

Abstract
Background
Previous studies have shown that lung cancer stem cells express CD133 and that certain cancer stem cells express cancer germline antigens (CGAs). The transcriptional regulation of CD133 is complicated and poorly understood. We investigated CD133 and CGA expression in a non-small cell lung cancer cell line.
Methods
The expression levels of CD133 and CGAs (MAGE-6, GAGE, SSX, and TRAG-3) were measured in an NCI-H292 lung cancer cell line. The methylation status of the CD133 gene promoter region was analyzed. The expression levels and promoter methylation statuses of CD133 and CGAs were confirmed by treatment with the demethylating agent 5-aza-2′-deoxycytidine (ADC).
Results
After treatment with ADC, CD133 expression was no longer detected. MAGE-6 and TRAG-3 were detected before ADC treatment, while GAGE and SSX were not detected. ADC treatment upregulated MAGE-6 and TRAG-3 expression, while GAGE expression was still undetected after treatment, and only weak SSX expression was observed. GAGE expression was not correlated with expression of CD133, while the levels of expression of MAGE-6, TRAG-3, and SSX were inversely correlated with CD133 expression.
Conclusion
These results showed that CD133 expression can be regulated by methylation. Thus, the demethylation of the CD133 promoter may compromise the treatment of lung cancer by inactivating cancer stem cells and/or activating CGAs.
Keywords: Lung neoplasms, Neoplastic stem cells, Demethylation
Introduction
The cancer stem cell (CSC) hypothesis suggests that intra-tumoral heterogeneity may be accounted for by a subclone of cells that are capable of self-renewal, new tumor formation, and repopulation of the heterogeneous original tumor [1]. Thus, therapeutic interventions should target these cells rather than the tumor mass itself, as CSCs may be an important cause of conventional therapeutic failure in various malignant tumors [1]. CD133, which is associated with organ-specific stem cells, has been described as a putative marker of cancer-initiating cells in various tumor types [2]. Cancer-initiating cells exhibit a low proliferative rate, as well as self-renewal capacity, differentiation potential among actively proliferating tumor cells, and resistance to chemotherapy and radiotherapy. Recently, CD133 overexpression in non-small-cell lung cancer (NSCLC) was found to be associated with neuroendocrine histological transformation and resistance to targeted therapy [3]. Tirino et al. [2] reported that CD133-positive (CD133+) cells isolated from NSCLC can form tumors and act as tumor-initiating cells. As such, the clinical relationship between CD133 expression and NSCLC has been an area of intense investigation in medical oncology. Hilbe et al. [4] noted that CD133+ cells may contribute to tumor angiogenesis in NSCLC. Furthermore, Bertolini et al. [5] reported that CD133 is involved in the response to chemotherapy in lung cancer. High CD133 expression is correlated with a tendency toward shorter progression-free survival in NSCLC patients treated with platinum-containing regimens, indicating the presence of chemoresistant, highly tumorigenic, and stem-like features [5]. The molecular features of such CD133+ NSCLC cells may provide a rationale for more specific therapeutic targeting and more well-defined predictive factors in these tumors. Indeed, Gedye et al. [6] reported that CD133+ melanoma cells exhibit increased expression of specific cancer germline antigens (CGAs), such as NY-ESO-1, and these in vitro results indicate the possibility that CSCs may express similar antigens in vivo and that the immune targeting of such antigens may be a clinical strategy for cancer adjuvant therapy. In addition, several reports have shown that CD133+ tumor cells are more effective in repairing radiation-induced DNA damage than CD133− tumor cells [7]. Therefore, the exact role of CD133 in lung cancer is not yet completely understood, indicating that further studies are needed.
