

Abstract
Background.
The failing right ventricle (RV) does not respond like the left ventricle (LV) to guideline directed medical therapy of heart failure, perhaps due to interventricular differences in their molecular pathophysiology.
Methods.
Using the canine tachypacing-induced biventricular heart failure (HF) model, we tested the hypothesis that interventricular differences in microRNAs (miRs) expression distinguish failing RV from failing LV.
Results.
Severe RV dysfunction was indicated by elevated end-diastolic pressure (11.3±2.5 versus 5.7±2.0 mmHg; P<0.0001) and diminished fractional area change (24.9±7.1 versus 48.0±3.6%; P<0.0001) relative to pre-pacing baselines. Microarray analysis of ventricular tissue revealed that miR-21 and miR-221, two activators of pro-fibrotic and proliferative processes, increased the most, at four- and two-fold, respectively, in RV-HF versus RV-Control. Neither miR-21 or miR-221 was statistically significant different in LV-HF versus LV-Control. These changes were accompanied by more extensive fibrosis in RV-HF than LV-HF. To test whether miR-21 and miR-221 upregulation is specific to RV cellular response to mechanical and hormonal stimuli associated with HF, we subjected fibroblasts and cardiomyocytes isolated from normal canine RV and LV to cyclic overstretch and/or aldosterone. These two stressors markedly upregulated miR-21 and miR-221 in RV fibroblasts but not in LV fibroblasts nor cardiomyocytes of either ventricle. Furthermore, miR-21/−221 knock-down significantly attenuated RV but not LV fibroblast proliferation.
Conclusions.
We identified a novel, biological difference between RV and LV fibroblasts that might underlie distinctions in pathological remodeling of the RV in biventricular heart failure.
Introduction
In patients with heart failure (HF), whether with preserved or reduced left ventricular (LV) ejection fraction, the development of secondary right ventricular (RV) dysfunction is a strong predictor of adverse outcomes.1 Besides its pathophysiological implications, biventricular failure poses a therapeutic challenge: the failing RV is less responsive than the failing LV to β-adrenergic blockers, angiotensin converting enzyme inhibitors, or angiotensin receptor blockers.2 RV biology is governed by molecular mechanisms that remain poorly understood and understudied, relative to that of the LV. Most experimental studies of RV failure have focused on animal models of pulmonary arterial hypertension. However, the most common cause of RV failure is not pulmonary arterial hypertension but rather secondary pulmonary hypertension due to LV disease. Only limited data exist regarding distinct structural and molecular alterations occurring in RV dysfunction secondary to LV failure. The primary goal of our study was to address this glaring knowledge gap. We tested the hypothesis that biventricular failure with secondary pulmonary hypertension may be characterized by interventricular differences in microRNA (miRNA or miR) expression that regulate distinct structural and functional derangements within each ventricle. Previous studies showed that hemodynamic stressors (either increased preload or afterload) induce certain miRNAs in the hypertrophied and failing RV.3–5 However, much less is known about miRNA changes in RV versus LV in other HF models. Therefore, we tested our hypothesis in dogs with tachypacing-induced HF, an established preclinical model of dilated cardiomyopathy with moderate secondary pulmonary hypertension.6 We characterized the progressive dysfunction of each ventricle and compared the miRNA expression profiles of failing RV versus failing LV. To explore inherent disparities between the biological stress responses of each ventricle, we isolated and cultured cardiomyocytes and fibroblasts from non-failing control RV and LV, subjected the cells to mechanical and neurohormonal HF-related pathological stimuli, and assessed whether the miRNAs identified by our tissue analysis were also differently expressed in this in vitro model.
Methods
The data that support the findings of this study are available from the corresponding authors upon reasonable request. Eighteen male mongrel dogs (age: 9–13 months; weight: 21–27 kg) were chronically instrumented and HF was induced in nine of them by pacing the LV as previously described.7 Control tissue was obtained from seven chronically instrumented dogs not subjected to cardiac pacing (non-failing controls). LV and RV were explanted after euthanasia and enzymatically dissociated to isolate cardiomyocytes and fibroblasts. The protocol was approved by the Institutional Animal Care and Use Committee (IACUC) of Temple University and conformed to the guiding principles for the care and use of laboratory animals published by the National Institutes of Health.
