

Abstract
Rationale:
The ubiquitin-editing protein A20 in dendritic cells (DCs) suppresses NF-ĸB signaling and constrains DC-mediated T cell stimulation, but the role of A20 in modulating the hypertensive response requires elucidation.
Objective:
Here, we tested the hypothesis that A20 in CD11c-expressing myeloid cells mitigates angiotensin II induced hypertension by limiting renal T cell activation.
Methods and Results:
Mice with heterozygous deletion of A20 in CD11c-expressing myeloid cells (Cd11c-Cre+ A20flox/wt = DC ACT) have spontaneous DC activation but have normal baseline blood pressures. In response to low dose chronic angiotensin (Ang) II infusion, DC ACT mice compared to wild-type (WT) controls had an exaggerated hypertensive response and augmented proportions of CD62LloCD44hi effector memory T lymphocytes in the kidney lymph node. After 10 days of Ang II, DC ACT kidneys had increased numbers of memory effector CD8+, but not CD4+ T cells, compared to WTs. Moreover, the expressions of TNF-α and IFN-γ were upregulated in the DC ACT renal CD8+ T cells but not CD4+ T cells. Saline challenge testing revealed enhanced renal fluid retention in the DC ACT mice. DC ACT kidneys showed augmented protein expression of epithelial sodium channel-gamma (γ-ENaC) and sodium-hydrogen antiporter 3 (NHE3). DC ACT mice also had greater reductions in renal blood flow following acute injections with Ang II and enhanced oxidant stress in the vasculature as evidenced by higher circulating levels of malondialdehyde (MDA) compared to WT controls. To directly test whether enhanced T cell activation in the DC ACT cohort was responsible for their exaggerated hypertensive response, we chronically infused Ang II into lymphocyte-deficient DC ACT Rag1−/− mice and WT (Cd11c-Cre− A20flox/wt) Rag1−/− controls. The difference in blood pressure elevation accruing from DC activation was abrogated on the Rag1−/− strain.
Conclusions:
Following stimulation of the renin angiotensin system, A20 suppresses DC activation and thereby mitigates T cell-dependent blood pressure elevation.
Keywords: Hypertension, A20, dendritic cells, cytokines, sodium transporters, sodium channels
Subject Terms: Hypertension
The ubiquitin editor A20 suppresses NF-kB signaling, but its role in the regulation of blood pressure requires elucidation. In the current study, we find that A20 in CD11c-expressing myeloid cells protects against hypertension by inhibiting the accumulation of effector T lymphocytes in the kidney and renal fluid retention during chronic Ang II infusion. We confirm that A20 in myeloid cells limits hypertension by restraining T cell activation during renin-angiotensin system (RAS) stimulation. Mechanistically, A20 in myeloid cells restricts the generation of pro-hypertensive cytokines, including TNF-α and IFN-γ, in CD8+ rather than CD4+ T cells that accumulate in the kidney during hypertension. This study thus identifies a novel role for myeloid A20 to regulate blood pressure responses, which should facilitate development of immune-directed therapies for patients with biologically resistant hypertension.
INTRODUCTION
Hypertension is the most prominent risk factor for cardiovascular disease, and over a billion individuals are afflicted with hypertension worldwide1. Stroke, heart failure, and chronic kidney disease are catastrophic complications of hypertension, and blood pressure remains poorly controlled in up to half of the hypertensive patients2. Novel anti-hypertensive therapies are therefore urgently needed.
Inappropriate immune activation is a major contributor to experimental hypertension, and cells of the immune system mediate blood pressure elevation in part via their actions in the kidney3–6. Studies with Rag1-deficient mice revealed that T lymphocytes, in particular, are absolutely required to mount a full hypertensive response to angiotensin (Ang) II7. For example, T cells can raise blood pressure by promoting oxidative stress and sodium reabsorption in the kidney8, 9. T cells mediate these effects through the elaboration of cytokines that influence renal epithelial function and even through direct contact with renal tubular cells10–12. Dendritic cells (DCs) are the most potent antigen presenting cells that activate T cells, and a full hypertensive response requires interactions between DCs and T cells to generate memory effector T cells characterized by CD44hiCD62lo expression13, 14.
A20 is a zinc finger protein and ubiquitin-editing enzyme that inhibits NF-ĸB activation15. Accordingly, deficiency of A20 allows unchecked inflammation and perinatal lethality16. In humans, conditions that distort A20’s function lead to autoimmune disease17–20. In mice, deletion of A20 restricted to DCs causes DC-dependent expansion of activated T cells21. Homozygous deletion of A20 in DCs causes inflammatory bowel disease (IBD), which could lead to hypotension from fluid losses in the gut. However, mice with heterozygous deletion of A20 in CD11c+ myeloid cells (DC ACT) have an enhanced propensity for DC-mediated T cell activation but are phenotypically normal at baseline. Moreover, DC ACT mice do not develop IBD, and thus have normal baseline blood pressures, allowing us to study the effects of myeloid cell A20 on the hypertensive response in these animals.
In the current studies, we test the hypothesis that A20 in CD11c+ myeloid cells protects against hypertension. Using murine conditional mutant lines, we demonstrate that A20 in CD11c+ myeloid cells limits activation of T cells in the kidney and its draining lymph node and mitigates the severity of hypertension in the chronic Ang II infusion model. Our results also suggest that cytokines including TNF-α and IFN-γ from the CD8+ but not CD4+ T cells in the DC ACT kidney act on renal tubular function and contribute to hypertension.
METHODS
Data, methods, and materials will be made available upon request.
Animal experiments.
All animal experiments were approved by the Durham Veterans’ Affairs Medical Center Institutional Animal Care and Use Committee. All mice were housed and bred in the animal facilities at the Durham Veterans’ Affairs Medical Center according to NIH guidelines. All animals were maintained on a 12:12-h light/dark cycle at temperatures ranging between 67–76 °F, 30–70 % relative humidity and free access to food and water.
Male C57BL/6 A20flox/flox mice were generously provided by Dr. Gianna Hammer and bred with female C57BL/6 Cd11c-Cre transgenic mice to generate male C57BL/6 Cd11c-Cre+ A20flox/wt (DC ACT) mice. These mice exhibit evidence of DC-mediated T cell activation but are otherwise phenotypically normal at baseline. Cd11c-Cre+ A20flox/wt mice were crossed with the recombination activating protein one (Rag1) whole body knockout (Rag 1−/−) strain to generate male Rag1−/− Cd11c-Cre+ A20flxo/wt (Rag1−/− DC ACT) mice for experiments. Within specific genotypes, mice were randomly assigned to experiments.
Chronic Ang II-induced hypertension,
10–12 week old male mice were subjected to our hypertension protocol as previously described22. Male mice were used to allow comparisons to our previously published hypertension studies. To reduce total nephron number and capacity for sodium excretion, all mice underwent left nephrectomy followed one week later by implantation of a pressure-sensing radiotelemetry catheter (Model#: PA-C10, DSI). Mice without an interpretable blood pressure tracing (2 WT and 3 DC ACT mice) were excluded from analysis. After 10 days of recovery, an osmotic minipump (Cat#: 0000298, ALZET) was implanted to infuse Ang II continuously for 28 days.
Cell preparations and flow cytometry.
Following Ang II infusion, the hypertensive kidneys were harvested and digested into single cell suspensions. For the T cell panel, cells were stained with fluorescently-labeled anti-CD45 (Cat#:103149, BioLegend), anti-CD3 (Cat#:100308, BioLegend), anti-CD4 (Cat#:100557, BioLegend), anti-CD8 (Cat#:100734, BioLegend), anti-CD62L (Cat#:104436, BioLegend), and anti-CD44 (Cat#:17-0441-82, Invitrogen) as described23 and subjected to flow cytometric analysis. In the DC panel, cells were stained with fluorescently-labeled anti-CD45 (Cat#:103138, BioLegend), anti-CD3 (Cat#:100355, BioLegend), anti-CD19 (Cat#:115543, BioLegend), anti-NK1.1(Cat#:108749, BioLegend), anti-MHCII (Cat#:107626, BioLegend), anti-CD11c (Cat#:117308, BioLegend), anti-CD40 (Cat#:124622, BioLegend), anti-CD80 (Cat#:104731, BioLegend), and anti-CD86(Cat#:105031, BioLegend) prior to analysis. Representative flow plots were chosen to reflect the means from the summary data. The numbers shown on the representative flow plots are exact percentages for the samples shown.
