubMed] [Google Scholar]
- 5.Bolhuis A, Aldrich-Wright J R. Bioorg Chem. 2014;55:51–59. doi: 10.1016/j.bioorg.2014.03.009. [DOI] [PubMed] [Google Scholar]
- 6.Banerjee S, Veale E B, Phelan C M, Murphy S A, Tocci G M, Gillespie L J, Frimannsson D O, Kelly J M, Gunnlaugsson T. Chem Soc Rev. 2013;42:1601–1618. doi: 10.1039/c2cs35467e. [DOI] [PubMed] [Google Scholar]
- 7.Pazos E, Mosquera J, Vázquez M E, Mascareñas J L. ChemBioChem. 2011;12:1958–1973. doi: 10.1002/cbic.201100247. [DOI] [PubMed] [Google Scholar]
- 8.Ihmels H, Thomas L. Intercalation of Organic Ligands as a Tool to Modify the Properties of DNA. In: Jin J-l, Grote J., editors. Materials Science of DNA Chemistry. Boca Raton, FL, USA: CRC Press; 2011. pp. 49–75. [Google Scholar]
- 9.Pett L, Hartley J, Kiakos K. Curr Top Med Chem. 2015;15(14):1293–1322. doi: 10.2174/1568026615666150413155431. [DOI] [PubMed] [Google Scholar]
- 10.Pommier Y. ACS Chem Biol. 2013;8:82–95. doi: 10.1021/cb300648v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carelle N, Piotto E, Bellanger A, Germanaud J, Thuillier A, Khayat D. Cancer. 2002;95:155–163. doi: 10.1002/cncr.10630. [DOI] [PubMed] [Google Scholar]
- 12.Sonis S T, Elting L S, Keefe D, Peterson D E, Schubert M, Hauer-Jensen M, Bekele B N, Raber-Durlacher J, Donnelly J P, Rubenstein E B. Cancer. 2004;100(Suppl 9):1995–2025. doi: 10.1002/cncr.20162. [DOI] [PubMed] [Google Scholar]
- 13.Ludwig H, Van Belle S, Barrett-Lee P, Birgegård G, Bokemeyer C, Gascón P, Kosmidis P, Krzakowski M, Nortier J, Olmi P, et al. Eur J Cancer. 2004;40(15):2293–2306. doi: 10.1016/j.ejca.2004.06.019. [DOI] [PubMed] [Google Scholar]
- 14.Albini A, Fagnoni M. The Greenest Reagent in Organic Synthesis: Light. In: Tundo P, Esposito V, editors. Green Chemical Reactions. Dordrecht, Netherlands: Springer; 2008. pp. 173–189. ((NATO Science for Peace and Security Series (Series C: Environmental Security))). [DOI] [Google Scholar]
- 15.Pianowski Z L. Chem – Eur J. 2019;25:5128–5144. doi: 10.1002/chem.201805814. [DOI] [PubMed] [Google Scholar]
- 16.Hüll K, Morstein J, Trauner D. Chem Rev. 2018;118:10710–10747. doi: 10.1021/acs.chemrev.8b00037. [DOI] [PubMed] [Google Scholar]
- 17.Szymański W, Beierle J M, Kistemaker H A V, Velema W A, Feringa B L. Chem Rev. 2013;113(8):6114–6178. doi: 10.1021/cr300179f. [DOI] [PubMed] [Google Scholar]
- 18.Velema W A, Szymanski W, Feringa B L. J Am Chem Soc. 2014;136:2178–2191. doi: 10.1021/ja413063e. [DOI] [PubMed] [Google Scholar]
- 19.Andersson J, Li S, Lincoln P, Andréasson J. J Am Chem Soc. 2008;130(36):11836–11837. doi: 10.1021/ja801968f. [DOI] [PubMed] [Google Scholar]
