

Abstract
Rationale:
Aging is one of the strongest risk factors for atherosclerosis. Yet whether aging increases the risk of atherosclerosis independently of chronic hyperlipidemia is not known.
Objective:
To determine if vascular aging prior to the induction of hyperlipidemia enhances atherogenesis.
Methods and Results:
We analyzed the aortas of young and aged normolipidemic wild type (WT), disease free mice and found that aging led to elevated IL-6 levels and mitochondrial dysfunction, associated with increased mitophagy and the associated protein Parkin. In aortic tissue culture, we found evidence that with aging mitochondrial dysfunction and IL-6 exist in a positive feedback loop. We triggered acute hyperlipidemia in aged and young mice by inducing liver-specific degradation of the LDL receptor combined with a 10-week western diet and found that atherogenesis was enhanced in aged WT mice. Hyperlipidemia further reduced mitochondrial function and increased the levels of Parkin in the aortas of aged mice but not young mice. Genetic disruption of autophagy in smooth muscle cells of young mice exposed to hyperlipidemia led to increased aortic Parkin and IL-6 levels, impaired mitochondrial function, and enhanced atherogenesis. Importantly, enhancing mitophagy in aged, hyperlipidemic mice via oral administration of spermidine prevented the increase in aortic IL-6 and Parkin, attenuated mitochondrial dysfunction, and reduced atherogenesis.
Conclusions:
Prior to hyperlipidemia, aging elevates IL-6 and impairs mitochondrial function within the aorta, associated with enhanced mitophagy and increased Parkin levels. These age-associated changes prime the vasculature to exacerbate atherogenesis upon acute hyperlipidemia. Our work implies that novel therapeutics aimed at improving vascular mitochondrial bioenergetics or reducing inflammation before hyperlipidemia may reduce age-related atherosclerosis.
Subject Terms: Animal Models of Human Disease, Atherosclerosis, Basic Science Research, Inflammation, Vascular Biology
Keywords: Atherosclerosis, aging, hyperlipidemia, mitochondria, animal model of cardiovascular disease, vascular biology, atherogenesis
Graphical Abstract
INTRODUCTION
Cardiovascular diseases are the leading cause of morbidity and mortality in older people, accounting for ~25% of all deaths in the United States. Importantly, 81% of cardiovascular patients who die are > 65-years old.1 Epidemiological studies have established that aging is a major risk factor for atherosclerosis.1, 2 However, it is not known whether this increased risk reflects chronic hypercholesterolemia, or other age-associated effects. For instance, macroautophagy (herein termed autophagy), including mitophagy, is thought to decline with age. This process removes damaged mitochondria from the cell,3 and is a key regulator of longevity.4 Autophagy packages dysfunctional or damaged organelles and delivers them to lysosomes for degradation. Dysfunctional autophagy results in the release of damage-associated molecular patterns, which induces inflammation to promote chronic vascular diseases such as atherosclerosis.5 Thus, autophagy prevents inflammation by clearing defective organelles.6
Several studies have linked autophagy to atherogenesis. For instance, a key adapter protein of the inner membrane of the autophagosome, p62, accumulates in atherosclerotic aortas of Apoe−/− mice but not in the aortas of wild type (WT) mice.7 Furthermore, deletion of key autophagy genes in myeloid cells in mice increases atherosclerotic plaque necrosis by reducing macrophage efferocytosis.8 Enhancing autophagy by feeding atherogenic mice the polyamine spermidine increases lifespan and reduces atherogenesis.9, 10 However, how autophagy and specifically mitophagy change with vascular aging is not known.
Current murine models of atherosclerosis fail to separate the effects of intrinsic aging from chronic hypercholesterolemia on atherogenesis. Specifically, Ldlr−/− and Apoe−/− mice exhibit hypercholesterolemia as early as 3-months of age and subsequent metabolic derangements from 12-months of age.11-13 In this study, to overcome these limitations, we induced acute hypercholesterolemia in young and aged WT mice by ectopically expressing “proprotein convertase subtilisin/kexin 9” (Pcsk9), which triggers the degradation of the LDL receptor. We used a liver-trophic adeno-associated virus to overexpress Pcsk9, and subsequently fed mice a high fat western-type diet (WD).14 This allowed us to directly examine the effect of intrinsic aging on the development of atherosclerosis.
Employing this inducible hyperlipidemia model, we used young and aged, male and female mice to investigate how aging impacts the vasculature prior to and in response to acute hyperlipidemia. We show that prior to hyperlipidemia, vascular aging leads to elevated IL-6 levels, impaired mitochondrial function with enhanced mitophagy that exacerbates atherogenesis arising from acute hyperlipidemia.
METHODS
The data that support the findings of this study are available from the corresponding author upon reasonable request.
See Online Supplemental Material for detailed methods and regulatory approval.
RESULTS
Aging impairs mitochondrial function within the aorta.
Mitochondrial dysfunction is a hallmark of aging,3, 15 although how this impacts the vasculature is not clear. We hypothesized that mitochondrial function within the vasculature also decreases with age, even prior to hyperlipidemia. To test this hypothesis, we measured maximal oxygen consumption rates (OCR) during coupled oxidative phosphorylation (OXPHOS) and uncoupled respiration in the aortas of young and aged WT mice on a low-fat diet (LFD, i.e., a normal laboratory diet). Maximal OCRs were determined by titrating a mitochondrial inner membrane uncoupler as well as electron transport chain substrates and inhibitors using a standard protocol (Figure 1A and methods).16 We observed a 40% reduction (P=0.026) in maximal complex I+II OXPHOS and a 50% (P=0.02) reduction in maximal complex II uncoupled OCR (Figure 1B) in the aortas from aged (i.e., 18-19 months of age) versus young (i.e., 2-3 months of age) male C57BL/6 WT mice. Despite the decline in aortic OCR with aging, aged and young WT mice on a LFD exhibited similar whole-body respiratory exchange ratio (Online Figure I). Thus, aging leads to a decline in mitochondrial function within the aorta of healthy mice.
Figure 1. Aging leads to mitochondrial dysfunction and increased aortic Parkin expression during normolipidemia.

WT mice were maintained on a LFD until either 3-months or 18-months of age. At this point, the thoracic aorta was harvested for oxygen consumption rate (OCR) measurements, western blots or immunofluorescence staining. (A) Representative protocol for measuring the maximal OCR using inhibitors, substrates, and uncoupler (see Methods). (B) Maximal aortic OCR with substrates for complex I and I+II coupled OXPHOS and maximal complex I+II and II uncoupled OCR. (C) Aortic lysates from LFD-fed young and aged WT C57BL/6 mice were immunoblotted against PINK1, Parkin, Complex V, Complex III, and Complex II and β-actin. (D-H) Quantification of immunoblot in C. (I) Ratio of mtDNA to nuclear DNA as measured by real time PCR. (J) Fixed frozen aortic sections (6μm) were stained with primary antibodies against Parkin and smooth muscle α-actin or secondary antibodies only and nuclei were stained with hoechst. Scale bars: 10μm. (K) Mean fluorescence intensity (MFI) of parkin signal within the smooth muscle α-actin positive media layer was quantified. 2-way ANOVA with Sidak’s multiple comparison test for B. Unpaired t-test for D and E. Mann-Whitney U-test for F-I and K. Each data point represents a biological replicate. All results are presented as mean ±SEM. A=aged, CI=complex I, CII=complex II, Y=young.
To determine if reductions in aortic OCR with aging correlated with increased mitochondrial DNA damage, we assessed mitochondrial DNA breaks via qPCR. We found no evidence of increased mitochondrial DNA breaks in the aged aortas as compared to young aortas (Online Figure II A-B), suggesting that the age-related mitochondrial dysfunction is not associated with mitochondrial DNA breaks.
Aging increases the levels of the mitophagy protein Parkin within the aorta.