Cell sorting.
Mouse kidneys were harvested from hypertensive animals and digested into single cell suspensions. Cells were then stained with fluorescently-labeled anti-CD45 (Cat#:103138, BioLegend), anti-CD3 (Cat#:100308, BioLegend), anti-CD4 (Cat#:100557, BioLegend), anti-CD8 (Cat#:100734, BioLegend), and subjected to fluorescent cell-sorting as described22. CD45+/CD3+/CD4+ and CD45+/CD3+/CD8+ cells were collected, and RNA was harvested from cells meeting these criteria for real-time PCR analysis.
RNA extraction and real-time quantitative PCR.
Total RNA was extracted from samples according to the manufacturer’s instructions of the RNeasy Plus Mini kit (Cat#: 74136, Qiagen) or RNeasy Micro kit (Cat#: 74004, Qiagen). RNA was reverse transcribed with a high capacity cDNA reverse transcriptase kit (Cat#: 4368813, Invitrogen), and real time quantitative PCR (RT-qPCR) was performed with Taqman assay (Cat#: 4370074) from Invitrogen. Taqman primers and probes were used for IL-1β, TNF-α, and IFN-γ.
Saline challenge test.
In brief, mice were anesthetized with isoflurane and then injected i.p. with a volume of warmed isotonic saline equivalent to 10% of their body weight. Mice then awoke and were placed immediately in metabolic cages for urine collection over 4 hours. Urine volumes were quantitated, and results were tabulated as the percentage of the volume load originally injected.
Immunoblotting.
Kidney tissues were lysed and subsequently sonicated in RIPA buffer that contained 250 μM phenylmethanesulfonyl fluoride (PMSF), 2 mM EDTA, and 5 mM dithiothrietol (DTT) (pH 7.5). Protein concentrations were determined by the use of Coomassie reagent. 40 μg of protein of each sample was denatured in boiling water for 10 min, then separated by SDS-PAGE, and transferred onto PVDF membranes. The blots were blocked with 5% nonfat dry milk in Tris-buffered saline (TBST) for 1 h, followed by overnight incubation with the primary antibody. After washing with TBST, blots were incubated with goat anti-rabbit horseradish peroxidase (HRP)-conjugated secondary antibody and visualized using Enhanced Chemiluminescence (ECL, Cat#: 32106, Invitrogen). The blots were quantitated by using ImageJ 1.38 (NIH, USA). Primary antibodies were as follows: rabbit anti-NHE3 antibody (Cat#: SPC-400D, Stressmarq Biosciences INC.), rabbit anti-α-ENaC antibody (Cat#: SPC-403D, Stressmarq Biosciences INC.), rabbit anti-β-ENaC antibody (Cat#: SPC-404D, Stressmarq Biosciences INC.), rabbit anti-γ-ENaC antibody (Cat#: SPC-405D, Stressmarq Biosciences INC.), rabbit anti-NCC antibody (Cat#: NBP1–44270, Novus Biologicals.), rabbit anti-NKCC2 antibody (Cat#: 38436S, Cell Signaling Technology), and rabbit anti- GAPDH (Cat#: 2118L, Cell Signaling Technology).
Statistical analysis.
The values of each parameter within a group are expressed as the mean ± the standard error of the mean (SEM). For the blood pressure measurement experiments, comparisons between the groups were performed using the two-way ANOVA. For comparisons between just 2 groups with normally distributed data, statistical significance was assessed using unpaired Student’ t-test. Normality was determined using the Shapiro-Wilks and the Kolmogorov-Smirnov tests. For comparisons between groups with non-normally distributed variables, the Mann-Whitney U test was employed. p<0.05 was considered statistically significant. No across-experiment multiple test corrections were performed. Graphpad Prism was used for statistical analysis. Sample sizes were chosen based on previous studies using our hypertension model.
RESULTS
A20 in CD11c-expressing myeloid cells limits Ang II-induced blood pressure elevation.
Mice with homozygous deletion of A20 in CD11c-expressing myeloid cells have elevated gene expression for inflammatory mediators in the kidney and reduced body weights at baseline attributable to their susceptibility to inflammatory bowel disease (Online Figure I). By contrast, Cd11c-Cre+ A20flox/wt (DC ACT) mice with heterozygous deletion of A20 in CD11c-expressing cells and Cre− A20flox/wt littermate controls (WT) have similar body weights, kidney weights, heart weights, and renal expression of inflammatory mediators and injury markers at baseline (Online Figure I), allowing us to study the effects of A20-dependent T cell activation on the hypertensive response while circumventing effects of baseline abnormalities. DC ACT and WT mice underwent unilateral nephrectomy to enhance salt sensitivity followed by implantation of pressure-sensing catheters. Baseline blood pressures measured by radio-telemetry were virtually identical in the WT and DC ACT mice (Figure 1A, Online Figure II). To induce hypertension, mice were infused subcutaneously with a low dose of Ang II, 300ng/kg/min, via osmotic mini-pump. During Ang II infusion, mean arterial blood pressures (MAPs) in the WT group rose approximately 15 mmHg and remained elevated throughout the infusion period. However, the DC ACT mice exhibited more severe hypertension compared to WT controls (Figure 1A, 143 ± 1.7 vs. 131 ± 1.5 mmHg, p<0.001). A similar pattern was appreciated with systolic and diastolic blood pressures (Online Figure II). As the C57BL/6 strain is relatively resistant to hypertensive kidney injury, low dose Ang II induced only minimal and similar levels of renal injury in the 2 groups with no increases in albuminuria or kidney weight seen in either Ang II-infused cohort (Online Figure III). However, consistent with their higher blood pressures, the DC ACT mice had exaggerated cardiac hypertrophy following 4 weeks of hypertension (Figure 1B, 7.28 ± 0.27 vs. 6.35 ± 0.33 mg heart weight/g body weight, p<0.05) and induction of β-myosin heavy chain (β-MHC), reflecting pathologic cardiac hypertrophy24 (Online Figure IV). The hearts from both groups showed increased levels of perivascular fibrosis and cardiac expression of fibrosis markers following Ang II compared to baseline controls (Online Figure IV). Nevertheless, at this low dose of Ang II, the higher blood pressure levels in the DC ACT animals were not sufficient to alter levels of cardiac fibrosis compared to WT controls. Moreover, cardiac T cell infiltration at this Ang II dose was mild and similar between the WT and DC ACT cohorts (Online Figure V). These data indicate that A20 in CD11c-expressing myeloid cells protects against Ang II-induced hypertension, but suggest that differences in renal injury do not underpin the exaggerated hypertensive response seen in the DC ACT cohort.
Figure 1. A20 in CD11c+ myeloid cells limit angiotensin (Ang) II-induced hypertension.
