

ABSTRACT
Lineage 2 (East Asian), which includes the Beijing genotype, is one of the most prevalent lineages of Mycobacterium tuberculosis (Mtb) throughout the world. The Beijing family is associated to hypervirulence and drug-resistant tuberculosis. The study of this genotype’s circulation in Latin America is crucial for achieving total control of TB, the goal established by the World Health Organization, for the American sub-continent, before 2035. In this sense, the present work presents an overview of the status of the Beijing genotype for this region, with a bibliographical review, and data analysis of MIRU-VNTRs for available Beijing isolates. Certain countries present a prevalent trend of <5%, suggesting low transmissibility for the region, with the exception of Cuba (17.2%), Perú (16%) and Colombia (5%). Minimum Spanning Tree analysis, obtained from MIRU-VNTR data, shows distribution of specific clonal complex strains in each country. From this data, in most countries, we found that molecular epidemiology has not been a tool used for the control of TB, suggesting that the Beijing genotype may be underestimated in Latin America. It is recommended that countries with the highest incidence of the Beijing genotype use effective control strategies and increased care, as a requirement for public health systems.
KEYWORDS: Mycobacterium tuberculosis, Beijing, Latin America, Molecular epidemiology, spoligotyping, MIRU-VNTR
Introduction
In 1993, tuberculosis (TB) was declared a global emergency by the World Health Organization (WHO) and continues to be. According to the WHO, TB is among the 10 leading causes of death in the world and is the greatest cause of death due to a single infectious agent. The resistance of Mycobacterium tuberculosis (Mtb) to antituberculous drugs is a global challenge and represents a serious public health issue. In 3.3% of the new cases of TB and in 18% of previously treated cases, the infection is multidrug-resistant (TB-MDR) or resistant to Rifampicin (TB-RR). In addition, about 1.7 billion people (23% of the world population) are latently infected with Mtb, and thus, represent a threat to the elimination of TB [1]. Despite all efforts to control its impact, which accompany the high number of people with latent TB, HIV infection, diabetes, cancer, autoimmune diseases, the application of biological therapy, malnutrition, poverty, etc., there has not been a significant global reduction of this disease. Strains that are resistant to first- and second-line drugs are being found with greater frequency. In 2017, the WHO estimated there were about 10 million incident cases of TB in the world, with 1.6 million deaths [1].
Bacterial genotype is determinant in the evolution and prognosis of active TB. Indeed, there are several Mtb lineages that affect human populations, suggesting that the tubercle bacillus has developed a high degree of adaptation to the host [2]. Using the latest global phylogenetic reconstructions of the Mtb complex by single point mutations (SNPs) in different countries, 34,167 SNPs were found in the whole genome of 220 clinical isolates [3,4]. There are 7 phylogeographic lineages denominated 1–7, divided into two groups; modern lineages, and ancient or ancestral lineages. Modern lineages are characterized by being monophyletic, they share a single, globally common ancestor and present a deletion in the Tuberculosis Differentiation Region 1 (TbD1), where a significant number of genes from the mycobacterial membrane protein large (mmpL) family are located. Mycobacterial membrane proteins are involved in the transport of cell wall components, such as PDIM (Phthiocerol Dimycocerosates) and specific polyketides, as well as in the expulsion of anti-TB drugs (efflux pumps). The mmpS6 and mmpL6 genes are located in this region, both code for a membrane transport protein [5,6]. The following lineages belong to the group of modern lineages; lineage 2 (East Asian) which includes the Beijing genotype; lineage 3 (East African and Central Asia); and lineage 4 (Euro American). The ancestral lineages are paraphyletic, they are lineage 1 (East Africa, Philippines and, Edge of the Indian Ocean); lineage 5 (West African 1); lineage 6 (West African 2); and lineage 7 (Ethiopia) [3,7,8]. Using large sequence polymorphisms ‘LSPs’ and multilocus sequencing, lineage 7 had never been previously detected. This lineage is now identified by whole genome sequencing, from 36 clinical isolates from the Weldiya region in Ethiopia. These strains are characterized by an atypical pattern of spoligotyping, which is the loss of spacers 4–34. These isolates show an intact TbD1 region, and according to the sequencing data, this new lineage is located between ancestral lineage 1 and modern lineages 2, 3, and 4 [9–11].
Of the aforementioned lineages, two of them are predominant worldwide: the Euro-American lineage, with wide distribution in Europe and America, which is less prevalent in South Africa and Southeast Asia [2]; and the Beijing lineage that has highest prevalence in Asia and Europe [12], but it is distributed worldwide [13].
For patients infected with Beijing strains, it is quite important to have an appropriate follow-up, since this particular genotype is associated with multi-resistant phenotype and increased pathogenicity [14].
The aim of the present study is to describe the distribution of the Beijing genotype in the Latin American and the Caribbean region, trying to give useful basis for the implementation of effective control and surveillance strategies in this region.
Generalities of M. tuberculosis complex
TB is caused by members of the Mycobacterium tuberculosis complex (MTBC), which are the species that most frequently infect humans. Some time ago, it was generally believed that TB was caused by a pathogen with uniform characteristics, since its genome was considered as highly conserved and without horizontal gene transfer. The only strain that showed horizontal gene transfer and recombination between different strains is M. canettii, a smooth variant of MTBC that is one of the oldest members of this complex and exhibit higher genetic variability in comparison with other members of the complex. So far, there is no evidence of human-to-human transmission of this strain and there are reports of about 60 unique cases in Africa, suggesting that the origin of this bacterium is environmental, and no specific reservoir has been identified [3,15]. Even though M. canettii can produce gene horizontal transfer, there is no evidence that other members of the MTBC can do it, preventing this strain as a source of genetic variability to other members.
The members from MTBC are classified into two groups: (i) those which have adapted to humans, like M. tuberculosis sensu stricto, that has subtle genomic differences to differentiate them [16], and M. africanum, which only circulates in Africa [17]. This latter strain is divided into two groups according to their biochemical and geographical characteristics. M. africanum subtype I with similar properties to M. bovis; and M. africanum subtype II, which has similar properties to M. tuberculosis [18], (ii) the second group of strains is those that have adapted to animals, such as Mycobacterium microti [19], Mycobacterium bovis [20], Mycobacterium pinnipedii [21], Mycobacterium mungi [22], M. orygis [23], Mycobacterium suricattae [24], Chimpanzee bacillus [25] and free-living Mycobacterium canettii [26]. Strains that are adapted to animals can cause disease in humans by zoonotic transmission [27]. In fact, the taxonomic classification of Mycobacterium species is subject to constant modifications and updates, genetic analysis, and variations in diverse findings. For example, Rojas MA et al. [28] performed whole genome sequencing of ATCC strains for each species of the complex, as well as subsequent DNA-DNA digital hybridization tests, identification of Average Nucleotide Identity, and calculation of phylogenomic distances to propose the conformation of MTBC, the species M. tuberculosis, M. africanum, M. bovis, M. caprae, M. microti, and M. pinnipedii, were considered as heterotypic synonyms of M. tuberculosis. The term var is recommended to differentiate them, for example, M. tuberculosis var africanum. Thus, species M. canettii, M. mungi, and M. orygis should all be considered to be strains of M. tuberculosis. Although this proposition is relevant, it should be validated by expert taxonomists [28].
M. tuberculosis began its clonal expansion along the African continent. Initially, it was believed that MTBC originated in animal hosts, but with the current and available evidence it is thought that the bacteria co-evolved with humans [3,7]. The availability of new generation sequencing methodologies (NGS), and whole genome sequencing (WGS) determined that the ancestral MTBC strains were phylogenetically separated from strains adapted to animals, before the Neolithic demographic transition from the African continent, and before human migration outside Africa [29]. This resulted in the dispersion of some members of MTBC and the acquisition of specificity, in terms of the predominance of genotypes in certain geographical regions. As such, bacterial distribution has been altered by the ability of human populations to navigate, trade, and interact with distant populations, and in consequence, there are bacterial adaptations to certain populations that have created lineages, sublineages, families, and genotypes [30,31]. In fact, the presence of the MTBC genetic material found in bony lesions in mummies from Egyptian, European, Asian, African, and pre-Columbian civilizations, dating to antiquity supports this claim. These observations are widely described [32,33], and suggest a possible co-evolution with humans [27,32,34–38].
M. tuberculosis shows low capacity of genetic material exchange in both, intra-species and inter-species [39], as well as limited transduction in the CRISPR (clustered regularly interspaced short palindromic repeats) locus or DR locus (Direct Repeats), which restrict the acquisition of bacteriophages´ DNA [40]. Even so, there is a large variation in its bacterial phenotype. Thus, small variations such as mutations (SNPs, deletions, and insertions) and changes at the regulatory level, often induced by selective environmental pressures or human influences contribute to induce variations in the bacterial phenotype. Different studies performed in animals [41–44], cellular models [45–48], and human studies [49–51] demonstrated the existence of hyper, hypo, and mild virulent strains [52–54], as well as variation in resistance to antibiotics [55–57].
Differences in virulence and in the drug-resistance profile have an overall negative impact on treatment efficacy, which makes the goal of decreasing mortality and incident TB cases in the world difficult to achieve, particularly by specific mycobacterial genotypes, such as Beijing [34]. Therefore, the aim of this work is to analyze the available published literature related to the circulation of the Beijing genotype of M. tuberculosis in Latin America and the Caribbean, with special emphasis on Colombia, in order to highlight the relevant findings of this genotype in America and its effect on the Latin-American population.
Beijing genotype: origin, description, classification and global presence
The Beijing genotype, which belongs to lineage 2 or East Asian lineage of M. tuberculosis, is a phylogeographic lineage responsible for TB in at least a quarter of the total cases worldwide [58]. This genotype was described for the first time in 1995 by Van Soolingen et al., from a population study of circulating TB genotypes in China, between 1992 and 1994. In this study, all the clinical isolates subsequently named as Bejing were found to share the same RFLP-IS6110 pattern, with 15 to 20 copies of this insertion-sequence, also these strains showed a significantly different spoligotype than isolates from other regions in the world that only demonstrated spacers 35 to 43 in the DR locus. These type of strains circulate not only in China, but also in neighboring countries such as Mongolia, South Korea, and Thailand [59]. Strains with these genotypic characteristics are known as typical or classical Beijing. Clinical isolates with differences in RFLP-IS6110 banding patterns and lacking spoligotyping spacers 35 to 43 are known as atypical Beijing or Beijing-like [60]. There is another group of strains that have been denominated ‘Pseudo-Beijing’, which show the lineage´s typical spoligotype, but they have a shorter deletion (100-bp or less) in the differentiation regions (RD) RD207 and RD105, than the deletion detected in typical Beijing strains [61]. Furthermore, strains of this genotype were identified in the 90s in New York City, in HIV+ patients, in hospitals and prisons. At that time, they were known as Multidrug-Resistant (MDR) ‘W’ strains. Later, it became evident that these strains were part of a small sublineage of the Beijing family. For strains in this phylogenetic group, the denomination W/Beijing is commonly used [62,63].