In recent years, the relationship between the immune system and cancer biology has become increasingly clear, and cancer immunotherapeutics with CGAs or cancer/testis antigens have been identified as potential strategies both for improving recognition and for selectively carrying out the effective elimination of tumor cells [8]. As described above, studies have reported potential relationships between CSCs and the expression of genes coding for cancer-specific antigens [7], with representative cancer-specific antigens including MAGE-6, GAGE, SSX, and TRAG-3. Therefore, we investigated the relationship between CSCs and cancer-specific antigen expression in lung cancer with the goal of applying our findings to the treatment of this disease.
In the present study, we investigated the expression of CD133 and its association with DNA methylation status in an NSCLC cell line. We also examined the relationship between CD133 expression and CGAs in lung cancer cells with the aim of furthering the identification of personalized therapies for lung cancer.
Methods
The study protocol was approved by the Institutional Review Board of Kosin University Gospel Hospital (IRB approval no., 13-008).
Cell lines and cell culture
Cells from the NCI-H292 lung cancer cell line were obtained from the Korean Cell Line Bank (Seoul, Korea). The cells were cultured in RPMI 1640 medium (Invitrogen, Mulgrave, Australia) supplemented with 10% fetal calf serum (CSL, Victoria, Australia), 2 mM glutamine, 25 mM HEPES, 100 U/mL penicillin, and 100 μg/mL streptomycin (RF10), passaged with 2 mM ethylenediaminetetraacetic acid (EDTA) in phosphate-buffered saline (pH 7.4). Cultures were maintained in humidified incubators at 37°C in a 5% CO2 and 95% air atmosphere. Routine monitoring confirmed that all cells repeatedly tested negative for mycoplasma infection, bacterial contamination, and genetic heterogeneity, the last of which was assessed with DNA fingerprinting analysis (AmpFlSTR Identifiler PCR Amplification Kit; Applied Biosystems, Foster City, CA, USA). Cell lines were used at passage 10 or less.
Cell proliferation assays
To assess the proliferation of cell populations, 96-well plates were seeded with 500 cells/well, and cell viability was measured at various time points by the conversion of methyl terazol sulfate to formazan (Promega, Madison, WI, USA) according to the manufacturer’s instructions.
Nucleic acid isolation and complementary DNA synthesis
To isolate nucleic acids, cells from the same isolation and passage were treated identically as follows. Cells were grown to subconfluence (70%), washed, and kept in serum-starved media for 2 hours. Cells were lysed using 10 mM Tris (pH 7.4), 0.1% sodium dodecyl sulfate, 0.1% Tween 20, 0.5% TritonX100, 150 mM sodium chloride, 10 mM EDTA, 1 M urea, 10 mM N-ethylmaleimide, 4 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride and collected by scraping. Both genomic DNA and total RNA were isolated from washed cell pellets. Genomic DNA was extracted using a G-DEX kit (Intron Biotechnology, Burlington, MA, USA) according to the manufacturer’s instructions. Total RNA was isolated from each of the samples after lysis in guanidinium isothiocyanate and phenol extraction using a commercial kit (Trizol; Invitrogen Laboratories, San Diego, CA, USA). For complementary DNA (cDNA) synthesis, 2 mg of total RNA was reverse transcribed using random hexamers, deoxynucleoside triphosphates, and 1 μL (200 units) of Superscript II reverse transcriptase (Invitrogen, Carlsbad, CA, USA) in a final volume of 20 μL for 80 minutes at 42°C after a 15-minute denaturation step at 80°C. Finally, 80 μL of distilled water was added to the reverse transcription (RT) reaction.