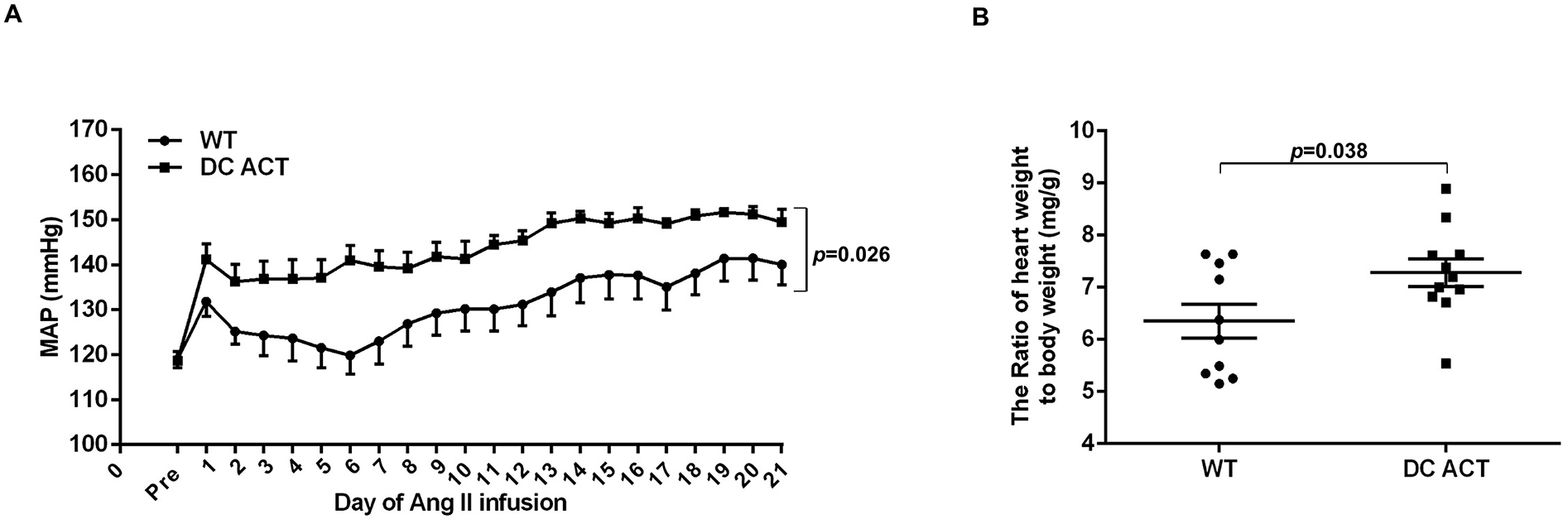
(A) Mean arterial pressures measured by radio-telemetry in the experimental groups at baseline (“pre”) and during chronic Ang II infusion. Wild-type (“WT”), circles. Cd11c-Cre+ A20flox/wt (“DC ACT”), squares. N=8 mice/group. Comparisons between the groups were performed using the two-way ANOVA. (B) The ratio of heart weight/body weight(mg/g) in the WT and DC ACT groups after 28 days of Ang II infusion. N=10–11 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
Figure 7. A20 in CD11c+ myeloid cells suppresses the hypertensive response via a T cell-dependent mechanism.

(A) Mean arterial pressures measured by radio-telemetry in Rag1−/−mice (“Rag1−/−”, open circles) and Rag1−/− Cd11c-Cre+ A20flox/wt (“Rag1−/− DC ACT”, open squares) mice at baseline (“pre”) and during chronic Ang II infusion. Comparisons between the groups were performed using the two-way ANOVA. (B) Ratio of heart weight/body weight (mg/g) in the Rag1−/− and Rag1−/−DC ACT groups after 28 days of Ang II infusion. N=5–6 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
A20 in CD11c-expressing myeloid cells inhibits T cell activation in the renal lymph node during hypertension.
Naïve T cells are marked by CD62LhiCD44lo expression, whereas effector memory T cells exhibit a CD62LloCD44hi profile25. Therefore, to determine the impact of A20 in DCs on the activation of T cells draining from the kidney, we measured the proportions of CD62LhiCD44lo naïve and CD62LloCD44hi effector memory T cells in the kidney lymph node by flow cytometry after 10 days of hypertension, a timepoint at which we have seen immune-dependent differences in renal sodium handling during hypertension26 (Figure 2A). In general, we saw a mild shift toward higher proportions of CD4+ T cells and lower proportions of CD8+ T cells in the DC ACT cohort (Figure 2B). Among these T cell subsets, the proportions of effector memory T cells were augmented in the kidney lymph nodes from the DC ACT cohort (Figure 2C–E). This pattern was stronger within the CD8+ than in the CD4+ T cell subsets and became even more evident by day 28 of Ang II (Online Figure VI). We also detected a similar pattern with splenic T cells isolated at day 10 of Ang II infusion (Online Figure VII). Thus, A20 in CD11c-expressing myeloid cells limits T cell activation in the kidney’s draining lymph node and spleen during hypertension.
Figure 2. A20 in CD11c+ myeloid cells inhibit T cell activation in the renal lymph node during hypertension.
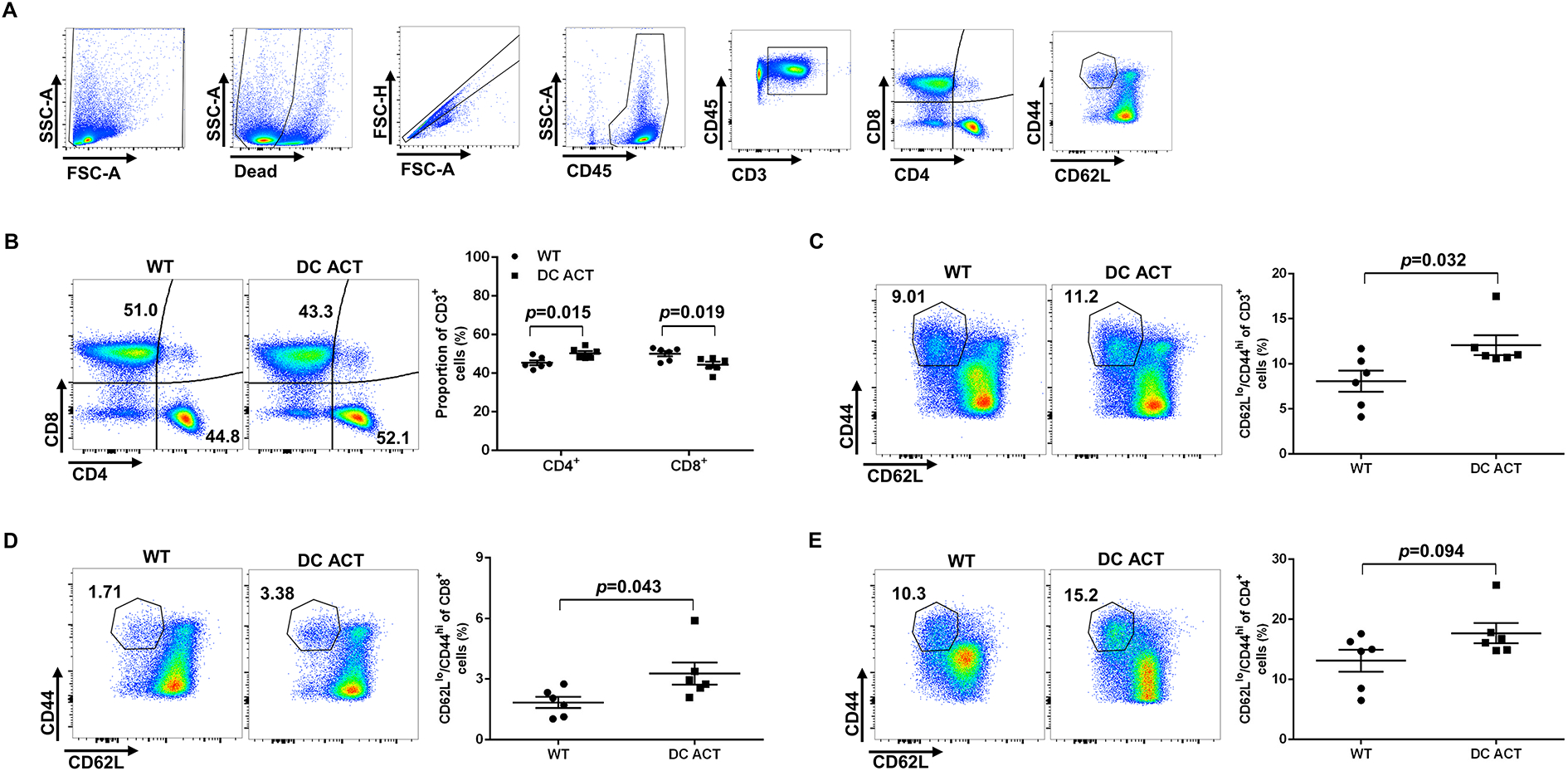
Flow cytometric analysis of renal lymph node cells after 10 days of Ang II-induced hypertension. (A) Gating strategy for parsing T cell populations in renal lymph node. (B) Representative flow plots and proportions of the CD4+ and CD8+ T lymphocytes from WT and DC ACT groups. (C) Representative flow plots and proportions of the CD44hiCD62Llo CD3+ T lymphocytes from WT and DC ACT groups. (D) Representative flow plots and proportions of the CD44hiCD62Llo CD8+ T cells from WT and DC ACT cohorts. (E) Representative flow plots and proportions of the CD44hiCD62Llo CD4+ T cells from WT and DC ACT mice. N=6 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
A20 deficiency in CD11c+ myeloid cells augments accumulation of activated T cell in the kidney during hypertension.
