

Abstract
The location of Pt nanoparticles was studied in Pt/zeolite Y/γ‐Al2O3 composite catalysts prepared by H2PtCl6 ⋅ 6H2O (CPA) or Pt(NH3)4(NO3)2 (PTA) as Pt precursors. The aim of this study is to validate findings from Transmission Electron Microscopy (TEM) by using characterization techniques that sample larger amounts of catalyst per measurement. Quantitative X‐ray Photoelectron Spectroscopy (XPS) showed that the catalyst prepared with CPA led to a significantly higher Pt/Al atomic ratio than the catalyst prepared with PTA confirming that the 1‐2 nm sized Pt nanoparticles in the former catalyst were located on the open and mesoporous γ‐Al2O3 component, whereas they were located in the micropores of zeolite Y in the latter. By using infrared spectroscopy, a shift in the absorption band maximum of CO chemisorbed on Pt nanoparticles was observed, which can be attributed to a difference in electronic properties depending on the support of the Pt nanoparticles. Finally, model hydrogenation experiments were performed using β‐phenylcinnamaldehyde, a reactant molecule with low diffusivity in zeolite Y micropores, resulting in a 5 times higher activity for the catalyst prepared by CPA compared to PTA. The combined use of these characterization techniques allow us to draw more robust conclusions on the ability to control the location of Pt nanoparticles by using either CPA or PTA as precursors in zeolite/γ‐Al2O3 composite catalyst materials.
Keywords: Bifunctional catalysts, Metal nanoparticle location, Quantitative XPS, CO infrared spectroscopy, Catalysis
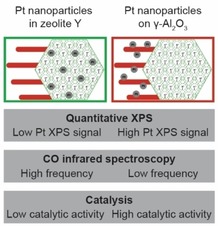
Catalysis Assessment: The location of metal nanoparticles in practical composite catalysts, known to impact their catalytic performance, is assessed using three common laboratory characterization techniques (Quantitative XPS, CO infrared spectroscopy and catalysis), that have the benefit of sampling multiple orders more catalyst per measurement than the commonly‐used TEM.
Introduction
The structural properties of supported metal catalysts, e. g.: metal loading, metal nanoparticle size and their distribution across the catalyst support, as well as the support acidity and porosity, strongly affect their selectivity, activity and stability in catalytic reactions.1, 2, 3 Bifunctional catalysts with zeolite acid sites and metal sites are a special class of solid catalysts that are used for the conversion of crude oil fractions or renewable hydrocarbon feedstocks into fuels and chemicals.4, 5, 6 For optimal catalytic performance of these catalysts, a close intimacy between the metal and acid sites is essential.7, 8, 9 When metal nanoparticles, responsible for dehydrogenation‐hydrogenation, are located inside zeolite micropores, a ‘closest’ intimacy with the zeolite acid sites, responsible for isomerization and cracking, is ensured. On the other hand, locating metal nanoparticles inside zeolite micropores can limit their accessibility for larger hydrocarbon reactants that have low diffusivity through such small pores.10, 11
Due to the strong impact of metal nanoparticle location on the catalytic performance, various synthetic approaches were developed over the last decades to improve control over location of metal nanoparticles within (micro)porous supports.12, 13, 14 For example, Kim et al. were able to control the location of Pt nanoparticles by using [Pt(NH3)4]2+ (aq) to ion‐exchange with zeolite protons to obtain Pt nanoparticles inside ZSM‐5 micropores, whereas impregnation with previously synthesized colloidal particles (∼1.4 nm in diameter) was used to deposit Pt nanoparticles on the outer surface of the ZSM‐5 crystallites (micropore width: 0.56 nm).15 Several papers report methods to locate metal clusters within zeolite micropores by adding stabilized metal colloids or metal complexes before complete crystallization of the zeolites.16, 17, 18, 19 Alternatively, applying specific thermal treatments have been used to control the distribution of metal nanoparticles in porous supports.20
In these and many other studies of supported metal catalysts, TEM is one of the most commonly used techniques to investigate the location of metal nanoparticles on the support.21, 22, 23, 24 However, the severe limitations of TEM are the low amount of catalyst that is sampled per measurement, and the projection of a catalyst's three dimensional structure in a two dimensional image. The latter issue can be overcome by the use of Electron tomography (or 3D TEM) that has been successfully applied to establish the location of metal particles within e. g. zeolite micropores.22, 24, 25, 26 Aside from TEM, several other characterization techniques have been sporadically employed to try to directly or indirectly determine the location of the metal nanoparticles on a support, including catalysis,16, 17, 18, 19, 27, 28, 29 CO infrared spectroscopy,18, 30 SAXS,21 XAS31, 32 and XPS.27, 28, 33
Zeolite based catalysts used in industrial processes are typically shaped as millimeter sized bodies using a porous metal oxide binder, and may consist of additional components to further improve catalyst performance.2, 3 While academic catalyst research focuses often on structurally‐simple model catalysts, a number of recent publications focuses on more practical catalysts and the interplay of all the structural features that determines the catalytic performance.34, 35 In a recent study, industrially relevant zeolite Y/γ‐Al2O3 extrudates were used as support for Pt nanoparticles, located either inside zeolite Y or on the γ‐Al2O3 binder.25 The resulting bifunctional catalysts were investigated by using a heavy hydrocarbon feedstock, and remarkable beneficial effects on product selectivity were observed for the catalyst with Pt nanoparticles on the γ‐Al2O3 binder compared to the catalysts with Pt nanoparticles inside zeolite Y. TEM was used to investigate the location of Pt nanoparticles in the zeolite Y/γ‐Al2O3 extrudates and to overcome the challenge of overlapping features in two dimensional TEM images, authors used ultramicrotomy to section the catalysts into 70 nm thick slices. Besides that, an electron tomography study was used to study the location of Pt nanoparticles. However, both of these approaches suffer from sampling issues, with an estimated amount of sample being imaged in the order of 10−14 g. In studies of supported metal catalysts, synthesis‐originated structural heterogeneities, such as: variation in local metal loading or metal nanoparticle size and location are regularly observed, either at the nanoscale between individual support particles24, 36 up to the microscale between individual support grains.37 It is therefore desirable to validate results of characterization techniques, especially those that sample low amounts of catalysts per measurement, with complementary characterization techniques. In this study, the location of Pt nanoparticles in Pt/zeolite Y/γ‐Al2O3 catalysts is assessed using a combination of commonly used laboratory techniques (quantitative XPS, CO infrared spectroscopy and catalysis) that sample significantly more catalyst than can be achieved by TEM analysis (Table 1, SI section 1.1, Table S1). The presented methods for assessment of the location of metal nanoparticles could be adopted for a wider range of catalysts, in particular catalyst with bimodal porosities, for example, to study the migration of metal nanoparticles during catalysis.38
Table 1.