It is believed that the success of this genotype at a global level is determined by diverse causes, such as vaccination with M. bovis Calmette–Guerin bacilli (BCG) [64], which is massively used in the Asian region. This hypothesis is based on several studies that have isolated this kind of strain in vaccinated patients from endemic countries [59,65,66]. Strains of Beijing genotype have been associated with outbreaks of multidrug-resistant tuberculosis (MDR-TB), and from Eastern Asia, this strain has spread to diverse regions throughout the world. Compared to other MTBC lineages, it seems that the Beijing lineage has selective advantages, such as higher ability to acquire drug resistance, high mutagenic capacity, and increased transmissibility and hypervirulence [12]. These last two characteristics are observed more frequently in strains with lesser degrees of ancestral evolution, mainly in areas whose presence is most recent; such as countries from Latin America, southern Africa and in Eastern Europe. Strains with higher ancestry, particularly from East Asia, show variable antibiotic susceptibility and mild virulence [58].
Lineage 2 or East Asian strains are characterized by the presence or deletion of diverse long sequence polymorphisms (LSP), whose size varies between 224 and 11,985-bp. LSPs contribute to the classification of this genotype. Thus, so far all these studied strains show RD105 and RD207 deletions, which are responsible for the lack of the first 34 spacers in the DR locus (Direct Repeats), present in all members of the MTBC [67]. RD181 is characteristic of almost all strains of the Beijing family, and is a marker of early evolution; RD150 and RD142 may or may not be present in such strains, and this is a characteristic of strains with more recent evolution [68].
The identification of the LSPs has been extended by PCR amplification, partial or total sequencing, or through a hybridization probe. Considering that these are large genomic deletions, they involve partial or total deletion of mycobacterial genes that produce virulence variations [69]. These major genomic deletions are: RD 105, 4267-bp, completely deletes Rv0072 genes coding part of an uncharacterized permease transport protein type ABC [70]; Rv0073 codes for an ATP-like glutamine ATP binding protein, and partially eliminates Rv0071 genes that encode a possible intron type II maturase, which is a gene not essential for the in vitro mycobacterial growth [71]. Rv0073 codes for an ATP-like glutamine ATP binding protein, and partially eliminates Rv0071 genes, that encodes for a possible intron type II maturase, a gene not essential for the in vitro bacterial growth [72].
Other significant deletions founded in Beijing strains are deletion RD207, 7399-bp, that is mainly responsible for the loss of spacers in the DR locus (Direct Repeats) of this genotype, and is responsible for the characteristic spoligotype of 9 spacers, 34 to 43; which produce partial deletion of 7 genes and loss of the insertion sequence IS6110. The genes deleted in this region are: Rv2814c codes for a transposase subunit required for the transposition of IS6110; Rv2815c is the gene that codifies a putative transposase for IS6110 that merges with Rv2814c, which is possibly responsible for the change in the reading frame Rv2816c, Rv2817c, Rv2818c, Rv2819c, and Rv2820c, which codify conserved hypothetical proteins and correspond to possible specific genomic islands for Mtb during translation [73]; Rv2816c, Rv2817c, Rv2818c, Rv2819c, and Rv2820c code for conserved hypothetical proteins, corresponding to possible specific genomic islands for Mtb [74,75]. Rv2816c, Rv2817c, Rv2818c, Rv2819c, and Rv2820c code for conserved hypothetical proteins, corresponding to possible specific Mtb genomic islands [75]. RD142 is a 2851-bp deletion, a region with four genes: Rv1190 and Rv1191, which code for hypothetical proteins; Rv1192 codes for a protein of unknown function; and for sigma factor L (sigL) [75–77]. RD 152 is a deletion of 11,985-bp, that involves the loss of 12 cutl, picD, and wag22 genes, which are antigenic members belonging to the PE-PGRS protein family [78]; Rv1754c codes for a conserved protein rich in proline; Rv1756c and Rv1757c codify a putative transposase subunit of IS6110; Rv1760 encodes a possible triacylglycerol synthetase, responsible for the accumulation of cell wall lipids; Rv1761c codifies a possible hydrophobic end transporter protein involved in cell wall lipid metabolism; Rv1762c encodes a protein of unknown function; Rv1763 and Rv1764 codify putative transposases for the insertion of IS6110; and Rv1765c encodes a hypothetical conserved protein from Mtb’s cytosol [72,77].
Another important marker for the identification of Mtb Beijing genotype isolates from Asia is an intact pks 15/1 gene [79]; this gene encodes enzymes that contribute in the synthesis of Phthiocerol Dimycocerosates (PDIM), a lipid virulence factor located in the cell wall [80–85], which is the most apolar lipid of the cell wall and is structurally related to phenolic glycolipid. Its structure is so dense that it has been called the cell wall CERA [77]. PDIM is present in all species of theMTBC, and in some non-tuberculous mycobacteria (MNT) that are pathogenic in humans and animals. All the strains that produce phthiocerol dimycocerosates or phthiocerol phthioceranates also synthesize structurally related substances, such as phenolphthiocerol [86]. In theory, virulent Mtb strains have a whole copy of this gene and produce PDIM. However, strains of H37Rv and CDC1551 have a deletion of seven base pairs in this gene, that affect the production of both PDIM and peptide PGLs; strains from lineage 2 characteristically produce both lipids, which are probably related to exacerbated virulence. This has been demonstrated in animal models (Mouse and Guinea Pig), and cellular models (macrophages), with virulent strains (HN878) deficient in pks 15/1, or strains transfected with this gene (CDC1551, H37Rv, Erdman), showing higher virulence, with respect to their wild counterpart [79,87].
In addition, other genetic markers have been used for the classification of Beijing strains in ancestral or modern sublineages. The presence of an intact NTF (Noise Transfer Function) locus, with an intact RD181, is characteristic of earlier or ‘atypical ancestral’ strains; when it is deleted these strains are classified as ‘ancestral’ or ‘classical ancestral’ [88]. The most modern strains of the Beijing family are characterized by the presence of one or two copies of IS6110 inserted in the NTF chromosomal region, and by single nucleotide mutations (SNPs) in the genes Mut2 and Mut4, that code for repair enzymes of DNA and are unique to the Beijing genotype [89]. In Mut2, the mutation occurs in codon 58, producing the change of glycine for arginine, and in the Mut4 gene, the change of arginine for glycine is in codon 48. Those Beijing strains that do not present SNPs in these genes are called modern strains [90,91].
According to recent phylogeographic studies, based on MIRU-VNTR analysis and identification of SNPs in the whole genome sequence of 4,987 Beijing strains from 99 countries [12], it was suggested that this lineage originated in the northeastern region of Asia, in northern China, Korea, and Japan, approximately 6,600 years ago. It was determined that this genotype has six sublineages, or main groups or clonal complexes (CC): CC1, CC2, CC3, CC4, CC5 and CC6, and a basal sublineage BL7. With these seven sublineages, a phylogenetic minimum spanning tree (MStree) was built and grouped in three main branches. Subsequently, the presence of IS-6110 was investigated in the NTF region in a subgroup of 337 isolates, and the clonal complexes 1 to 5 (CC1-CC5) were classified as typical modern strains. The presence of such insertion-sequences in CC6 and BL7 was classified as atypical Beijing ancestral variants, due to the absence of one or two copies of IS6110 in the NTF region [12]. In addition, these sublineages have different dispersion patterns worldwide: CC1 (Central Asia around the Black Sea), CC2 (Russia and Eastern Europe), CC3 (East Asia, Pacific Ocean, and Americas), CC4 (Eastern and Southern Africa, and Pacific Ocean), CC5 (Pacific Ocean, Micronesia, and Polynesia), CC6, and BL7 (East Asia, North America, and Mexico) [12,92]. Thus, depending on the genetic markers, different results for lineage 2 phylogeny are obtained, which means that there is no consensus regarding the whole characterization of this group. With the aim of unifying the classification of this lineage, an evolutionary and comparative analysis was recently carried out with all the markers used in various studies [93]. In this analysis, the genome of 1,398 lineage 2 strains from 32 countries in 13 independent studies was included. After excluding repetitive regions, mobile elements, genes from the PE-PPE family, genes associated with drug resistance, and artifactual SNPs associated with InDels, 39,786 SNPs were used to reconstruct a maximum likelihood phylogeny. The obtained results divided lineage 2 into two phylogenetic clades: proto-Beijing and ‘classical’ Beijing. Proto-Beijing clade strains are characterized by RD105 deletion and atypical spoligotyping pattern, containing spacers 1–43 [93]. The ‘classical’ Beijing clade strains are characterized by RD207 deletion; an early branch within the clade that contains the intact mutT2, mutT4, and ogt genes. Subsequent evolutionary stages consist of acquiring deletion RD181, and a mutation in the mutT4 gene (codon 48). Another important monophyletic group consists of strains with mutations in the mutT4 (codon 48) and ogt (codon 37) genes. All these branches are considered ‘ancestral’ Beijing, as such, within the Beijing clade. The largest monophyletic group (1,212 strains from the aforementioned study) contains strains with mutations in mutT2 (codon 58), mutT4 (codon 48), ogt (codon 12), and the presence of the insertion-sequence IS6110 in the NTF region. These strains are considered ‘modern’ Beijing. Finally, the Beijing clade is divided into eight groups, two to five subgroups, depending on the study´s characteristics.
Shitikov et al. proposed another classification of lineage 2, which includes proto-Beijing and Beijing clades, and has a greater discriminatory power (HGDI 0.79) compared with other methods of genotyping [93]. This classification considers 10 groups within the Beijing clade, three of them belong to the ‘ancestral’ Beijing group (ancestral Asia 1, ancestral Asia 2, ancestral Asia 3), and seven belong to the ‘modern’ Beijing group (Asian African 1, Asian African 2, Asian African 2/RD142, Asian African 3, Pacific RD150, Europe/Russia B0/W148 outbreak, and Central Asia).
Presence of the Beijing genotype in Latin America and the Caribbean
As previously mentioned, the Beijing genotype is widely dispersed in the world, and due to the genotyping studies typing MTBC clinical isolates, the identification of this genotype is more frequent.
The countries that form Latin America, or the ‘New World,’ were settled by people from European countries like Spain, France, Portugal, England and Holland [94]. The entire American region is divided into North America, Central America, and South America. The Latin American region, which is characterized by Spanish migration and settlement, is divided into North America (Mexico), Central America (Guatemala, Belize, El Salvador, Honduras, Nicaragua, Costa Rica, and Panama), South America (Colombia, Venezuela, Peru, Ecuador, Bolivia, Paraguay, Uruguay, Chile, Argentina, Brazil, Guyana, Guyana, and Suriname), and the Caribbean islands. The most representative islands are The Greater Antilles (Cuba, Dominican Republic, and Puerto Rico), the Dutch and French Antilles, Trinidad and Tobago, among others.