Mitophagy removes dysfunctional mitochondria.17 The E3 ubiquitin ligase Parkin, a key mitophagy protein, is recruited to impaired mitochondria, and this recruitment requires PINK1 expression.18 The relationship between aging and mitophagy within the vasculature is unknown. To address this question, we measured the levels of Parkin and PINK1 within the aorta of young and aged mice maintained on a LFD. We found that the aortas from aged mice exhibited significantly higher (i.e., greater than two-fold, P =0.0001) levels of Parkin but not PINK1 (Figure 1C-E). Importantly, baseline levels of the mitochondrial electron transport chain proteins complex V, complex III, and complex II, and the mitochondrial-to-nuclear DNA ratio, were similar between young and aged aortas (Figure 1F-I). Thus, increased Parkin was not due to an increase in mitochondrial content. Fluorescence imaging indicated that the age-related increase in Parkin occurred in all vascular layers of the aorta, but particularly in smooth muscle cells (SMCs) (Figure 1J-K). Together, with the data above, our findings suggest that aging leads to dysfunctional mitochondria and the accumulation of Parkin within the aorta.
Aging increases mitophagy within the aorta despite preserved autophagy.
Given the increases in Parkin within the aged aorta, we examined potential differences in the autophagic flux within the aorta in young and aged mice. Specifically, we measured autophagic proteins LC3I, LC3II, the LC3 II:I ratio as a measure of autophagic flux, the autophagy substrate/autophagosome marker p62, and the lysosomal protein Lamp1.19 We measured these in aortas ex vivo with serum-free media (i.e., serum-starved) and with complete media (i.e., with 10% FBS). Serum-starvation, which induces autophagy,20 significantly increased the LC3II:I ratio regardless of aging (Online Figure III A-B), suggesting that autophagic flux in response to serum-starvation is preserved in the aorta of normolipidemic hosts with aging. p62 and Lamp1 exhibited similar expression regardless of aging and was reduced by starvation (Online Figure III C-D). Additionally, fluorescence imaging revealed that p62 and Lamp1 exhibited a similar co-localized expression pattern within aged and young aortas (Online Figure III G-H).
Aging was associated with increased levels of Parkin and of Nix, a protein that tethers mitochondria to the autophagic machinery (Online Figure III A and E-F).21 Parkin and Nix levels were unaffected by serum-starvation, suggesting they are not degraded during autophagic flux (Online Figure III A and III E-F). Overall, these data suggest that autophagy in the aorta is not affected by aging.
To assess basal mitophagy in young and aged aortas, we monitored the colocalization of mitochondria with lysosomes by fluorescence microscopy. The frequency of mitochondria-lysosome colocalization was significantly higher in aortas from aged mice than in those from young mice, both maintained on a LFD (Figure 2A-C, P=0.029). Additionally, we isolated aortic mitochondria and measured poly-ubiquitination, which tags mitochondria for removal by mitophagy,22 and observed a two-fold increase with aging (Figure 2D-E, P=0.002). Finally, we employed young and aged mitophagy reporter mtKeima mice. These mice express the pH-sensitive and lysosome resistant-protein Keima fused to the mitochondrial targeting sequence from Cox VIII.23 As a result, mitochondria that are in the lysosomes emit a red signal in these mice.23 By coupling this signal with mitotracker green, a mitochondrial marker, the ratio of red to green emission can be objectively calculated as a mitophagy index that signifies the proportion of mitochondria within the lysosomes. Aortas from 4 month and 16-month old mtKeima mice indicated that the aged mtKeima mice exhibit a 30% increased mitophagy index within the aorta as compared to young mtKeima mice (Figure 2F-G, P=0.029). Overall, these data suggest that aging leads to an increase in dysfunctional mitochondria and mitophagy within the aorta.
Figure 2. Aging leads to increased measures of aortic mitophagy and mitochondrial ubiquitination.

(A) Aortas from young or aged WT, LFD-fed mice were harvested and incubated with mitotracker and lysotracker then stained with hoechst and imaged by confocal microscopy. Arrows denote colocalized mitochondrial puncta and lysosomal puncta. Scale bars=10μm. (B) Fluorescence histograms of mitotracker and lysotracker signal of puncta denoted by arrows in (A) showing colocalized signal. (C) Percent of total mitochondrial puncta MFI that colocalizes with lysosome puncta (see methods). (D-E) Mitochondria were isolated from the aortas of young and aged mice and were immuoblotted against ubiquitin, β-actin, and CoxIV. Isolated mitochondria lanes were normalized against CoxIV. (F-G) Aortas were harvested from young (4 months of age) and aged (16 months of age) mtKiema mice (see methods). Aortas were incubated with mitotracker green. The mitotracker green signal (488nm excitation) and mtKeima red signal (561nm excitation) were assessed by fluorescence microscopy (see methods) and the ratio of 561:488 (mitophagy index) is shown in G. Scale bars in L= 10μm. Each data point represents a biological replicate. All results are presented as mean ±SEM. Mann-Whitney U-test for C, E, G. A=aged, Y=young.
Aging leads to increased expression of TLR9-MyD88 IL-6 pathway within the aorta.
Mitochondrial damage-associated molecular patterns activate the Toll-Like Receptor 9 (TLR9)-MyD88 pathway to produce inflammatory cytokines such as IL-6.24, 25 Because our data suggest that aging reduces mitochondrial function and increases mitophagy within the aorta of normolipidemic mice, we evaluated the levels of TLR9-MyD88-IL-6 pathway. We found that aortas from aged mice had 2-3-fold higher levels of TLR9 (P=0.0008), MyD88 (P=0.02), and IL-6 (P=0.0047) compared to those from young mice (Figure 3A). Thus, aging is associated with elevated levels of TLR9-MyD88-IL-6 within the aorta.
Figure 3. Aging leads to increased IL-6 aortic expression during normolipidemia. IL-6 impairs OCR and increases Parkin in aortic tissue culture.

(A) Thoracic aortas from young or aged WT, LFD-fed mice were harvested and frozen for immunoblots and blotted against TLR9, MyD88, IL-6, and β-actin. Quantification of immunoblots is to the right. (B) Thoracic aortas from young WT mice were incubated in DMEM+10%FBS with either 0 or 10ng/mL of IL-6 for 2h and then OCR was assessed per Figure 1A. (C) Thoracic aortas from young WT mice were incubated in DMEM+10%FBS with either 0 or 10ng/mL of IL-6 and then the lysate was immunoblotted against Parkin, Nix, and β-actin. (D) Thoracic aortas from aged (18 months of age) WT mice were incubated in DMEM+10%FBS with 5μg/ml anti-IL-6 antibody or isotype control antibody for 2h and then OCR measured. (E) Thoracic aortas from aged (18months of age) WT mice were incubated in DMEM+10%FBS with 5μg/ml anti-IL-6 antibody or isotype control for 2h and then the lysate was immunoblotted against Parkin, Nix, and β-actin. Each data point represents a biological replicate. All results are presented as mean ±SEM. Mann-Whitney U-test for A, C, and E. 2-way ANOVA with Sidak’s post-hoc test for B and D. A=aged, CI=complex I, CII=complex II, Y=young,
IL-6 impairs mitochondrial function and increases Parkin levels in aortic tissue culture.
IL-6 is an inflammatory cytokine that impacts mitochondrial function in a variety cells.26,27,28 Therefore, we reasoned that elevated IL-6 in the aged aorta could impair mitochondrial function and elevate Parkin levels. To determine if IL-6 treatment is sufficient to impair mitochondrial function in aortas, we cultured young aortas with recombinant IL-6. We found that this treatment reduced maximal coupled OXPHOS (P=0.02) and uncoupled (P=0.0004) respiration within the young aorta by 20% (Figure 3B), and also increased the levels of both Parkin and Nix two-fold (Figure 3C), implying that IL-6 treatment compromises mitochondrial function and induces mitophagy.
To determine if Parkin was critical for the IL-6-dependent decrease in OCR, we cultured aortas from young Parkin-deficient mice with IL-6. We found that OCR declined similarly in both the aortas of young WT and Parkin-deficient aortas (compare Figure 3B to Online Figure IV A). Aortic OCR measurements were similar between young WT and Parkin-deficient aortas cultured in standard conditions without IL-6 (Online Figure IV B). These data suggest that Parkin elevation is a consequence, rather than a cause, of mitochondrial dysfunction within the aorta, likely reflecting increased mitophagy.