Employed characterization techniques and the respective amount of catalyst that is sampled per measurement, estimated using the parameters employed in this study.
|
Characterization technique |
Amount [g] |
|---|---|
|
TEM |
∼10−14 |
|
Quantitative XPS |
∼10−9 |
|
CO infrared spectroscopy |
∼10−2 |
|
Catalysis |
5 ⋅ 10−2 |
Results and Discussion
Pt/zeolite Y/γ‐Al2O3 catalysts were prepared with the aim to generate Pt nanoparticles onto either the zeolite Y or the γ‐Al2O3 component of a zeolite Y/γ‐Al2O3 composite support (50/50 wt.), using a previously reported methodology.25 H2PtCl6 ⋅ 6H2O or Pt(NH3)4(NO3)2 precursors dissolved in water yield the anionic [PtCl6]2− (aq) complex or the cationic [Pt(NH3)4]2+ (aq) complex, respectively, whereafter these solutions were added to an aqueous suspension of the zeolite Y/γ‐Al2O3 composite support. In acidic conditions (pH∼3.0), [PtCl6]2− (aq) adsorbs on the positively charged γ‐Al2O3 surface (PZC∼8.5) support due to
electrostatic attraction, while interactions with the zeolite Y component are minimal.39, 40 On the other hand, the [Pt(NH3)4]2+ (aq) complex ion‐exchanges with protons of the zeolite Y component, which is rather independent of pH.40 The ion‐exchange was performed at mildly acidic conditions (pH∼5.0) whereby γ‐Al2O3 is slightly positively charged to restrict adsorption of the cationic Pt complex. After Pt deposition, samples were dried and calcined/reduced to convert the adsorbed Pt complexes into Pt nanoparticles of 1–2 nm in diameter. The properties of the prepared catalysts are provided in Table 2.
Table 2.
Properties of the catalysts used for XPS and HAADF‐STEM measurements. Pt weight loadings were determined by ICP whereas Pt nanoparticle sizes were determined from HAADF‐STEM images.
|
Sample |
Pt precursor |
Pt adsorption mechanism |
Pt wt. loading [%] |
Pt nanoparticle size [nm] |
|---|---|---|---|---|
|
Pt−Y/A−NH3 |
Pt(NH3)4(NO3)2 |
Ion‐exchange |
0.5[a] |
1.4±0.4 |
|
Pt−Y/A−Cl |
H2PtCl6 ⋅ 6H2O |
Strong electrostatic adsorption |
0.5[a] |
1.8±0.4 |
[a] Pt wt. loading indicated corresponds to catalysts characterized with HAADF‐STEM and quantitative XPS. Catalysts wt. loadings used for CO infrared spectroscopy and catalysis are between 0.3–0.4 wt.% Pt.
High‐Angle Annular Dark Field Scanning Transmission Electron Microscopy (HAADF‐STEM) analysis of 70 nm thick microtomed sections of the samples provided local information about the size and location of Pt nanoparticles. It was observed that in the Pt−Y/A−Cl catalyst Pt nanoparticles were mostly present on γ‐Al2O3 (Figure 1a), whose highly irregular porous structure of aggregated ∼10 nm long platelets can be easily distinguished from large (200–1000 nm) mesoporous zeolite crystals. In Pt−Y/A−NH3 (Figure 1b), Pt nanoparticles were almost exclusively present in the zeolite Y component. Previous research using electron tomography has shown that Pt nanoparticles in zeolite Y are located inside zeolite crystalline domains even though their size exceeds that of the zeolite micropores.24 To further aid distinguishing the zeolite Y from the γ‐Al2O3, HAADF‐STEM was combined with EDX elemental mapping as can be observed in Figure S1.
Figure 1.

HAADF‐STEM images of microtomed sections of Pt−Y/A−Cl (a) and Pt−Y/A‐NH3 (b). The γ‐Al2O3 and zeolite Y components of the samples can be identified by their different morphology and are indicated in the images with red and green, respectively. 1–2 nm Pt nanoparticles can be identified as bright white dots, almost exclusively present in the γ‐Al2O3 for Pt−Y/A−Cl and almost exclusively present in zeolite Y for Pt−Y/A−NH3.
In both composite catalysts, a small number of Pt nanoparticles close to the interface between γ‐Al2O3 and zeolite Y appeared to be located on the ‘opposite’ component, as can be evidenced in Figure S2. In such cases, electron tomography could be used to determine the exact location of individual Pt nanoparticles. However, considering the low amount of catalyst sampled in a TEM measurement, it is desirable to investigate the severity of these heterogeneities (Pt nanoparticles located on the ‘opposite’ component) within larger amounts of the Pt/zeolite Y/γ‐Al2O3 composite catalysts.