Reverse transcription-polymerase chain reaction
To analyze CD133 expression, cDNA was amplified in 25 μL of a polymerase chain reaction (PCR) mixture, together with 1 μL of the RT reaction mixture, primers, and 1 unit of Taq DNA polymerase. RT-PCR was carried out using RT-specific primers (located +343 to +679 bases from the translation start site in the messenger RNA [mRNA] sequence): a CD133 RT sense primer (5′-CTGGGGCTGCT GTTTATTATTCTG-3′) and a CD133 RT antisense primer (5′-ACGCCTTGTCCTTGGTAGTGTTG-3′) (Table 1). The PCR protocol consisted of 5 minutes at 94°C for the initial denaturation; 35 cycles of 94°C (30 seconds), 55°C (30 seconds), and 72°C (60 seconds); and a final elongation step lasting 7 minutes at 72°C. PCR amplification was performed with a programmable thermal cycler (PCR System 9700; Applied Biosystems). Primers for β-actin were used to confirm RNA integrity. cDNA from the same synthesis reaction was used for RT-PCR reactions for both CD133 and β-actin. Amplified DNA fragments were visualized on a 2% agarose gel stained with ethidium bromide. To detect CGA genes, RT-PCR was performed with DNA primers designed by the National Center for Biotechnology Information (National Institutes of Health, Bethesda, MD, USA) (Table 1).
Table 1.
Primer sequences for detecting cancer germline antigens used in this study
| Primer | Category | Sequence |
|---|---|---|
| CD133 | Sense | TATTTGGTTATGTTTTTAGTTTTTT |
| Antisense | CCTAATCAACAAATACCTCTCTC | |
| MAGE-6 | Sense | TGGAGGACCAGAGGCCCCC |
| Antisense | CAGGATGATTATCAGGAAGCCTGT | |
| GAGE | Sense | GACCAAGGCGCTATGTAC |
| Antisense | GTCCATCTCCTGCCCCTG | |
| SSX | Sense | GTGCCATGAACGGAGACGA |
| Antisense | GTCTGTGGGTCCAGGCATGT | |
| TRAG-3 | Sense | TGGGGGAGGCTTGGAGAGACC |
| Antisense | TGCCCTTGTGGTCCCGCTATG |
A, adenine; C, cytosine; G, guanine; T, thymine.
Methylation-specific polymerase chain reaction
To analyze methylation, 2 mg of genomic DNA obtained from the lung cancer cell line was modified using the EZ DNA Methylation Kit (Zymo Research, Orange, CA, USA). Primers specific for bisulfite-modified DNA were designed using MethPrimer software (http://www.urogene.org/methprimer/index1.html). The primers (located −8061 to −7782 bases from the transcription start site) used in this study were as follows: CD133 M primer sense (5′-TTCGGG ATAGAGGAAGTCGTAA-3′), CD133 M primer antisense (5′-CTCCCGCCCTAATCACCGCT-3′), CD133 U primer sense (5′-TTTGGGATAGAGGAAGTTGTAA-3′), and CD133 U primer antisense (5′-CTCCCACCCTAATCACC ACT-3′). The PCR conditions were as follows: 94°C for 5 minutes; then 43 cycles of 94°C for 30 seconds, 60°C (methylation-specific PCR [MS-PCR]) or 62°C (non-MS PCR) for 30 seconds, and 72°C for 60 seconds; and finally 72°C for 7 minutes. The amplified DNA fragments were visualized on a 2% agarose gel stained with ethidium bromide.
Bisulfite sequencing analysis
Sequencing primers recognizing both methylated and unmethylated sites (MU)—CD133 MU sense (5′-TATTTGG TTATGTTTTTAGTTTTTT-3′) and CD133 MU antisense (5′-CCTAATCAACAAATACCTCTCTC-3′)—were designed using MethPrimer software. These primers were located approximately −8103 to −7708 bases from the transcription start site. The PCR conditions used were as follows: 5 minutes at 94°C for initial denaturation; 40 amplification cycles of 94°C (30 seconds), 59°C (30 seconds), and 72°C (60 seconds); and a final elongation step lasting 7 minutes at 72°C. The PCR products obtained with bisulfite sequencing primers were inserted into the pGEM-T Easy vector (Promega, Madison, WI, USA) for cloning. Sequences of 5 individual colonies for each analyzed cell line were determined using universal pUC/M13 primers, and each sequence was analyzed using a Taq dideoxy terminator cycle sequencing kit on an ABI 3730 DNA sequencer (Applied Biosystems).