Following activation in the lymphoid organs, these effector memory T cells can recirculate to the kidney to have profound effects on blood pressure regulation14. We therefore posited that these activated T cells could accumulate in the kidney to influence the hypertensive response. Thus, to quantify effector memory T cells in the hypertensive kidney, we performed flow cytometric analysis on single cell suspensions from the kidney after 10 days of Ang II. As illustrated by our gating strategy (Figure 3A), cells were stained with fluorescently-labeled anti-CD45, anti-CD3, anti-CD4, Anti-CD8, Anti-CD62L, and Anti-CD44. We found greater numbers of CD3+ T lymphocytes in the kidneys from our DC ACT mice compared to WT controls (Figure 3B). Additionally, the absolute number of CD4+ and CD8+ T cells were increased in the DC ACT group (Figure 3C). Numbers of effector memory T cells marked by CD62LloCD44hi expression were markedly increased within the CD8+ T cell subset, but not in the CD4+ subset from the DC ACT kidneys (Figure 3D–E). As CD8+ T cells are known to play a key role in T cell-dependent blood pressure elevation27, these data suggest that A20 in CD11c-expressing myeloid cells may limit blood pressure elevation by reducing the renal accumulation of activated CD8+ T cells.
Figure 3. A20 deficiency in CD11c+ myeloid cells enhances accumulation of effector memory T cells in the kidney during hypertension.
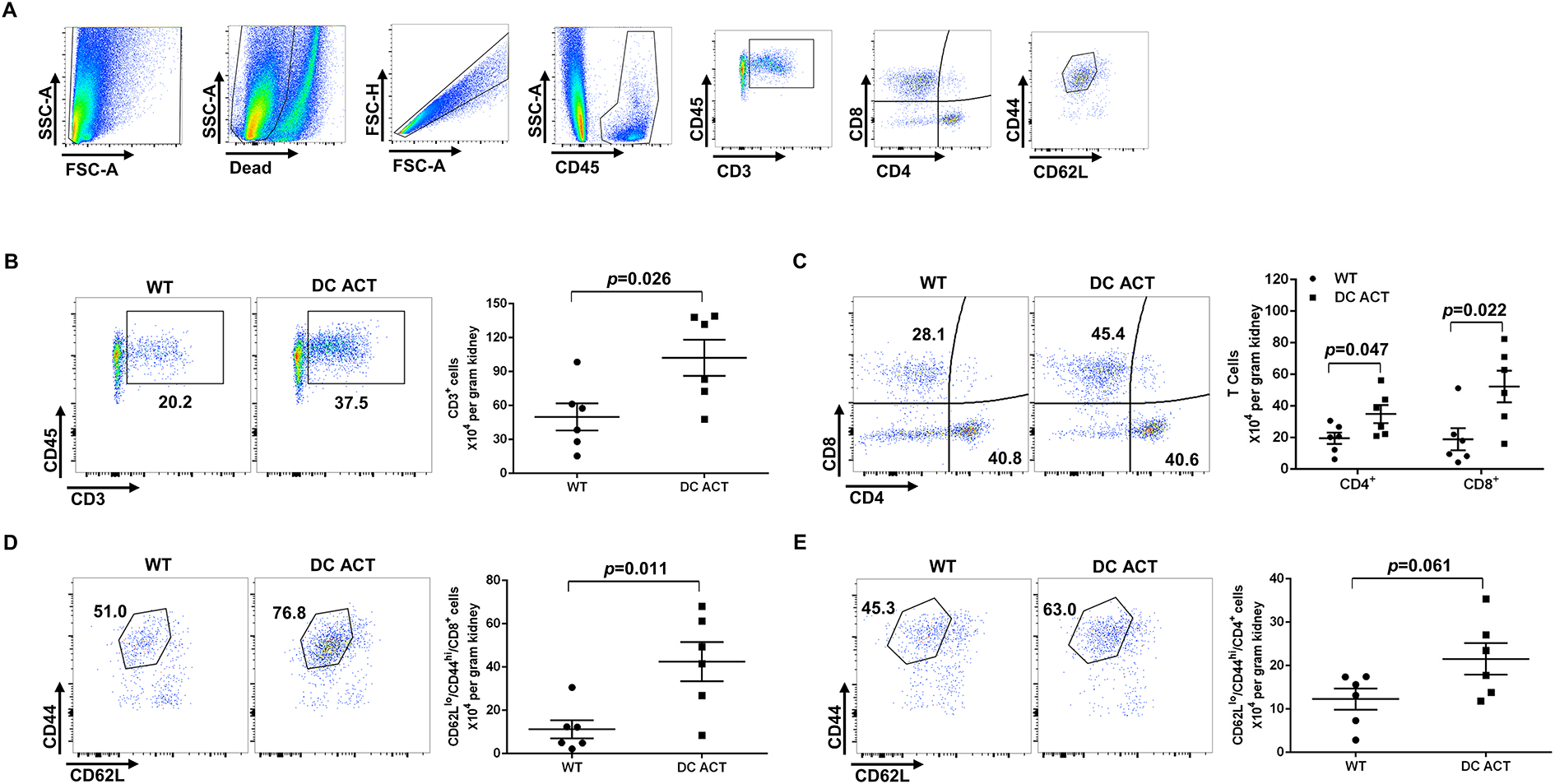
Flow cytometric analysis of single cell suspensions from the kidney harvested after 10 days of Ang II infusion. (A) Gating strategy for parsing T cell populations in kidney. (B-E) Representative flow plots and absolute number of the (B) CD3+ T lymphocytes, (C) CD4+ and CD8+ T cells, (D) CD44hiCD62Llo CD8+ T cells, and (E) CD44hiCD62Llo CD4+ T cells from the WT and DC ACT kidney. N=6 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
A20 suppresses renal DC activation during Ang II-dependent hypertension.
A20 in DCs has been reported to limit T cell activation in part by suppressing the expression of the DC maturation marker CD4021. Thus, to determine whether A20 regulates DC activation in our hypertension model, we examined CD40 expression on DCs isolated from the kidney after 10 days of Ang II infusion. To this end, we selected for CD45+ live hematopoietic cells that did not express markers for T cells (CD3), B cells (CD19), or NK cells (NK1.1) based on a “fluorescence minus one (FMO)” gate, and then analyzed CD40 expression on the remaining cells co-expressing the DC markers CD11c and MHCII (Figure 4A). We found that the absolute numbers of CD11c+ MHCIIhi DCs in the hypertensive kidney from WT and DC ACT mice were similar (Figure 4B). The absolute numbers of CD40+ DCs were elevated in the DC ACT kidneys compared to the WT cohort whereas the numbers of CD80 and CD86 DCs were numerically but not significantly higher in the DC ACT cohort (Figure 4C–E). Whereas absolute numbers of CD11c+ MHCIIhi DCs were increased in the DC ACT spleens compared to WT controls, the pattern of DC co-stimulatory molecule expression on splenic DCS in the 2 groups was similar to that seen in the kidney (Online Figure VIII). Thus, A20 in DCs constrains their expression of the co-stimulatory molecule CD40, providing a possible mechanism through which A20 in DCs limits renal T cell activation.
Figure 4. A20 suppresses renal DC activation during Ang II-dependent hypertension.

Assessment of expression of co-stimulatory molecules CD40, CD80 and CD86 on CD11c+MHCIIhi DCs isolated from the kidney after 10 days of Ang II. (A) In gating strategy, T cells, B cells, and NK cells were first excluded via negative staining for CD3, CD19, and NK1.1, respectively, based on “fluorescence minus one” (FMO) threshold. Then DCs were identified by co-expression of CD11c and MHCII and parsed for CD40, CD80, or CD86 positivity. (B) Representative flow plots and absolute number of CD11c+MHCIIhi Cells from WT and DC ACT groups. (C-E) Representative flow plots and absolute number of (C) CD40+, (D) CD80+, and (E) CD86+ DCs from WT and DC ACT mice. N=6 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
Pro-hypertensive cytokines are upregulated in CD8+ T cells in the kidneys of Ang II-infused DC ACT mice.