Quantitative XPS
XPS can provide quantitative information about atomic composition of the outer few nanometers of solid samples. It has been used by Wang et al. to determine if Pd nanoparticles were successfully encapsulated by a crystalline silica shell (silicalite‐1),27 whereas it was used by Winter et al. to determine if metal particles were located in the inner core or on the external surface of carbon nanofibers.33
Here XPS was used to determine the location of Pt nanoparticles in Pt/zeolite Y/γ‐Al2O3 composite catalysts, by relying on the probability of which part of the sample is studied in an XPS measurement. The sampling depth for XPS is only a few nanometres due to the low photoelectron escape depth (electron mean free path, EMFP), that is a function of the kinetic energy of the photoelectrons and the matrix material.41 For the γ‐Al2O3 platelets or nanometer‐sized zeolite crystallites, it can be reasonably assumed that every orientation angle of these components within the X‐ray beam (ø=400 μm) is of equal probability and all orientations are present within the irradiated area (Figure 2). Therefore, in both cases, the resulting XPS signal will be an average of the of the outer surface of the sample.42 Because the thickness of the γ‐Al2O3 platelets is similar to the EMFP in Al2O3 (kinetic energy: 1410–1415 eV), this implies that every part of the sample has equal probability of being sampled and thus, for 1–2 nm sized Pt nanoparticles supported on γ‐Al2O3 the XPS signal is an average of the entire sample.43 The zeolite Y crystallites are between 200 and 1000 nm in size while the EMFP for zeolite Y is approximately 4 nm (kinetic energy: 1410–1415 eV). As a consequence, only the outer layer of zeolite Y crystallites is studied, with a thickness determined by the EMFP, whereas the part below this layer does not contribute to the XPS signal (Figure 2). For more details about the XPS measurements and the approximation of EMFP in the samples, we refer to SI section 1.2. To assess the location of Pt nanoparticles in Pt−Y/A−Cl and Pt−Y/A−NH3, samples were used with similar Pt weight loading and Pt particle size (Table S2). XPS spectra of the fitted Pt 4 f core lines can be observed in Figure 3, while for the full regional spectra we refer to Figure S5.
Figure 2.
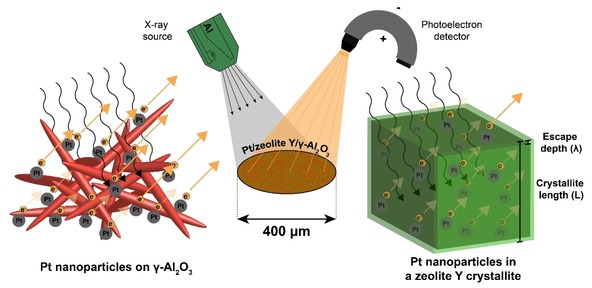
Scheme illustrating an XPS measurement of Pt/zeolite Y/γ‐Al2O3, either with Pt nanoparticles supported on γ‐Al2O3 (left) or Pt nanoparticles located in a zeolite Y crystallite (right). For Pt nanoparticles on γ‐Al2O3, the signal of Pt 4f photoelectrons (kinetic energy: 1410–1415 eV) consists of contributions from Pt nanoparticles located on the external surfaces of γ‐Al2O3 platelets. In zeolite Y crystallites, here schematically depicted as a cube with side length L, photoelectrons from Pt can only escape from the outer layer (thickness: λ) of the crystallite while the volume ‘below’ this layer cannot be assessed with XPS. The resulting XPS signal thus only consists of contributions of this outer layer of the zeolite Y crystals, while for γ‐Al2O3 every part of the sample has equal probability of being sampled.
Figure 3.
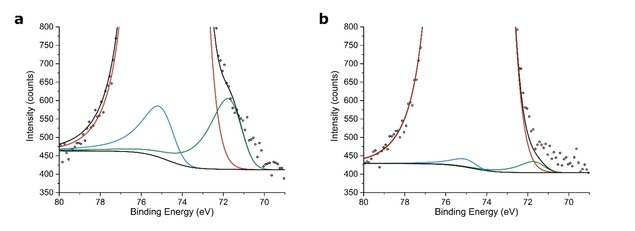
High resolution regional XPS spectra of Pt−Y/A−Cl (a) and Pt−Y/A−NH3 (b) showing different Pt signal intensities depending on the location of the Pt nanoparticles. A Shirley background is applied to remove the contribution of the inelastically scattered electrons. Color scheme: Pt 4f5/2 (light blue), Pt 4f7/2 (dark green), Al 2p (red), Shirley background and overall fit (black).
Table 3 shows the surface atomic ratios obtained by XPS, obtained after correction with atomic sensitivity factors, while bulk atomic ratios based on ICP analysis for Pt and manufacturers specifications for Si and Al. Furthermore, physical mixtures of Pt−Y−NH3 with γ‐Al2O3 (Pt−Y−NH3+A) and Pt−A−Cl with zeolite Y (Pt−A−Cl+Y) were measured, that have similar bulk composition to the studied Pt/zeolite Y/γ‐Al2O3 catalysts. For other relevant reference samples that were used for the fitting and standard deviations in the XPS measurements, we refer to Table S2.
Table 3.
Bulk and surface elemental composition of the Pt/zeolite Y/γ‐Al2O3 catalysts and physical mixtures with similar bulk composition. Bulk atomic ratios were calculated based on ICP measurements for Pt and specifications from manufacturer for Si and Al, whereas the surface atomic ratios were obtained by XPS.
|
|
Bulk |
Surface (XPS) |
||
|---|---|---|---|---|
|
Sample |
Si/Al [at/at] |
Pt/Al [at/at] |
Si/Al [at/at] |
Pt/Al [at/at] |
|
Pt−Y/A−NH3 |
0.80 |
0.0024 |
0.18 |
0.0003 |
|
Pt−Y/A−Cl |
0.80 |
0.0025 |
0.18 |
0.0021 |
|
Pt−Y−NH3+A |
0.78 |
0.0024 |
0.79 |
0.0001 |
|
Pt−A−Cl+Y |
0.69 |
0.0025 |
0.83 |
0.0009 |
The Si/Al surface ratios obtained by XPS were lower for the Pt/zeolite Y/γ‐Al2O3 composite catalysts (Pt−Y/A−Cl and Pt−Y/A−NH3) than their bulk Si/Al ratios, that can be attributed to coating of zeolite Y crystallites by γ‐Al2O3 platelets thus attenuating the XPS signal of Si, as was also evidenced by TEM imaging (Figure S6).25, 44 Another factor that could contribute to the higher aluminum concentration as observed by XPS, is the preferential breaking at void‐rich γ‐Al2O3 domains during the grinding prior XPS measurements, whereas the more dense zeolite Y crystals stay intact.