Hemodynamic, echocardiographic, histological and molecular analysis in cardiac tissue and isolated cells are described in detail in Supplements.
Statistical Analysis
miRNA microarray data was analyzed for differential miRNA expression between pre-specified groups (RV-HF vs. RV-Ctrl, LV-HF vs. LV-Ctrl, and RV-HF vs. LV-HF) by two-tailed Student’s t-test using GraphPad Software (Prism 7.0). To minimize false negative results, multiple comparisons correction was not performed. Given the small microarray sample size (n=3 per group), P<0.10 was considered statistically significant.
All other data were analyzed using SigmaStat 4.0, with P<0.05 considered statistically significant. Given that sample sizes were <10 per cohort, data normality was assessed by visual inspection. Non-normally distributed was subsequently analyzed using ANOVA on Ranks followed by Dunn’s multiple comparison tests. All other data for more than two groups were analyzed using either repeated measures ANOVA (for longitudinal echocardiographic data) or one-way ANOVA followed by Tukey post-hoc multiple comparisons test. Data for two groups were analyzed by two-tailed Student’s t-test.
Results
Cardiac remodeling and function
In end-stage HF, mean pulmonary artery pressure increased, while mean aortic pressure decreased (Figures 1A and 1B), reflecting the development of secondary pulmonary hypertension and systemic hypotension. End-diastolic pressure increased significantly in both ventricles, along with a decrease in RV and LV dP/dtmax (Supplemental Figure 1). During the time course of cardiac pacing, the fractional area change of both LV and RV displayed a progressive decline that became statistically significant by day 7 in LV and day 21 in RV, both persisting through day 28 (Figures 1C and 1D). Fibrosis in LV and RV tissue sections of control and failing hearts was quantified histologically by measuring the collagen surface area and was more pronounced in failing RV than in failing LV (Figures 1 E and 1F).
Figure 1. Tachypacing induced biventricular dysfunction with secondary pulmonary hypertension and disproportionate right ventricular fibrosis.
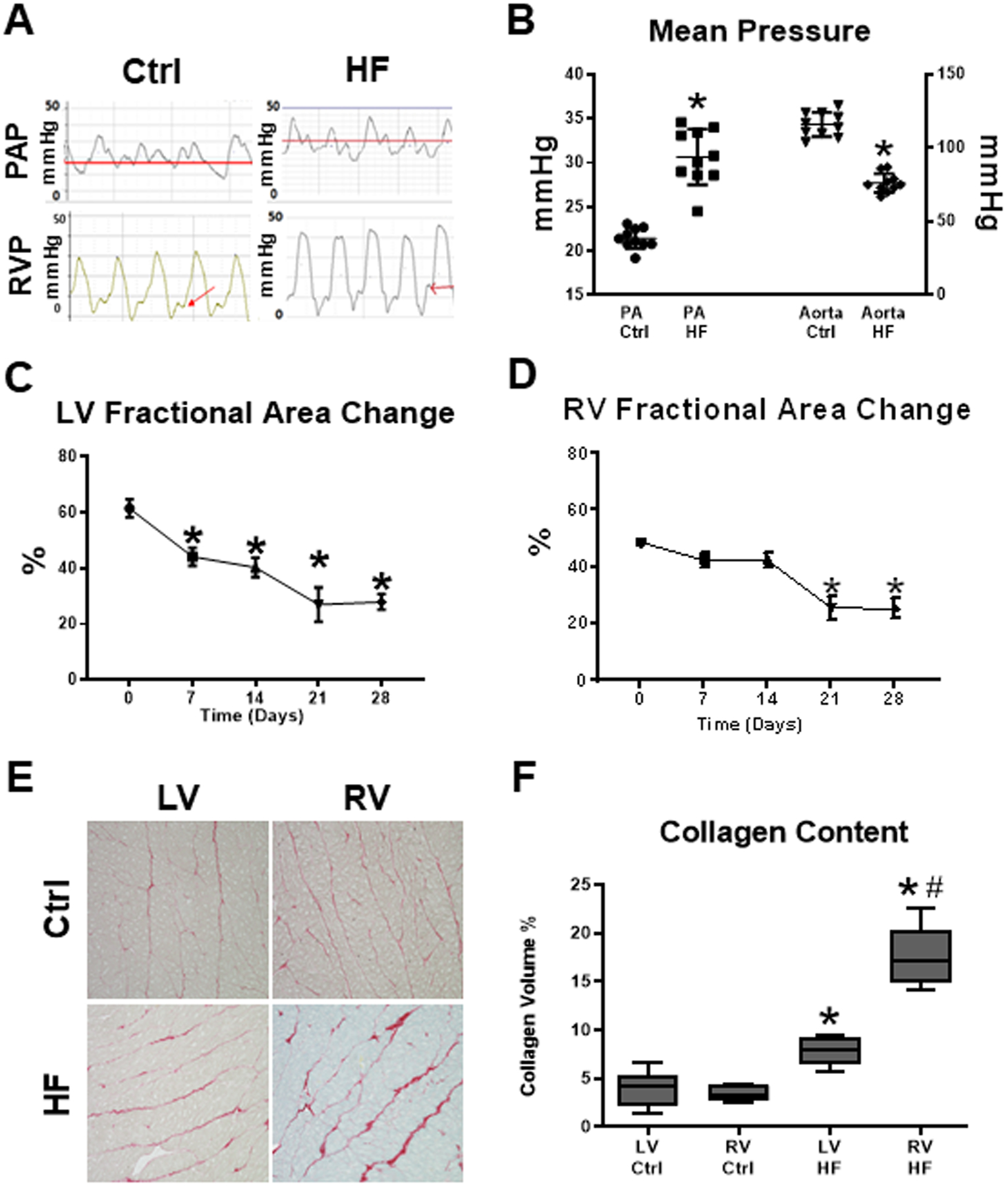
(A)Pulmonary artery and right ventricular pressure tracings in tachypacing (HF) and control (Ctrl) dogs. (B)Mean pulmonary artery pressure increased, while mean aortic pressure decreased, reflecting the secondary pulmonary hypertension and systemic hypotension that ensue in advanced heart failure (HF). RV end-diastolic pressure was significantly increased in HF. n=9 per group, *P<0.0001 vs time 0, Control (Ctrl). (C)Fractional area change of LV and (D)RV during the time course of cardiac pacing protocol; *P<0.05 vs Ctrl, analyzed by repeated measures ANOVA. (E)Sirius red staining of ventricular tissue. (F)Histological analysis indicated that collagen surface area was greater in failing RV than in failing LV. n= 5 per group. *P<0.01 vs respective Ctrl; #P<0.05 vs LV HF, analyzed by ANOVA on Ranks.
Myocardial molecular alterations
Microarray analysis determined the expression profile of 289 miRNAs in the ventricles of failing and control animals (Supplemental Table 1). A total of 139 distinct miRNAs were detected at significant levels in the canine ventricles, with 26 miRNAs differentially expressed between the failing RV (RV- HF) versus non-failing control RV (RV-Ctrl) (Figure 2A, P<0.10). Of those 26 miRNAs, 4 were also differentially expressed between failing LV (LV-HF) and non-failing control LV (LV-Ctrl), while 6 were also differentially expressed between RV-HF and LV-HF (Figure 2B, P<0.10). Of the 26 miRNAs that were differentially expressed in RV-HF versus RV-Ctrl, miR-21 and −221 exhibited at least a 2-fold change (FC) relative to RV-Ctrl (log2(FC)>1.0, P=0.07 for miR-21, P=0.01 for miR-221), consistent with the known association between upregulation of these miRs and cardiac fibrosis.6–8 We confirmed these transcriptional changes by quantitative RT-PCR (Figure 2C). Upregulation of miR-21 and miR-221 in the failing RV was accompanied by protein downregulation of their shared target phosphatase and tensin homologue (PTEN) and miR-21 target programmed cell death-4 (PDCD4) (Supplemental Figure 2).
Figure 2. miR-21 and miR-221 are upregulated in the failing RV of tachypacing canine model and in isolated adult canine fibroblasts subjected to cyclic stretch and/or aldosterone stimulation.
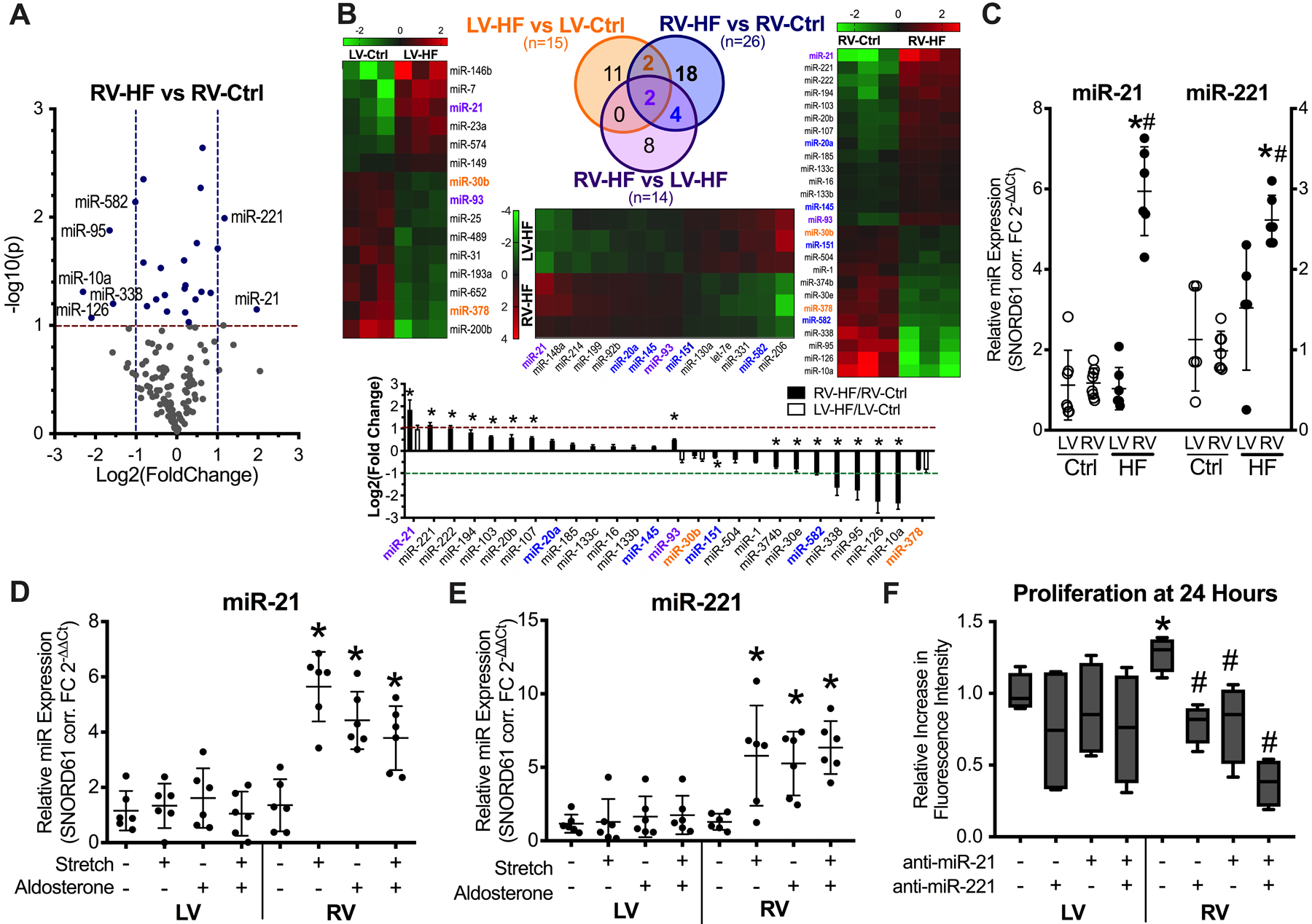
(A)Volcano plot of miRNA expression in RV-HF vs RV-Ctrl. Blue dots, miRNAs that were differentially expressed at P<0.10. Labeled dots, miRNAs that were differentially expressed at a minimum 2-fold change in either direction (n=3 per group). (B)Heat maps, Venn diagram, and summary bar graph of differentially expressed miRNAs. Orange font, differentially expressed in RV-HF vs RV-Ctrl and in LV-HF vs LV-Ctrl but not statistically significantly different in RV-HF vs LV-HF. Blue font, differentially expressed in RV-HF vs RV-Ctrl and in RV-HF vs LV-HF, but not statistically significantly different in LV-HF vs LV-Ctrl. Purple font, differentially expressed across all three comparisons: LV-HF vs LV-Ctrl, RV-HF vs RV-Ctrl, and RV-HF vs LV-HF. *P<0.05 vs respective LV-HF/LV-Ctrl. (C)Quantitative RT-PCR analysis of miR-21 and miR-221 in ventricular tissue, n= 6 per group. *P<0.01 vs respective Ctrl; #P<0.01 vs LV HF. (D)Cyclic overstretch and/or aldosterone induced a marked increase in miR-21 (*P<0.01 vs unstimulated) and (E)miR-221 (*P<0.05 vs unstimulated) only in RV fibroblasts. (F)Inhibition of miR-21/−221 attenuated proliferation in RV but not LV fibroblasts. n= 4 per experimental condition. *P<0.05 vs respective LV, #P<0.05 vs RV without antimir, analyzed by ANOVA on Ranks.
Response of RV and LV cardiomyocytes and fibroblasts to mechanical and hormonal stimuli
The ensuing question was whether the increase in miR-21 and miR-221 in failing RV was attributable to their upregulation in cardiomyocytes, fibroblasts, or both. Moreover, it was necessary to rule out the influence of ventricular afterload on miR-21 and miR-221 expression. Therefore, cardiomyocytes and fibroblasts were isolated from LV and RV of normal hearts. Cardiomyocytes could be unequivocally recognized under the microscope given their unique rod-like shape (Supplemental Figure 3A). The relative purity of isolated fibroblasts was thereby confirmed by immunohistochemistry (Supplemental Figure 3B) and FACS analysis (Supplemental Figures 3C–F) based on the presence of specific fibroblast markers vimentin, FSP1 and PDGF-α, and the absence of markers of myofibroblasts/smooth muscle cells, endothelial cells and CD45-positive leukocytes.
Cardiomyocytes and fibroblasts were separately cultured and subjected to cyclic overstretch and/or exposed to aldosterone. Neither stretch nor aldosterone caused significant changes in miR-21 and miR- 221 expression in cardiomyocytes (Supplemental Figure 4A–D). However, they induced a marked increase in miR-21 and miR-221 in RV fibroblasts but not LV fibroblasts (Figures 2D and 2E). Transcript expression of miR-21/−221 targets Pten and Spry1 in fibroblasts were consistent with miR-21/−221 expression (Supplemental Figures 4E and 4F). Proliferation was significantly higher in RV compared to LV fibroblasts and knockdown of miR-21/221 significantly attenuated proliferation only in RV fibroblasts (Figure 2F).
Discussion
Our study shows a remarkable and previously unknown biological difference between RV and LV fibroblasts. In an established preclinical model of dilated cardiomyopathy, 14 miRNAs were differentially expressed in failing RV compared to failing LV, despite a similar temporal development of dysfunction in both ventricles. We focused on two miRNAs that were upregulated in failing RV relative to non-failing control RV, namely miR-21 and miR-221. The upregulation of miR-21, a known enhancer of pro-fibrotic intracellular pathways,3–5, 8 was associated with more pronounced fibrosis in the RV, as previously reported in models of afterload/preload-induced RV hypertrophy and failure.6–8 One possible factor that could have caused such structural and molecular differences between the two ventricles in dogs is the disparate afterload, decreased for the LV (systemic hypotension) and increased for the RV (secondary pulmonary hypertension). We could not measure sufficient parameters to calculate systolic wall stress with reasonable accuracy. However, it is evident that the combined effect of increased internal diameter and pulmonary arterial pressure necessarily led to stress augmentation in the RV, a chamber that is disadvantaged by its thinner wall compared to the LV. Other complex neurohormonal factors might also have unevenly affected the failing heart. Therefore, to exclude confounding factors and assess whether and which cardiac cells display ventricle-specific differences in miRNA regulation, we tested LV and RV cardiomyocyte and fibroblast responses to HF-associated pathological stimuli in vitro, under controlled and matched conditions. Cyclic overstretch and/or high concentrations of aldosterone, two stressors widely utilized to simulate, respectively, important mechanical and neurohormonal stresses in HF, induced marked overexpression of miR-21 and miR-221 and downregulation of their target mRNAs in RV fibroblasts alone (Supplemental Figure 2A). These surprising findings suggest that RV fibroblasts are characterized by a specific response to insults, a feature that might render the RV more vulnerable to increased afterload and neurohormonal activation and more refractory to therapeutic interventions that otherwise benefit the LV. Interestingly, a previous study in left atrial and LV tissue collected from tachypaced dogs showed that fibroblasts are more predisposed than cardiomyocytes to changes in miRNA expression.9 Fibroblasts display a heterogeneous phenotype that depends on their anatomical site,10 probably due to the local tissue milieu. Interestingly, fibroblasts retain a “memory” of their specific gene expression patterns when harvested and cultured in vitro,10 as we also found in our study. During embryonic development, the majority of cardiac fibroblasts derive from the proepicardial organ and later migrate and colonize the two ventricles. Due to their own distinct embryological origins of the first and second heart fields respectively, LV and RV cardiomyocytes might confer onto later colonizing fibroblasts different features that persist throughout post-natal life. One of them might be the overexpression of specific miRNAs in response to pathophysiological stimuli. Upregulation of miR-21 has been demonstrated in pathologic, pressure overload-induced hypertrophy (both LV and RV), ischemia/reperfusion injury, and especially fibrosis.3–5, 8 However, pathological cardiac remodeling in mice can also occur in the absence of miR-21.11
While miR-21 and −221 have been linked to epithelial-to-mesenchymal transition (EMT), our in vitro experiments showed no statistically significant difference in expression of EMT genes VIM, FN1, CDH1, or CDH2 in fibroblasts of either ventricle in response to miR-21/−221 knock-down. However, knockdown of miR-21/221 attenuated proliferation of adult RV but not LV fibroblasts, suggesting that miR-21 and miR-221 are critical for only RV fibroblast proliferation. Less is known about miR-221 than miR-21, and the handful of studies report conflicting findings. In contrast to our data, a recent study of fibrotic myocardium from HF patients attributed an anti-fibrotic function to miR-221/222.12 Major methodological differences preclude a direct comparison of their findings with ours.
An obvious limitation of our study is the unfeasibility to directly test, in vivo, whether preventing upregulation of miR-21 and miR-221 can limit RV fibrosis and/or preserve RV function. This would require RV-selective miR antagonism or knockdown or conditional knockout, which are not possible at this time in our large animal model.
The translational significance of our finding is in the therapeutic potential of targeting miR-21/−221 as an RV-selective anti-fibrotic strategy. RV fibrosis has long been implicated as a pathogenic factor underlying RV failure in patients with pulmonary arterial hypertension. The central role of RV fibrosis in reduced RV systolic function has been demonstrated in HF patients and animal models. In HF patients who underwent cardiac transplantation, the worse the RV systolic performance as measured by RV free wall longitudinal strain, the worse the patient’s exercise capacity on cardiopulmonary stress test and the greater the extent of RV fibrosis.13 In contrast, LV function did not correlate with extent of LV fibrosis or exercise capacity. Moreover, experimental animal models have revealed that even established RV fibrosis is reversible and its reversibility is associated with RV functional recovery.14, 15
In conclusion, we provide evidence that the molecular pathophysiology of RV failure is distinct from that of LV failure. Thus, we believe that our findings might contribute to a deeper understanding of the molecular mechanisms underlying the development of RV failure and spur further investigation of therapeutically targetable biological stress responses of RV cardiac fibroblasts.
Supplementary Material
Sources of Funding
This work was supported by the NIH grants R01 HL129120 (F.A.R.), K08HL109159 (E.J.T.) and R01 HL135177 (M.K.) and the American College of Cardiology - Presidential Career Development Award (E.J.T.).
Footnotes
Disclosures
Nothing to disclose.
References
- 1.Bosch L, Lam CSP, Gong L, Chan SP, Sim D, Yeo D, Jaufeerally F, Leong KTG, Ong HY, Ng TP, et al. Right ventricular dysfunction in left-sided heart failure with preserved versus reduced ejection fraction. Eur J Heart Fail. 2017;19:1664–1671. [DOI] [PubMed] [Google Scholar]
- 2.Konstam MA, Kiernan MS, Bernstein D, Bozkurt B, Jacob M, Kapur NK, Kociol RD, Lewis EF, Mehra MR, Pagani FD, et al. , American Heart Association Council on Clinical Cardiolog, Council on Cardiovascular Disease in the Young, Council on Cardiovascular Surgery and Anesthesia. Evaluation and Management of Right-Sided Heart Failure: A Scientific Statement From the American Heart Association. Circulation. 2018;137:e578–e622. [DOI] [PubMed] [Google Scholar]
- 3.Batkai S, Bar C and Thum T. MicroRNAs in right ventricular remodelling. Cardiovasc Res. 2017;113:1433–1440. [DOI] [PubMed] [Google Scholar]
- 4.Reddy S, Zhao M, Hu DQ, Fajardo G, Hu S, Ghosh Z, Rajagopalan V, Wu JC and Bernstein D. Dynamic microRNA expression during the transition from right ventricular hypertrophy to failure. Physiol Genomics. 2012;44:562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reddy S, Hu DQ, Zhao M, Blay E Jr, Sandeep N, Ong SG, Jung G, Kooiker KB, Coronado M, G Fajardo, et al. miR-21 is associated with fibrosis and right ventricular failure. JCI Insight. 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pagnamenta A, Dewachter C, McEntee K, Fesler P, Brimioulle S and Naeije R. Early right ventriculo-arterial uncoupling in borderline pulmonary hypertension on experimental heart failure. J Appl Physiol (1985). 2010;109:1080–5. [DOI] [PubMed] [Google Scholar]
- 7.Woitek F, Zentilin L, Hoffman NE, Powers JC, Ottiger I, Parikh S, Kulczycki AM, Hurst M, Ring N, Wang T, et al. Intracoronary Cytoprotective Gene Therapy: A Study of VEGF-B167 in a Pre-Clinical Animal Model of Dilated Cardiomyopathy. J Am Coll Cardiol. 2015;66:139–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et al. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. [DOI] [PubMed] [Google Scholar]
- 9.Chen Y, Wakili R, Xiao J, Wu CT, Luo X, Clauss S, Dawson K, Qi X, Naud P, Shi YF, et al. Detailed characterization of microRNA changes in a canine heart failure model: Relationship to arrhythmogenic structural remodeling. J Mol Cell Cardiol. 2014;77:113–24. [DOI] [PubMed] [Google Scholar]
- 10.Chang HY, Chi JT, Dudoit S, Bondre C, van de Rijn M, Botstein D and Brown PO. Diversity, topographic differentiation, and positional memory in human fibroblasts. Proc Natl Acad Sci U S A. 2002;99:12877–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Patrick DM, Montgomery RL, Qi X, Obad S, Kauppinen S, Hill JA, van Rooij E and Olson EN. Stress-dependent cardiac remodeling occurs in the absence of microRNA-21 in mice. J Clin Invest. 2010;120:3912–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Verjans R, Peters T, Beaumont FJ, van Leeuwen R, van Herwaarden T, Verhesen W, Munts C, Bijnen M, Henkens M, Diez J, et al. MicroRNA-221/222 Family Counteracts Myocardial Fibrosis in Pressure Overload-Induced Heart Failure. Hypertension. 2018;71:280–288. [DOI] [PubMed] [Google Scholar]
- 13.Lisi M, Cameli M, Righini FM, Malandrino A, Tacchini D, Focardi M, Tsioulpas C, Bernazzali S, Tanganelli P, Maccherini M, et al. RV Longitudinal Deformation Correlates With Myocardial Fibrosis in Patients With End-Stage Heart Failure. JACC Cardiovasc Imaging. 2015;8:514–522. [DOI] [PubMed] [Google Scholar]
- 14.Gomez-Arroyo J, Sakagami M, Syed AA, Farkas L, Van Tassell B, Kraskauskas D, Mizuno S, Abbate A, Bogaard HJ, Byron PR and Voelkel NF. Iloprost reverses established fibrosis in experimental right ventricular failure. Eur Respir J. 2015;45:449–62. [DOI] [PubMed] [Google Scholar]
- 15.McKellar SH, Javan H, Bowen ME, Liu X, Schaaf CL, Briggs CM, Zou H, Gomez AD, Abdullah OM, Hsu EW, et al. Animal model of reversible, right ventricular failure. J Surg Res. 2015;194:327–33. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.