Due to European migration, several diseases were spread, such as smallpox, measles, and possibly tuberculosis, which were of devastating proportions. However, we know that TB was already present in America before the European migration, due to the isolation of Mtb DNA in pre-Columbian mummies [38]. Thus, it is reasonable to think that the variety of genotypes that we know today in the American continent are a product not only of European migration but also of the arrival of African slaves that brought with them their respective Mtb genotypes, resulting in wide genetic variability. More recently, due to wide intercontinental travel, genotypes from the Asian continent, such as the Beijing genotype, have been introduced into the American population, and are the subject of the present review [1]. According to the estimated data produced by the WHO, in 2017, the American continent contributed to about 3% of the global caseload of TB, which corresponds to around 300,000 cases per year. The countries with the highest rate of incidence are Haiti, Peru, and Bolivia, with 100–199 cases per 100,000 inhabitants. The rest of the countries in the subcontinent have average incidence rates of 25–99 cases per 100,000 inhabitants, with the exception of Chile, which is a low incidence country with 0–24 cases per 100,000 inhabitants [1].
Methodology
In the present study, a non-systematic search of published articles located on the internet was carried out looking for reported genotypes of M. tuberculosis, as well as for articles whose subject was the Beijing–lineage 2 genotype from East Asia, that considered the fields of classical molecular epidemiology typing, and/or modern typing methodologies such as complete genome sequencing. All available results for Beijing strains genotyping were considered in the context of each country´s genetic variability. Other documents, such as dissertations, graduate theses, and research posters were also considered (Figure 1). It should be mentioned that in addition to the search in databases using keywords, a document search of articles and reports of interest was also carried out.
Figure 1.
Search Methodology.
Initially, only those documents that reported Beijing genotype were included. Then, even those articles that did not report Beijing strains were included, in order to get the full view of each country´s context of the disease. January 2019 was the time limit of this bibliographic search. Previous to this date, all published articles have been consulted, the consolidated data is shown in Supplementary Table 1. Furthermore, we performed a Minimum Spanning Tree based in the MIRU-VNTR classification of 24 loci, which were found in the articles that reported results in Beijing strains, in order to determine how these strains were related to each other. Considering that the number of strains with 24 loci was very low, we consulted MIRU data reported by Merker et al. [12], and used as has been reported for Latin American countries (Supplementary Table 2).
Results and discussion
A total of 86 articles published in Latin America were detected and reviewed in order to get information about Mtb genotypes. According to the methodology criteria, the summation of reports of the Beijing genotype corresponds to the percentage for each lineage. Due to the view obtained exclusively from the summation of collected data, temporal variations over certain time periods cannot be identified.
A relevant bibliography from articles, academic dissertations, or official reports in Belize, El Salvador, Nicaragua, and Costa Rica was not found since these countries do not have available data. The countries with available information are shown in Table 1. The number of genotyped isolates, included the Beijing family, in each country from reliable sources are included. Mexico is the country with the greatest number of available sources (16 articles), followed by Brazil (14 manuscripts), Colombia (13 articles) and Peru (10 papers), among others.
Table 1.
| Country | N° papers | N° typed samples | Beijing per country n (%) | References |
|---|---|---|---|---|
| México | 16 | 1901 | 25 (1.31%) | [95–103] |
| [104–107,167] | ||||
| Guatemala | 2 | 553 | 13 (2.3%) | [169,176] |
| Honduras | 1 | 206 | 1 (0.5%) | [108] |
| Panamá | 3 | 351 | 4 (1.13%) | [109,110,170] |
| Haití | 1 | 157 | 1 (0.63%) | [111] |
| Islas del Caribe | 4 | 1091 | 8 (0.73%) | [171,179–181] |
| Cuba | 4 | 649 | 112 (17.2%) | [112,159–161] |
| Colombia | 13 | 1989 | 100 (5.0%) | [113–118,183] |
| [119–122,168,174,175] | ||||
| Venezuela | 5 | 2790 | 8 (0.30%) | [123–127] |
| Perú | 10 | 6206 | 634 (16%) | [128–132,163,164] |
| [133–135] | ||||
| Ecuador | 3 | 512 | 9 (1.17%) | [136,177] |
| Bolivia | 1 | 100 | 0 (0%) | [137] |
| Chile | 4 | 645 | 4 (0.62%) | [138–141] |
| Paraguay | 1 | 220 | 1 (0.45%) | [142] |
| Argentina | 5 | 3375 | 23 (0.83%) | [143–147] |
| Brasil | 14 | 4439 | 37 (0.87%) | [90,137,148–152] |
| [153–158,172] | ||||
| Total Sub-Continental | 87 | 25,184 | 980 (3.9%) |
The information in Table 1, along with information from other genotypes in the region obtained from the bibliographic review (Supplementary File 1), are shown by country in Figure 2. N.D (not determined) corresponds to strains that were not typified by either spoligotyping or MIRU-VNTR. It is also important to mention that the Beijing genotype is the second most frequent in Cuba after the T family. In Peru, it is the third most frequent after the LAM and Haarlem genotypes. In Colombia, Beijing genotype is the fourth most frequent after the LAM, Haarlem, and T family. In Guatemala, Beijing is the most frequent genotype. Figure 3 shows the timeline of the appearance of Beijing genotype by country, as described by bibliographical sources.
Figure 2.
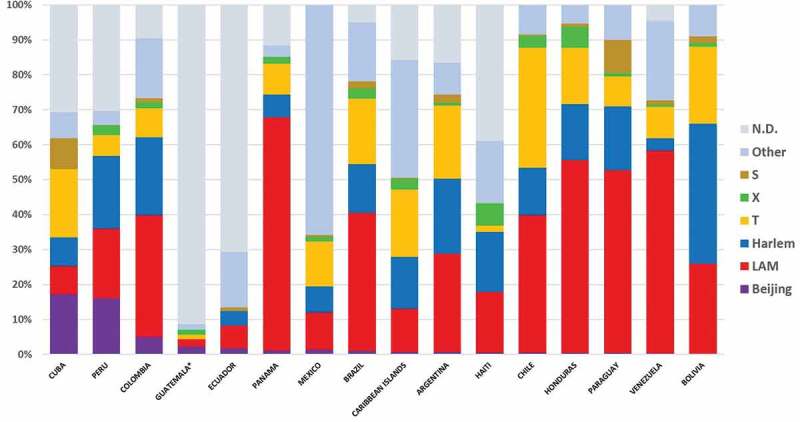
M. tuberculosis genotypes distribution by country in Latin America.
Figure 3.
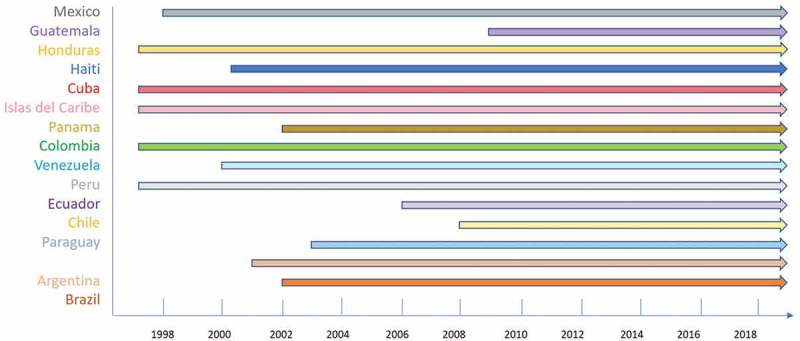
Timeline of Beijing in Latin American countries.
According to the information for the Latin American region, the lineage 2/Asian represented by Beijing genotype corresponds to 3.9% of the identified isolates (980/25,184), being 91.4% (896/980) SIT1/Beijing ‘classic’ genotype, characterized by spoligotyping by the absence of the first 34 spacer sequences in the CRISPR region. Interestingly, 8.1% (80/896) of the strains were SIT190/Beijing-like, which are characterized by the lack of spacer 40; being the most common strains within the Beijing variant. Less than 1% total cases of other Beijing-like genotypes were found in Mexico, Guatemala, Ecuador, and Cuba.
The countries with the higher number of Beijing genotype isolates were Cuba (17.26%, n = 112), Peru (16%, n = 634), and Colombia (5%, n = 100) (Table 1, Figure 2). The genotypes present in Cuba are SIT1/Beijing classic, and only one strain is SIT190/Beijing-like [159]. Even though Cuba has one of the lowest TB incidence rates in Latin America [160], this country has the greatest presence of the Beijing genotype compared to other countries in the region, as has been reported since 1995. It is important to highlight that the presence of the Beijing genotype in Cuba has increased from 11.3% to 25.6%, between 1995 and 2010 [160].
The high frequency of Beijing genotype in Cuba can be explained by the diplomatic/commercial exchange that this country has held with the ex-Soviet Union, which could export these strains to the island with a high resistance to first-line antibiotics, especially to streptomycin due to the high circulation of resistant strains in Russia [161]. However, more studies are needed to confirm this hypothesis. The most common genotype in Cuba is the T genotype (19.5%), which is the most common family in any Latin American country.
Peru is the country with the second largest isolates of Beijing genotype in Latin America, 16% (634/25,123 strains). The Beijing genotype is usually found in patients from Lima and its surroundings, where almost a third of the country’s population is concentrated (30.3%, census 2017). There were no reports of the Beijing genotype in other regions in the country. The first Beijing genotype isolate was reported in 1999, in Lima [162,163]. Nevertheless, the arrival of this genotype in Peru may have been produced by Chinese and Japanese migrations to Peru during the seventeenth century, which is much earlier than any anti-tuberculous therapy, resulting in a founder effect and susceptible strains [164]. The later manifestation of drug resistance in Peru is the consequence of an inappropriate TB control program, but its impact has varied among genotypes. The emergence of the Beijing genotype’s immunity did not achieve any impact; LAM strains underwent different mutations that allowed them to become resistant and to have a greater impact [164]. All reported Beijing genotypes in Peru were SIT1.
Colombia is the third country with the greatest presence of Beijing genotype (5%, n = 100), which has been reported since 1997, specifically in the port city of Buenaventura, on the pacific coast [165]. Reports in Colombia are produced from national studies in many regions of the country. It is interesting that Buenaventura is the only city with reported Beijing genotype isolates. Unlike other countries in the region, the SIT190/Beijing-like is the most common genotype in Colombia (79%); and it is almost exclusively present here, with the exception of Cuba, which has reported one case caused by this genotype. SIT406/Beijing-like is the other Beijing-like genotype recently reported in Colombia, isolated in an indigenous patient [166], and which has also been detected in a patient in Guadalajara, Mexico [167]. Another consideration to recognize is that Beijing-like strains cover the whole chemotherapy-resistant range, from monoresistant to extensively resistant strains (XDR) [169–172,187], differently from phenotypes reported in other countries where a correlation to resistance is not seen, except in Cuba where the correlation is identified. As so, a study made in Cali (Colombia) with 10 cases of XDR-TB found 5 to be carrying the SIT190/Beijing genotype. Three were new cases; one was a relapse; one was a treatment failure; four of the patients passed away [170].
A recent study undertaken in Colombia, by our research group, along with the Instituto Nacional de Salud, genotyped 201 isolates of multidrug-resistant M. tuberculosis; 34.32% (25 cases) came from Antioquia; 24.37% (21 cases) came from Valle del Cauca (where Buenaventura is located). These regions show the greatest number of new cases annually, and the greatest number of TB-MDR cases in the country. As high as 4.9% (10 cases) come from Atlántico. The genotypes most frequently found were SIT42/LAM9 with 25 isolates (12.4%); SIT190/Beijing with 21 isolates (10.4%); and SIT62 (H1) with 21 isolates (10.4%) [173]. An interesting feature of the present study is the genotypic and phenotypic differences among Beijing genotype strains isolated from Peru, Colombia, and Cuba, which can be explained from a historical, social, and biological perspective. In Cuba, probably the Beijing genotype has been brought by the ex-soviet immigration, which has introduced resistant strains to the island. In Peru, the existence of Beijing genotype is probably the consequence of massive Japanese and Chinese immigration, which started many years ago and continues until today. There is not a historical explanation of how Beijing genotyped was introduced to Colombia. One possibility is that Beijing strains were introduced through Buenaventura, which is the most active port for commercial and migratory exchange with Asia, located on Colombia´s pacific coast; unlike the port of Barranquilla, which is located on the Caribbean coast where this genotype is not found [165,174] (Llerena, Rodriguez et al., manuscript in preparation). Another characteristic of the Colombian strains is the relation with antibiotic resistance that can be explained by two hypotheses. One hypothesis, proposed by Lasserson et al. [165], attributes this event to a failure in implementing TB control programs in the region, as well as by flaws in applying DOTS strategy. It is also possible that some already resistant Beijing genotype strains are coming directly from Asia through Buenaventura.
From an anthropological point of view, it seems that the initial population infected with the Beijing genotype, in Peru, is mainly Chinese and Japanese, while in Colombia, the main population infected with this genotype is Afro-Colombian. In Cuba, the infected population is mestizo and the dominant strains are drug-resistant; brought by Eastern European individuals. This suggests that the Beijing genotype does not infect selectively a specific human population.
According to the data collected in Colombia, the increase of the Beijing genotype has been seen from 1999 until today [175], reaching 5% of total TB patients (Table 1 and Figure 1). The data from the dissertation mentioned above were not taken into account for the calculations made in this review since it is a study designed for MDR strains and might modify the results.
The information obtained from Guatemala comes from two articles. In an article from 2015, which is the most recent [176], 5 out of 11 Beijing strains were characterized by genome sequencing. The obtained results suggest that the strains circulating in Guatemala are derived from different founding events, one of them being ancestral. The contribution to TB cases is low, with 13 Beijing isolates from 553 reported cases (2.3%).
In spite of having open borders with countries where Beijing genotype is relatively common, such as Peru (16%) and Colombia (5%); Ecuador reported scare isolates Beijing genotype, only 1.17% (Table 1, Figure 2), with a strong correlation to drug immunity [177].
From the consulted bibliography, Mexico is the country with the highest mycobacterial genetic variability, with a total of 16 different genotypes, being LAM family the most prevalent genotype (lineage 4 Euromerican), followed by the Harlem, T, S, and X families; all of which belong to the same lineage. The Beijing genotype is poorly represented, with 25/1901 isolates (1.3%); mainly the SIT1/Beijing classic genotype. Only one SIT406/Beijing-Like isolate was found, characterized by lacking 1–36 spacers, typified by locus DR by spoligotyping, different from the Beijing Classic, which is characterized by the lack of spacers 1–34, and the presence of spacers 35–43. The rest of the countries in the region present an incidence rate lower than 1%, even for countries with large populations, like Brazil. Countries from the southern cone region (Chile, Argentina, and Paraguay) have had historically very low incidence of Beijing genotype, suggesting lesser immigration, either from Asia or Eastern Europe. Eastern Caribbean islands, unlike Cuba, have different epidemiological data, which is possibly influenced by migration, mainly from France and the Netherlands [178–181]. Finally, it is important to mention that information about the Beijing genotype in Honduras, Haiti, Bolivia, and Paraguay is limited, considering that there is just one article available per country.
Various studies suggest that the M. Tuberculosis SIT1/Beijing genotype strains have an increased virulence compared to other genotypes [163], even compared to ancestral Beijing strains [164], which is a genotype found in only one strain, in Guatemala [176]. SIT1/Beijing genotype strains are considered highly transmissible and multi-drug resistant. However, it seems that it is not completely true in America, because since 1994, when the first Beijing isolate was reported until today, this genotype has kept in general a low profile in the majority of the Latin America countries [162]. This low frequency of Beijing family strains in this region also suggests low transmissibility and virulence, as was suggested by Garzón-Chávez in Ecuador [182]. In contrast, in Colombia, another hypothesis has been proposed, based on experimental data of in-vivo infections, using a murine model in BALB/c mice infected with Colombian strains (Cerezo et al., unpublished data). The results show that mice infected with the Beijing-like strains die at a faster rate than mice infected with LAM control, and Beijing classic strains, circulating in Colombia. These results agree with clinical findings since most of the patients infected with the SIT190/Beijing-like genotype have died because of tuberculosis [183]. Furthermore, using an experimental model of transmission show that mice infected with this strain do not infect cohoused healthy mice, because infected mice die before transmitting the infection, supporting the high virulence of this strain and its low transmissibility (Cerezo et al., unpublished data). This could explain why in Colombia, despite the increase of Beijing genotype isolates, there has not been any drastic increase in TB cases.
MST analysis
With the aim of performing a more profound analysis of Beijing genotype strains circulating in Latin America, the data from the 24-locus MIRU-VNTR typing used in this study were compared with the data obtained from the 24-locus MIRU-VNTR typing of isolates from other countries in the region, reported by Merker et al. [12]. Supplementary File 2 provides a graph of the corresponding database. Our review included information from 300 strains, another 157 strains were added from Merker [12]. Thus, a total of 457 strains were analyzed with the MIRU-VNTRplus platform (www.miru-vntrplus.org) [184]. The distribution of Beijing strains is Bolivia n = 1, Brazil n = 9, Colombia n = 12, Cuba n = 10, Ecuador n = 14, El Salvador n = 3, Guatemala n = 5, Haiti n = 2, Honduras n = 6, Jamaica n = 2, Mexico n = 89, Panamá n = 2, Peru n = 268, Puerto Rico n = 27, Venezuela n = 8. Minimum spanning tree (MST), clonal complex, and dendrogram are represented in Figure 4, Supplementary File 3, and Supplementary Figure 1, respectively.
Figure 4.
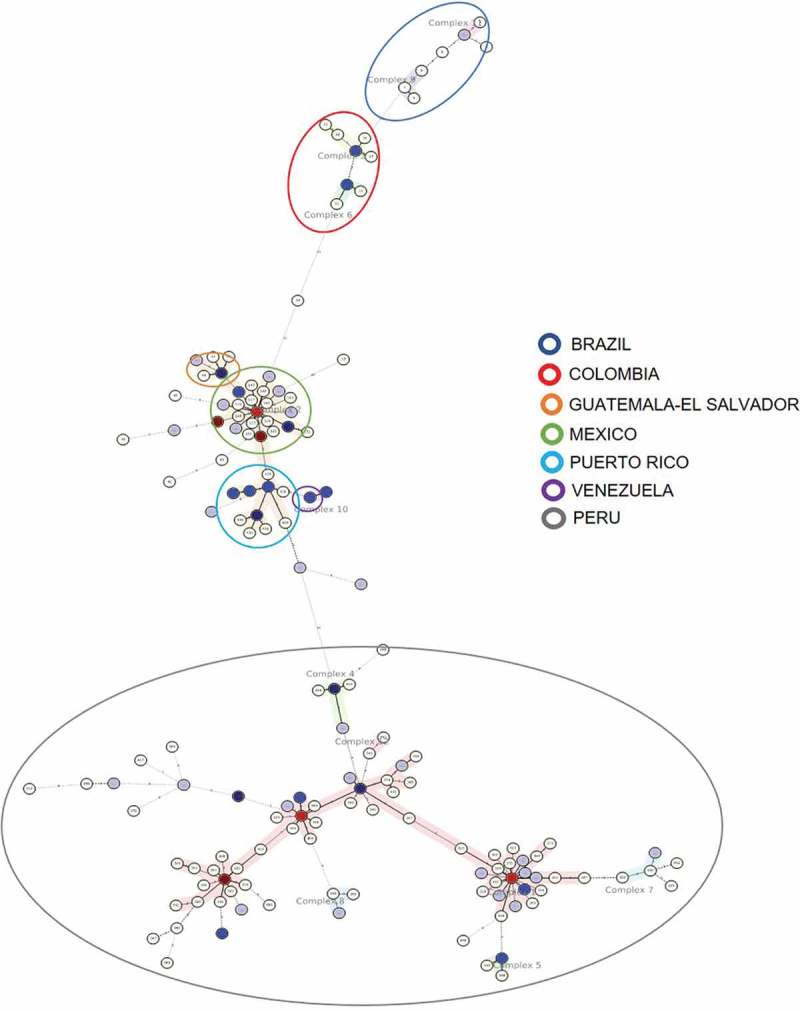
MST of Beijing genotype in Latin America.
According to the Minimum spanning tree (Figure 4), it can be noted that each country forms its own clonal complex; except Ecuador, which is presenting a high heterogeneity among its own strains, but presenting no clonal complex. Therefore, it is not possible to infer the background of the Beijing genotype in this country. However, some of the strains are closer to Mexican complex 2 strains; one of the strains is distantly related to the Colombian complex 6 strains. These findings partially agree with Garzón-Chavez [182], informing that Ecuadorian strains are diverse, do not conform a complex and are closer to Brazilian strains. In contrast, this study suggests that they are closely related to Mexican strains [182]
Colombian strains conform clonal complexes 3 and 6, which are related to Brazilian strains making up both clonal complexes 11 and 9. This is an unexpected result, even considering that both countries share common borders, that there is no direct relation between both populations, possibly due to the existence of geographical barriers. One Colombian strain from complex 6 is related to another strain from Ecuador, which is itself correlated to Mexican strains from complex 2.
Mexican strains conform clonal complex 2, including strains from Guatemala, El Salvador, Honduras, Cuba, and Puerto Rico. This result shows a close relation between the populations sharing the same geographical area, Central American and Western Caribbean zones. It is worth highlighting that Venezuelan strains conform complex 10, which is related to complex 2, where Puerto Rican strains are found.
Peruvian strains form six complexes, closely related to each other, being complex 1 the one with the highest number of strains. There are no similar relations with the strains from Ecuador, Colombia, Brazil, and Bolivia, even as these countries share common borders with Peru. This indicates that once Japanese and Chinese immigrants introduced new strains, as mentioned previously, these strains circulated within Lima’s population, and from then onwards they started spreading to other regions in the country. This hypothesis has not been confirmed due to the lack of information about the Beijing genotype in other regions of Peru. The fact is that these strains are contained within Peru, and suggest that there has not been any migration of infected individuals to other bordering countries.
In brief, when the Beijing genotype is introduced into a country, it co-evolves with the population that it infects, and it is transmitted over time to neighboring populations, creating specific clonal strains in each country. Variables like migration among countries can alter clonality and generate more diversity, as seen in countries like Mexico and Peru.
It can be seen from this study that molecular epidemiology is not considered an important tool for controlling TB, since there are no routine studies genotyping clinical isolates, and determining the TB transmission dynamic within the whole population of countries in the region. Selected studies performed in specific regions, generally in big cities, can be found. Hence, the Beijing genotype may be underestimated in the Latin American region. Finally, we suggest that in order to achieve the TB eradication goal for 2035 in Latin America, it is important to study the lineages circulating throughout the region, especially the Beijing genotype, which is correlated in some countries with drug resistance and hypervirulent phenotype, as is the case in Colombia. The implementation of effective strategies of control by governments’ health-care systems, where the Beijing genotype has a great presence, is of vital importance.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].WHO Global tuberculosis report. Geneva: World Health Organization; 2018. WHO Publication; 2018. [Google Scholar]
- [2].Mokrousov I, Vyazovaya A, Iwamoto T, et al. Latin-American-Mediterranean lineage of Mycobacterium tuberculosis: human traces across pathogen’s phylogeography. Mol Phylogenet Evol. 2016;99:133–143. [DOI] [PubMed] [Google Scholar]
- [3].Brites D, Gagneux S.. Co-evolution of Mycobacterium tuberculosis and Homo sapiens. Immunol Rev. 2015;264(1):6–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Coscolla M, Gagneux S.. Consequences of genomic diversity in Mycobacterium tuberculosis. Semin Immunol. 2014;26(6):431–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Brosch R, Gordon SV, Marmiesse M, et al. A new evolutionary scenario for the Mycobacterium tuberculosis complex. Proc Natl Acad Sci. 2002. March 19;99(6):3684–3689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Jain M, Cox JS. Interaction between polyketide synthase and transporter suggests coupled synthesis and export of virulence lipid in M. tuberculosis. PLoS Pathog. 2005;1(1):0012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Gagneux S. Host-pathogen coevolution in human tuberculosis. Philos Trans R Soc Lond B Biol Sci. 2012;367(1590):850–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Orgeur M, Brosch R. Evolution of virulence in the Mycobacterium tuberculosis complex. Curr Opin Microbiol. 2018;41:68–75. [DOI] [PubMed] [Google Scholar]
- [9].Gagneux S, PM S. Global phylogeography of Mycobacterium tuberculosis and implications for tuberculosis product development. Lancet Infect Dis. 2007;7(5):328–337. [DOI] [PubMed] [Google Scholar]
- [10].Firdessa R, Berg S, Hailu E, et al. Mycobacterial lineages causing pulmonary and extrapulmonary tuberculosis, Ethiopia. Emerg Infect Dis. 2013;19(3):460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Gagneux S. Ecology and evolution of Mycobacterium tuberculosis. Nat Rev Microbiol. 2018;16(4):202–213. [DOI] [PubMed] [Google Scholar]
- [12].Merker M, Blin C, Mona S, et al. Evolutionary history and global spread of the Mycobacterium tuberculosis Beijing lineage. Nat Genet. 2015;47(3):242–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Wiens KE, Woyczynski LP, Ledesma JR, et al. Global variation in bacterial strains that cause tuberculosis disease: A systematic review and meta-analysis. BMC Med. 2018. 30;16(1):196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Buu TN, Huyen MN, Lan NTN, et al. The Beijing genotype is associated with young age and multidrug-resistant tuberculosis in rural Vietnam. Int J Tuberc Lung Dis. 2009;13(7):900–906. [PubMed] [Google Scholar]
- [15].Koeck JL, Fabre M, Simon F, et al. Clinical characteristics of the smooth tubercle bacilli 'Mycobacterium canettii' infection suggest the existence of an environmental reservoir. Clin Microbiol Infect. 2011;17(7):1013–1019. [DOI] [PubMed] [Google Scholar]
- [16].Koch R. Die Aetiologie der Tuberkulose. Klin Wochenschr. 1932;11(12):490–492. [Google Scholar]
- [17].Castets MN, Rist HB. La variété africaine du bacille tuberculeux humain. Med d’Afrique Noire. 1969;16:321–322. [Google Scholar]
- [18].Mostowy S, Onipede A, Gagneux S, et al. Genomic Analysis Distinguishes. Society. 2004;42(8):3594–3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Wells AQ, DMO . Tuberculosis in wild voles. Nature. 1937;139:917. [Google Scholar]
- [20].Karlson A, Lessel E. Mycobacterium bovis nom. nov. Int J Syst Bacteriol. 1970;20(3):273–282. [Google Scholar]
- [21].Cousins DV, Bastida R, Cataldi A, et al. Tuberculosis in seals caused by a novel member of the Mycobacterium tuberculosis complex: Mycobacterium pinnipedii sp nov. Int J Syst Evol Microbiol. 2003;53(5):1305–1314. [DOI] [PubMed] [Google Scholar]
- [22].Alexander KA, Laver PN, Michel AL, et al. Novel Mycobacterium tuberculosis complex pathogen, M Mungi. Emerg Infect Dis. 2010;16(8):1296–1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].van Ingen J, Rahim Z, Mulder A, et al. Characterization of Mycobacterium orygis as M. tuberculosis complex subspecies. Emerg Infect Dis. 2012;18(4):653–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Parsons SDC, Drewe JA, van Pittius NCG, et al. Novel cause of tuberculosis in meerkats, South Africa. Emerg Infect Dis. 2013;19(12):2004–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Coscolla M, Lewin A, Metzger S, et al. Novel Mycobacterium tuberculosis complex isolate from a wild chimpanzee. Emerg Infect Dis. 2013;19(6):969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Van Soolingen D, Hoogenboezem T, De Haas PEW, et al. A novel pathogenic taxon of the Mycobacterium tuberculosis complex, Canetti: characterization of an exceptional isolate from Africa. Int J Syst Bacteriol. 1997;47(4):1236–1245. [DOI] [PubMed] [Google Scholar]
- [27].Bos KI, Harkins KM, Herbig A, et al. Pre-Columbian mycobacterial genomes reveal seals as a source of New World human tuberculosis. Nature. 2014;514(7523):494–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Rojas MA, McGough KJ, Rider-Riojas CJ, et al. Phylogenomic analysis of the species of the Mycobacterium tuberculosis complex demonstrates that Mycobacterium africanum, Mycobacterium bovis, Mycobacterium caprae, Mycobacterium microti and Mycobacterium pinnipedii are later heterotypic synonyms of Mycobacterium tuberculosis. Int J Syst Evol Microbiol. 2018;68(1):324–332. [DOI] [PubMed] [Google Scholar]
- [29].Comas I, Coscolla M, Luo T, et al. Out-of-Africa migration and Neolithic co-expansion of Mycobacterium tuberculosis with modern humans. Nat Genet. 2013;45(10):1176–1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].de Jong BC, Antonio M, Gagneux S. Mycobacterium africanum-review of an important cause of human tuberculosis in West Africa. PLoS Negl Trop Dis. 2010;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Galagan JE. Genomic insights into tuberculosis. Nat Rev Genet. 2014;15(5):307–320. [DOI] [PubMed] [Google Scholar]
- [32].Zink AR, Sola C, Reischl U, et al. Characterization of Mycobacterium tuberculosis complex DNAs from Egyptian mummies by spoligotyping characterization of Mycobacterium tuberculosis complex DNAs from Egyptian Mummies by spoligotyping. J Clin Microbiol. 2003;41(1):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Konomi N, Lebwohl E, Mowbray K, et al. Detection of mycobacterial DNA in andean mummies. J Clin Microbiol. 2002;40(12):4738–4740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baron H, Hummel S, Herrmann B. Mycobacterium tuberculosis complex DNA in ancient human bones. J Archaeol Sci. 1996;23(5):667–671. [Google Scholar]
- [35].Donoghue HD, Spigelman M, Zias J, et al. Mycobacterium tuberculosis complex DNA in calcified pleura from remains 1400 years old. Lett Appl Microbiol. 1998;27(5):265–269. [PubMed] [Google Scholar]
- [36].Zink AR, Sola C, Reischl U, et al. Characterization of Mycobacterium tuberculosis complex DNAs from Egyptian mummies by spoligotyping. J Clin Microbiol. 2003;41(1):359–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mays S, Taylor GM, Legge AJ, et al. Paleopathological and biomolecular study of tuberculosis in a medieval skeletal collection from England. Am J Phys Anthropol. 2001;114(4):298–311. [DOI] [PubMed] [Google Scholar]
- [38].Salo WL, Aufderheide AC, Buikstra J, et al. Identification of Mycobacterium tuberculosis DNA in a pre-Columbian Peruvian mummy. Proc Natl Acad Sci. 1994. 15;91(6):2091–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Da Silva Rabello MC, Matsumoto CK, de Almeida LGP, et al. First description of natural and experimental conjugation between mycobacteria mediated by a linear plasmid. PLoS One. 2012;7(1):e29884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11(3):181–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Aguilar LD, Hanekom M, Mata D, et al. Mycobacterium tuberculosis strains with the Beijing genotype demonstrate variability in virulence associated with transmission. Tuberculosis (Edinb). 2010;90(5):319–325. [DOI] [PubMed] [Google Scholar]
- [42].Palanisamy GS, Smith EE, Shanley CA, et al. Disseminated disease severity as a measure of virulence of Mycobacterium tuberculosis in the guinea pig model. Tuberculosis. 2008;88(4):295–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Barczak AK, Domenech P, Boshoff HIM, et al. In vivo phenotypic dominance in mouse mixed infections with Mycobacterium tuberculosis clinical isolates. J Infect Dis. 2005;192(4):600–606. [DOI] [PubMed] [Google Scholar]
- [44].Shamputa IC, Rigouts L, Eyongeta LA, et al. Genotypic and phenotypic heterogeneity among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients. J Clin Microbiol. 2004;42(12):5528–5536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Van Laarhoven A, Mandemakers JJ, Kleinnijenhuis J, et al. Low induction of proinflammatory cytokines parallels evolutionary success of modern strains within the Mycobacterium tuberculosis beijing genotype. Infect Immun. 2013;81(10):3750–3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Chen YY, Chang JR, Huang WF, et al. The pattern of cytokine production in vitro induced by ancient and modern Beijing Mycobacterium tuberculosis strains. PLoS One. 2014. 11;9(4):e94296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wang C, Peyron P, Mestre O, et al. Innate immune response to Mycobacterium tuberculosis Beijing and other genotypes. PLoS One. 2010. 25;5(10):e13594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Chacón-Salinas R, Serafín-López J, Ramos-Payán R, et al. Differential pattern of cytokine expression by macrophages infected in vitro with different Mycobacterium tuberculosis genotypes. Clin Exp Immunol. 2005;140(3):443–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Rakotosamimanana N, Raharimanga V, Andriamandimby SF, et al. Variation in gamma interferon responses to different infecting strains of Mycobacterium tuberculosis in acid-fast bacillus smear-positive patients and household contacts in Antananarivo, Madagascar. Clin Vaccine Immunol. 2010;17(7):1094–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Finan C, Ota MOC, Marchant A, et al. Natural variation in immune responses to neonatal Mycobacterium bovis Bacillus Calmette-Guerin (BCG) vaccination in a cohort of Gambian infants. PLoS One. 2008;3(10):e3485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].De Jong BC, Hill PC, Brookes RH, et al. Mycobacterium africanum elicits an attenuated T cell response to early secreted antigenic target, 6 kDa, in patients with tuberculosis and their household contacts. J Infect Dis. 2006. 1;193(9):1279–1286. [DOI] [PubMed] [Google Scholar]
- [52].Lopez B, Aguilar D, Orozco H, et al. A marked difference in pathogenesis and immune response induced by different Mycobacterium tuberculosis genotypes. Clin Exp Immunol. 2003;133(1):30–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Aguilar D, Hanekom M, Mata D, et al. Mycobacterium tuberculosis strains with the Beijing genotype demonstrate variability in virulence associated with transmission. Tuberculosis (Edinb). 2010;90(5):319–325. [DOI] [PubMed] [Google Scholar]
- [54].González-Pérez M, Mariño-Ramírez L, Parra-López CA, et al. Virulence and immune response induced by Mycobacterium avium complex strains in a model of progressive pulmonary tuberculosis and subcutaneous infection in BALB/c mice. Infect Immun. 2013;81(11):4001–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Zhang J, Mi L, Wang Y, et al. Genotypes and drug susceptibility of Mycobacterium tuberculosis Isolates in Shihezi, Xinjiang Province, China. BMC Res Notes. 2012;5(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Casali N, Nikolayevskyy V, Balabanova Y, et al. Microevolution of extensively drug-resistant tuberculosis in Russia. Genome Res. 2012;22(4):735–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Machado D, TS C, Perdigão J, et al. Interplay between mutations and efflux in drug resistant clinical isolates of Mycobacterium tuberculosis. Front Microbiol. 2017;27(8):711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Luo T, Comas I, Luo D, et al. Southern East Asian origin and coexpansion of Mycobacterium tuberculosis Beijing family with Han Chinese. Proc Natl Acad Sci U S A. 2015;112(26):8136–8141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Van Soolingen D, Qian L, De Haas PEW, et al. Predominance of a single genotype of Mycobacterium tuberculosis in countries of East Asia. J Clin Microbiol. 1995;33(12):3234–3238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mokrousov I, Narvskaya O, Otten T, et al. Phylogenetic reconstruction within Mycobacterium tuberculosis Beijing genotype in northwestern Russia. Res Microbiol. 2002;153(10):629–637. [DOI] [PubMed] [Google Scholar]
- [61].Fenner L, Malla B, Ninet BB, et al. “Pseudo-Beijing”: evidence for convergent evolution in the direct repeat region of Mycobacterium tuberculosis. PLoS One. 2011;6(9):8–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Parwati I, van Crevel R, van Soolingen D. Possible underlying mechanisms for successful emergence of the Mycobacterium tuberculosis Beijing genotype strains. Lancet Infect Dis. 2010;10(2):103–111. [DOI] [PubMed] [Google Scholar]
- [63].Glynn JR, Whiteley J, Bifani PJ, et al. Worldwide occurrence of Beijing/W strains of Mycobacterium tuberculosis a systematic review. Emerg Infect Dis. 2002;8(8):843–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kremer K, Van Der Werf MJ, Au BKY, et al. Vaccine-induced immunity by typical Mycobacterium tuberculosis Beijing strains. Emerg Infect Dis. 2009;15(2):335–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Jeon BYBY, Derrick SC, Lim J, et al. Mycobacterium bovis BCG immunization induces protective immunity against nine different Mycobacterium tuberculosis strains in mice. Infect Immun. 2008;76(11):5173–5180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Hermans PWM, Messadi F, Guebrexabher H, et al. Analysis of the population structure of Mycobacterium tuberculosis in ethiopia, tunisia, and the netherlands: usefulness of DNA typing for global tuberculosis epidemiology. J Infect Dis. 1995;171(6):1504–1513. [DOI] [PubMed] [Google Scholar]
- [67].Comas I, Homolka S, Niemann S, et al. Genotyping of genetically monomorphic bacteria: DNA sequencing in Mycobacterium tuberculosis highlights the limitations of current methodologies. PLoS One. 2009;4:11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Gagneux S, DeRiemer K, Van T, et al. Variable host-pathogen compatibility in Mycobacterium tuberculosis. Proc Natl Acad Sci. 2006. 21;103(8):2869–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Tsolaki AG, Gagneux S, Pym AS, et al. Genomic deletions classify the Beijing/W Strains as a distinct genetic lineage of Mycobacterium tuberculosis genomic deletions classify the Beijing/W Strains as a distinct genetic lineage of Mycobacterium tuberculosis. J Clin Microbiol. 2005;43(7):3185–3191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Bespyatykh J, Shitikov E, Butenko I, et al. Proteome analysis of the Mycobacterium tuberculosis Beijing B0/W148 cluster. Sci Rep. 2016;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Cole ST, Brosch R, Parkhill J, et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998. 11;393(6685):537–544. [DOI] [PubMed] [Google Scholar]
- [72].de Souza GA, Leversen NA, Målen H, et al. Bacterial proteins with cleaved or uncleaved signal peptides of the general secretory pathway. J Proteomics. 2011. 21;75(2):502–510. [DOI] [PubMed] [Google Scholar]
- [73].Tsolaki AG, Hirsh AE, DeRiemer K, et al. Functional and evolutionary genomics of Mycobacterium tuberculosis: insights from genomic deletions in 100 strains. Proc Natl Acad Sci U S A. 2004. 6;101(14):4865–4870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Mazandu GK, Mulder NJ. Function prediction and analysis of Mycobacterium tuberculosis hypothetical proteins. Int J Mol Sci. 2012;13(6):7283–7302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dejesus MA, Gerrick ER, Xu W, et al. Comprehensive essentiality analysis of the Mycobacterium tuberculosis genome via saturating transposon mutagenesis. MBio. 2017. 17;8(1):ii: e02133-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Sachdeva P, Misra R, Tyagi AK, et al. The sigma factors of Mycobacterium tuberculosis: regulation of the regulators. FEBS J. 2010;277(3):605–626. [DOI] [PubMed] [Google Scholar]
- [77].Forrellad MA, Klepp LI, Gioffré A, et al. Virulence factors of the Mycobacterium tuberculosis complex. Virulence. 2013. January 1;4(1):3–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Brennan MJ, Delogu G. The PE multigene family: A “molecular mantra” for mycobacteria. Trends Microbiol. 2002;10(5):246–249. [DOI] [PubMed] [Google Scholar]
- [79].Reed MB, Domenech P, Manca C, et al. A glycolipid of hypervirulent tuberculosis strains that inhibits the innate immune response. Nature. 2004;431(7004):84–87. [DOI] [PubMed] [Google Scholar]
- [80].Camacho LR, Ensergueix D, Perez E, et al. Identification of a virulence gene cluster of Mycobacterium tuberculosis by signature-tagged transposon mutagenesis. Mol Microbiol. 1999;34(2):257–267. [DOI] [PubMed] [Google Scholar]
- [81].Cox JS, Chess B, McNeil M, et al. Complex lipid determines tissue-specific replication of Mycobacterium tuberculosis in mice. Nature. 1999;402(6757):79–83. [DOI] [PubMed] [Google Scholar]
- [82].Astarie-Dequeker C, Le Guyader L, Malaga W, et al. Phthiocerol dimycocerosates of M. tuberculosis participate in macrophage invasion by inducing changes in the organization of plasma membrane lipids. PLoS Pathog. 2009;5(2):e1000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Cambier CJ, Takaki KK, Larson RP, et al. Mycobacteria manipulate macrophage recruitment through coordinated use of membrane lipids. Nature. 2014. 9;505(7482):218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Barczak AK, Avraham R, Singh S, et al. Systematic, multiparametric analysis of Mycobacterium tuberculosis intracellular infection offers insight into coordinated virulence. PLoS Pathog. 2017;13(5):e1006363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Quigley J, Hughitt VK, Velikovsky CA, et al. The cell wall lipid PDIM contributes to phagosomal escape and host cell exit of Mycobacterium tuberculosis. MBio. 2017. 7;8(2):ii: e00148-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Giovannini D, Cappelli G, Jiang L, et al. A new Mycobacterium tuberculosis smooth colony reduces growth inside human macrophages and represses PDIM Operon gene expression. Does an heterogeneous population exist in intracellular mycobacteria? Microb Pathog. 2012;53(3–4):135–146. [DOI] [PubMed] [Google Scholar]
- [87].Velayati AA, Masjedi MR, Farnia P, et al. Emergence of new forms of totally drug-resistant tuberculosis Bacilli. Chest. 2009;136(2):420–425. [DOI] [PubMed] [Google Scholar]
- [88].de Keijzer J, de Haas PE, de Ru AH, et al. Disclosure of selective advantages in the “Modern” Sublineage of the Mycobacterium tuberculosis Beijing genotype family by quantitative proteomics. Mol Cell Proteomics. 2014;13(10):2632–2645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Ebrahimi-Rad M, Bifani P, Martin C, et al. Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing family. Emerg Infect Dis. 2003;9(7):838–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ribeiro SCM, Gomes LL, Amaral EP, et al. Mycobacterium tuberculosis strains of the modern sublineage of the Beijing family are more likely to display increased virulence than strains of the ancient sublineage. J Clin Microbiol. 2014;52(7):2615–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Yin QQ, Liu HC, Jiao WW, et al. Evolutionary history and ongoing transmission of phylogenetic sublineages of Mycobacterium tuberculosis Beijing genotype in China. Sci Rep. 2016;6:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hanekom M, Van Der Spuy GD, Streicher E, et al. A recently evolved sublineage of the Mycobacterium tuberculosis Beijing strain family is associated with an increased ability to spread and cause disease. J Clin Microbiol. 2007;45(5):1483–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Shitikov E, Kolchenko S, Mokrousov I, et al. Evolutionary pathway analysis and unified classification of East Asian lineage of Mycobacterium tuberculosis. Sci Rep. 2017. 23;7(1):9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Quijada M. Sobre el origen y difusion del nombre “América latina” (o una variacion heterodoxa en torno al tema de la construccion social de la verdad. Revista de Indias. 1998;58:214. [Google Scholar]
- [95].Pérez-Guerrero L, Milián-Suazo F, Arriaga-Díaz C, et al. Epidemiología molecular de las tuberculosis bovina y humana en una zona endémica de Querétaro, México. Salud Publica Mex. 2008;50(4):286–291. [DOI] [PubMed] [Google Scholar]
- [96].Martinez-Gamboa A, Ponce-de-Leon AG-FA, Bobadilla-del-Valle M, et al. Molecular analysis of Mycobacterium tuberculosis strains with an intact pks15/1 gene in a rural community of Mexico. Arch Med Res. 2008;39:809e814. [DOI] [PubMed] [Google Scholar]
- [97].Molina-Torres CA, Moreno-Torres E, Ocampo-Candiani J, et al. Mycobacterium tuberculosis spoligotypes in Monterrey, Mexico. J Clin Microbiol. 2010;48(2):448–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Nava-Aguilera E, López-Vidal Y, Harris E, et al. Clustering of Mycobacterium tuberculosis cases in Acapulco: spoligotyping and risk factors. Clin Dev Immunol. 2011;2011:1–12 Article ID 408375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Macías Parra M, Kumate Rodríguez J, Arredondo García JL, et al. Mycobacterium tuberculosis complex genotype diversity and drug resistance profiles in a pediatric population in Mexico. Tuberc Res Treat. 2011;2011:239042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Zenteno-Cuevas R, Silva-Hernández FM-DF, Ramírez-Hernández M, et al. Characterisation of pks15/1 in clinical isolates of Mycobacterium tuberculosis from Mexico. Mem Inst Oswaldo Cruz. 2013;108(6):718–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Martinez-Guarneros A, Rastogi N, Couvin D, et al. Genetic diversity among multidrug-resistant Mycobacterium tuberculosis strains in Mexico. Infect Genet Evol. 2013;14:434–443. [DOI] [PubMed] [Google Scholar]
- [102].López-Rocha E, Juárez-Álvarez J, Riego-Ruiz L, et al. Genetic diversity of the Mycobacterium tuberculosis complex in San Luis Potosí, México. BMC Res Notes. 2013;6:172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Zenteno-Cuevas R, Mendoza-Damián F, Muñoz IC, et al. Description of the population structure and genetic diversity of tuberculosis in Estado de México, a low prevalence setting from Mexico. APMIS. 2015;123(2):116–122. [DOI] [PubMed] [Google Scholar]
- [104].Vazquez-Chacon CA, Martínez-Guarneros A, Couvin D, et al. Human multidrug-resistant Mycobacterium bovis infection in Mexico. Tuberculosis (Edinb). 2015;95(6):802–809. [DOI] [PubMed] [Google Scholar]
- [105].Juarez-Eusebio DM, Munro-Rojas D, Muñiz-Salazar R, et al. Molecular characterization of multidrug-resistant Mycobacterium tuberculosis isolates from high prevalence tuberculosis states in Mexico. Infect Genet Evol. 2017;55:384–391. [DOI] [PubMed] [Google Scholar]
- [106].Flores-López CA, Zenteno-Cuevas R, Laniado-Laborín R, et al. Molecular epidemiology of Mycobacterium tuberculosis in Baja California, Mexico: a result of human migration? Infect Genet Evol. 2017;55:378–383. [DOI] [PubMed] [Google Scholar]
- [107].Guevara-Méndez A, Perez-Navarro M, Almaraz-Velasco R, et al. A first insight into the genetic diversity of Mycobacterium tuberculosis in Veracruz, Mexico. Int J Mycobacteriol. 2017;6(1):14–20. [DOI] [PubMed] [Google Scholar]
- [108].Rosales S, Pineda-García L, Ghebremichael S, et al. Molecular diversity of Mycobacterium tuberculosis isolates from patients with tuberculosis in Honduras. BMC Microbiol. 2010. 3;10:208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Lanzas F, Karakousis PC, Sacchettini JC, et al. Multidrug-resistant tuberculosis in panama is driven by clonal expansion of a multidrug-resistant Mycobacterium tuberculosis strain related to the KZN extensively drug-resistant m. tuberculosis strain from South Africa. J Clin Microbiol. 2013. October;51(10):3277–3285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Sambrano D, Correa R, Almengor P, et al. Mycobacterium tuberculosis isolates from single outpatient clinic in Panama City exhibit wide genetic diversity. Am J Trop Med Hyg. 2014;91(2):310–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Ferdinand S, Sola C, Verdol B, et al. Molecular characterization and drug resistance patterns of strains of Mycobacterium tuberculosis isolated from patients in an AIDS counseling center in Port-au-Prince, Haiti: A 1-year study. J Clin Microbiol. 2003;41(2):694–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Díaz R, Gómez RI, García N, et al. Molecular epidemiological study on transmission of tuberculosis in a hospital for mentally handicapped patients in Havana, Cuba. J Hosp Infect. 2001;49(1):30–36. [DOI] [PubMed] [Google Scholar]
- [113].Laserson KF, Osorio L, Sheppard JD, et al. Clinical and programmatic mismanagement rather than community outbreak as the cause of chronic, drug-resistant tuberculosis in Buenaventura, Colombia, 1998. Int J Tuberc Lung Dis. 2000;4(7):673–683. [PubMed] [Google Scholar]
- [114].Ferro BE, Nieto LM, Rozo JC, et al. Multidrug-resistant Mycobacterium tuberculosis, southwestern Colombia. Emerg Infect Dis. 2011;17(7):1259–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Cerezo I, Jiménez Y, Hernandez J, et al. A first insight on the population structure of Mycobacterium tuberculosis complex as studied by spoligotyping and MIRU-VNTRs in Bogotá, Colombia. Infect Genet Evol. 2012;12(4):657–663. [DOI] [PubMed] [Google Scholar]
- [116].Nieto LM, Ferro BE, Villegas SL, et al. Characterization of extensively drug-resistant tuberculosis cases from Valle del Cauca, Colombia. J Clin Microbiol. 2012;50(12):4185–4187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Villegas SL, Ferro BE, Perez-Velez CM, et al. High initial multidrug-resistant tuberculosis rate in Buenaventura, Colombia: a public-private initiative. Eur Respir J. 2012;40(6):1569–1572. [DOI] [PubMed] [Google Scholar]
- [118].Realpe T, Correa N, Rozo JC, et al. Population structure among Mycobacterium tuberculosis isolates from pulmonary tuberculosis patients in Colombia. PLoS One. 2014;9(4):e93848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Pino C, Tauch A, Murcia MI. Complete genome sequence of the clinical Beijing-like strain Mycobacterium tuberculosis 323 using the PacBio Real-Time sequencing platform. Genome Announcements (GenomeA). 2015;3(2):3–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Castro C, Ricardo A, Zabaleta A, et al. Caracterización de aislamientos clínicos de Mycobacterium tuberculosis obtenidos de individuos positivos para HIV en Colombia, 2012. Biomedica. 2017;37:86–95. [DOI] [PubMed] [Google Scholar]
- [121].Alvarez N, Hurtado UA, Haft D, et al. Whole-genome sequence of a Beijing extensively drug-resistant Mycobacterium tuberculosis clinical isolate from Buenaventura, Colombia. Genome Announc. 2016. 14;4(1):ii: e01549-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Puerto D, Erazo L, Zabaleta A, et al. Caracterización de aislamientos clínicos de Mycobacterium tuberculosis de pueblos indígenas de Colombia. Biomédica Inst Nac Salud. 2019;39:3. [DOI] [PubMed] [Google Scholar]
- [123].Aristimuño L, Armengol R, Cebollada A, et al. Molecular characterisation of Mycobacterium tuberculosis isolates in the first national survey of anti-tuberculosis drug resistance from Venezuela. BMC Microbiol. 2006;10(6):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Aristimuño L, España M, Guilarte A, et al. Multidrug-resistant Mycobacterium tuberculosis Beijing/W genotype in Venezuela. J Med Microbiol. 2007;56(Pt 12):1707–1708. [DOI] [PubMed] [Google Scholar]
- [125].Sequera CM, Delgado SV, Araque MW, et al. Mycobacterium tuberculosis: espoligotipos en el Estado Carabobo, Venezuela TT - Mycobacterium tuberculosis: spoligotypes in the Carabobo State, Venezuela. Rev Chil Infect. 2008;25(5):362–367. [PubMed] [Google Scholar]
- [126].Abadía E, Sequera M, Ortega D, et al. Mycobacterium tuberculosis ecology in Venezuela: epidemiologic correlates of common spoligotypes and a large clonal cluster defined by MIRU-VNTR-24. BMC Infect Dis. 2009;9(6):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Méndez MV, León C, Escalona A, et al. Evaluación del poder discriminatorio de técnicas de epidemiología molecular en aislados Venezolanos de Mycobacterium tuberculosis. Invest Clin. 2016;57(1):25–37. [PubMed] [Google Scholar]
- [128].de Viedma DG, Chaves F, Inigo J. New Route of importation of Mycobacterium tuberculosis Beijing Genotype. Emerg Infect Dis. 2006;12(1):169–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Iwamoto T, Grandjean L, Arikawa K, et al. Genetic diversity and transmission characteristics of Beijing family strains of Mycobacterium tuberculosis in Peru. PLoS One. 2012;7(11):e49651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Barletta F, Otero L, Collantes J, et al. Genetic variability of Mycobacterium tuberculosis complex in patients with no known risk factors for MDR-TB in the North-eastern part of Lima, Peru. BMC Infect Dis. 2013;13(28):397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Cáceres O, Rastogi N, Bartra C, et al. Characterization of the genetic diversity of extensively-drug resistant Mycobacterium tuberculosis clinical isolates from pulmonary tuberculosis patients in Peru. PLoS One. 2014;9(12):e112789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Barletta F, Otero L, De Jong BC, et al. Predominant Mycobacterium tuberculosis families and high rates of recent transmission among new cases are not associated with primary multidrug resistance in Lima, Peru. J Clin Microbiol. 2015;53(6):1854–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Tarazona D, Galarza M, Levano KS, et al. Análisis genómico comparativo de cepas peruanas de Mycobacterium tuberculosis. Rev Perú Med Exp Salud Publica. 2016;3(2):256–263. [PubMed] [Google Scholar]
- [134].Zelner JL, Murray MB, Becerra MC, et al. Identifying hotspots of multidrug-resistant tuberculosis transmission using spatial and molecular genetic data. J Infect Dis. 2016. 15;213(2):287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Monteserin J, Charchaflie A, Gravina E, et al. Genetic diversity of Mycobacterium tuberculosis in Lima, Peru. 2011; 32nd Annual Congress of the European Society of Mycobacteriology. Conference Paper. [Google Scholar]
- [136].Jiménez P, Calvopiña K, Herrera D, et al. Identification of the Mycobacterium tuberculosis Beijing lineage in Ecuador. Biomedica. 2017;37:2. [DOI] [PubMed] [Google Scholar]
- [137].Monteserin J, Camacho M, Barrera L, et al. Genotypes of Mycobacterium tuberculosis in patients at risk of drug resistance in Bolivia. Infect Genet Evol. 2013;17:195–201. [DOI] [PubMed] [Google Scholar]
- [138].Mancilla EM, Martínez HA, Palavecino BC, et al. Variantes genéticas de Mycobacterium tuberculosis aisladas de pacientes de la Xa Región de Chile. Rev Chil Infectología. 2006;23(3):220–225. [DOI] [PubMed] [Google Scholar]
- [139].Miranda C, Wozniaky A, García P, et al. Presencia del genotipo Beijing entre cepas del complejo Mycobacterium tuberculosis en dos centros de salud de la Región Metropolitana-Chile. Rev Chil Infectología. 2014;31(1):21–27. [DOI] [PubMed] [Google Scholar]
- [140].Balcells ME, García P, Meza P, et al. A first insight on the population structure of Mycobacterium tuberculosis complex as studied by spoligotyping and miru-vntrs in Santiago, Chile. PLoS One. 2015;11;10(2):e0118007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Lagos J, Couvin D, Arata L, et al. Analysis of Mycobacterium tuberculosis genotypic lineage distribution in Chile and neighboring countries. PLoS One. 2016. 12;11(8):e0160434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Candia N, Lopez B, Zozio T, et al. First insight into Mycobacterium tuberculosis genetic diversity in Paraguay. BMC Microbiol. 2007;7(8):75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Morcillo N, Di Giulio B, Chirico C, et al. First description of Mycobacterium tuberculosis Beijing genotype in Argentina. Rev Argent Microbiol. 2005;37(2):92–95. [PubMed] [Google Scholar]
- [144].Gonzalo X, Ambroggi M, Cordova E, et al. Molecular epidemiology of Mycobacterium tuberculosis, Buenos Aires, Argentina. Emerg Infect Dis. 2011;17(3):528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [145].Ritacco V, López B, Ambroggi M, et al. HIV infection and geographically bound transmission of drug-resistant Tuberculosis, Argentina. Emerg Infect Dis. 2012;18(11):1802–1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Monteserin J, Mazzeo E, López B, et al. Genotypic diversity of Mycobacterium tuberculosis in two distinct areas of Argentina. Abstracts/Int J Infec Dis. 2018;73S:3–398. [DOI] [PubMed] [Google Scholar]
- [147].Monteserin J, Paul R, Gravina E, et al. Genotypic diversity of Mycobacterium tuberculosis in Buenos Aires, Argentina. Infect Genet Evol. 2018;62:1–7. [DOI] [PubMed] [Google Scholar]
- [148].Lazzarini LCO, Huard RC, Boechat NL, et al. Discovery of a novel Mycobacterium tuberculosis lineage that is a major cause of tuberculosis in Rio de Janeiro, Brazil. J Clin Microbiol. 2007;45(12):3891–3902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Von Groll A, Martin A, Felix C, et al. Fitness study of the RDRio lineage and Latin American-Mediterranean family of Mycobacterium tuberculosis in the city of Rio Grande, Brazil. FEMS Immunol Med Microbiol. 2010;58(1):119–127. [DOI] [PubMed] [Google Scholar]
- [150].Mendes NH, Melo FA, Santos AC, et al. Characterization of the genetic diversity of Mycobacterium tuberculosis in São Paulo city, Brazil. BMC Res Notes. 2011;4(29):269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Spies FS, Ribeiro AW, Ramos DF, et al. Streptomycin resistance and lineage-specific polymorphisms in Mycobacterium tuberculosis gidB gene. J Clin Microbiol. 2011;49(7):2625–2630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Cardoso Oelemann M, Gomes HM, Willery E, et al. The forest behind the tree: phylogenetic exploration of a dominant Mycobacterium tuberculosis strain lineage from a high tuberculosis burden country. PLoS One. 2011. 25;6(3):e18256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Gomes HM, Elias AR, Oelemann MAC, et al. Spoligotypes of Mycobacterium tuberculosis complex isolates from patients residents of 11 states of Brazil. Infect Genet Evol. 2012;12(4):649–656. [DOI] [PubMed] [Google Scholar]
- [154].Luiz R Dos SS, Suffys P, Barroso EC, et al. Genotyping and drug resistance patterns of Mycobacterium tuberculosis strains observed in a tuberculosis high-burden municipality in Northeast, Brazil. Braz J Infect Dis. 2013;17(3):338–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Costa ERD, Lazzarini LCO, Perizzolo PF, et al. Mycobacterium tuberculosis of the RDRio genotype is the predominant cause of tuberculosis and associated with multidrug resistance in Porto Alegre City, South Brazil. J Clin Microbiol. 2013;51(4):1071–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Martins MC, Giampaglia CMS, Oliveira RS, et al. Population structure and circulating genotypes of drug-sensitive and drug-resistant Mycobacterium tuberculosis clinical isolates in São Paulo state, Brazil. Infect Genet Evol. 2013;14:39–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Vasconcellos SEG, Acosta CC, Gomes LL, et al. Strain classification of Mycobacterium tuberculosis isolates in brazil based on genotypes obtained by spoligotyping, mycobacterial interspersed repetitive unit typing and the presence of large sequence and single nucleotide polymorphism. PLoS One. 2014. 14;9(10):e107747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Gomes LL, Marin MA, Lasunskaia E, et al. Genome comparison of an ancestral isolate and a modern isolate of Mycobacterium tuberculosis of the Beijing lineage from São Paulo, Brazil. Genome Announc. 2015. 1;3(5):ii: e01129-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Diaz R, Kremer K, De Haas PEW, et al. Molecular epidemiology of tuberculosis in Cuba outside of Havana, July 1994-June 1995: utility of spoligotyping versus IS6110 restriction fragment length polymorphism. Int J Tuberc Lung Dis. 1998;2(9):743–750. [PubMed] [Google Scholar]
- [160].Herrera Avila YM, Fonseca Gómez CM, Gozá Valdés R, et al. Tipificación con oligonucleótidos espaciadores de Mycobacterium tuberculosis en Cuba. Spacer oligonucleotide typing Mycobacterium Tuberculosis en Cuba. Rev Cubana Med Trop. 2015;67(1):85–96. [Google Scholar]
- [161].European concerted action on new generation genetic markers and techniques for the epidemiology and control of tuberculosis RIVM, Bilthoven, the Netherlands. Beijing/W genotype Mycobacterium tuberculosis and drug resistance. Emerg Infect Dis. 2006;12(5):736–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Ritacco V, López B, Cafrune PI, et al. Mycobacterium tuberculosis strains of the Beijing genotype are rarely observed in tuberculosis patients in South America. Mem Inst Oswaldo Cruz. 2008;103(5):489–492. [DOI] [PubMed] [Google Scholar]
- [163].Taype CA, Agapito JC, Accinelli RA, et al. Genetic diversity, population structure and drug resistance of Mycobacterium tuberculosis in Peru. Infect Genet Evol. 2012;12(3):577–585. [DOI] [PubMed] [Google Scholar]
- [164].Grandjean L, Iwamoto T, Lithgow A, et al. The Association between Mycobacterium tuberculosis genotype and drug resistance in Peru. PLoS One. 2015;10(5):e0126271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Laserson KF, Osorio L, Sheppard JD, et al. Clinical and programmatic mismanagement rather than community outbreak as the cause of chronic, drug-resistant tuberculosis in Buenaventura, Colombia, 1998. Int J Tuberc Lung Dis. 2000;4(7):673–683. [PubMed] [Google Scholar]
- [166].Puerto D, Erazo L, Zabaleta A, et al. Characterization of clinical isolates of Mycobacterium tuberculosis from indigenous peoples of Colombia. Biomédica. 2019;39(2):78–92. [DOI] [PubMed] [Google Scholar]
- [167].Flores-Treviño S, Morfín-Otero R, Rodríguez-Noriega E, et al. Genetic diversity of Mycobacterium tuberculosis from Guadalajara, Mexico and identification of a rare multidrug resistant Beijing genotype. PLoS One. 2015. 19;10(2):e0118095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Villegas SL, Ferro BE, Perez-Velez CM, et al. High initial multidrug-resistant tuberculosis rate in Buenaventura, Colombia: A public-private initiative. Eur Respir J. 2012;40(6):1569–1572. [DOI] [PubMed] [Google Scholar]
- [169].Pereira CRC. Caracterización de cepas de Mycobacterium tuberculosis a través de la técnica de espoligotipificación. Guatemala: USAC; 2011tesis. [Google Scholar]
- [170].Rosas S, Bravo J, Gonzalez F, et al. High clustering rates of multidrug-resistant Mycobacterium tuberculosis genotypes in Panama. BMC Infect Dis. 2013;13:442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Gallagher J, Muirhead RH, Brain L. Tuberculosis in cattle and badgers. Vet Rec. 1999. 3;145(1):26. [PubMed] [Google Scholar]
- [172].Santos ACB, Gaspareto RM, Viana BHJ, et al. Mycobacterium tuberculosis population structure shift in a 5-year molecular epidemiology surveillance follow-up study in a low endemic agro-industrial setting in São Paulo, Brazil. Int J Mycobacteriol. 2013;2(3):156–165. [DOI] [PubMed] [Google Scholar]
- [173].Polo CL. Prevalencia de la Resistencia a Fármacos de Segunda Línea en una Cohorte de Pacientes con Tuberculosis Farmacorresistente, y Descripción de los Genotipos de Mycobacterium tuberculosis Multirresistentes Circulantes en Colombia, Años 2012 a 2013. Bogotá-Colombia: Pontificia universidad Javeriana, Facultad de Ciencias Básicas; Maestría en Ciencias Biológicas; 2015. [Google Scholar]
- [174].Rodríguez-Castillo JG, Pino C, Niño LF, et al. Comparative genomic analysis of Mycobacterium tuberculosis Beijing-like strains revealed specific genetic variations associated with virulence and drug resistance. Infect Genet Evol. 2017;54:314–323. [DOI] [PubMed] [Google Scholar]
- [175].Puerto G, Erazo L, Wintaco M, et al. Mycobacterium tuberculosis genotypes determined by spoligotyping to be circulating in Colombia between 1999 and 2012 and their possible associations with transmission and susceptibility to first-line drugs. PLoS One. 2015;10(6):e0124308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Saelens JW, Lau-Bonilla D, Moller A, et al. Whole genome sequencing identifies circulating Beijing-lineage Mycobacterium tuberculosis strains in Guatemala and an associated urban outbreak. Tuberculosis (Edinb). 2015;95(6):810–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [177].Zurita J, Espinel N, Barba P, et al. Genetic diversity and drug resistance of Mycobacterium tuberculosis in Ecuador. Int J Tuberc Lung Dis. 2019;23(2):166–173. [DOI] [PubMed] [Google Scholar]
- [178].Sola C, Devallois A, Horgen L, et al. Tuberculosis in the caribbean: using spacer oligonucleotide typing to understand strain origin and transmission. Emerg Infect Dis. 1999;5(3):404–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Brudey K, Filliol I, Ferdinand S, et al. Long-term population-based genotyping study of Mycobacterium tuberculosis complex isolates in the French departments of the Americas. J Clin Microbiol. 2006;44(1):183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Baboolal S, Millet J, Akpaka PE, et al. First insight into Mycobacterium tuberculosis epidemiology and genetic diversity in Trinidad and Tobago. J Clin Microbiol. 2009;47(6):1911–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Millet J, Baboolal S, Streit E, et al. A first assessment of Mycobacterium tuberculosis genetic diversity and drug-resistance patterns in Twelve Caribbean Territories. Biomed Res Int. 2014;2014:11 Article ID 718496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Ecuador FI, Garzon-Chavez D, Zurita J, et al. Prevalence, drug resistance, and genotypic diversity of the Mycobacterium tuberculosis Beijing Family in Ecuador. Microb Drug Resist. 2019;25(6):931–937. [DOI] [PubMed] [Google Scholar]
- [183].Murcia MI, Manotas M, Jiménez YJ, et al. First case of multidrug-resistant tuberculosis caused by a rare “Beijing-like” genotype of Mycobacterium tuberculosis in Bogotá, Colombia. Infect Genet Evol. 2010;10(5):678–681. [DOI] [PubMed] [Google Scholar]
- [184].Weniger T, Krawczyk J, Supply P, et al. MIRU-VNTRplus: A web tool for polyphasic genotyping of Mycobacterium tuberculosis complex bacteria. Nucleic Acids Res. 2010;38(WebServer issue):W326–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.