To determine if IL-6 is required for mitochondrial dysfunction in the aged aorta, we cultured the aortas of aged mice with a neutralizing monoclonal antibody to IL-6. We found that this antibody treatment, as compared to isotype control, resulted in a 20% increase in maximal coupled OXPHOS (P=0.002) and uncoupled (P=0.011) respiration (Figure 3D) and in a two-fold reduction in Parkin (P=0.016) and Nix levels (P=0.06) (Figure 3E). These data suggest that IL-6, which is increased within the aged aorta, is necessary and sufficient to impair mitochondrial function and elevate Parkin. Overall, our data suggest that a positive feedback loop of mitochondrial dysfunction and elevated IL-6 exists within the aorta during aging.
FCCP treatment enhances mitophagy and reduces Parkin, TLR9-MyD88 and IL-6 levels in aged aortas in vitro.
FCCP is a potent uncoupler of oxidative phosphorylation, which results in impaired mitochondrial ATP synthesis.16 FCCP also enhances mitophagy15 by reducing cytosolic pH independently of mitochondrial membrane potential.29 To verify that FCCP treatment increased mitophagy in the aorta, we used the mitophagy reporter mtKeima mice described above. We employed aortas from aged (16 months of age) mtKeima mice and observed an increase in mitophagy upon FCCP treatment (Figure 4A-B). In addition, FCCP treatment reduced the levels of aortic Cox IV, a mitochondrial protein that decreases with increased mitochondrial degradation or removal,30 consistent with increased mitophagy (Figure 4C-D).
Figure 4. FCCP enhances mitophagy in the aorta of aged mice in tissue culture and reduces levels of Parkin, TLR9, MyD88, and IL-6.

(A-B) Thoracic aortas from aged (16 months of age) mtKiema mice were incubated with mitotracker green and also 10μM of FCCP or vehicle control. The mitotracker green signal (488nm excitation) and mtKeima red signal (561nm excitation) were assessed by fluorescence microscopy (see methods) and the ratio of 561:488 (mitophagy index) is shown in B. (C) Thoracic aortas from 18-month old WT mice were harvested and divided into 5 equal parts and cultured in DMEM+10% FBS supplemented with the indicated concentrations of FCCP for 2h. Lysates were immunoblotted against Parkin, TLR9, MyD88, IL-6, CoxIV, and β-actin. (D-H) Quantification of immunoblots. Each point is a biological replicate. All results are presented as mean ±SEM. Kruskal-Wallis with Dunn’s post-hoc test for D-H. Mann-Whitney U-test for B.
Next, we examined whether FCCP treatment can mitigate the age-associated elevations of Parkin, TLR9, MyD88 and IL-6 within the aged aorta. We found that culturing aortas with 1μM FCCP for 2h reduced the maximal uncoupled aortic respiration (Online Figure V), as expected. Importantly, administering increasing doses of FCCP reduced the levels of Parkin l TLR9, MyD88, and IL-6 within the aorta (Figure 4C-H). These data suggest that mitophagy in the aged aorta limits the clearance of damaged mitochondria, resulting in mitochondrial dysfunction and elevated levels of Parkin, TLR-9, IL-6 and MyD88.
Young and aged WT mice display similar metabolic profiles.
Our data from WT mice fed the LFD indicate that aging leads to elevated IL-6 levels and decreased mitochondrial function, concomitant with an increase in mitophagy and the associated proteins, Parkin and Nix, within the aorta. To determine if these changes in aged mice reflect potentially different metabolisms,31 we examined a number of metabolic parameters, including body weight, body composition, serum cholesterol, and glucose metabolism of young and aged WT mice maintained on LFD.
Aged WT mice had a higher body weight than young WT mice maintained on a LFD (Week 0 data, Online Figure VI A). Non-invasive measurements revealed that, on average, an aged mouse had 16% increase in lean mass compared to a young mouse, but no difference in fat mass (Week 0, Online Figure VI B-C). Aged and young mice maintained on a LFD exhibited similar, low levels of fasting total serum cholesterol, averaging between 100-110 mg/dL (Week 0, Online Figure VI D) and behaved similarly in glucose tolerance and insulin tolerance tests (Week 0, Online Figure VI E-F, left panels). Aged and young mice fed a LFD also had similar levels of fasting insulin, leptin, and adiponectin (Online Figure VII A-C). Thus, young and aged WT mice fed a LFD have similar metabolic readouts based on multiple criteria. These findings suggest that the age-associated changes within the aorta in aged mice fed the LFD do not arise from systemic metabolic alterations.
Similar induction of acute hypercholesterolemia in young and aged WT mice.
We hypothesized that the age-associated changes in the aorta during normolipidemia “prime” the aorta to enhance atherogenesis. To test this hypothesis, we induced acute hypercholesterolemia in young and aged male and female WT C57BL/6 mice that had been maintained on a LFD. Specifically, we induced degradation of the LDL receptor via intraperitoneal (i.p.) injection of an adeno-associated virus (AAV) that expresses Pcsk9 (denoted as PCSK9-AAV), as previously published.14 We concurrently switched the diet of the mice to a 10-week high fat, WD (see methods for diet description). We found that acute overexpression of Pcsk9 coupled with a WD induced a durable elevation in circulating cholesterol levels, which were initially similar between young and aged mice but ~15% higher in young mice than aged mice after the WD-feeding period (Online Figure VI D, P=0.043). The elevated cholesterol levels we detected in young PCSK9-AAV treated young and aged mice were similar to those observed in contemporaneously WD fed, young Ldlr−/− mice, a common murine model to study atherosclerosis (Online Figure VI D).
Young and aged PCSK9-AAV mice gained weight similarly over the WD-feeding period, with respect to both lean mass and fat mass (Online Figure VI A-C). Young and aged mice had no difference in food intake at baseline or at the end of the WD-feeding period (Online Figure VIII A-B). Although young mice were more active than aged mice prior to hypercholesterolemia, there was no difference in activity levels between groups after the WD-feeding period (Online Figure VIII C-D). Feeding C57BL/6 mice a western diet induces insulin resistance.32 Indeed, the WD led to glucose and insulin intolerance in young and aged mice, and young mice showed reduced glucose tolerance compared to aged mice by the end of the WD-feeding period (Online Figure VI E-F). Overall, young and aged mice were equally susceptible to the induction of hypercholesterolemia, although young mice exhibited slightly increased cholesterol levels and impaired glucose handling after 10-weeks on the WD (Online Figure VI D-E).
Aged mice show increased atherosclerosis in response to acute hypercholesterolemia.
To investigate atherosclerosis, we harvested the aortic roots from young and aged male and female C57BL/6 PCSK9-AAV mice after the WD-feeding and quantified atherosclerotic lesion size and composition in the aortic root (Figure 5A [male], Online Figure IX A [female]). Total aortic root lesion size was similar in young and aged atherosclerotic male and female mice (Figure 5A-B, Online Figure IX A-B) but lesions within the brachiocephalic artery were larger in aged mice compared to young mice (Figure 5D-E). We next analyzed the necrotic area of aortic root atherosclerotic lesions, which are defined as acellular/without intact cells and can be visualized using Masson’s trichrome stain (Online Figure X).33 Aortic root atherosclerotic lesions of aged mice had an increased necrotic area within the atherosclerotic lesion (Figure 5C, Online Figure IX C) (see methods) and greater Mac2 staining suggesting higher macrophage content (Figure 5F-G), consistent with plaque instability.
Figure 5. Atherosclerotic lesions show an age-associated increased size of necrosis induced by acute hyperlipidemia.