The surface Pt/Al ratio reported in Table 3 for Pt−Y/A−Cl is by a factor 7 higher than for Pt−Y/A−NH3, while a similar difference in Pt/Al ratio is observed for physical mixtures. For Pt−Y/A−NH3 and Pt−Y−NH3, we expect that the majority of Pt nanoparticles was located in zeolite Y ‘underneath’ the outer surface layer invisible to the XPS signal, while for Pt−Y/A−Cl and Pt−A−Cl the Pt nanoparticles are located on the γ‐Al2O3 component where they can contribute to the XPS signal. When the zeolite Y crystallites are approximated as cubes with side length L (Figure 2), we anticipate that the difference in the XPS Pt/Al ratio depending on Pt nanoparticle location inside versus outside the zeolite crystals can be described by equation (1):
| (1) |
Wherein IPt and IAl correspond to the Pt and Al peak areas, respectively, after correction for atomic sensitivity factors. λzeolite Y is the EMFP of Pt 4 f/Al 2p photoelectrons in zeolite Y. Assuming a uniform distribution of crystallites between 200–1000 nm and an EMFP of 4 nm, this should correspond to a difference of a factor ∼50, that is a significantly larger difference than is observed here. A complicating factor for the studied composite catalysts, is that Pt nanoparticles outside zeolite Y are located on the γ‐Al2O3 binder, with a significantly higher specific surface area of 314 m2/g compared to the external surface of zeolite Y crystallites of ∼8 m2/g45 leading to a lower surface coverage of Pt nanoparticles. This results in a lower Pt/Al surface ratio as observed by XPS and, consequently, a smaller difference between the Pt/zeolite Y/γ‐Al2O3 composite catalysts based on the location of Pt nanoparticles.43 Due to the structural complexity of the composite catalysts, the differences in XPS Pt/Al ratio cannot be accurately described by the ‘Pt nanoparticle inside versus outside zeolite Y’ approximation.
The quantification of the spectra of the Pt/zeolite Y/γ‐Al2O3 composite catalysts was challenging, due to the low intensity of the Pt 4 f signals and overlap in the Pt 4 f and Al 2p core levels, while the isolated Pt 4d core levels could not be used because of a too low signal/noise ratio. Nevertheless, regarding the standard deviations in Pt/Al ratios indicated in Table S2, there is a significant difference in the XPS Pt/Al ratio between Pt−Y/A−NH3 and Pt−Y/A−Cl, that is also observed for the physical mixture reference samples. The uncertainties in the measurements as a consequence of low Pt weight loading and the overlap in Al 2p and Pt 4 f do therefore not affect the outcome of this study.
The accurate quantification of XPS spectra, proved to be a useful tool that is able to differentiate the location of Pt nanoparticles in Pt/zeolite Y/γ‐Al2O3 composite catalysts. A further quantification of XPS signals calls for a more elaborate study (i. e. the intensity of the Pt 4 f signal could be significantly enhanced in resonant photoemission experiments at a synchrotron) and modelling efforts. Alternatively, another metal could be used that does not overlap strongly with core lines of the support.
CO Infrared Spectroscopy
Fourier‐transform infrared spectroscopy (FTIR), transmission IR spectra combined with CO chemisorption can provide information on the electronic properties of Pt nanoparticles. Typically, uniformly sized supported Pt particles, results in a CO absorption band between 2100–2000 cm−1.46 Due to metal‐support interactions, the electronic properties of Pt particles (1–2 nm) are affected by the support and therefore CO infrared spectroscopy can be used to determine Pt nanoparticle location.47, 48 Stakheev et al. previously studied metal‐support interactions using CO‐FTIR to determine the location of Pt metal clusters in K‐L zeolite and was able to distinct between Pt particles inside or outside zeolite crystallites, whereas Liu et al. was able to describe differences in electronic properties between Pt/MCM‐22 catalysts with different Pt dispersion and Pt nanoparticle location.18, 29
Here we have used samples with 1–2 nm Pt nanoparticles on a composite support and used references with Pt nanoparticles supported on either γ‐Al2O3 (Pt−A−Cl) or zeolite Y (Pt−Y−NH3) to obtain the CO band maximum corresponding to Pt nanoparticles on these supports. The CO surface coverage of Pt nanoparticles and temperature influences the degree of dipolar coupling between CO molecules.46, 49 All samples had similar Pt nanoparticle sizes that resulted in a similar CO coverage, although Pt nanoparticles located inside zeolite micropores could suffer from geometrical constraints making it more difficult for CO molecules to access the Pt sites. Figure 4 shows the FT‐IR spectra of chemisorbed CO on the samples after subtraction of a background spectrum taken prior to CO adsorption. Both the Pt−Y/A−Cl and Pt−A−Cl samples, displayed in red in Figure 4 have a band maximum around ∼2060 cm−1 that has been reported for CO coordinated to Pt particles that do not have extended crystal faces on Pt/Al2O3 at room temperature.50
Figure 4.
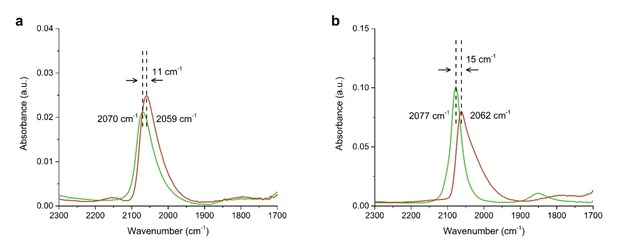
CO‐FTIR of Pt−Y/A−NH3 (a, green) and Pt−Y/A−Cl (a, red) showing a shift in band maximum likely as a results of metal‐support interaction indicating a different Pt nanoparticle location. Pt−Y−NH3 (b, green) and Pt−A−Cl (b, red) are provided as references.