- 20.Hammarson M, Nilsson J R, Li S, Lincoln P, Andréasson J. Chem – Eur J. 2014;20(48):15855–15862. doi: 10.1002/chem.201405113. [DOI] [PubMed] [Google Scholar]
- 21.Brieke C, Heckel A. Chem – Eur J. 2013;19:15726–15734. doi: 10.1002/chem.201302640. [DOI] [PubMed] [Google Scholar]
- 22.Czerwinska I, Juskowiak B. Int J Biol Macromol. 2012;51:576–582. doi: 10.1016/j.ijbiomac.2012.06.027. [DOI] [PubMed] [Google Scholar]
- 23.Fortuna C G, Mazzucato U, Musumarra G, Pannacci D, Spalletti A. J Photochem Photobiol, A. 2010;216:66–72. doi: 10.1016/j.jphotochem.2010.09.007. [DOI] [Google Scholar]
- 24.Dohno C, Uno S-n, Nakatani K. J Am Chem Soc. 2007;129(39):11898–11899. doi: 10.1021/ja074325s. [DOI] [PubMed] [Google Scholar]
- 25.Dohno C, Yamamoto T, Nakatani K. Eur J Org Chem. 2009:4051–4058. doi: 10.1002/ejoc.200900323. [DOI] [Google Scholar]
- 26.Basak A, Mitra D, Kar M, Biradha K. Chem Commun. 2008:3067–3069. doi: 10.1039/b801644e. [DOI] [PubMed] [Google Scholar]
- 27.Wang X, Huang J, Zhou Y, Yan S, Weng X, Wu X, Deng M, Zhou X. Angew Chem. 2010;122:5433–5437. doi: 10.1002/ange.201002290. [DOI] [PubMed] [Google Scholar]
- 28.Bergen A, Rudiuk S, Morel M, Le Saux T, Ihmels H, Baigl D. Nano Lett. 2016;16:773–780. doi: 10.1021/acs.nanolett.5b04762. [DOI] [PubMed] [Google Scholar]
- 29.Linares M, Sun H, Biler M, Andréasson J, Norman P. Phys Chem Chem Phys. 2019;21(7):3637–3643. doi: 10.1039/c8cp05326j. [DOI] [PubMed] [Google Scholar]
- 30.Mammana A, Carroll G T, Areephong J, Feringa B L. J Phys Chem B. 2011;115:11581–11587. doi: 10.1021/jp205893y. [DOI] [PubMed] [Google Scholar]
- 31.Pace T C S, Müller V, Li S, Lincoln P, Andréasson J. Angew Chem, Int Ed. 2013;52(16):4393–4396. doi: 10.1002/anie.201209773. [DOI] [PubMed] [Google Scholar]
- 32.Presa A, Barrios L, Cirera J, Korrodi-Gregório L, Pérez-Tomás R, Teat S J, Gamez P. Inorg Chem. 2016;55(11):5356–5364. doi: 10.1021/acs.inorgchem.6b00362. [DOI] [PubMed] [Google Scholar]
- 33.Paramonov S V, Lokshin V, Ihmels H, Fedorova O A. Photochem Photobiol Sci. 2011;10:1279–1282. doi: 10.1039/c1pp05094j. [DOI] [PubMed] [Google Scholar]
- 34.Ihmels H, Mattay J, May F, Thomas L. Org Biomol Chem. 2013;11:5184–5188. doi: 10.1039/c3ob40930a. [DOI] [PubMed] [Google Scholar]
- 35.Juskowiak B, Chudak M. Photochem Photobiol. 2004;79(2):137–144. doi: 10.1111/j.1751-1097.2004.tb00003.x. [DOI] [PubMed] [Google Scholar]
- 36.Ihmels H, Otto D, Dall'Acqua F, Faccio A, Moro S, Viola G. J Org Chem. 2006;71(22):8401–8411. doi: 10.1021/jo0612271. [DOI] [PubMed] [Google Scholar]
- 37.Ramamurthy V, Sivaguru J. Chem Rev. 2016;116:9914–9993. doi: 10.1021/acs.chemrev.6b00040. [DOI] [PubMed] [Google Scholar]