5-Aza-2′-deoxycytidine treatment
RT-PCR was used to evaluate the expression of CD133, common-GAGE (1, 2, 8, and b3–7), TRAG-3, common SSX (1–9), and MAGE A6 mRNA in NCI-H292 cells that received treatment with 1 μM 5-aza-2′-deoxycytidine (ADC) for 24 hours, 48 hours, and 72 hours. Briefly, 2×105 cells were seeded in two 75-cm2 culture flasks on day 0. The cells were treated with a control or 5 mmol/L of ADC (Sigma-Aldrich, St. Louis, MO, USA) for 24 hours on day 2. The culture was replenished with medium containing ADC every 48 hours (on days 4 and 6), and the medium was changed 24 hours after adding ADC. The cells were harvested on day 8 to obtain RNA, which was used for cDNA synthesis and the analysis of CD133 expression, as described above.
Direct DNA sequencing of polymerase chain reaction products
The Wizard Plus SV Minipreps kit (Promega, Madison, WI, USA) was used to prepare template DNA for sequencing after the subcloning of RT-PCR products. An automatic DNA sequencer (ABI 3700; Macrozen, Seoul, Korea) was used for sequencing. The sequence data were analyzed with the National Center for Biotechnology Information (National Institutes of Health) Blast search program.
Results
Morphological changes of NCI-H292 cells treated with ADC
The morphology of tumor cells treated with ADC did not change. However, the cell number was slightly decreased by ADC treatment (Fig. 1).
Fig. 1.

Morphology of NCI-H292 cells before (A) and after (B) ADC treatment (×100). Morphology did not change after ADC treatment, but cell number decreased upon ADC treatment. ADC, 5-aza-2′-deoxycytidine.
Expression of CD133 in NCI-H292 cells
The gene expression of CD133 in NCI-H292 cells as determined by RT-PCR analysis was significantly lower after ADC treatment than in controls (Figs. 2, 3). DNA sequencing was used to confirm the specificity of the RT-PCR products.
Fig. 2.

CD133 hypermethylation status of NCI-H292 cells. These results show that the expression of CD133 is regulated by a hypermethylated promoter region. (A) Methylated CD133. (B) Unmethylated CD133.
Fig. 3.
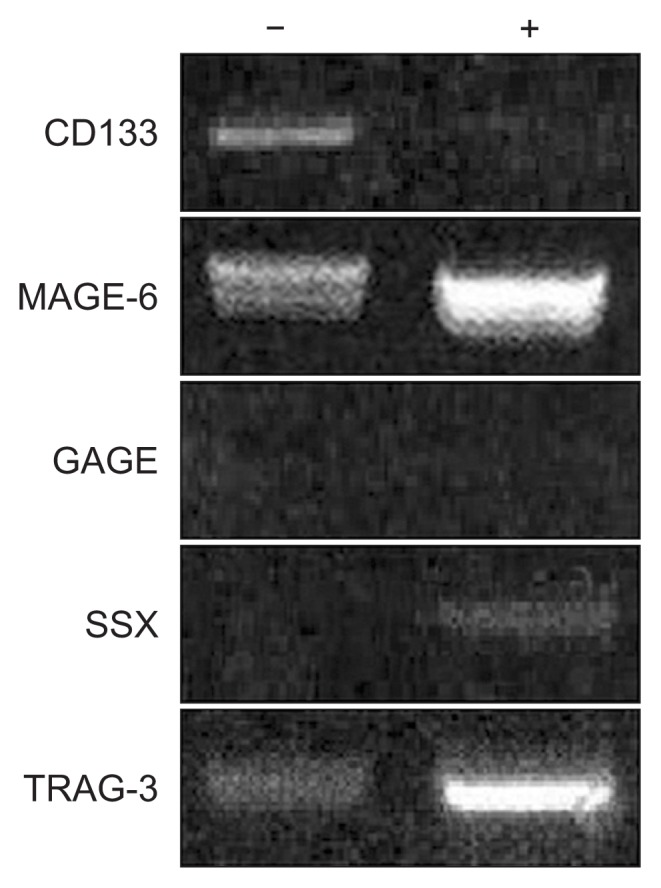
Expression of CD133, MAGE-6, GAGE, SSX, and TRAG-3 mRNA before and after treatment with a 5-aza′-2 demethylating agent (ADC). ADC treatment resulted in loss of CD133 expression. The expression of MAGE-6 and TRAG-3 was enhanced after ADC treatment. GAGE expression was not detectable before or after ADC treatment. Weak SSX expression was observed after ADC treatment. These findings explain the efficacy of demethylating agents such as ADC for inhibiting lung cancer stem cells, with prominent targeting against cancer germline antigens, and suggest that cell-based immunotherapy for lung cancer may be promising. −, before ADC treatment; +, after ADC treatment; ADC, 5-aza-2′-deoxycytidine.