This is a clear indication that for Pt−Y/A−Cl monodisperse Pt nanoparticles are located on the γ‐Al2O3 binder. The band maximum of linearly coordinated CO on Pt−Y−NH3 is present at 2077 cm−1 shown in green in Figure 4b, in accordance with earlier studies reporting band maxima of 2083 cm−1 for Pt nanoparticles of≤1 nm supported on H−Y zeolite.51 The band maximum of Pt−Y/A−NH3 is present at 2070 cm−1, that is lower than for Pt−Y−NH3 but significantly higher than for Pt−A−Cl. This could indicate that Pt nanoparticles could be mainly located in the zeolite Y component with a small fraction located on the γ‐Al2O3 and the CO band maximum forms therefore an average of these populations of Pt nanoparticles. The downside of CO infrared spectroscopy is that differences between band maxima were small and a large overlap existed between relatively broad peaks. Overall, CO infrared spectroscopy data indicates Pt nanoparticles were located on γ‐Al2O3 for Pt−Y/A−Cl and predominantly located inside zeolite Y micropores for Pt−Y/A−NH3.
Catalysis
In a model reaction using 50 mg of catalyst, the catalytic activity of Pt/zeolite Y/γ‐Al2O3 composite catalysts is determined for hydrogenation of β‐phenylcinnamaldehyde (BPCMA, 0.81×1.0 nm).28 Due to its relatively large size, it has low diffusivity in zeolite Y micropores (0.74×0.74 nm)52 and therefore can hardly reach Pt nanoparticles located inside the zeolite Y. If Pt nanoparticles are located on ‘accessible’ locations such as the zeolite Y mesopores or the external surface or the γ‐Al2O3 binder, this would lead to fast conversion of the reactant. Similar approaches have been reported, either based on gas phase or liquid phase reactions, to determine the location of metal nanoparticles in microporous catalysts.17, 18, 19, 27, 28
Catalytic experiments were performed in a batch reactor loaded with equal amounts of BPCMA and catalyst. Figure 5 shows rate constants calculated from concentration profiles obtained by GC analysis of samples taken during the catalytic experiments. The references are used to study the catalytic activity of catalysts for which the location of Pt nanoparticles is known. For the concentration profiles that were used to calculate rate constants by fitting, we refer to Figure S7 and Table S3 and for product identification we refer to Figure S8,9.
Figure 5.
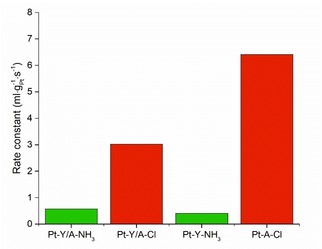
Catalytic activity of the different catalysts in the hydrogenation of β‐phenylcinnamaldehyde (BPCMA). First order rate constants were obtained by fitting the reaction profiles with first order kinetic over 20 h of reaction. The composite catalysts have a Pt weight loading of 0.3–0.5 wt.%, whilst Pt‐A−Cl and Pt−Y−NH3 have weight loadings of 0.7–0.8 wt.%. Conditions: 135 mmol BPCMA/l, BPCMA/Pt (mol/mol)=950, i‐pro 6 ml, H2O 1 ml, H2 20 bar, 70 °C, 500 rpm.
The catalytic activity of Pt−Y/A−NH3 and Pt−Y−NH3 in this reaction was low, as could be expected for catalysts with Pt nanoparticles located inside zeolite micropores. Pt−Y/A−NH3 had a factor 1.5 higher activity with respect to Pt−Y−NH3 that can be contributed to a small fraction of Pt nanoparticles that are located on the γ‐Al2O3 binder. Pt−Y/A−Cl on the other hand, had a factor 5 higher catalytic activity compared to Pt−Y/A−NH3 that forms a strong indication that Pt nanoparticles are located on the γ‐Al2O3 component for the former sample. The catalytic activity of Pt−A−Cl in the same reaction was also studied and was, as the activity of Pt−Y/A−Cl, significantly higher than Pt−Y/A−NH3 and Pt−Y−NH3. The difference in activity between Pt−Y/A−Cl and Pt−A−Cl could be attributed to Pt nanoparticles located in the zeolite, but more likely it is caused by undesired side reactions due to the reactivity of BPCMA on Brønsted acid sites. In the supporting information data is provided of a catalytic test performed with a mixture of Pt−A−Cl and zeolite Y powder, having a catalytic activity very similar to Pt−Y/A−NH3. Overall, the catalytic experiments performed here proved to be a very useful tool in assessment of the Pt nanoparticle location in the Pt/zeolite Y/γ‐Al2O3 composite catalysts.
Conclusions
The location of metal nanoparticles may have great impact on the performance of supported metal catalysts, but for structurally complex, practical catalysts used in industrial processes and consisting of multiple components, assessment of the location of metal particles presents great challenges.25, 53, 54
TEM has proven to be an accurate technique for determining the size and the location of metal nanoparticles in such catalysts, but a limitation is the amount of catalyst that can be sampled per measurement. In this work we explored quantitative XPS, CO infrared spectroscopy and catalysis as techniques that can offer information on metal location, while being able to sample larger amounts of catalysts. Quantitative XPS analysis of catalysts with equal Pt loading, equally sized Pt nanoparticles, but with Pt nanoparticles located either inside zeolite Y crystals or on the γ‐Al2O3 binder of Pt/zeolite Y/γ‐Al2O3 catalysts, lead to a significant differences in Pt/Al atomic ratios as a consequence of a different Pt nanoparticle location. By using CO infrared spectroscopy, a systematic shift in CO band maximum between catalysts was observed depending on the location of Pt nanoparticles, while by performing model reactions using a reactant molecule with low diffusivity in zeolite Y, large differences in activity were observed between catalysts with Pt nanoparticle located either inside zeolite Y or on the more accessible γ‐Al2O3 binder. Therefore, the results of this study show that Pt/zeolite Y/γ‐Al2O3 composite catalysts can be prepared with great control over Pt nanoparticle location and without significant variations within samples, simply by using either CPA or PTA as Pt precursor during catalyst synthesis.
Considering the techniques used in this study (quantitative XPS, CO infrared spectroscopy, catalysis) are more common laboratory techniques compared to TEM, the methods used in this study, and combinations thereof, can be a valuable asset in screening practical catalysts. Furthermore, they can be used to ensure that the results obtained from TEM imaging are not compromised by local variations in structure.