WT C57BL/6 mice were maintained on a LFD until either 3-months or 18-months and were then transfected with PCSK9-AAV and fed a WD for 10 weeks. At this point, aortic roots were obtained, paraffin-embedded, and stained with H&E. (A) Cross sections of aortic roots from male C57BL/6 mice show total lesion area, outlined in dashed lines, and necrotic core area, denoted by asterisks. Higher magnification shows the presence of necrotic core in aged mice. Scale bars: 100μm. (B and C) Quantification of total lesion area and necrotic core. (D) Cross sections of brachiocephalic artery from male C57BL/6 mice showing total lesion area, outlined in dashed lines. Scale bars: 100μm and 10μm for magnified image. (E) Quantification of total lesion area. (F) Cross sections of the aortic root were stained with Mac2 monoclonal antibody as described in detailed methods. Scale bar: 100μm. (G) Quantification of Mac2 positive staining area as a percentage of the total plaque area. Each data point represents a biological replicate. For atherosclerotic plaque and necrotic area, each biological replicate is the sum of 30 serial sections, 6μm per section. All results are presented as mean ±SEM. Student’s t-test for B, C, and G. Mann-Whitney U-test for E. A=aged, Y=young.
The age-enhanced effect of atherogenesis was not limited to the C57BL/6 strain of mice, as female mice that were outcrossed onto 4 different genetic backgrounds (designated as UM-HET334) exhibited increased necrotic core area with aging (Online Figure IX E-H). All mice from this point are male unless otherwise indicated.
As a high fat diet induces a peripheral monocytosis, which can contribute to macrophage accumulation within atherosclerotic lesions,35-37 we assessed the number of circulating monocytes, both patrolling (i.e., surface expression of Ly6clo) and inflammatory (i.e., surface expression of Ly6chi), in mice on a LFD as well as after a WD and PCSK9-AAV treatment. During the LFD, the numbers of circulating monocytes were similar in young and aged mice. After treatment with the WD, the frequency of inflammatory monocytes increased in both young and aged mice, with the aged mice exhibiting a significant (P=0.011) increase in inflammatory but not patrolling monocytes (Online Figure XI A-B). Additionally, Parkin levels within the monocytes, both during the LFD and after the induction of hyperlipidemia, were similar between young and aged mice (Online Figure XI D-E). Similar results were noted with other myeloid cells such as neutrophils (Online Figure XI C and F).
Finally, we did not observe vascular lesions in young and aged WT mice fed a LFD (Online Figure XII), consistent with prior studies that WT, normolipidemic mice are resistant to atherosclerosis.38 Overall, these results indicate that, despite their slightly lower cholesterol levels at the end of the PCSK9-AAV, WD protocol, aged mice exhibit increased atherogenesis in response to acute hypercholesterolemia compared to young mice.
Atherogenesis increased aortic Parkin levels and further impaired mitochondrial function in aged mice.
To determine if atherosclerosis affected aortic mitochondrial function in young and aged mice, we examined the levels of Parkin, and PINK1, as well as aortic OCRs. We found that the levels of both PINK1 and Parkin significantly increased during atherosclerosis in aged mice, but not young mice (Figure 6A-C). The levels of autophagy protein 5 (ATG5), a protein involved in autophagosome-lysosome fusion,20 within the aorta were unaltered by diet and were similar in young and aged mice (Figure 6D). During normolipidemia, Nix was elevated in aged mice as compared to young mice (Figure 6E), although there was no significant elevation in Nix between age groups during atherosclerosis (Figure 6E). Moreover, atherosclerosis reduced the maximal coupled and uncoupled respiration within the aorta in aged but not young mice (Figure 6F-I), specifically a decline in complex I OXPHOS (Figure 6F) and complex I and II uncoupled respiration (Figure 6H). Overall, these data suggest that atherosclerosis exacerbates the age-associated mitochondrial dysfunction within the aorta.
Figure 6. Aged but not young mice show reduced mitochondrial function and increased levels of Parkin and PINK1 during atherogenesis.

(A) Thoracic aortas were harvested from 3-month or 18-month WT, LFD-fed mice, and WT mice maintained on a low-fat diet until either 3-months or 18-months which were then transfected with PCSK9-AAV and fed a WD for 10 weeks. Lysates were blotted against PINK1, Parkin, ATG5, Nix and β-actin (Note, that Nix immunoblot was run separately to other proteins and has its own loading β-actin control, below). (B-E) Quantification of immunoblots from A. (F) Maximal thoracic aorta OCR with substrates for complex I OXPHOS. (G): Maximal aortic OCR with substrates for complex I+II OXPHOS. (H) Maximal aortic complex I+II uncoupled OCR. (I) Maximal aortic complex II uncoupled OCR. Each data point represents a biological replicate. Note, that the normolipidemic data (i.e., WT LFD denotation) are the same data shown in Figure 1. Data from Figure 1 and Figure 6 were run contemporaneously. All results are presented as mean ±SEM. 2-way ANOVA with Sidak’s post-hoc test for B-C and F-I. Kruskal-Wallis with Dunn’s post-hoc test for E. A=aged, Y=young.
Reduced vascular autophagy combined with hyperlipidemia increases IL-6 and impairs mitochondria in young mice.
To determine if autophagy, which includes mitophagy, is critical for aortic IL-6 levels and mitochondrial function, we inhibited autophagy within VSMCs of young mice by deleting ATG5 an essential protein for autophagosome formation and thus for removing damaged organelles, including mitochondria.39 We bred homozygous floxed Atg5 (Atg5fl/fl) mice with mice expressing tamoxifen-inducible Cre in VSMCs (Myh11-creERT2) and treated young (~2-4 months-old) Atg5fl/flMyh11-cre/ERT2 mice with tamoxifen to generate VSMC-ATG5−/− mice or with vehicle to generate SMC-ATG5+/+ littermate controls.
We confirmed that ATG5 levels are reduced upon tamoxifen treatment of Atg5fl/flMyh11-cre/ERT2 mice, consistent with loss of ATG5 in VSMCs (Online Figure XIII A). During normolipidemia and two months of Atg5 gene deletion within VSMCs, there was neither an alteration in the TLR9-MyD88-IL-6 pathway nor Parkin and PINK1 levels in young, normolipidemic VSMC-ATG5−/− mice compared to littermate controls (Figure 7A-B). These data suggest that in young normolipidemic mice, impaired vascular autophagy alone does not alter Parkin levels or the TLR9-IL-6-MyD88 pathway.
Figure 7: Conditional loss of ATG5 in VMSCs leads to reduced mitochondrial function with increased levels of Parkin, TLR9, MyD88 and IL-6 levels during atherosclerosis.

Inducible Atg5fl/flMyh11-cre/ERT2 mice were treated with either tamoxifen (ATG5−/−) or vehicle (ATG5+/+) and some were transfected with PCSK9-AAV and fed a WD for 10 weeks before measuring proteins, mitochondrial function and aortic root sections. (A) Thoracic aortas from LFD-fed Atg5fl/flMyh11-cre/ERT2 mice treated with tamoxifen or littermate controls treated with vehicle were harvested and blotted against PINK1, Parkin, TLR9, MyD88, IL-6, and β-actin. (B) Quantification of immunoblots. (C) Atg5fl/flMyh11-cre/ERT2 mice were treated with tamoxifen or littermate controls treated with vehicle and all mice were then transfected with PCSK9-AAV and fed a WD for 10-weeks. Aortas were harvested and blotted against PINK1, Parkin, TLR9, MyD88, IL-6, and β-actin. (D) Quantification of immunoblots. (E) Maximal thoracic aorta OCR with substrates for complex I and I+II coupled OXPHOS and maximal complex I+II and II uncoupled OCR. (F) Cross sections of aortic root show total lesion area, outlined in dashed lines; and necrotic core area, outlined in dotted lines and denoted by asterisks. Scale bars: 100μm. (G and H) Quantification of total lesion area and necrotic core. Each data point represents a biological replicate. For atherosclerotic plaque and necrotic area, each biological replicate is the sum of 30 serial sections, 6μm per section. All results are presented as mean ±SEM. Mann-Whitney U-test for B, D, G, and H. 2-way ANOVA with Sidak’s multiple comparison test for E. CI=complex I, CII=complex II, Tam=tamoxifen, Veh=vehicle
We next determined whether impaired autophagy within VSMC coupled with hyperlipidemia would lead to increased IL-6 and Parkin levels, impaired mitochondrial function and accelerated atherosclerosis in the aorta of young mice. We induced acute hyperlipidemia in tamoxifen-treated Atg5fl/flMyh11-cre/ERT2 mice and their littermate controls (Online Figure XIII B-C). Hyperlipidemic, VSMC-ATG5−/− mice exhibited an increase in aortic TLR9, MyD88, and IL-6 levels compared to atherosclerotic littermate controls, accompanied by increased levels of aortic Parkin, but not PINK1 (Figure 7C-D). We also measured mitochondrial function within the aorta of hyperlipidemic VSMC-ATG5−/− and littermate control mice and found that maximal complex I+II OXPHOS and maximal complex I+II uncoupled OCR were significantly reduced in hyperlipidemic VSMC-ATG5−/− mice compared to littermate controls (Figure 7E). Thus, young mice with VSMC deletion of ATG5 exhibit increased IL-6 aortic levels with decreased mitochondrial function and elevated Parkin during atherogenesis. Importantly, the total atherosclerotic lesion area within the aortic root was doubled in VSMC-ATG5−/− mice (Figure 7F-G, P=0.0001) compared to littermate controls, in agreement with similar studies that conditionally deleted the autophagy gene Atg7 in SMCs.40, 41
In sum, neither impairing autophagy alone (Figure 7A-B) nor inducing atherosclerosis in young mice (Figure 6A-C) is sufficient to increase aortic IL-6, PINK1, Parkin, or mitochondrial dysfunction. However, disrupting autophagy within the VSMC of young atherosclerotic mice phenocopies some of the aortic features of aged atherosclerotic mice, including increased levels of IL-6 and Parkin, and reduced mitochondrial function. Together, these data support that at least two insults (impaired autophagy and atherogenesis) are required to increase IL-6, impair mitochondrial function and elevate Parkin levels within the aorta of young mice.