Analysis of CD133 promoter methylation status with methylation-specific-polymerase chain reaction
To assess whether CD133 gene expression was influenced by the methylation of its promoter and whether promoter CpG islands of the gene were methylated or unmethylated in NCI-H292 cancer cells, we used MS-PCR with primers specific for methylated and unmethylated DNA. Only methylated DNA bands were evident; non-methylated DNA bands were not detected (Figs. 2, 3).
Analysis of promoter methylation status of the CD133 gene by bisulfite sequencing revealed that the promoter P1 and exon 1A were hypermethylated with a high (65%–70%) content of methylated CpG dinucleotides in NCI-H292 cells. Representative sequence graphs of the cell lines were created. To confirm whether CD133 expression was related to DNA demethylation status, treatment with ADC was performed. We observed a loss of expression of CD133 after treatment with ADC, suggesting that the methylation status of promoter CpG dinucleotides was correlated with CD133 expression (Figs. 2–4).
Fig. 4.

Results of direct sequencing of CD133.
RT-PCR analysis identified significant expression of MAGE-6 and TRAG-3 before ADC treatment, whereas common GAGE (1, 2, 8, and b3–7) and SSX were not expressed (Fig. 3). After treatment with ADC, upregulation of MAGE-6 and TRAG-3 expression was observed, while expression of SSX increased slightly. Expression of GAGE was not altered by treatment with ADC (Fig. 3).
Discussion
Recently, CD133 has become a marker of interest in lung CSC research [2]. CSCs are thought to maintain tumor bulk because of their ability to self-renew and differentiate into various cell types. Interestingly, CD133+ cells in lung cancer exhibit a high tumorigenic ability [2] and can undergo histological transformation [3].
Promoter methylation is the essential feature of the epigenetic modulation of gene expression [9,10]. The importance of DNA methylation is supported by the finding that many human diseases, such as chronic inflammation, are associated with abnormal patterns of methylation. Moreover, aberrant methylation of CpG islands is characteristic of various human cancers and is particularly evident during early oncogenesis [9,10].
CD133 methylation status is cancer cell-specific. Furthermore, the regulation of CD133 expression by demethylating agents also differs among cell types and displays tissue-specificity. Since abnormal DNA methylation of the gene encoding CD133 is related to CD133 expression in tumors [11], CD133 expression is thought to be regulated by a hypermethylated promoter region. Therefore, we analyzed the methylation status of the CD133 gene promoter with MS-PCR after sodium-bisulfite modification. Primers for promoter P2 sequences for MS-PCR and bisulfite sequencing analysis, as well as sequencing primers for a 32 CpG dinucleotide region, were previously described by Jeon et al. [11]. In this study, CD133 gene promoter methylation was observed in NCI-H292 lung cancer cells. In a previous report, these cells exhibited significant methylation bands, indicating that hypermethylation occurred at more than 10% of the total CpG islands [12].
DNA methylation in the promoter region of a gene is associated with the loss of gene expression and plays an important role in gene silencing, especially in CGAs [9,10]. Inactivation of the CSC marker CD133 by aberrant promoter methylation may be useful for chemotherapy for lung cancer. However, the loss of CD133 expression in early lung cancer is regulated differently than disruptions in the expression of other CGA genes [9]. Indeed, the inverse correlation between CD133 transcription and methylation can explain the loss of cell surface CD133 expression in differentiated cells. This observation is consistent with a report that cell differentiation is accompanied by epigenetic changes that can account for the future phenotypic profile of progeny [13]. In this study, the bisulfite sequencing results indicated differences in the methylation status among subclones of the same cell line. This variance suggests multiple origins of different clones from different alleles of the gene.
In some reports, CD133 was re-expressed after demethylation with ADC in some cell lines [9]. These results confirm that inactivation of CD133 expression is related to epigenetic modification, which in lung cancer cells is due to promoter demethylation. In some cases, CD133+ cancer cells exhibit increased expression of certain CGAs [6,14]. Specifically, NY-ESO-1 is expressed in CD133+ melanoma cells, and these cells can be targeted for killing by NY-ESO-1-specific CD8+ T-lymphocytes [6]. Thus, if CGAs express cancer germline antigens in vivo, immune targeting for CGAs may be an effective clinical strategy for the adjuvant treatment of cancers. In this study, we evaluated the expression of MAGE, GAGE, SSX, and TRAG-3 in NCI-H292 cells in response to altered CD133 expression. GAGE and SSX were not expressed in untreated cells, while the expression of MAGE-6 and TRAG-3 was detectable. These results indicated that CGA expression is cell-type dependent and that different CGAs can be expressed in different cell types. After treatment with ADC, the expression levels of MAGE-6, TRAG-3, and SSX were upregulated, while only GAGE remained unaffected, suggesting that the mechanism of GAGE expression may not be demethylation in NCI-H292 cells. To design novel therapeutic agents against CSCs, it is desirable to seek targets for CSCs for which CGAs become activated following demethylation. In this study, MAGE-6 and TRAG-3 were identified as potential targets of immunochemotherapy in CD133+ tumor cells. Some recent studies have shown that cancer cell lines acquire epigenetic and phenotypic changes over time [15]. To minimize this problem, we performed assays with freshly derived early passage (<10) cells. Our examination of immune targeting in the CD133+ lung cancer cell line was based upon 1 early passage cell line in which CGA antigen expression and CD133 expression fortuitously intersected. These results therefore raise the possibility that CGA antigens may be immunological targets for CD133+ lung cancer cells. This study helps enhance our understanding of the relationship between CSCs and CGAs and may shed light on the causes of therapeutic failure in various malignant tumors.
In conclusion, the NCI-H292 cell line expressed CD133 with hypermethylation, as confirmed by MS-PCR, bisulfite sequencing analysis, and treatment with ADC. ADC treatment resulted in the loss of CD133 expression and the upregulation of expression of 3 CGAs (MAGE-6, TRAG-3, and SSX). These results may contribute to our understanding of the role of CD133 in the pathogenesis of lung cancer associated with CGA expression and explain the efficacy of demethylating agents in the inhibition of lung CSCs with prominent targeting against CGAs. In turn, this may provide new knowledge for the further development of cancer and cell-based immunotherapy. The overall findings of this study demonstrated that epigenetic changes in a lung cancer cell line with maintenance of hypermethylation are capable of restoring CD133 expression.
Footnotes
Conflict of interest
No potential conflict of interest relevant to this article was reported.
References
- 1.Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer, and cancer stem cells. Nature. 2001;414:105–11. doi: 10.1038/35102167. [DOI] [PubMed] [Google Scholar]
- 2.Tirino V, Camerlingo R, Franco R, et al. The role of CD133 in the identification and characterisation of tumour-initiating cells in non-small-cell lung cancer. Eur J Cardiothorac Surg. 2009;36:446–53. doi: 10.1016/j.ejcts.2009.03.063. [DOI] [PubMed] [Google Scholar]
- 3.Koyama K, Katsurada N, Jimbo N, et al. Overexpression of CD 133 and BCL-2 in non-small cell lung cancer with neuroendocrine differentiation after transformation in ALK rearrangement-positive adenocarcinoma. Pathol Int. 2019;69:294–9. doi: 10.1111/pin.12782. [DOI] [PubMed] [Google Scholar]
- 4.Hilbe W, Dirnhofer S, Oberwasserlechner F, et al. CD133 positive endothelial progenitor cells contribute to the tumour vasculature in non-small cell lung cancer. J Clin Pathol. 2004;57:965–9. doi: 10.1136/jcp.2004.016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bertolini G, Roz L, Perego P, et al. Highly tumorigenic lung cancer CD133+ cells display stem-like features and are spared by cisplatin treatment. Proc Natl Acad Sci U S A. 2009;106:16281–6. doi: 10.1073/pnas.0905653106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gedye C, Quirk J, Browning J, et al. Cancer/testis antigens can be immunological targets in clonogenic CD133+ melanoma cells. Cancer Immunol Immunother. 2009;58:1635–46. doi: 10.1007/s00262-009-0672-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bao S, Wu Q, McLendon RE, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature. 2006;444:756–60. doi: 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 8.Cebon J. Cancer vaccines: where are we going? Asia Pac J Clin Oncol. 2010;6(Suppl 1):S9–15. doi: 10.1111/j.1743-7563.2010.01270.x. [DOI] [PubMed] [Google Scholar]
- 9.Huang J, Plass C, Gerhauser C. Cancer chemoprevention by targeting the epigenome. Curr Drug Targets. 2011;12:1925–56. doi: 10.2174/138945011798184155. [DOI] [PubMed] [Google Scholar]
- 10.Taberlay PC, Jones PA. DNA methylation and cancer. Prog Drug Res. 2011;67:1–23. doi: 10.1007/978-3-7643-8989-5_1. [DOI] [PubMed] [Google Scholar]
- 11.Jeon YK, Kim SH, Choi SH, et al. Promoter hypermethylation and loss of CD133 gene expression in colorectal cancers. World J Gastroenterol. 2010;16:3153–60. doi: 10.3748/wjg.v16.i25.3153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pellacani D, Packer RJ, Frame FM, et al. Regulation of the stem cell marker CD133 is independent of promoter hypermethylation in human epithelial differentiation and cancer. Mol Cancer. 2011;10:94. doi: 10.1186/1476-4598-10-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gan Q, Yoshida T, McDonald OG, Owens GK. Concise review: epigenetic mechanisms contribute to pluripotency and cell lineage determination of embryonic stem cells. Stem Cells. 2007;25:2–9. doi: 10.1634/stemcells.2006-0383. [DOI] [PubMed] [Google Scholar]
- 14.Looijenga LH, Hersmus R, Gillis AJ, et al. Genomic and expression profiling of human spermatocytic seminomas: primary spermatocyte as tumorigenic precursor and DMRT1 as candidate chromosome 9 gene. Cancer Res. 2006;66:290–302. doi: 10.1158/0008-5472.CAN-05-2936. [DOI] [PubMed] [Google Scholar]
- 15.Lee J, Kotliarova S, Kotliarov Y, et al. Tumor stem cells derived from glioblastomas cultured in bFGF and EGF more closely mirror the phenotype and genotype of primary tumors than do serum-cultured cell lines. Cancer Cell. 2006;9:391–403. doi: 10.1016/j.ccr.2006.03.030. [DOI] [PubMed] [Google Scholar]