Experimental Section
Catalyst Synthesis. Cylindrical extrudates consisting of 50 wt.% of mesoporous zeolite Y (Zeolyst CBV 760) and 50 wt. % of γ‐Al2O3 (designated as Y/A) were obtained from Shell Projects and Technology. Mesoporous zeolite Y was purchased from Zeolyst (Si/Al=30, CBV760) and HMPA pseudo‐boehmite as precursor for γ‐Al2O3 was obtained from Shell Projects and Technology. Extrudates were crushed and sieved to particles of 0.2–0.5 mm in diameter. Textural analysis of the Y/A support, as well as γ‐Al2O3 and zeolite Y was performed using N2 physisorption at liquid nitrogen temperature on a Micromeritics TriStar II Plus, after drying the samples at 300 °C for 10 hours in an N2 flow. The isotherms and textural properties can be found in Figure S10–12 and Table S4.
Pt−Y/A−NH3 and Pt−Y−NH3 catalysts. 1 g of 0.2–0.5 mm Y/A particles were suspended in 300 ml Milli‐Q water at room temperature and stirred for 1 hour, after which the pH of the suspension was 6.4. ∼50 ml of aqueous solution containing 8.8 mg of Pt(NH3)4(NO3)2 (Sigma‐Aldrich, 99.995 % purity) was added dropwise to the suspension (aiming at nominal Pt loading of 0.5 wt.%), after which the pH dropped to 6.0. After addition of the Pt precursor, the suspension was stirred for another 3 hours, after which the pH dropped to 5.6. The suspension was filtered and washed with Milli‐Q water and dried in air overnight at 120 °C. The dried sample was calcined in a flow of 20 % O2/N2 at 350 °C with a ramp of 0.2 °C ⋅ min−1 (GHSV ∼16.700 h−1), where after it was reduced in a flow of H2 (GHSV ∼3.300 h−1) for 3 hours at 300 °C, using a ramp of 5 °C ⋅ min−1. Pt−Y−NH3 reference catalyst was prepared using the method described above, but using only zeolite Y (CBV 760) in powder form as support. Prior to the calcination/reduction the powder was pressed into a pellet, crushed and sieved into a sieve fraction of 0.2–0.5 mm in diameter while gas flows were kept constant.
Pt−Y/A−Cl and Pt−A−Cl catalysts. A suspension containing 1 g of 0.2–0.5 mm Y/A particles in 300 ml Milli‐Q water was stirred for 1 hour at room temperature, after which the pH of the suspension was lowered from 5.5 to 3.0 by adding a few drops of a 1 M HCl solution. ∼50 ml of aqueous solution containing 13.5 mg of H2PtCl6⋅6H2O (Sigma‐Aldrich, ∼38 wt.% Pt) was added dropwise to the suspension (aiming at nominal Pt loading of 0.5 wt%). After 3 hours of stirring (pH increased to 3.8), the suspension was filtered and washed with Milli‐Q water, and dried in air overnight at 120 °C. Dried catalyst precursor was reduced in a flow of H2 for 3 hours at 500 °C, with a ramp of 5 °C ⋅ min−1 (GHSV ∼3300 h−1). Pt−A−Cl reference catalyst was prepared using the method described above, but using γ‐Al2O3 in powder form as support. The γ‐Al2O3 was obtained by calcination of HMPA pseudo‐boehmite at 550 °C for 2 hours, using a ramp of 5 °C/min. Prior to the reduction the powder was pressed into a pellet, crushed and sieved into a sieve fraction of 0.2–0.5 mm in diameter, while gas flows were kept constant.
Elemental analysis. Pt elemental analysis was performed at Kolbe Mikroanalytisches Laboratorium, Oberhausen using an ICP‐optical emission spectrometer (Perkin Elmer) after sample dissolution according to standard in‐house procedures.
Transmission Electron Microscopy. High‐Angle Annular Dark‐Field Scanning Transmission Electron Microscopy (HAADF‐STEM) imaging was performed on an FEI Talos F200X transmission electron microscope, equipped with a high‐brightness field emission gun (X‐FEG) and operated at 200 kV. For these analyses, catalysts were embedded in Epofix resin, left to cure in air overnight at 60 °C, and cut to 70 nm sections using a Reichert‐Jung Ultracut E ultramicrotome with Diatome Ultra 35° diamond knife. Sections were deposited on carbon‐coated copper TEM grids.
X‐ray Photoelectron Spectroscopy. XPS was performed using a Thermo Scientific K‐Alpha spectrometer equipped with a monochromated Al Kα (hν=1486.6 eV) X‐ray source. The peak binding energies (BE) were calibrated against the sp3 C 1s peak of adventitious carbon at 284.8 eV. Sample charging was compensated by low energy e’ and Ar+ ion flooding during measurements. Quantitative analysis of XPS data was performed using Casa XPS software based on high resolution regional spectra covering the Al 2 s, Si 2p, Al 2p and Pt 4 f core levels in the BE range of 60–130 eV. The ratio between the Al 2p peak and Al 2 s peak was obtained from a reference Al2O3 sample and used during the fitting of the Pt containing samples to determine the area of the Al 2p in the overlapping Pt 4 f/Al 2p region. The remaining area was then fitted to two components with asymmetric (metallic) line shape for Pt 4f5/2 and Pt 4f7/2 core levels, whereas the line shape and the FWHM were kept identical for both peaks of the spin‐orbit split doublet (splitting was fixed to 3.35 eV). Atomic ratios were computed using atomic sensitivity factors. Values were averaged for two locations of the sample using a spot size of 400 μm. More information about the fitting of XPS spectra can be found in SI section 1.2.
CO infrared spectroscopy. CO infrared spectroscopy experiments were performed on a Perkin Elmer 2000 FTIR using a transmission in‐situ cell with CaF2 windows. Spectra were recorded with a resolution of 4 cm−1 by coaddition of 25 scans. For each measurement, a self‐supporting pellet was made of ∼10 mg of sample. The pellet was dried overnight at 120 °C (p<10−5 mbar). Thereafter, the sample was reduced in a flow of H2 (Linde Gas, quality 5.0) at ∼1 bar whilst heating up to 300 °C (1 h, 5 °C/min) followed by evacuation of the cell for 1 h. Then, the temperature was lowered to 50 °C and a reference spectrum was measured in vacuum (p<10−5 mbar). A 10 % CO/He gas mixture (Linde Gas, 99.998 % purity) of 200 mbar was introduced in the cell for 30 min followed by evacuation for 30 min. After evacuation of gaseous CO (p<10−5 mbar), a spectrum was measured of the sample with remaining chemisorbed CO. The final spectra were obtained by subtraction of the reference spectrum from the spectrum of the sample with chemisorbed CO.