- 38.Bibal B, Mongin C, Bassani D M. Chem Soc Rev. 2014;43:4179–4198. doi: 10.1039/c3cs60366k. [DOI] [PubMed] [Google Scholar]
- 39.MacGillivray L R, Papaefstathiou G S, Friščić T, Hamilton T D, Bučar D-K, Chu Q, Varshney D B, Georgiev I G. Acc Chem Res. 2008;41(2):280–291. doi: 10.1021/ar700145r. [DOI] [PubMed] [Google Scholar]
- 40.Nagarathinam M, Peedikakkal A M P, Vittal J J. Chem Commun. 2008:5277–5288. doi: 10.1039/b809136f. [DOI] [PubMed] [Google Scholar]
- 41.Mishra A, Behera R K, Behera P K, Mishra B K, Behera G B. Chem Rev. 2000;100(6):1973–2012. doi: 10.1021/cr990402t. [DOI] [PubMed] [Google Scholar]
- 42.Fedorova O A, Saifutiarova A E, Gulakova E N, Guskova E O, Aliyeu T M, Shepel N E, Fedorov Y V. Photochem Photobiol Sci. 2019;18:2208–2215. doi: 10.1039/c9pp00028c. [DOI] [PubMed] [Google Scholar]
- 43.Wei P, Zhang J-X, Zhao Z, Chen Y, He X, Chen M, Gong J, Sung H H-Y, Williams I D, Lam J W Y, et al. J Am Chem Soc. 2018;140:1966–1975. doi: 10.1021/jacs.7b13364. [DOI] [PubMed] [Google Scholar]
- 44.Berdnikova D V, Aliyeu T M, Delbaere S, Fedorov Y V, Jonusauskas G, Novikov V V, Pavlov A A, Peregudov A S, Shepel' N E, Zubkov F I, et al. Dyes Pigm. 2017;139:397–402. doi: 10.1016/j.dyepig.2016.11.053. [DOI] [Google Scholar]
- 45.Budyka M F, Gavrishova T N, Potashova N I, Chernyak A V. Mendeleev Commun. 2015;25:106–108. doi: 10.1016/j.mencom.2015.03.008. [DOI] [Google Scholar]
- 46.Ushakov E N, Vedernikov A I, Lobova N A, Dmitrieva S N, Kuz’mina L G, Moiseeva A A, Howard J A K, Alfimov M V, Gromov S P. J Phys Chem A. 2015;119(52):13025–13037. doi: 10.1021/acs.jpca.5b10758. [DOI] [PubMed] [Google Scholar]
- 47.Berdnikova D V, Sosnin N I, Fedorova O A, Ihmels H. Org Biomol Chem. 2018;16:545–554. doi: 10.1039/c7ob02736b. [DOI] [PubMed] [Google Scholar]
- 48.Botti V, Cesaretti A, Ban Ž, Crnolatac I, Consiglio G, Elisei F, Piantanida I. Org Biomol Chem. 2019;17(35):8243–8258. doi: 10.1039/c9ob01186b. [DOI] [PubMed] [Google Scholar]
- 49.Wang M-Q, Liu S, Tang C-P, Raza A, Li S, Gao L-X, Sun J, Guo S-P. Dyes Pigm. 2017;136:78–84. doi: 10.1016/j.dyepig.2016.08.041. [DOI] [Google Scholar]
- 50.Narayanaswamy N, Narra S, Nair R R, Saini D K, Kondaiah P, Govindaraju T. Chem Sci. 2016;7:2832–2841. doi: 10.1039/c5sc03488d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Berdnikova D V, Fedorova O A, Tulyakova E V, Li H, Kölsch S, Ihmels H. Photochem Photobiol. 2015;91(3):723–731. doi: 10.1111/php.12405. [DOI] [PubMed] [Google Scholar]
- 52.Narayanaswamy N, Das S, Samanta P K, Banu K, Sharma G P, Mondal N, Dhar S K, Pati S K, Govindaraju T. Nucleic Acids Res. 2015;43:8651–8663. doi: 10.1093/nar/gkv875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Xie X, Choi B, Largy E, Guillot R, Granzhan A, Teulade-Fichou M-P. Chem – Eur J. 2013;19(4):1214–1226. doi: 10.1002/chem.201203710. [DOI] [PubMed] [Google Scholar]
- 54.Mazzoli A, Carlotti B, Consiglio G, Fortuna C G, Miolo G, Spalletti A. Photochem Photobiol Sci. 2014;13(6):939–950. doi: 10.1039/c4pp00023d. [DOI] [PubMed] [Google Scholar]
- 55.Mazzoli A, Carlotti B, Bonaccorso C, Fortuna C G, Mazzucato U, Miolo G, Spalletti A. Photochem Photobiol Sci. 2011;10:1830–1836. doi: 10.1039/c1pp05214d. [DOI] [PubMed] [Google Scholar]
- 56.Fortuna C G, Barresi V, Berellini G, Musumarra G. Bioorg Med Chem. 2008;16:4150–4159. doi: 10.1016/j.bmc.2007.12.042. [DOI] [PubMed] [Google Scholar]
- 57.Fortuna C G, Barresi V, Bonaccorso C, Consiglio G, Failla S, Trovato-Salinaro A, Musumarra G. Eur J Med Chem. 2012;47:221–227. doi: 10.1016/j.ejmech.2011.10.060. [DOI] [PubMed] [Google Scholar]
- 58.Barresi V, Bonaccorso C, Consiglio G, Goracci L, Musso N, Musumarra G, Satriano C, Fortuna C G. Mol BioSyst. 2013;9(10):2426–2429. doi: 10.1039/c3mb70151d. [DOI] [PubMed] [Google Scholar]
- 59.Schmidt D, Rodat T, Heintze L, Weber J, Horbert R, Girreser U, Raeker T, Bußmann L, Kriegs M, Hartke B, et al. ChemMedChem. 2018;13:2415–2426. doi: 10.1002/cmdc.201800531. [DOI] [PubMed] [Google Scholar]
- 60.Granzhan A, Ihmels H. Synlett. 2016;27:1775–1793. doi: 10.1055/s-0035-1561445. [DOI] [Google Scholar]
- 61.Ihmels H, Karbasiyoun M, Löhl K, Stremmel C. Org Biomol Chem. 2019;17:6404–6413. doi: 10.1039/c9ob00809h. [DOI] [PubMed] [Google Scholar]
- 62.Xie X, Zuffo M, Teulade-Fichou M-P, Granzhan A. Beilstein J Org Chem. 2019;15:1872–1889. doi: 10.3762/bjoc.15.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Das A K, Ihmels H, Kölsch S. Photochem Photobiol Sci. 2019;18:1373–1381. doi: 10.1039/c9pp00096h. [DOI] [PubMed] [Google Scholar]
- 64.Chang L, Liu C, He S, Lu Y, Zhang S, Zhao L, Zeng X. Sens Actuators, B. 2014;202:483–488. doi: 10.1016/j.snb.2014.05.089. [DOI] [Google Scholar]
- 65.Yao H, Chang L, Liu C, Jiao X, He S, Liu H, Zeng X. J Fluoresc. 2015;25:1637–1643. doi: 10.1007/s10895-015-1650-x. [DOI] [PubMed] [Google Scholar]
- 66.Zacharioudakis E, Cañeque T, Custodio R, Müller S, Cuadro A M, Vaquero J J, Rodriguez R. Bioorg Med Chem Lett. 2017;27(2):203–207. doi: 10.1016/j.bmcl.2016.11.074. [DOI] [PubMed] [Google Scholar]
- 67.Maçoas E, Marcelo G, Pinto S, Cañeque T, Cuadro A M, Vaquero J J, Martinho J M G. Chem Commun. 2011;47:7374–7376. doi: 10.1039/c1cc12163d. [DOI] [PubMed] [Google Scholar]
- 68.Richards A, Stevens T S. J Chem Soc. 1958:3067–3073. doi: 10.1039/jr9580003067. [DOI] [Google Scholar]
- 69.Marcelo G, Pinto S, Cañeque T, Mariz I F A, Cuadro A M, Vaquero J J, Martinho J M G, Maçôas E M S. J Phys Chem A. 2015;119(11):2351–2362. doi: 10.1021/jp507095b. [DOI] [PubMed] [Google Scholar]
- 70.Pithan P M, Decker D, Druzhinin S I, Ihmels H, Schönherr H, Voß Y. RSC Adv. 2017;7:10660–10667. doi: 10.1039/c6ra27684a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jones G, Jackson W R, Choi C-y, Bergmark W R. J Phys Chem. 1985;89:294–300. doi: 10.1021/j100248a024. [DOI] [Google Scholar]
- 72.Sirajuddin M, Ali S, Badshah A. J Photochem Photobiol, B. 2013;124:1–19. doi: 10.1016/j.jphotobiol.2013.03.013. [DOI] [PubMed] [Google Scholar]
- 73.Stootman F H, Fisher D M, Rodger A, Aldrich-Wright J R. Analyst. 2006;131:1145–1151. doi: 10.1039/b604686j. [DOI] [PubMed] [Google Scholar]
- 74.Šmidlehner T, Piantanida I, Pescitelli G. Beilstein J Org Chem. 2018;14:84–105. doi: 10.3762/bjoc.14.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Norden B, Rodger A, Dafforn T. Linear Dichroism and Circular Dichroism. Cambridge, U.K.: Royal Society of Chemistry; 2010. [Google Scholar]
- 76.Nordén B, Kurucsev T. J Mol Recognit. 1994;7(2):141–155. doi: 10.1002/jmr.300070211. [DOI] [PubMed] [Google Scholar]
- 77.Holzmann N, Bernasconi L, Callaghan K M, Bisby R H, Parker A W. Chem Phys Lett. 2018;692:146–151. doi: 10.1016/j.cplett.2017.12.028. [DOI] [Google Scholar]
- 78.Amjaour H, Wang Z, Mabin M, Puttkammer J, Busch S, Chu Q R. Chem Commun. 2019;55:214–217. doi: 10.1039/c8cc08017h. [DOI] [PubMed] [Google Scholar]
- 79.Ihmels H, Luo J. J Photochem Photobiol, A. 2008;200:3–9. doi: 10.1016/j.jphotochem.2008.04.008. [DOI] [Google Scholar]
- 80.Mondal B, Zhang T, Prabhakar R, Captain B, Ramamurthy V. Photochem Photobiol Sci. 2014;13:1509–1520. doi: 10.1039/c4pp00221k. [DOI] [PubMed] [Google Scholar]
- 81.Yamada S, Azuma Y, Aya K. Tetrahedron Lett. 2014;55:2801–2804. doi: 10.1016/j.tetlet.2014.03.036. [DOI] [Google Scholar]
- 82.Mondal B, Captain B, Ramamurthy V. Photochem Photobiol Sci. 2011;10:891–894. doi: 10.1039/c1pp05070b. [DOI] [PubMed] [Google Scholar]
- 83.Hill Y, Linares M, Briceño A. New J Chem. 2012;36:554–557. doi: 10.1039/c2nj20855e. [DOI] [Google Scholar]
- 84.Kole G K, Tan G K, Vittal J J. J Org Chem. 2011;76:7860–7865. doi: 10.1021/jo201268p. [DOI] [PubMed] [Google Scholar]
- 85.Peedikakkal A M P, Peh C S Y, Koh L L, Vittal J J. Inorg Chem. 2010;49:6775–6777. doi: 10.1021/ic100853h. [DOI] [PubMed] [Google Scholar]
- 86.Horner M, Hünig S. Liebigs Ann Chem. 1982:1183–1210. doi: 10.1002/jlac.198219820619. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional spectroscopic data, detailed experimental procedures, 1H NMR spectra, and crystallographic data.