As the pro-inflammatory cytokines, TNF-α, IFN-γ and IL-17A can all contribute to blood pressure elevation through diverse actions in the kidney11, 28–30, we measured mRNA expression for these cytokines in CD4+ and CD8+ cells isolated directly from the kidney of the Ang II-infused WT and DC ACT animals (Figure 5A). By real-time qPCR analysis, the mRNA levels of TNF-α, and IFN-γ were significently increased in CD8+ T cells the DC ACT kidney whereas IL-17A expression was not significantly different between the CD8+ T cells from the 2 groups (Figure 5B). By contrast, we saw no significant alterations in the expression of those pro-hypertensive cytokines within the CD4+ T cells from the DC ACT kidneys (Figure 5C). At the whole kidney level, DC ACT kidneys showed enhanced mRNA expression for TNF-α, IFN-γ, IL-17A, and the chemokine CCL5 (Online Figure IX). We also detected elevated circulating levels of TNF-α protein in the Ang II-infused DC ACT cohort (Online Figure IX). These data suggest that A20 in CD11c+ myeloid cells limit blood pressure elevation by broadly constraining expression of pro-hypertensive cytokines in CD8+ T cells.
Figure 5. Pro-hypertensive cytokines are upregulated in CD8+ T cells from the kidneys of Ang II-infused DC ACT mice.

After 10 days of Ang II infusion, the CD4+ and CD8+ T cells were isolated from the kidney and sorted by flow, and mRNA expression for cytokines was quantitated via qPCR. (A) Gating strategy for renal T cell isolation. (B-C) Relative mRNA expression for TNF-α, IFN-γ, and IL-17a in (B) CD8+ and (C) CD4+ T cells. N=5 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
A20 in CD11c+ myeloid cells facilitates diuresis following induction of hypertension.
Cytokines including IFN-γ, TNF-α and IL-17A can promote blood pressure elevation during activation of the renin angiotensin system by modulating renal tubular function10–12, 31. To determine if A20 in DCs controls blood pressure by limiting volume expansion, uni-nephrectomized WT and DC ACT mice at baseline or day 10 of Ang II were challenged intraperitoneally with the saline equivalent of 10% of their body weight and placed in metabolic cages for urine collection. The ratio of urine volume to the injected volume at each time point was quantitated as a measure of diuresis. Body weights in the 2 groups were not different (Online Figure X). At baseline, WT and DC ACT mice excreted similar proportions of the injected volumes (Figure 6A). Following 10 days of Ang II, both groups excreted lower proportions of the injected volume compared to the baseline condition, consistent with Ang II-induced volume retention. However, in comparison to the Ang II-infused WT cohort, the Ang II-infused DC ACT animals excreted an even lower volume, suggesting an exaggerated tendency for Ang II-mediated volume retention (Figure 6A). Consistent with this finding, the protein expressions of sodium transporters including the cleaved epithelial sodium channel-gamma (γ-ENaC) and sodium-hydrogen antiporters 3 (NHE3) were significantly increased in the DC ACT kidneys compared to WT controls (Figure 6B–6C). By contrast, the levels of α-ENaC, β-ENaC, and NKCC2 were similar in the 2 groups whereas levels of full length γ-ENaC and NCC were decreased in the DC ACT group (Figure 6B–6C). Despite the exaggerated diuresis in the DC ACT group, our assay could not detect differences in sodium excretion (Online Figure X). Consistent with a role to regulate NF-ĸB activation rather than gene expression, we could not detect differences between the WT and DC ACT cohorts in mRNA levels for NF-kB subunits in whole kidney or in cultures of splenic CD11c+ myeloid cells exposed to Ang II (Online Figure XI).
Figure 6. A20 in CD11c+ myeloid cells impairs diuresis following induction of hypertension.

(A) At baseline and after 10 days of Ang II infusion, WT and DC ACT mice were challenged with an i.p. bolus of saline equivalent to 10% of their body weights and placed in metabolic cages for 4 hours of urine collection. Ratio of urine volumes to injected volumes are shown. (B) After 3 weeks of Ang II infusion, the kidney protein levels of the α-ENaC, β-ENaC, γ-ENaC, NHE3, NKCC2, and NCC was determined by immunoblotting. (C) Densitometry of these blots normalized to GAPDH. N=9 mice/group. Statistical significance was assessed using an unpaired Student’ t-test. Data are mean ± SE.
We also considered the possibility that activated T cells in the DC ACT kidney could impair diuresis by reducing renal blood flow. Consistent with this notion, we detected a greater, dose-dependent reduction in renal blood flow in DC ACT animals acutely injected with Ang II (Online Figure XII). T cells cause vascular dysfunction by raising local oxidant stress. Enhanced oxidant stress in the DC ACT vasculature was evidenced by higher circulating levels of malondialdehyde (MDA) in these mice vs. WT controls (Online Figure XII). Thus, ablation of A20 in CD11c+ myeloid cells contributes to attenuated renal blood flow, sodium transporter activation, and volume expansion, which could promote blood pressure elevation.
A20 in CD11c+ myeloid cells suppresses the hypertensive response via a T cell-dependent mechanism.
To discriminate whether A20 in CD11c+ myeloid cells regulates blood pressure through effects on DC-mediated T cell activation, we crossed the DC ACT mice onto the Rag1−/− strain that lacks lymphocytes. We then subjected Rag1−/− DC ACT mice and Rag1−/− WT mice with normal A20 expression to our hypertension model. In contrast to our original DC ACT mice experiment (Figure 1A), Rag1−/− DC ACT mice had a similar hypertensive response to Rag1−/− WT mice (Figure 7A). Consistent with their similar blood pressures, the Rag1−/− DC ACT mice developed similar levels of cardiac hypertrophy compared to Rag1−/− mice (Figure 7B). Thus, lymphocytes are required to mediate exaggerated hypertension that accrues from deficiency of A20 in CD11c-expressing myeloid cells during RAS activation.
DISCUSSION
Hypertension is the major modifiable risk factor for cardiovascular disease that now causes one-third of deaths worldwide32. Blood pressure elevation can result from dysfunction in any of the cardiovascular control centers including the kidney, heart, vasculature, and nervous system, and inappropriate immune responses promote hypertension by acting on any of these control centers33. Activated dendritic cells (DCs) augment the hypertensive response by stimulating T lymphocytes to alter vascular and renal epithelial functions34, 35. In the current study, we find that the ubiquitin editor A20 in CD11c+ myeloid cells attenuates the hypertensive response during RAS activation by limiting T cell activation in the kidney.
A20 restricts the maturation of the DCs by blunting activation of NF-κB signaling pathway, and deficiency of A20 in CD11c-expressing myeloid cells causes systemic autoimmunity and inflammatory bowel disease, a condition that could actually reduce blood pressure by causing volume losses in the gut36,21. As mice with heterozygous deletion of A20 in CD11c+ myeloid cells (DC ACT) have normal baseline blood pressure, we employed the DC ACT line to understand the role of A20 in DCs to regulate RAS-dependent hypertension. The similar blood pressures in the WT and DC ACT mice indicate that A20 in CD11c+ myeloid cells does not control normal blood pressure homeostasis. Nevertheless, during chronic low dose Ang II infusion, the DC ACT mice developed exaggerated hypertension and cardiac hypertrophy, consistent with a role for A20 in CD11c+ myeloid cells to limit the susceptibility to Ang II-induced hypertension.
The capacity of T lymphocytes to exacerbate the hypertensive response has been established in multiple experimental models, and T cells accumulate within the vascular wall and kidney during hypertension7, 37. Consistent with a role for A20 in DCs to temper immune-mediated hypertension via actions in the kidney, we find augmented accumulation of both CD4+ and CD8+ T cells within the kidneys from the Ang II-infused DC ACT cohort. Interactions of T cells with DCs can cause differentiation of T cells to a CD44hiCD62Llo effector memory phenotype that is critical to hypertension pathogenesis14, and we find an increased accumulation of effector memory T cells in both the kidney and its draining lymph node from the hypertensive DC ACT animals. CD8+ T lymphocytes, in particular, are thought to mediate the pro-hypertensive effects of T cells in hypertension27, and we find that the enhancement in renal T cell accumulation in the DC ACT cohort is more striking in the CD8+ subset than the CD4+ subset. Moreover, within the DC ACT kidney, we found dramatically increased numbers of CD8+ but not CD4+ effector memory T cells compared to WT controls, consistent with the augmented hypertension in the DC ACT cohort.
DCs activate T cells via the presentation of antigen in the cleft of a major histocompatibility complex. Upregulation of co-stimulatory molecule receptors including CD80, CD86, and/or CD40 on the surface of the maturing DC potentiates maximal T cell activation38. A20 on DCs has been reported to constrain expression of CD40 and CD80 on the DC21. In the hypertensive kidneys and spleen from the DC ACT animals, we detected significantly increased numbers of CD40+ DCs but only non-significant increases in CD80+ and CD86+ DCs in the DC ACT kidney and spleen versus WT controls. These data suggest that A20 on CD11c+ myeloid cells constrains T cell activation by suppressing renal DC expression of CD40.
Following activation by DCs, T cells can modulate blood pressure through the elaboration of inflammatory cytokines that influence renal epithelial cell function and/or vascular tone. Among these cytokines, IFN-γ, TNF-α, and several Interleukins have all been shown to promote hypertension through actions in the kidney6, 7, 26, 31. We find that A20 in DCs suppresses the expression of several of these cytokines in renal CD8+ T cells, which may underpin the protective effect of DC A20 in RAS-dependent hypertension. These cytokines can instigate blood pressure elevation by promoting renal fluid retention with consequent volume expansion per Ohm’s law. Consistent with that notion, we detected a blunted diuresis in response to an IP saline challenge in the Ang II-infused DC ACT cohort compared to WT controls. Moreover, the DC ACT kidneys showed upregulated protein expression for the active form of multiple sodium channels including γ-ENaC and NHE3. While A20 in CD11c+ myeloid cells appears to attenuate Ang II-induced fluid retention, our experiments could not confirm alterations in sodium excretion and cannot discriminate whether the effects on sodium transporter expression are cytokine-mediated or rather accrue from direct interactions of CD8+ T cells with the renal epithelium as has been reported39. Nevertheless, the sodium channel reportedly impacted by direct CD8 cell-tubular interactions is the NCC transporter in the distal convoluted tubule, and protein levels of NCC were down- rather than up-regulated in the DC ACT kidneys, possibly reflecting pressure natriuresis in response to the higher blood pressure pressures in this group. Moreover, T cells accumulate around the renal blood vessels rather than the renal tubules in our hypertension model22, 40, and we detected more severe reductions in renal blood flow in the DC ACT cohort in response to Ang II. Thus, A20 in CD11c+ myeloid cells can limit volume expansion and blood pressure elevation both through effects on sodium transporter function and through effects on the renal vasculature.
Finally, to test whether A20 in CD11c+ myeloid cells mitigates blood pressure elevation via effects on T cell activation, we compared the hypertensive response in DC ACT and WT mice crossed to the lymphocyte-deficient Rag1−/− strain. Both groups developed a low level of hypertension, suggesting that our Rag1−/− strain may have lost some of its resistance to hypertension as has been reported in the literature41. Here, we have not investigated compensatory mechanisms permitting hypertension in our Rag1−/− animals. Nevertheless, the difference in the hypertensive response between the groups was abrogated in the absence of lymphocytes. Thus, the capacity of A20 in CD11c+ myeloid cells to limit hypertension depends on the curtailing of lymphocyte activation. While the Rag1-deficient strain lacks both B and T cells, the role of DCs in presenting antigens to T lymphocytes implicates T cells rather than B cells in the protective effect of DC A20 on blood pressure regulation.
In sum, we find that the ubiquitin-editing protein A20 in CD11c+ myeloid cells affords protection from RAS-mediated hypertension by limiting renal T cell activation. This blunted T cell activation has consequent effects on the elaboration of pro-hypertensive cytokines within the kidney that limits volume retention accruing from chronic Ang II infusion (Figure 8). These studies identify a new target within CD11c+ myeloid cells for the amelioration of immune-mediated blood pressure elevation and should lead to novel therapeutics for those patients with truly biologically resistant hypertension.
Figure 8. A role for A20 in dendritic cells (DCs) to protect against Ang II-induced hypertension.
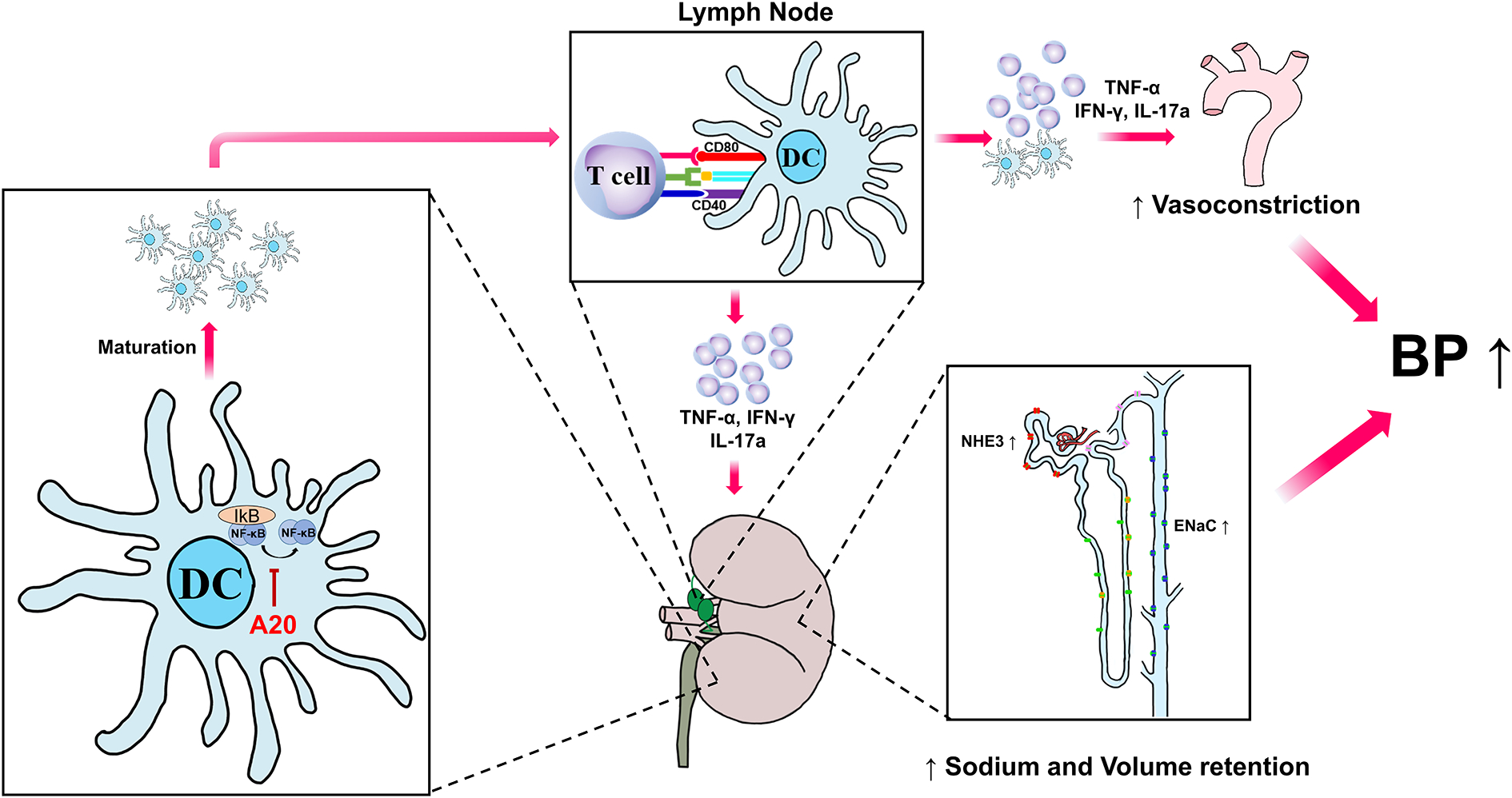
A20 in DCs inhibit IkB kinase and NF-ĸB activation, which limits the maturation of DCs. In response to Ang II, DCs are recruited to the kidney where they mature and are exposed to antigens. In the renal lymph nodes, activated DCs expressing co-stimulatory molecules prime T lymphocytes that further circulate in the kidney and release pro-hypertensive cytokines. Pro-hypertensive cytokines such as TNF-α, IFN-γ, and IL-17A increase sodium transporter expression and reduce blood flow in the kidney to drive volume retention and blood pressure elevation.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
A20 is a ubiquitin editor that inhibits NF-ĸB signaling.
Loss of A20 in CD11c+ myeloid cells causes autoimmunity.
50% loss of A20 in CD11c+ myeloid cells permits enhanced T lymphocyte activation without other baseline abnormalities.
What New Information Does This Article Contribute?
A20 in CD11c+ myeloid cells protects against Angiotensin (Ang) II-induced hypertension.
A20 in CD11c+ myeloid cells limits activation of T cells in the hypertensive kidney and restricts their capacity to produce pro-hypertensive cytokines including TNF-α and IFN-γ.
A20 in CD11c+ myeloid cells blunts renal sodium transporter expression and volume retention that contribute to blood pressure elevation.
ACKNOWLEDGMENTS
We thank Taylor Robinette for her technical and administrative assistance. We thank Junyi Zhang for her technical support.
SOURCES OF FUNDING
This work was supported by NIH grants DK118019, HL128355; Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development Grant BX000893; American Heart Association Grants 19POST34380480 and 18TPA34170047.
Nonstandard Abbreviations and Acronyms:
- DCs
dendritic cells
- MAP
mean arterial blood pressures
- WT
wild-type
- DC ACT
CD11c-Cre+ A20flox/wt
- TNF-α
tumor necrosis factor alpha
- IFN-γ
interferon gamma
- IL-17A
interleukin 17A
- IL-1β
interleukin 1beta
- Rag1
recombination activating protein 1
- Ang II
angiotensin II
- IBD
inflammatory bowel disease
- β-MHC
beta-myosin heavy chain
- MDA
malondialdehyde
- RBF
renal blood flow
- ENaC
epithelial sodium channel
- NHE3
sodium-hydrogen antiporter 3
- NKCC2
sodium-potassium-chloride cotransporter
- NCC
sodium-chloride cotransporter
Footnotes
DISCLOSURES
None.
REFERENCES
- 1.Collaboration NCDRF. Worldwide trends in blood pressure from 1975 to 2015: A pooled analysis of 1479 population-based measurement studies with 19.1 million participants. Lancet. 2017;389:37–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bromfield SG, Bowling CB, Tanner RM, Peralta CA, Odden MC, Oparil S, Muntner P. Trends in hypertension prevalence, awareness, treatment, and control among us adults 80 years and older, 1988–2010. J Clin Hypertens (Greenwich). 2014;16:270–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rodriguez-Iturbe B, Pons H, Quiroz Y, Gordon K, Rincon J, Chavez M, Parra G, Herrera-Acosta J, Gomez-Garre D, Largo R, Egido J, Johnson RJ. Mycophenolate mofetil prevents salt-sensitive hypertension resulting from angiotensin ii exposure. Kidney Int. 2001;59:2222–2232 [DOI] [PubMed] [Google Scholar]
- 4.Mattson DL, James L, Berdan EA, Meister CJ. Immune suppression attenuates hypertension and renal disease in the dahl salt-sensitive rat. Hypertension. 2006;48:149–156 [DOI] [PubMed] [Google Scholar]
- 5.Xiao L, Kirabo A, Wu J, Saleh MA, Zhu L, Wang F, Takahashi T, Loperena R, Foss JD, Mernaugh RL, Chen W, Roberts J, 2nd, , Osborn JW, Itani HA, Harrison DG. Renal denervation prevents immune cell activation and renal inflammation in angiotensin ii-induced hypertension. Circulation research. 2015;117:547–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Norlander AE, Saleh MA, Kamat NV, Ko B, Gnecco J, Zhu L, Dale BL, Iwakura Y, Hoover RS, McDonough AA, Madhur MS. Interleukin-17a regulates renal sodium transporters and renal injury in angiotensin ii-induced hypertension. Hypertension. 2016;68:167–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Guzik TJ, Hoch NE, Brown KA, McCann LA, Rahman A, Dikalov S, Goronzy J, Weyand C, Harrison DG. Role of the t cell in the genesis of angiotensin ii induced hypertension and vascular dysfunction. J Exp Med. 2007;204:2449–2460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Crowley SD, Song Y-S, Lin EE, Griffiths R, Kim H-S, Ruiz P. Lymphocyte responses exacerbate angiotensin ii-dependent hypertension. Am J Physiol Regul Integr Comp Physiol. 2010;298:R1089–1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Miguel C, Guo C, Lund H, Feng D, Mattson DL. Infiltrating t lymphocytes in the kidney increase oxidative stress and participate in the development of hypertension and renal disease. Am J Physiol Renal Physiol. 2011;300:F734–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li K, Guo D, Zhu H, Hering-Smith KS, Hamm LL, Ouyang J, Dong Y. Interleukin-6 stimulates epithelial sodium channels in mouse cortical collecting duct cells. Am J Physiol Regul Integr Comp Physiol. 2010;299:R590–595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kamat NV, Thabet SR, Xiao L, Saleh MA, Kirabo A, Madhur MS, Delpire E, Harrison DG, McDonough AA. Renal transporter activation during angiotensin-ii hypertension is blunted in interferon-gamma−/− and interleukin-17a−/− mice. Hypertension. 2015;65:569–576 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hashmat S, Rudemiller N, Lund H, Abais-Battad JM, Van Why S, Mattson DL. Interleukin-6 inhibition attenuates hypertension and associated renal damage in dahl salt-sensitive rats. Am J Physiol Renal Physiol. 2016;311:F555–561 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vinh A, Chen W, Blinder Y, Weiss D, Taylor WR, Goronzy JJ, Weyand CM, Harrison DG, Guzik TJ. Inhibition and genetic ablation of the b7/cd28 t-cell costimulation axis prevents experimental hypertension. Circulation. 2010;122:2529–2537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Itani HA, Xiao L, Saleh MA, Wu J, Pilkinton MA, Dale BL, Barbaro NR, Foss JD, Kirabo A, Montaniel KR, Norlander AE, Chen W, Sato R, Navar LG, Mallal SA, Madhur MS, Bernstein KE, Harrison DG. Cd70 exacerbates blood pressure elevation and renal damage in response to repeated hypertensive stimuli. Circ Res. 2016;118:1233–1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lee EG, Boone DL, Chai S, Libby SL, Chien M, Lodolce JP, Ma A. Failure to regulate tnf-induced nf-kappab and cell death responses in a20-deficient mice. Science. 2000;289:2350–2354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Turer EE, Tavares RM, Mortier E, Hitotsumatsu O, Advincula R, Lee B, Shifrin N, Malynn BA, Ma A. Homeostatic myd88-dependent signals cause lethal inflammation in the absence of a20. J Exp Med. 2008;205:451–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plenge RM, Cotsapas C, Davies L, Price AL, Bakker PIW, Maller J, Pe’er I, Burtt NP, Blumenstiel B, DeFelice M, Parkin M, Barry R, Winslow W, Healy C, Graham RR, Neale BM, Izmailova E, Roubenoff R, Parker AN, Glass R, Karlson EW, Maher N, Hafler DA, Lee DM, Seldin MF, Remmers EF, Lee AT, Padyukov L, Alfredsson L, Coblyn J, Weinblatt ME, Gabriel SB, Purcell S, Klareskog L, Gregersen PK, Shadick NA, Daly MJ, Altshuler D. Two independent alleles at 6q23 associated with risk of rheumatoid arthritis. Nat Genet. 2007;39:1477–1482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graham RR, Cotsapas C, Davies L, Hackett R, Lessard CJ, Leon JM, Burtt NP, Guiducci C, Parkin M, Gates C, Plenge RM, Behrens TW, Wither JE, Rioux JD, Fortin PR, Graham DC, Wong AK, Vyse TJ, Daly MJ, Altshuler D, Moser KL, Gaffney PM. Genetic variants near tnfaip3 on 6q23 are associated with systemic lupus erythematosus. Nat Genet. 2008;40:1059–1061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Musone SL, Taylor KE, Lu T, Nititham J, Ferreira RC, Ortmann W, Shiffrin N, Petri MA, Kamboh MI, Manzi S, Seldin MF, Gregersen PK, Behrens TW, Ma A, Kwok PY, Criswell LALA. Multiple polymorphisms in the tnfaip3 region are independently associated with systemic lupus erythematosus. Arthritis Rheum. 2008;58:S622–S622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nair RP, Duffin KC, Helms C, Ding J, Stuart PE, Goldgar D, Gudjonsson JE, Li Y, Tejasvi T, Feng BJ, Ruether A, Schreiber S, Weichenthal M, Gladman D, Rahman P, Schrodi SJ, Prahalad S, Guthery SL, Fischer J, Liao W, Kwok PY, Menter A, Lathrop GM, Wise CA, Begovich AB, Voorhees JJ, Elder JT, Krueger GG, Bowcock AM, Abecasis GR, Collaborative Association Study of P. Genome-wide scan reveals association of psoriasis with il-23 and nf-kappab pathways. Nat Genet. 2009;41:199–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hammer GE, Turer EE, Taylor KE, Fang CJ, Advincula R, Oshima S, Barrera J, Huang EJ, Hou B, Malynn BA, Reizis B, DeFranco A, Criswell LA, Nakamura MC, Ma A. Expression of a20 by dendritic cells preserves immune homeostasis and prevents colitis and spondyloarthritis. Nat Immunol. 2011;12:1184–1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang JD, Patel MB, Song YS, Griffiths R, Burchette J, Ruiz P, Sparks MA, Yan M, Howell DN, Gomez JA, Spurney RF, Coffman TM, Crowley SD. A novel role for type 1 angiotensin receptors on t lymphocytes to limit target organ damage in hypertension. Circ Res. 2012;110:1604–1617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang JD, Patel MB, Griffiths R, Dolber PC, Ruiz P, Sparks MA, Stegbauer J, Jin H, Gomez JA, Buckley AF, Lefler WS, Chen D, Crowley SD. Type 1 angiotensin receptors on macrophages ameliorate il-1 receptor-mediated kidney fibrosis. J Clin Invest. 2014;124:2198–2203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Crowley SD, Gurley SB, Herrera MJ, Ruiz P, Griffiths R, Kumar AP, Kim H-S, Smithies O, Le TH, Coffman TM. Angiotensin ii causes hypertension and cardiac hypertrophy through its receptors in the kidney. Proc Natl Acad Sci U S A. 2006;103:17985–17990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gerberick GF, Cruse LW, Miller CM, Sikorski EE, Ridder GM. Selective modulation of t cell memory markers cd62l and cd44 on murine draining lymph node cells following allergen and irritant treatment. Toxicol Appl Pharmacol. 1997;146:1–10 [DOI] [PubMed] [Google Scholar]
- 26.Zhang J, Rudemiller NP, Patel MB, Karlovich NS, Wu M, McDonough AA, Griffiths R, Sparks MA, Jeffs AD, Crowley SD. Interleukin-1 receptor activation potentiates salt reabsorption in angiotensin ii-induced hypertension via the nkcc2 co-transporter in the nephron. Cell Metab. 2016;23:360–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Trott DW, Thabet SR, Kirabo A, Saleh MA, Itani H, Norlander AE, Wu J, Goldstein A, Arendshorst WJ, Madhur MS, Chen W, Li CI, Shyr Y, Harrison DG. Oligoclonal cd8+ t cells play a critical role in the development of hypertension. Hypertension. 2014;64:1108–1115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Chae CU, Lee RT, Rifai N, Ridker PM. Blood pressure and inflammation in apparently healthy men. Hypertension. 2001;38:399–403 [DOI] [PubMed] [Google Scholar]
- 29.Bautista LE, Vera LM, Arenas IA, Gamarra G. Independent association between inflammatory markers (c-reactive protein, interleukin-6, and tnf-alpha) and essential hypertension. J Hum Hypertens. 2005;19:149–154 [DOI] [PubMed] [Google Scholar]
- 30.Sriramula S, Haque M, Majid DS, Francis J. Involvement of tumor necrosis factor-alpha in angiotensin ii-mediated effects on salt appetite, hypertension, and cardiac hypertrophy. Hypertension. 2008;51:1345–1351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Saleh MA, McMaster WG, Wu J, Norlander AE, Funt SA, Thabet SR, Kirabo A, Xiao L, Chen W, Itani HA, Michell D, Huan T, Zhang Y, Takaki S, Titze J, Levy D, Harrison DG, Madhur MS. Lymphocyte adaptor protein lnk deficiency exacerbates hypertension and end-organ inflammation. The Journal of clinical investigation. 2015;125:1189–1202 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim SS, Vos T, Flaxman ADea. A comparative risk assessment of burden of disease and injury attributable to 67 risk factors and risk factor clusters in 21 regions, 1990–2010: A systematic analysis for the global burden of disease study 2010. Lancet (London, England). 2012;380:2224–2260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Coffman TM. Under pressure: The search for the essential mechanisms of hypertension. Nat Med. 2011;17:1402–1409 [DOI] [PubMed] [Google Scholar]
- 34.Kirabo A, Fontana V, de Faria APC, Loperena R, Galindo CL, Wu J, Bikineyeva AT, Dikalov S, Xiao L, Chen W, Saleh MA, Trott DW, Itani HA, Vinh A, Amarnath V, Amarnath K, Guzik TJ, Bernstein KE, Shen XZ, Shyr Y, Chen S-c, Mernaugh RL, Laffer CL, Elijovich F, Davies SS, Moreno H, Madhur MS, Roberts J, 2nd, Harrison DG. Dc isoketal-modified proteins activate t cells and promote hypertension. The Journal of clinical investigation. 2014;124:4642–4656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Barbaro NR, Foss JD, Kryshtal DO, Tsyba N, Kumaresan S, Xiao L, Mernaugh RL, Itani HA, Loperena R, Chen W, Dikalov S, Titze JM, Knollmann BC, Harrison DG, Kirabo A. Dendritic cell amiloride-sensitive channels mediate sodium-induced inflammation and hypertension. Cell Rep. 2017;21:1009–1020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kool M, van Loo G, Waelput W, De Prijck S, Muskens F, Sze M, van Praet J, Branco-Madeira F, Janssens S, Reizis B, Elewaut D, Beyaert R, Hammad H, Lambrecht BN. The ubiquitin-editing protein a20 prevents dendritic cell activation, recognition of apoptotic cells, and systemic autoimmunity. Immunity. 2011;35:82–96 [DOI] [PubMed] [Google Scholar]
- 37.Rudemiller N, Lund H, Jacob HJ, Geurts AM, Mattson DL, PhysGen Knockout P. Cd247 modulates blood pressure by altering t-lymphocyte infiltration in the kidney. Hypertension. 2014;63:559–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Gool SW, Vandenberghe P, de Boer M, Ceuppens JL. Cd80, cd86 and cd40 provide accessory signals in a multiple-step t-cell activation model. Immunol Rev. 1996;153:47–83 [DOI] [PubMed] [Google Scholar]
- 39.Liu Y, Rafferty TM, Rhee SW, Webber JS, Song L, Ko B, Hoover RS, He B, Mu S. Cd8(+) t cells stimulate na-cl co-transporter ncc in distal convoluted tubules leading to salt-sensitive hypertension. Nat Commun. 2017;8:14037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Crowley SD, Frey CW, Gould SK, Griffiths R, Ruiz P, Burchette JL, Howell DN, Makhanova N, Yan M, Kim HS, Tharaux PL, Coffman TM. Stimulation of lymphocyte responses by angiotensin ii promotes kidney injury in hypertension. Am J Physiol Renal Physiol. 2008;295:F515–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ji H, Pai AV, West CA, Wu X, Speth RC, Sandberg K. Loss of resistance to angiotensin ii-induced hypertension in the jackson laboratory recombination-activating gene null mouse on the c57bl/6j background. Hypertension. 2017;69:1121–1127 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.