Catalytic experiments. Hydrogenation of β‐phenylcinnamaldehyde (BPCMA, Sigma‐Aldrich) was performed in stainless steel autoclaves with a total volume of 15 ml. 50 mg of Pt−Y/A catalysts (or 25 mg of Pt−A−Cl or Pt−Y−NH3 reference catalysts) with a sieve fraction of 0.2–0.5 mm, was suspended in 7 ml of 2‐propanol (Merck, ≥99.5 %) containing 200 mg of dissolved BPCMA and 100 μl of n‐tetradecane (Sigma‐Aldrich, 99 %) as internal standard. Reactors were heated to 70 °C and continuously stirred at 500 rpm before starting the reaction by pressurizing the reactor to 20 bar with H2 (Linde Gas, quality 6.0). Samples were taken from the reaction mixture, diluted with 2‐propanol and analyzed on a VARIAN GC (Agilent VF‐5 column, FID detector). First order k values normalized to reactor volume and the amount of Pt present were obtained by fitting ln([BPCMA]t/[BPCMA]t=0) versus time (0–20 h).
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
Mark Meijerink and Lars van der Wal (both UU) are acknowledged for TEM analysis of the microtomed samples. Nikos Nikolopoulos (UU) is acknowledged for performing N2 physisorption measurements. Fouad Soulimani (UU) is acknowledged for providing the training for CO infrared spectroscopy measurements and Pascal Wijten (UU) is acknowledged for the training for the autoclave setup and GC. Shell Projects and Technology is acknowledged for supplying Zeolite Y/γ‐Al2O3 extrudates and HMPA pseudo‐boehmite.
J. Oenema, J. P. Hofmann, E. J. M. Hensen, J. Zečević, K. P. de Jong, ChemCatChem 2020, 12, 615.
Contributor Information
Dr. Jovana Zečević, Email: J.Zecevic@uu.nl.
Prof. Krijn P. de Jong, Email: K.P.dejong@uu.nl.
References
- 1. Munnik P., de Jongh P. E., de Jong K. P., Chem. Rev. 2015, 115, 6687–6718. [DOI] [PubMed] [Google Scholar]
- 2. Murzin D. Y., Catal. Lett. 2017, 147, 613–621. [Google Scholar]
- 3. Mitchell S., Michels N. L., Pérez-Ramírez J., Chem. Soc. Rev. 2013, 42, 6094–6112. [DOI] [PubMed] [Google Scholar]
- 4. Martens J. A., Verboekend D., Thomas K., Vanbutsele G., Gilson J. P., Pérez-Ramírez J., ChemSusChem 2013, 6, 421–425. [DOI] [PubMed] [Google Scholar]
- 5. Weitkamp J., ChemCatChem 2012, 4, 292–306. [Google Scholar]
- 6. Peng B., Yao Y., Zhao C., Lercher J. A., Angew. Chem. Int. Ed. 2012, 51, 2072–2075; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 2114–2117. [Google Scholar]
- 7. Weisz P. B., Adv. Catal. 1962, 13, 137–190. [Google Scholar]
- 8. Batalha N., Pinard L., Bouchy C., Guillon E., Guisnet M., J. Catal. 2013, 307, 122–131. [Google Scholar]
- 9. Francis J., Guillon E., Bats N., Pichon C., Corma A., Simon L. J., Appl. Catal. A 2011, 409, 140–147. [Google Scholar]
- 10. Vandegehuchte B. D., Thybaut J. W., Marin G. B., Ind. Eng. Chem. Res. 2014, 53, 15333–15347. [Google Scholar]
- 11. Poursaeidesfahani A., de Lange M. F., Khodadadian F., Dubbeldam D., Rigutto M., Nair N., Vlugt T. J. H., J. Catal. 2017, 353, 54–62. [Google Scholar]
- 12. Sachtler W. M. H., Zhang Z., Adv. Catal. 1993, 39, 129–220. [Google Scholar]
- 13. Wang N., Sun Q., Yu J., Adv. Mater. 2018, 31, 1803966. [DOI] [PubMed] [Google Scholar]
- 14. Kosinov N., Liu C., Hensen E. J. M., Pidko E. A., Chem. Mater. 2018, 30, 3177–3198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kim J., Kim W., Seo Y., Kim J. C., Ryoo R., J. Catal. 2013, 301, 187–197. [Google Scholar]
- 16. Altwasser S., Gläser R., Lo A. S., Liu P. H., Chao K. J., Weitkamp J., Microporous Mesoporous Mater. 2006, 89, 109–122. [Google Scholar]
- 17. Goel S., Wu Z., Zones S. I., Iglesia E., J. Am. Chem. Soc. 2012, 134, 17688–17695. [DOI] [PubMed] [Google Scholar]
- 18. Liu L., Díaz U., Arenal R., Agostini G., Concepción P., Corma A., Nat. Mater. 2017, 16, 132–138. [DOI] [PubMed] [Google Scholar]
- 19. Moliner M., Gabay J. E., Kliewer C. E., Carr R. T., Guzman J., Casty G. L., Serna P., Corma A., J. Am. Chem. Soc. 2016, 138, 15743–15750. [DOI] [PubMed] [Google Scholar]
- 20. Prieto G., Zečević J., Friedrich H., de Jong K. P., de Jongh P. E., Nat. Mater. 2012, 12, 34–39. [DOI] [PubMed] [Google Scholar]
- 21. Gommes C. J., Prieto G., Zečević J., Vanhalle M., Goderis B., de Jong K. P., de Jongh P. E., Angew. Chem. Int. Ed. 2015, 127, 11970–11974. [DOI] [PubMed] [Google Scholar]
- 22. Ben Moussa O., Tinat L., Jin X., Baaziz W., Durupthy O., Sayag C., Blanchard J., ACS Catal. 2018, 8, 6071–6078. [Google Scholar]