Pharmacological induction of autophagy/mitophagy during hyperlipidemia attenuates the elevation in IL-6, aortic mitochondrial dysfunction, and atherosclerosis in aged mice.
We hypothesized that enhancing autophagy and mitophagy in aged mice would reduce IL-6 and Parkin levels within the aorta. We augmented autophagy/mitophagy in WT aged mice maintained on a LFD by administering spermidine orally (see methods; Figure 8A). Spermidine is a polyamine drug that increases mitophagy, autophagy,42, 43 extends murine lifespan,9 and reduces atherogenesis in young Apoe−/− mice in an SMC-dependent manner.10 Using the aortas of young mtKeima mice that were maintained on a LFD, we verified that spermidine enhanced mitophagy within the aorta in tissue culture (Online Figure XIV A-B). However, 10-weeks of oral spermidine treatment administered to 18-month old mice on a LFD did not alter the levels of Parkin, PINK1, or TLR9-MyD88-IL-6 signaling in the aorta (Figure 8B-C).
Figure 8: Spermidine treatment in aged mice mitigates increased Parkin and IL-6 levels, and mitochondrial dysfunction during atherogenesis.

WT aged mice were maintained on a LFD until 18-months old and some were then transfected with PCSK9-AAV and fed a WD for 10 weeks. During the WD feeding period, a subset of randomly selected aged mice were supplemented with spermidine in the drinking water. Some aged mice were maintained on a LFD from 18-months and treated with spermidine for 10-weeks. (A) Schematic of experimental design for B and C. (B) Thoracic aortas from aged WT LFD-fed mice either treated with spermidine or vehicle were harvested and blotted against PINK1, Parkin, TLR9, MyD88, IL-6, and β-actin. (C) Quantification of immunoblots from B. (D) Schematic of experimental design for E-J. (E) Aortas from aged WT mice treated with PCSK9-AAV and fed a WD were either treated with spermidine or vehicle and blotted against PINK1, Parkin, TLR9, MyD88, IL-6, and β-actin. (F) Quantification of immunoblots from E. (G) Maximal thoracic aortic OCR with substrates for complex I and I+II coupled OXPHOS and maximal complex I+II and II uncoupled OCR. (H) Cross sections of aortic root show total lesion area, outlined in dashed lines, and necrotic core area, outlined in dotted lines. Scale bars: 100μm. (I and J) Quantification of total lesion area and necrotic core based on H&E. Scale bars: 100μm. Each data point represents a biological replicate. For atherosclerotic plaque and necrotic area, each biological replicate is the sum of 30 serial sections, 6μm per section. All results are presented as mean ±SEM. Mann-Whitney U-test for C, F, I, and J. 2-way ANOVA with Sidak’s multiple comparison test for G. CI=complex I, CII=complex II, Sp=spermidine, Veh=vehicle.
Next, we examined whether spermidine treatment can prevent the increase in IL-6 and Parkin levels and the further declines in mitochondrial function during hyperlipidemia in aged mice. Hence, we induced acute hyperlipidemia in aged WT mice, and supplemented the drinking water with spermidine or vehicle for 10-weeks during the WD feeding period (Figure 8D). Although spermidine treatment did not impact the induction of hypercholesterolemia or body weight gain (Online Figure XIV C-D), it mitigated the increase in the levels of IL-6 and Parkin in aged atherosclerotic mice (Figure 8E-F). In addition, compared to aortas from vehicle-treated hypercholesterolemic aged mice, spermidine-treated hypercholesterolemic aged mice had a 25-35% increase in maximal complex I+II OXPHOS (P=0.0132) and maximal complex I+II uncoupled OCR (P=0.0009) (Figure 8G). Finally, the atherosclerotic lesion size and necrotic core area within the aortic root were significantly smaller with spermidine treatment compared to control treated aged mice (Figure 8H-J). Spermidine did not, however, impact the number of lesional macrophages (Online Figure XIV E-F). Thus, spermidine treatment mitigates the hyperlipidemia-related increases in IL-6 and Parkin in aged mice. Spermidine also enhanced vascular mitochondrial function, and attenuated atherogenesis due to acute hypercholesterolemia in aged mice.
DISCUSSION
Our work reveals that, prior to hyperlipidemia, aging elevates IL-6 within the aorta and impairs mitochondrial function. This is accompanied by increased Parkin levels and elevated mitophagy within the aorta, which is likely due the increased removal of dysfunctional mitochondria. Our study suggests that the age-related elevation in IL-6 and impaired mitochondrial function “prime” the vasculature to promote atherogenesis: aged mice with a similar degree of hyperlipidemia as young mice showed more severe atherogenesis. Hence, our study identified a pathway of vascular aging prior to hyperlipidemia and linked this pathway to atherogenesis.
Although clinical epidemiological studies have identified aging as a strong risk factor for atherosclerosis, these clinical studies fail to separate biological aging from chronic exposure to atherogenic risk factors, notably hyperlipidemia. Regrettably, currently employed murine atherogenic models, e.g., Apoe−/− and Ldlr−/− mice, have also failed to separate biological effects of aging from chronic hyperlipidemia, as these mice exhibit hyperlipidemia from early adulthood. A study of ancient mummies indicates that atherosclerosis occurred before the modern era,44 a time before many common atherogenic risk factors such as hypercholesterolemia likely were evident. In this study, the presence of atherosclerosis, documented by X-ray computed tomography scanning, was correlated with age up to the 5th decade, although vascular lesions reduced during the 6th decade of life,44 complicating the contribution of aging to vascular disease. Based upon these studies, there is a lack of evidence directly linking biological aging to atherosclerosis. Our study has resolved this issue via our approach to induce hyperlipidemia in WT young and aged normolipidemic mice. The PCSK9-AAV approach, combined with a WD, induced a similar degree of hyperlipidemia in both young and aged mice, and unambiguously shows that biological aging, prior to hyperlipidemia enhances atherogenesis.
IL-6 is a pleiotropic cytokine that impacts mitochondrial function in a variety cells including reducing oxygen consumption in skeletal muscle,26 reducing mitochondrial membrane potential in islets,27 and elevating mitochondrial calcium levels in CD4+ T cells.28 Aging also leads to the secretion of inflammatory proteins, including IL-6, likely via cellular senescence.45 Impaired mitochondrial function within the aorta with aging could occur in other cell types beside VSMCs, such as endothelial cells. Importantly, recent studies have shown that cells in the aorta exhibit dynamic phenotypes, as smooth muscle cells can take on macrophage or fibroblast like phenotypes.46-48 Ultimately studies that employ aged mice that permit lineage tracing of specific cell populations48 will be required to determine how aging impacts the cell fate decision within the aorta during atherogenesis.
Declining mitochondrial function may lead to the production of reactive oxygen species49 which could impact the inflammatory status and metabolism of a cell. As mitochondria employ oxidative phosphorylation in oxygen replete conditions for energy generation, we measured the OCR of the aorta as a marker mitochondrial function in our study. However, aging may lead to a shift to anaerobic metabolism via glycolysis. Furthermore, dysfunctional mitochondria may release their contents (e.g., mtDNA) into the cytosol. Both reactive oxygen species and mitochondrial contents may synergize to activate innate immune signaling pathways,50, 51 leading to further increases in IL-6, which could exacerbate mitochondrial dysfunction. Our study establishes that there is a positive feedback loop between elevated IL-6 and mitochondrial dysfunction within the aorta with aging. Other inflammatory mediators, besides IL-6, may be released during mitochondrial dysfunction and could include TNFα, IL-1β, and chemokines such as CCL-2.45 These mediators may also be secreted in the aged aorta during normolipidemia and could also contribute to impairing mitochondrial function. Furthermore, IL-6 exhibits athero-protective effects although these effects were in young mice.52 Given the redundancy between inflammatory pathways and pleotropic function of certain cytokines, future investigation will be required to identify the key pathways that could be targeted to improve bioenergetics and vascular health with aging.
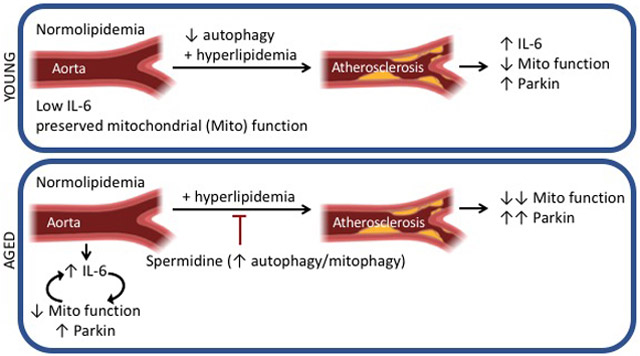
Our study reveals that young mice, prior to hyperlipidemia, exhibit low IL-6 levels and preserved mitochondrial function within the aorta (Online Figure XV A). In young mice, at least two insults are required to elevate IL-6 and impair mitochondrial function: impaired autophagy in smooth muscle cells combined with hyperlipidemia. Our study has revealed that neither alone is sufficient to increase IL-6 or Parkin, and impair mitochondrial function within the aorta of young mice. Furthermore, in our model (Online Figure XV B), aged mice already exhibit elevated IL-6 in the aorta during normolipidemia and dysfunction mitochondria, which triggers mitophagy. The elevated IL-6 and dysfunctional mitochondria could operate in a positive feedback loop. Spermidine treatment at the time of hyperlipidemia in aged mice, reduced IL-6 levels, improved mitochondrial function with reduced Parkin levels within the aorta. These effects were accompanied by reduced atherogenesis. Spermidine enhances autophagy and mitophagy.42, 43 However, it is possible that spermidine may be mediating its effects via multiple pathways such as inhibiting inflammatory pathways or reducing oxidative stress.43 Furthermore, it is possible that prolonged treatment of more than several months of spermidine in normolipidemic conditions could have beneficial effects on vascular aging. Whether other agents that increase autophagy and lifespan, such as rapamycin, which reduces atherogenesis in young Apoe−/− mice 53, ameliorate atherogenesis in aged mice will be investigated in future work.
A variety of immune cells and pathways, from both the innate and adaptive arms of the immune system, have been implicated in atherosclerosis pathogenesis.54, 55 Additionally, aging exerts complex effects on the immune system, including dysregulated inflammation, impaired innate immunity, and declining adaptive immunity.56, 57 Aging also impacts the stem cell niche and is known to lead to myeloid skewing,58 which could contribute to the recently appreciated role of clonal hematopoiesis and atherosclerosis.59, 60 How aging impacts specific immune cells to enhance atherosclerosis will require future investigation. Our study indicates that alterations within the vasculature, in addition to the potential effects of aging on the immune system, contribute to atherosclerosis.
Prior work has shown that VSMCs derived from human atherosclerotic lesions from subjects in their 8th decade exhibit reduced OCR with enhanced mitophagy compared to VSMCs from disease-free arteries.15 It will be important in future clinical studies to determine if the findings of our study, in particular elevated IL-6 and dysregulated mitophagy, occur within the aorta during human aging prior to the onset of vascular diseases. There are already clinically approved agents that target IL-6 to treat rheumatological diseases,61 so if our findings translate to humans then it may be possible to target specific inflammatory pathways, such as IL-6, to increase mitochondrial function and mitigate the effects of vasculature aging.
In conclusion, our study has definitively linked biological aging to atherogenesis. Specifically, our work has revealed that aging, independent of chronic hyperlipidemia, leads to elevated IL-6 levels, impaired vascular mitochondrial function and enhanced mitophagy. These alterations within the vasculature prior to hyperlipidemia “prime” the vasculature to enhance atherogenesis during hyperlipidemia. Our study suggests that novel therapies that mitigate against the effects of vascular aging prior to hyperlipidemia may reduce atherogenesis and improve healthspan in the elderly.
Supplementary Material
NOVELTY AND SIGNIFICANCE.
What Is Known?
Aging is a risk factor for atherosclerosis.
Mitochondrial function declines with aging.
Mitochondrial dysfunction contributes to atherosclerosis.
What New Information Does This Article Contribute?
Aging within the aorta during normolipidemia leads to mitochondrial dysfunction and elevations in the inflammatory cytokine Interleukin (IL)-6, with each positively enhancing the other.
Aging leads to elevations in mitophagy, a process to remove damaged or dysfunctional mitochondria within the aorta during normolipidemia.
The above aortic alterations with aging prime for enhanced atherosclerosis during hyperlipidemia.
Enhancing mitophagy pharmacologically during hyperlipidemia increases aortic mitochondrial function, reduces IL-6, and atherosclerosis with aging.
Although aging is a known epidemiological risk factor for atherosclerosis, most humans age with chronic hyperlipidemia. In established models of atherosclerosis (e.g., Ldlr−/− or Apoe−/− mice), mice also age with chronic hyperlipidemia. Hence, the true biological impact of aging on atherosclerosis has not been separated from chronic hyperlipidemia (high cholesterol levels). As mitochondrial function is a hallmark of aging, we sought to determine whether aging impairs mitochondrial function within the aorta prior to hyperlipidemia, and whether any identified age-dependent alterations impact atherosclerosis. We employed young and aged wild type mice and found that in a low cholesterol environment, aging leads to mitochondrial dysfunction with elevations in the proinflammatory cytokine IL-6 within the aorta. IL-6 and mitochondrial dysfunction co-exist within a positive feedback loop in the aorta with aging. The mitochondrial dysfunction is also accompanied by increased mitophagy, a process to remove damage mitochondria. Upon inducing hyperlipidemia in wild type mice, we found that aged mice display a higher degree of atherosclerosis than young mice. Importantly, enhancing mitophagy by adding spermidine to drinking water during hyperlipidemia restrained aortic mitochondrial dysfunction, IL-6 levels, and reduced atherosclerosis with aging. Taken together, our study indicates that therapies that improve vascular bioenergetics may reduce the burden of atherosclerosis with aging.
ACKNOWLEDGEMENTS
We thank Richard Miller (University of Michigan) for the UM-HET3 mice. We thank Life Science Editors for editorial assistance.
SOURCES OF FUNDING
These studies used the resources of the University of Michigan Animal Phenotyping Core (supported by NIH grants P30-DK020572, P30-DK089503, and U2CDK110678). This work was supported by NIH NRSA training grant F32-HL1400728 (to DJT), Multidisciplinary Cardiovascular Research Training Program T32-HL007853 (to DJT), R01-HL127687 and R01-AI138347 (both to DRG).
Nonstandard Abbreviations and Acronyms:
- AAV
adeno associated virus
- αSMa
smooth muscle α-actin
- ATG5
autophagy protein 5
- CCCP
Carbonyl cyanide m-chlorophenyl hydrazine
- LFD
Low fat diet
- MyD88
Myeloid differentiation factor 88
- mtDNA
mitochondrial DNA
- nucDNA
nuclear DNA
- Park2−/−
Parkin deficient
- PCSK9
Proprotein convertase subtilisin/kexin 9
- OCR
oxygen consumption rate
- OXPHOS
oxidative phosphorylation
- SMC
smooth muscle cell
- TLR
Toll-like receptor
- VSMC
vascular smooth muscle cell
- WD
Western Diet
- WT
wild type
Footnotes
DISCLOSURES
The authors have declared that no conflict of interest exists.
REFERENCES
- 1.Benjamin EJ, Virani SS, Callaway CW et al. Circulation. 2018;137. [Google Scholar]
- 2.Sturlaugsdottir R, Aspelund T, Bjornsdottir G, Sigurdsson S, Thorsson B, Eiriksdottir G and Gudnason V. Prevalence and determinants of carotid plaque in the cross-sectional REFINE-Reykjavik study. BMJ Open. 2016;6:e012457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopez-Otin C, Blasco MA, Partridge L, Serrano M and Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hsieh PN, Zhou G, Yuan Y, Zhang R, Prosdocimo DA, Sangwung P, Borton AH, Boriushkin E, Hamik A, Fujioka H, Fealy CE, Kirwan JP, Peters M, Lu Y, Liao X, Ramirez-Bergeron D, Feng Z and Jain MK. A conserved KLF-autophagy pathway modulates nematode lifespan and mammalian age-associated vascular dysfunction. Nat Commun. 2017;8:914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sliter DA, Martinez J, Hao L, Chen X, Sun N, Fischer TD, Burman JL, Li Y, Zhang Z, Narendra DP, Cai H, Borsche M, Klein C and Youle RJ. Parkin and PINK1 mitigate STING-induced inflammation. Nature. 2018;561:258–262. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6.Saitoh T, Fujita N, Jang MH, Uematsu S, Yang B-G, Satoh T, Omori H, Noda T, Yamamoto N, Komatsu M, Tanaka K, Kawai T, Tsujimura T, Takeuchi O, Yoshimori T and Akira S. Loss of the autophagy protein Atg16L1 enhances endotoxin-induced IL-1β production. Nature. 2008;456:264–268. [DOI] [PubMed] [Google Scholar]
- 7.Razani B, Feng C, Coleman T, Emanuel R, Wen H, Hwang S, Ting JP, Virgin HW, Kastan MB and Semenkovich CF. Autophagy links inflammasomes to atherosclerotic progression. Cell Metab. 2012;15:534–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liao X, Sluimer JC, Wang Y, Subramanian M, Brown K, Pattison JS, Robbins J, Martinez J and Tabas I. Macrophage autophagy plays a protective role in advanced atherosclerosis. Cell Metab. 2012;15:545–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Eisenberg T, Abdellatif M, Schroeder S et al. Cardioprotection and lifespan extension by the natural polyamine spermidine. Nat Med. 2016;22:1428–1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Michiels CF, Kurdi A, Timmermans JP, De Meyer GRY and Martinet W. Spermidine reduces lipid accumulation and necrotic core formation in atherosclerotic plaques via induction of autophagy. Atherosclerosis. 2016;251:319–327. [DOI] [PubMed] [Google Scholar]
- 11.Du W, Wong C, Song Y, Shen H, Mori D, Rotllan N, Price N, Dobrian AD, Meng H, Kleinstein SH, Fernandez-Hernando C and Goldstein DR. Age-associated vascular inflammation promotes monocytosis during atherogenesis. Aging cell. 2016;15:766–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Collins AR, Lyon CJ, Xia X, Liu JZ, Tangirala RK, Yin F, Boyadjian R, Bikineyeva A, Pratico D, Harrison DG and Hsueh WA. Age-accelerated atherosclerosis correlates with failure to upregulate antioxidant genes. Circ Res. 2009;104:e42–54. [DOI] [PubMed] [Google Scholar]
- 13.Vendrov AE, Stevenson MD, Alahari S, Pan H, Wickline SA, Madamanchi NR and Runge MS. Attenuated Superoxide Dismutase 2 Activity Induces Atherosclerotic Plaque Instability During Aging in Hyperlipidemic Mice. J Am Heart Assoc. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjorklund MM, Hollensen AK, Hagensen MK, Dagnaes-Hansen F, Christoffersen C, Mikkelsen JG and Bentzon JF. Induction of atherosclerosis in mice and hamsters without germline genetic engineering. Circ Res. 2014;114:1684–9. [DOI] [PubMed] [Google Scholar]
- 15.Yu EPK, Reinhold J, Yu H, Starks L, Uryga AK, Foote K, Finigan A, Figg N, Pung Y-F, Logan A, Murphy MP and Bennett M. Mitochondrial Respiration Is Reduced in Atherosclerosis, Promoting Necrotic Core Formation and Reducing Relative Fibrous Cap Thickness. Arteriosclerosis, Thrombosis, and Vascular Biology. 2017;37:2322–2332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doerrier C, Garcia-Souza LF, Krumschnabel G, Wohlfarter Y, Meszaros AT and Gnaiger E. High-Resolution FluoRespirometry and OXPHOS Protocols for Human Cells, Permeabilized Fibers from Small Biopsies of Muscle, and Isolated Mitochondria. Methods Mol Biol. 2018;1782:31–70. [DOI] [PubMed] [Google Scholar]
- 17.Fang EF, Hou Y, Palikaras K, Adriaanse BA, Kerr JS, Yang B, Lautrup S, Hasan-Olive MM, Caponio D, Dan X, Rocktäschel P, Croteau DL, Akbari M, Greig NH, Fladby T, Nilsen H, Cader MZ, Mattson MP, Tavernarakis N and Bohr VA. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nature Neuroscience. 2019;22:401–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N and Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol. 2010;189:211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Klionsky DJ. et al. Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy. 2016;12:1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mizushima N, Yoshimori T and Levine B. Methods in Mammalian Autophagy Research. Cell. 2010;140:313–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kanki T Nix: A receptor protein for mitophagy in mammals. Autophagy. 2010;6:433–435. [DOI] [PubMed] [Google Scholar]
- 22.Durcan TM and Fon EA. The three ‘P’s of mitophagy: PARKIN, PINK1, and post-translational modifications. Genes & Development. 2015;29:989–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sun N, Yun J, Liu J, Malide D, Liu C, Rovira Ilsa I, Holmström Kira M, Fergusson Maria M, Yoo Young H, Combs Christian A and Finkel T. Measuring In Vivo Mitophagy. Molecular Cell. 2015;60:685–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang Q, Raoof M, Chen Y, Sumi Y, Sursal T, Junger W, Brohi K, Itagaki K and Hauser CJ. Circulating mitochondrial DAMPs cause inflammatory responses to injury. Nature. 2010;464:104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.West AP and Shadel GS. Mitochondrial DNA in innate immune responses and inflammatory pathology. Nat Rev Immunol. 2017;17:363–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.VanderVeen BN, Fix DK, Montalvo RN, Counts BR, Smuder AJ, Murphy EA, Koh H-j and Carson JA. The regulation of skeletal muscle fatigability and mitochondrial function by chronically elevated interleukin-6. Experimental Physiology. 2019;104:385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Marasco MR, Conteh AM, Reissaus CA, Cupit JE, Appleman EM, Mirmira RG and Linnemann AK. Interleukin-6 Reduces β-Cell Oxidative Stress by Linking Autophagy With the Antioxidant Response. Diabetes. 2018;67:1576–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Yang R, Lirussi D, Thornton TM, Jelley-Gibbs DM, Diehl SA, Case LK, Madesh M, Taatjes DJ, Teuscher C, Haynes L and Rincón M. Mitochondrial Ca2+ and membrane potential, an alternative pathway for Interleukin 6 to regulate CD4 cell effector function. Elife. 2015;4:e06376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berezhnov AV, Soutar MPM, Fedotova EI, Frolova MS, Plun-Favreau H, Zinchenko VP and Abramov AY. Intracellular pH modulates autophagy and mitophagy. Journal of Biological Chemistry. 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Li Y, D’Aurelio M, Deng J-H, Park J-S, Manfredi G, Hu P, Lu J and Bai Y. An Assembled Complex IV Maintains the Stability and Activity of Complex I in Mammalian Mitochondria. Journal of Biological Chemistry. 2007;282:17557–17562. [DOI] [PubMed] [Google Scholar]
- 31.López-Otín C, Galluzzi L, Freije JMP, Madeo F and Kroemer G. Metabolic Control of Longevity. Cell. 2016;166:802–821. [DOI] [PubMed] [Google Scholar]
- 32.Zhang D, Christianson J, Liu ZX, Tian L, Choi CS, Neschen S, Dong J, Wood PA and Shulman GI. Resistance to high-fat diet-induced obesity and insulin resistance in mice with very long-chain acyl-CoA dehydrogenase deficiency. Cell Metab. 2010;11:402–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Daugherty A, Tall AR, Daemen M, Falk E, Fisher EA, Garcia-Cardena G, Lusis AJ, Owens AP 3rd, Rosenfeld ME, Virmani R, American Heart Association Council on Arteriosclerosis T, Vascular B and Council on Basic Cardiovascular S. Recommendation on Design, Execution, and Reporting of Animal Atherosclerosis Studies: A Scientific Statement From the American Heart Association. Arterioscler Thromb Vasc Biol. 2017;37:e131–e157. [DOI] [PubMed] [Google Scholar]
- 34.Miller RA and Chrisp C. Lifelong treatment with oral DHEA sulfate does not preserve immune function, prevent disease, or improve survival in genetically heterogeneous mice. J Am Geriatr Soc. 1999;47:960–6. [DOI] [PubMed] [Google Scholar]
- 35.Hilgendorf I, Swirski FK and Robbins CS. Monocyte Fate in Atherosclerosis. Arterioscler Thromb Vasc Biol. 2015;35:272–279. [DOI] [PubMed] [Google Scholar]
- 36.Swirski FK, Libby P, Aikawa E, Alcaide P, Luscinskas FW, Weissleder R and Pittet MJ. Ly-6Chi monocytes dominate hypercholesterolemia-associated monocytosis and give rise to macrophages in atheromata. J Clin Invest. 2007;117:195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tacke F, Alvarez D, Kaplan T, Jakubzick C, Spanbroek R, Llodra J, Garin A, Liu J, Mack M and van Rooijen N. Monocyte subsets differentially employ CCR2, CCR5, and CX3CR1 to accumulate within atherosclerotic plaques. J Clin Invest. 2007;117:185 – 94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Morrisett JD, Kim HS, Patsch JR, Datta SK and Trentin JJ. Genetic susceptibility and resistance to diet-induced atherosclerosis and hyperlipoproteinemia. Arteriosclerosis. 1982;2:312–24. [DOI] [PubMed] [Google Scholar]
- 39.Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H and Mizushima N. Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature. 2006;441:885–9. [DOI] [PubMed] [Google Scholar]
- 40.Grootaert MO, da Costa Martins PA, Bitsch N, Pintelon I, De Meyer GR, Martinet W and Schrijvers DM. Defective autophagy in vascular smooth muscle cells accelerates senescence and promotes neointima formation and atherogenesis. Autophagy. 2015;11:2014–2032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Nahapetyan H, Moulis M, Grousset E, Faccini J, Grazide M-H, Mucher E, Elbaz M, Martinet W and Vindis C. Altered mitochondrial quality control in Atg7-deficient VSMCs promotes enhanced apoptosis and is linked to unstable atherosclerotic plaque phenotype. Cell Death & Disease. 2019;10:119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Eisenberg T, Knauer H, Schauer A et al. Induction of autophagy by spermidine promotes longevity. Nature Cell Biology. 2009;11:1305–1314. [DOI] [PubMed] [Google Scholar]
- 43.Madeo F, Eisenberg T, Pietrocola F and Kroemer G. Spermidine in health and disease. Science. 2018;359:eaan2788. [DOI] [PubMed] [Google Scholar]
- 44.Thompson RC, Allam AH, Lombardi GP et al. Atherosclerosis across 4000 years of human history: the Horus study of four ancient populations. The Lancet. 2013;381:1211–1222. [DOI] [PubMed] [Google Scholar]
- 45.Tchkonia T, Zhu Y, van Deursen J, Campisi J and Kirkland JL. Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest. 2013;123:966–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gomez D and Owens GK. Smooth muscle cell phenotypic switching in atherosclerosis. Cardiovascular Research. 2012;95:156–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wirka RC, Wagh D, Paik DT, Pjanic M, Nguyen T, Miller CL, Kundu R, Nagao M, Coller J, Koyano TK, Fong R, Woo YJ, Liu B, Montgomery SB, Wu JC, Zhu K, Chang R, Alamprese M, Tallquist MD, Kim JB and Quertermous T. Atheroprotective roles of smooth muscle cell phenotypic modulation and the TCF21 disease gene as revealed by single-cell analysis. Nature Medicine. 2019;25:1280–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shankman LS, Gomez D, Cherepanova OA et al. KLF4-dependent phenotypic modulation of smooth muscle cells has a key role in atherosclerotic plaque pathogenesis. Nature Medicine. 2015;21:628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ježek J, Cooper KF and Strich R. Reactive Oxygen Species and Mitochondrial Dynamics: The Yin and Yang of Mitochondrial Dysfunction and Cancer Progression. Antioxidants , . 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zhang JZ, Liu Z, Liu J, Ren JX and Sun TS. Mitochondrial DNA induces inflammation and increases TLR9/NF-kappaB expression in lung tissue. Int J Mol Med. 2014;33:817–24. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 51.Mittal M, Siddiqui MR, Tran K, Reddy SP and Malik AB. Reactive oxygen species in inflammation and tissue injury. Antioxidants & redox signaling. 2014;20:1126–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Madan M, Bishayi B, Hoge M and Amar S. Atheroprotective role of interleukin-6 in diet- and/or pathogen-associated atherosclerosis using an ApoE heterozygote murine model. Atherosclerosis. 2008;197:504–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Elloso MM, Azrolan N, Sehgal SN, Hsu PL, Phiel KL, Kopec CA, Basso MD and Adelman SJ. Protective effect of the immunosuppressant sirolimus against aortic atherosclerosis in apo E-deficient mice. Am J Transplant. 2003;3:562–9. [DOI] [PubMed] [Google Scholar]
- 54.Tabas I and Lichtman AH. Monocyte-Macrophages and T Cells in Atherosclerosis. Immunity. 2017;47:621–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wolf D and Ley K. Immunity and Inflammation in Atherosclerosis. Circulation Research. 2019;124:315–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Montgomery R, Goldstein D and Shaw A. Age-dependent dysregulation of innate immunity. Nat Rev Immunol. 2013;13:875–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Nikolich-Žugich J The twilight of immunity: emerging concepts in aging of the immune system. Nature Immunology. 2018;19:10–19. [DOI] [PubMed] [Google Scholar]
- 58.Kovtonyuk LV, Fritsch K, Feng X, Manz MG and Takizawa H. Inflamm-Aging of Hematopoiesis, Hematopoietic Stem Cells, and the Bone Marrow Microenvironment. Frontiers in immunology. 2016;7:502–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jaiswal S, Natarajan P, Silver AJ, Gibson CJ, Bick AG, Shvartz E, McConkey M, Gupta N, Gabriel S, Ardissino D, Baber U, Mehran R, Fuster V, Danesh J, Frossard P, Saleheen D, Melander O, Sukhova GK, Neuberg D, Libby P, Kathiresan S and Ebert BL. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. NEJM. 2017;377:111–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Fuster JJ and Walsh K. Somatic Mutations and Clonal Hematopoiesis: Unexpected Potential New Drivers of Age-Related Cardiovascular Disease. Circulation research. 2018;122:523–532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Calabrese LH and Rose-John S. IL-6 biology: implications for clinical targeting in rheumatic disease. Nat Rev Rheumatol. 2014;10:720–727. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.