- 23. Mendes P. S. F., Taleb A. L., Gay A. S., Daudin A., Bouchy C., Silva J. M., Ribeiro M. F., J. Mater. Chem. A 2017, 5, 16822–16833. [Google Scholar]
- 24. Zečević J., van der Eerden A. M. J., Friedrich H., de Jongh P. E., de Jong K. P., ACS Nano 2013, 7, 3698–3705. [DOI] [PubMed] [Google Scholar]
- 25. Zečević J., Vanbutsele G., de Jong K. P., Martens J. A., Nature 2015, 528, 245–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Koster A. J., Ziese U., Verkleij A. J., Janssen A. H., de Jong K. P., J. Phys. Chem. B 2000, 104, 9368–9370. [Google Scholar]
- 27. Wang N., Sun Q., Bai R., Li X., Guo G., Yu J., J. Am. Chem. Soc. 2016, 138, 7484–7487. [DOI] [PubMed] [Google Scholar]
- 28. Liu C., Liu J., Yang S., Cao C., Song W., ChemCatChem 2016, 8, 1279–1282. [Google Scholar]
- 29. Cho H. J., Kim D., Li J., Su D., Xu B., J. Am. Chem. Soc. 2018, 140, 13514–13520. [DOI] [PubMed] [Google Scholar]
- 30. Stakheev A. Y., Shpiro E. S., Jaeger N. I., Schulz-Ekloff G., Catal. Lett. 1995, 32, 147–158. [Google Scholar]
- 31. Guzman J., Gates B. C., Dalton Trans. 2003, 3303–3318. [Google Scholar]
- 32. Kulkarni A., Lobo-Lapidus R. J., Gates B. C., Chem. Commun. 2010, 46, 5997–6015. [DOI] [PubMed] [Google Scholar]
- 33. Winter F., Bezemer G. L., van der Spek C., Meeldijk J. D., van Dillen A. J., Geus J. W., de Jong K. P., Carbon 2005, 43, 327–332. [Google Scholar]
- 34. Whiting G. T., Nikolopoulos N., Nikolopoulos I., Chowdhury A. D., Weckhuysen B. M., Nat. Chem. 2019, 11, 23–31. [DOI] [PubMed] [Google Scholar]
- 35. Michels N. L., Mitchell S., Pérez-Ramírez J., ACS Catal. 2014, 4, 2409–2417. [Google Scholar]
- 36. van der Wal L. I., de Jong K. P., Zečević J., ChemCatChem 2019, 11, 4081–4088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Plessers E., van den Reijen J. E., de Jongh P. E., de Jong K. P., Roeffaers M. B. J., ChemCatChem 2017, 9, 4562–4569. [Google Scholar]
- 38. Munnik P., de Jongh P. E., de Jong K. P., J. Am. Chem. Soc. 2014, 136, 7333–7340. [DOI] [PubMed] [Google Scholar]
- 39. Hao X., Spieker W. A., Regalbuto J. R., J. Colloid Interface Sci. 2003, 267, 259–264. [DOI] [PubMed] [Google Scholar]
- 40. Schreier M., Teren S., Belcher L., Regalbuto J. R., Miller J. T., Nanotechnology 2005, 16, S582-S591. [DOI] [PubMed] [Google Scholar]
- 41. Seah M. P., Dench W. A., Surf. Interface Anal. 1979, 1, 2–11. [Google Scholar]
- 42. Kuipers H. P. C. E., Solid State Ionics 1985, 16, 15–22. [Google Scholar]
- 43. Kerkhof F. P. J. M., Moulijn J. A., J. Phys. Chem. 1979, 83, 1612–1619. [Google Scholar]
- 44. Whiting G. T., Chung S., Stosic D., Chowdhury A. D., van der Wal L. I., Fu D., Zečević J., Travert A., Houben K., Baldus M., Weckhuysen B. M., ACS Catal. 2019, 4792–4803. [Google Scholar]
- 45. Janssen A. H., Koster A. J., de Jong K. P., Angew. Chem. Int. Ed. 2001, 40, 1102–1104; [PubMed] [Google Scholar]; Angew. Chem. 2001, 113, 1136–1138. [Google Scholar]
- 46. Thibault-Starzyk F., Maugé F., in Charact. Solid Mater. Heterog. Catal. From Struct. to Surf. React. (Eds.: M. Che, J. Vedrine), 2012, pp. 1–48. [Google Scholar]
- 47. Frenkel A. I., Small M. W., Smith J. G., Nuzzo R. G., Kvashnina K. O., Tromp M., J. Phys. Chem. C 2013, 117, 23286–23294. [Google Scholar]
- 48. Elsen A., Jung U., Vila F., Li Y., Safonova O. V., Thomas R., Tromp M., Rehr J. J., Nuzzo R. G., Frenkel A. I., J. Phys. Chem. C 2015, 119, 25615–25627. [Google Scholar]
- 49. Koningsberger D. C., Ramaker D. E., Miller J. T., de Graaf J., Mojet B. L., Top. Catal. 2001, 15, 35–42. [Google Scholar]
- 50. Barth R., Pitchai R., Anderson R. L., Verykios X. E., J. Catal. 1989, 116, 61–70. [Google Scholar]
- 51. Visser T., Nijhuis T. A., Van Der Eerden A. M. J., Jenken K., Ji Y., Bras W., Nikitenko S., Ikeda Y., Lepage M., Weckhuysen B. M., J. Phys. Chem. B 2005, 109, 3822–3831. [DOI] [PubMed] [Google Scholar]
- 52.C. Baerlocher, L. B. McCusker, W. M. Meier, D. H. Olson, “Database of Zeolite Structures,” can be found under http://www.iza-structure.org/databases, 2019.
- 53. Meirer F., Kalirai S., Morris D., Soparawalla S., Liu Y., Mesu G., Andrews J. C., Weckhuysen B. M., Sci. Adv. 2015, 1, e1400199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Meirer F., Morris D. T., Kalirai S., Liu Y., Andrews J. C., Weckhuysen B. M., J. Am. Chem. Soc. 2015, 137, 102–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary