

Abstract
The synaptic vesicle protein, synaptotagmin, is the principle Ca2+ sensor for synaptic transmission. Ca2+ influx into active nerve terminals is translated into neurotransmitter release by Ca2+ binding to synaptotagmin’s tandem C2 domains, triggering the fast, synchronous fusion of multiple synaptic vesicles. Two hydrophobic residues, shown to mediate Ca2+-dependent membrane insertion of these C2 domains, are required for this process. Previous research suggested that one of its tandem C2 domains (C2B) is critical for fusion, while the other domain (C2A) plays only a facilitatory role. However, the function of the two hydrophobic residues in C2A have not been adequately tested in vivo. Here we show that these two hydrophobic residues are absolutely required for synaptotagmin to trigger vesicle fusion. Using in vivo electrophysiological recording at the Drosophila larval neuromuscular junction, we found that mutation of these two key C2A hydrophobic residues almost completely abolished neurotransmitter release. Significantly, mutation of both hydrophobic residues resulted in more severe deficits than those seen in synaptotagmin null mutants. Thus, we report the most severe phenotype of a C2A mutation to date, demonstrating that the C2A domain is absolutely essential for synaptotagmin’s function as the electrostatic switch.
Author summary
The postulated role of synaptotagmin’s C2A domain in triggering neurotransmitter release has fluctuated wildly over the years. Early biochemical experiments suggested that the C2A domain was essential, while the C2B domain was superfluous. Then, functional experiments measuring neurotransmitter release in vivo following disruptions in Ca2+ binding suggested that C2B was essential, while C2A was superfluous. Subsequently, the use of more refined mutations to disrupt Ca2+ binding indicated that C2A played a facilitatory role. Here we show two hydrophobic residues of the C2A domain are absolutely required for synaptotagmin-triggered neurotransmitter release. Thus, after over twenty years of research, we now demonstrate that the C2A domain of synaptotagmin is an essential component of the Ca2+ sensor for triggering synaptic transmission in vivo.
Introduction
Ca2+ binding by synaptotagmin triggers the fast, synchronous fusion of maximally-primed synaptic vesicles thereby releasing neurotransmitter onto the postsynaptic cell [1, 2]. In the absence of this Ca2+ sensor, evoked release of neurotransmitter is dramatically decreased [3–6]. Synaptotagmin is an integral membrane protein found on synaptic vesicles whose cytosolic domain is composed of two Ca2+-binding C2 domains, C2A and C2B [Fig 1A and 1B, [7]]. One end of each C2 domain contains three loops of amino acids, two of which form a Ca2+-binding pocket: loops 1 and 3 contain five negatively-charged aspartate residues that coordinate Ca2+[8, 9]. In addition, there are two, highly-conserved, hydrophobic residues at the tips of each pocket: one in loop 1, adjacent to the first aspartate residue, and one in loop 3, between the 4th and 5th aspartate residues [Fig 1A,[8, 9]]. Prior to Ca2+ influx, the net negative charge of each Ca2+-binding pocket results in electrostatic repulsion of the negatively-charged presynaptic membrane, preventing fusion. Upon Ca2+ influx, Ca2+ binding to the pockets now results in a net positive charge. Accordingly, the electrostatic repulsion of the presynaptic membrane is changed to electrostatic attraction. Thus, synaptotagmin operates as an electrostatic switch [10–13]. Importantly, this electrostatic attraction now brings the hydrophobic residues located at the tips of the pockets into contact with the membrane. Modeling predicts, and in vitro studies confirm, that these hydrophobic residues insert into lipid bilayers in a Ca2+-dependent manner [14–16], resulting in positive curvature of the membrane that is theorized to promote vesicle fusion [17, 18].
Fig 1. Synaptotagmin structure and C2A mutations.

A, Protein alignment of loops 1 and 3 of the C2A domain of synaptotagmin 1 from Human, Mouse, Rat, and Drosophila (* = Ca2+ binding aspartates, boxes = loop 1 and loop 3 hydrophobic tip residues) B, Crystal structure of synaptotagmin and the SNARE complex showing a postulated role of the C2 domains in triggering fusion, adapted from [19]. Negatively charged residues of the Ca2+ binding pockets are shown as sticks in red, the hydrophobic residues at the tips of these pockets are shown as sticks in gray, and Ca2+ ions are shown as green spheres. VM = vesicle membrane and PM = presynaptic membrane. C, A cartoon depiction of the C2A domain. Colors as in panel B. D, Hydrophilic glutamic acid substitutions are indicated in white. Sequential mutation of C2A hydrophobic tip residues to hydrophilic residues is predicted to increasingly disrupt synaptotagmin’s ability to penetrate, warp and disorder lipids of the presynaptic membrane.
C2A is currently postulated to function as a secondary domain that is merely supportive of C2B, the primary functional domain of synaptotagmin. Importantly, mutation of a hydrophobic tip residue in loop 3 of C2B, which penetrates negatively-charged membranes, is embryonic lethal and causes a decrease in evoked release that is more severe than that seen in sytnull mutants. In comparison, mutating the analogous residue in the C2A domain does not impact viability and only inhibited neurotransmitter release by 50% [19]. However, the functional impact of mutations of the C2A hydrophobic residue in loop 1 has not been studied in vivo, nor has the impact of tandem mutations in both loops 1 and 3 simultaneously.
Since replacing the loop 3 hydrophobic residue in C2B with a large, polar glutamate prevented phospholipid-binding in vitro [19], we made homologous mutations of the loop 1 and loop 3 residues in C2A (Fig 1D). Electrophysiological recordings at the Drosophila neuromuscular junction revealed that mutation at either the loop 1 (this report) or loop 3 site [19] in isolation resulted in an ~50% reduction in evoked transmitter release, again suggesting C2A plays only a facilitatory role. Surprisingly, mutation of both the loop 1 and loop 3 sites simultaneously resulted in an almost complete abolishment of evoked release. This reduction in transmission in the tandem mutation was actually more severe than that observed in synaptotagmin null mutants. The current study establishes that these two hydrophobic residues of C2A, which have been shown to mediate Ca2+-dependent effector interactions in vitro [17, 18, 20, 21], are absolutely required for evoked transmitter release in vivo. Thus, we report the most severe deficits caused by any C2A domain mutation to date and demonstrate that the C2A domain is an essential component for translating synaptotagmin’s electrostatic switch function into vesicle fusion with the presynaptic membrane.
Materials and methods
Drosophila lines
We generated mutants with hydrophobic to hydrophilic substitutions of two residues. A transgenic wild type (P[sytWT]) was used as a positive control across all experiments. For a direct comparison of the level of evoked transmitter release in the most severe mutation, a previously characterized synaptotagmin null line (sytnull) was used as a negative control [22]. Using the Drosophila syt1 coding sequence [7, 23], a wild type control, a M224E, and a M224E/F286E mutant cDNA were synthesized by GeneWiz (South Plainfield, New Jersey) (Fig 1A and 1D). The cDNA was flanked by unique 5’ EcoRI and 3’ BglII restriction sites for directional subcloning into the pUAST-attB vector to place them under the control of the UAS promoter. The transgenes were injected into Drosophila embryos by BestGene (Chino Hills, California) where they were inserted into the attP2 landing site on the third chromosome using the PhiC31 targeted insertion system [24]. These syt 1 transgenes were driven pan-neuronally by the UAS/Gal4 system [25] using the elav promoter [26]. All transgenes were expressed in the absence of endogenous synaptotagmin 1 by crossing them into a synaptotagmin 1 null mutant background, sytAD4 [22, 23]. As no sex selection was employed, both males and females were used across all experiments. This study used the following genotypes: yw; sytAD4elavGal4/ sytAD4; P[UASsyt1WT]/+ (referred to as P[sytWT] or control), yw; sytAD4elavGal4/ sytAD4; P[UASsyt1C2A-M224E]/+ (referred to as P[sytA-ME]), yw; sytAD4elavGal4/ sytAD4; P[UASsyt1 C2A-M224E,F286E]/+ (referred to as P[sytA-ME,FE]) and yw; sytAD4elavGal4/ sytAD4 (referred to as sytnull). All experiments used 3rd instar larvae (L3).
Solutions
HL3.1 saline [70 mM NaCl, 5 mM KCl, 4 mM MgCl2, 10 mM NaHCO3, 5 mM Trehalose, 115 mM sucrose, 5 mM HEPES, pH 7.2 [27]], with the indicated Ca2+ concentrations, was used in all experiments. Phosphate buffered saline (PBS) consisted of [137 mM NaCl, 1.5 mM KH2PO4, 2.7 mM KCl, 8.1 mM Na2HPO4].
Immunoblotting
Synaptotagmin expression levels were determined using western blot analysis with actin levels serving as a loading control. The CNSs of L3s were dissected in HL3.1 saline where the Ca2+ was omitted to decrease vesicle fusion events during dissection. Individual CNSs were placed in Laemmli buffer (Bio-Rad, Hercules, CA) containing 5% β-mercaptoethanol, sonified with five 0.3 sec pulses at 1 Hz using a Branson Sonifier 450 (VWR Scientific, Winchester, PA), and separated by SDS-PAGE with 15% acrylamide. They were then transferred to Immobilon membranes (Millipore, Bedford, MA), and washed in blocking solution [5% milk, 4% normal goat serum (NGS, Fitzgerald Industries International, Acton, MA), 1% bovine serum albumin (BSA, Millipore-Sigma, Burlington, MA), and 0.02% NaN3 in PBS containing 0.05% Tween 20 (PBS-Tween, Fisher BioReagents, Fair Lawn, NJ)]. The membranes were then incubated overnight at 4°C with a 1:2,500 dilution of anti-synaptotagmin antibody, Dsyt-CL1 [2] and 1:10,000 dilution of anti-actin antibody, (MAB 1501, Millipore Bioscience Research Reagents, Billerica, MA) in PBS-Tween containing 10% NGS and 0.02% NaN3, washed in PBS-Tween for 1–3 hours, and probed with secondary antibodies at a 1:5,000 dilution of peroxidase-conjugated AffiniPure Goat Anti-Rabbit IgG (Jackson ImmunoResearch, West Grove, PA) and a 1:5,000 dilution of peroxidase-conjugated AffiniPure Donkey Anti-Mouse IgG (Jackson ImmunoResearch, West Grove, PA) in PBS-Tween containing 10% NGS for 1 hour at room temperature, and washed in PBS-Tween for 30 min. Protein bands were visualized on an Epichemi3 Darkroom with Labworks Imaging Software (UVP BioImaging, Upland, CA). To quantify expression levels within each blot, synaptotagmin/actin ratios were calculated and normalized to the mean synaptotagmin/actin ratio of the transgenic WT control lanes. This permitted comparison of synaptotagmin expression levels between blots. Outliers in loading amount, based on actin levels, were excluded from analysis. The analysis included at least 11 individual CNSs per genotype.
Immunolabeling
The localization of the transgenic synaptotagmin protein was visualized by immunohistochemistry. L3s were dissected in Ca2+-free HL3.1, fixed in PBS containing 4% formaldehyde for 1 hour, incubated with a 1:400 dilution of Dsyt-CL1 in dilution media [PBS with 0.1% Triton (PBST), 1% BSA, and 1% NGS] overnight at 4°C, washed in PBST for 1–3 hours, incubated in dilution media containing a 1:400 dilution of Alexa Fluor 488 goat anti-rabbit antibody (Invitrogen, Carlsbad, CA) for 1 hour at room temperature, washed in PBST for one hour, and mounted on microscope slides in Citifluor (Ted Pella, Redding, CA). Confocal images of the neuromuscular junction on muscle fibers 6 and 7 were taken on a Zeiss 880 light-scanning microscope (Zeiss, White Plains, NY), with a 40x objective and Zeiss Zen 2.1 acquisition software, version 11.0.3.190.
Electrophysiological recording and analyses
Electrophysiological recordings were made with an Axoclamp 2B amplifier (Molecular Devices, Sunnyvale, CA), a Powerlab 4/30 A/D converter (ADInstruments, Sydney, Australia), using LabChart software (ADInstruments, Sydney, Australia). L3s were dissected in Ca2+-free HL3.1 saline to expose the body wall musculature and the CNSs were removed. The saline was then changed to HL3.1 with 1mM Ca2+. Intracellular recordings were made from muscle fiber 6 of abdominal segments 3 and 4 using 10–20 MΩ intracellular electrodes that were pulled using a Sutter model P-97 micropipette puller (Novato, CA) and filled with 3 parts 2 M K3C6H5O7 to 1 part 3 M KCl. The resting potential was held at -65 mV by applying no more than ±1 nA of current. The nerve fiber was stimulated using an A360 stimulus isolator (World Precision Instruments, Sarasota, FL) through a glass suction electrode filled with HL3.1 containing 1mM Ca2+ and broken to have an ~1 micron tip.
Evoked release
Ten excitatory junction potentials (EJPs) were stimulated at 0.04 Hz and averaged for each muscle fiber. Mean responses are reported for 12–14 muscle fibers per genotype.
Spontaneous release
Spontaneous miniature EJPs (mEJPs) were recorded for 3 min prior to any external stimulation. Recordings were blinded and randomized and mEJPs were identified manually. mEJP frequency was determined by counting the number of events that occurred during the second minute of recording. The average amplitude of the first 100 of this population of mEJPs was also calculated. Mean responses are reported for 12–14 muscle fibers per genotype.
Paired pulse
For each muscle fiber, the nerve was stimulated with pairs of pulses having interpulse intervals of 10ms, 20ms, 50ms, and 100ms. For each interpulse interval, five pairs of pulses separated by 5 sec were averaged. Each interpulse interval test was also separated by 5 sec. The amplitude of the first EJP was calculated from the baseline to the first peak. The amplitude of the second EJP was calculated from the trough following the first EJP to the peak of the second EJP. The amplitude of the second EJP was divided by the amplitude of the first EJP to yield a paired pulse ratio (PPR). The mean PPRs of 12–14 fibers per genotype are reported.
Ca2+ curves
Five EJPs evoked at 0.5 Hz were recorded from an individual muscle fiber across at least 3 different Ca2+ concentrations between 0.05 mM and 5 mM (0.05 mM, 0.25 mM, 0.5 mM, 1 mM, 1.5 mM, 2.5 mM, 5 mM) and averaged, yielding a mean EJP amplitude at each Ca2+ level. The first 5 EJPs were always recorded in HL3.1 with 1.5 mM Ca2+ and, following stimulation in at least 2 other Ca2+ levels, 5 more EJPs were recorded in HL3.1 with 1.5 mM Ca2+. Recordings were only considered for analysis if the mean amplitude of the final EJPs in 1.5 mM Ca2+ was ≥ 90% of the initial mean EJP amplitude. Mean responses are reported for at least 12 muscle fibers per Ca2+ concentration. Lines of best fit were calculated using a nonlinear regression analysis, which provided EC50s and their 95% confidence intervals for each genotype. Each response level was normalized to the maximum value predicted by the line of best fit. Additionally, the hillslope for each curve was calculated to compare cooperativity of Ca2+-dependent release.
Experimental design and statistical analysis
Statistical analyses were performed using Prism 8 (GraphPad software, La Jolla, CA). All datasets included a minimum of 11 samples per genotype. In all electrophysiological experiments, recordings of mutants and controls were interspersed. Direct comparisons were only made between recordings done within the same time period, as absolute responses can be impacted by minor variations in solutions. All experiments requiring manual analysis of events had the genotypes blinded to the researcher. For comparison of two genotypes with Gaussian distributions of their datasets, we used unpaired student’s t-tests. For comparisons of three genotypes with Gaussian distribution of their datasets, we used one-way ANOVAs with Tukey post hoc tests of multiple comparisons to determine significance between all three genotypes. In the paired pulse experiments, data were analyzed with a repeated measures two-way ANOVA with Tukey post hoc tests. When datasets showed non-gaussian distributions, Kruskal-Wallis tests were used to compare the 3 genotypes, with Dunn’s post hoc tests of multiple comparisons. An alpha p-value of 0.05 was considered significant for all of the above tests. To compare Ca2+ curves, a line of best fit was determined using a nonlinear regression model and the 95% confidence intervals of the EC50 of each genotype were compared. If the confidence intervals didn’t overlap, the genotypes were considered significantly different.
Results
Synaptotagmin transgenes
P[sytWT], P[sytA-ME], and P[sytA-ME,FE]
Mutation of the hydrophobic residue at the tip of loop 3 of the C2A Ca2+-binding pocket inhibits evoked release by 50% [19], yet the in vivo function of the hydrophobic residue at the tip of loop 1 is unknown (see Fig 1A, Loop 1, M). Since both of these hydrophobic residues have been shown to mediate Ca2+-dependent interactions in vitro [16, 18, 20], we tested whether the loop 1 hydrophobic residue is required for efficient neurotransmitter release. We generated two lines containing mutations of this loop 1 methionine. In the first, only the loop 1 methionine was mutated to a hydrophilic glutamic acid (Fig 1D, P[sytA-ME]). In the second, both the loop 1 and loop 3 hydrophobic tip residues of C2A were mutated to glutamic acids (Fig 1D, P[sytA-ME,FE]). In all experiments, transgenic synaptotagmin was expressed in the sytnull background such that the only source of synaptotagmin 1 was from the transgene [22].
C2A hydrophobic residues are required for synaptotagmin function
Ca2+-evoked neurotransmitter release in the single P[sytA-ME] mutant was decreased to a similar extent as that seen previously in the single P[sytA-FE] mutant [19]. Electrophysiological recording of excitatory junction potentials (EJPs) from larval muscle fibers revealed an ~50% decrease in EJP amplitude in P[sytA-ME] compared to P[sytWT] (Fig 2A and 2B). EJP amplitude in P[sytWT] was 30.8 ± 1.8 mV (mean ± SEM, n = 12). Whereas in P[sytA-ME], it was significantly reduced at only 14.6 ± 1.3 mV (mean ± SEM, n = 14; one-way ANOVA F(2,37) = 137.7, p < 0.0001, Tukey post hoc p < 0.0001). This partial block of fast, synchronous neurotransmitter release is consistent with the idea that C2A plays only a facilitatory role in synaptotagmin function [19, 28, 29].
Fig 2. Mutation of the hydrophobic tip residues disrupts evoked transmitter release.
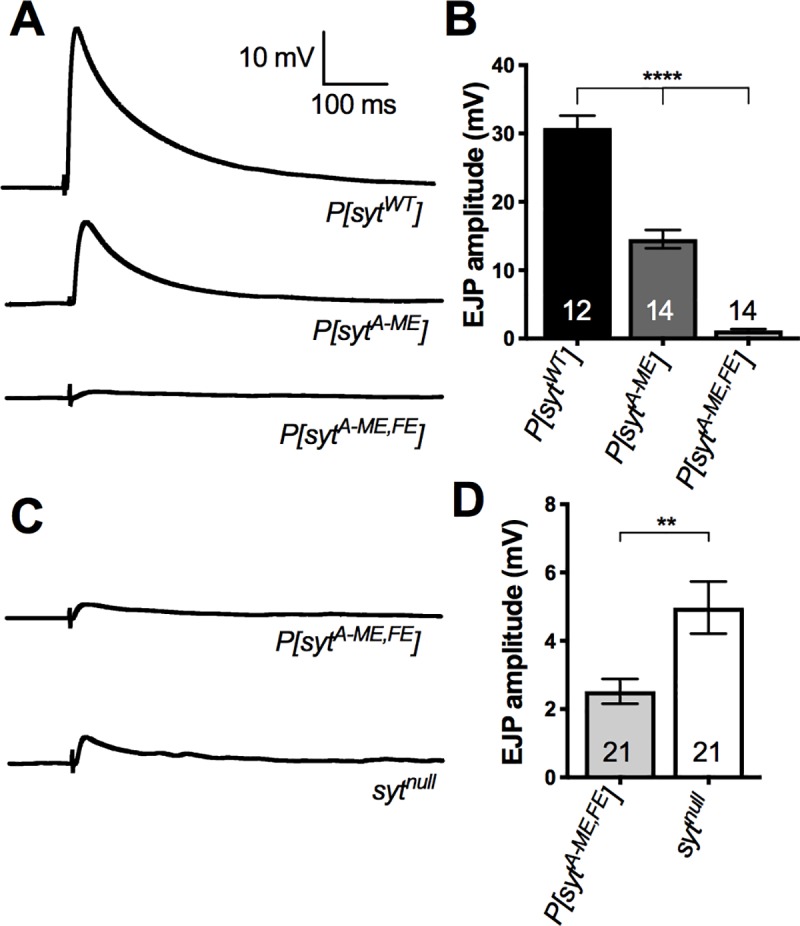
The single hydrophobic mutation decreased neurotransmitter release by 50% while the double mutation inhibited release to a greater extent than that seen in sytnull mutants. A, Representative traces of EJPs for P[sytWT], P[sytA-ME], and P[sytA-ME,FE]. B, Mean EJP amplitude ± SEM for P[sytWT], P[sytA-ME], and P[sytA-ME,FE] (Tukey multiple comparisons, p < 0.0001 = ****). C, Representative traces of EJPs for P[sytA-ME,FE] and sytnull. D, Mean EJP amplitude ± SEM for P[sytA-ME,FE] and sytnull (Tukey multiple comparisons, p < 0.01 = **).
Surprisingly, the double mutant, P[sytA-ME,FE], nearly abolished Ca2+-evoked neurotransmitter release (Fig 2A and 2B). EJP amplitude in P[sytA-ME,FE] mutants was only 1.2 ± 0.2 mV (mean ± SEM, n = 14). Since the double mutant showed such a dramatic decrease in EJP amplitude compared to both P[sytWT] and P[sytA-ME] (one-way ANOVA F(2,37) = 137.7, p < 0.0001, Tukey post hoc p < 0.0001), we also compared evoked transmitter release in P[sytA-ME,FE] mutants and sytnull larvae, which express no synaptotagmin [22]. Importantly, we found that the P[sytA-ME,FE] double mutant had a significantly decreased EJP amplitude even when compared to sytnull larvae (Fig 2C and 2D; mean ± SEM: P[sytA-ME,FE] = 2.5 ± 0.4 mV, n = 21 and sytnull = 5 ± 0.8 mV, n = 21; unpaired t-test t(40) = 2.896, p = 0.0061). Thus, residues required for Ca2+-dependent membrane penetration by both C2 domains are absolutely required for synaptotagmin to function as the Ca2+ sensor for fast, synchronous neurotransmitter release. While we’ve known for many years the critical nature of the C2B domain for triggering neurotransmitter release [2, 4, 19, 30, 31], no previous C2A mutation has resulted in synaptic deficits more severe than those in sytnull mutants in vivo.
Expression and localization of transgenic synaptotagmin
The deficits in evoked release are not a result of mis-expression or mis-localization of the transgenic proteins. Western analysis was performed on single CNSs from larvae using our anti-synaptotagmin antibody [DsytCL1, [2]] and an anti-actin antibody (MAB 1501) as a loading control. Synaptotagmin expression in P[sytA-ME] and in P[sytA-ME,FE] was 114.4 ± 24.0% and 78.7 ± 17.6% (respectively, mean ± SEM, n = 13 and n = 11) of that in P[sytWT] (n = 13). There were no significant differences in transgenic synaptotagmin expression levels between the control and either mutant line (Fig 3A and 3B, one-way ANOVA F(2,34) = 0.8165, p = 0.4505). Immunohistochemical labeling of the larval body wall musculature with anti-synaptotagmin antibody was used as a non-quantitative measure of protein localization, which demonstrated that the transgenic synaptotagmin was highly concentrated in synaptic boutons at the neuromuscular junction in both transgenic mutants and the transgenic WT control (Fig 3C). Thus, the transgenic protein was appropriately targeted to synaptic sites.
Fig 3. Expression and localization of synaptotagmin are unaffected by hydrophobic mutations.
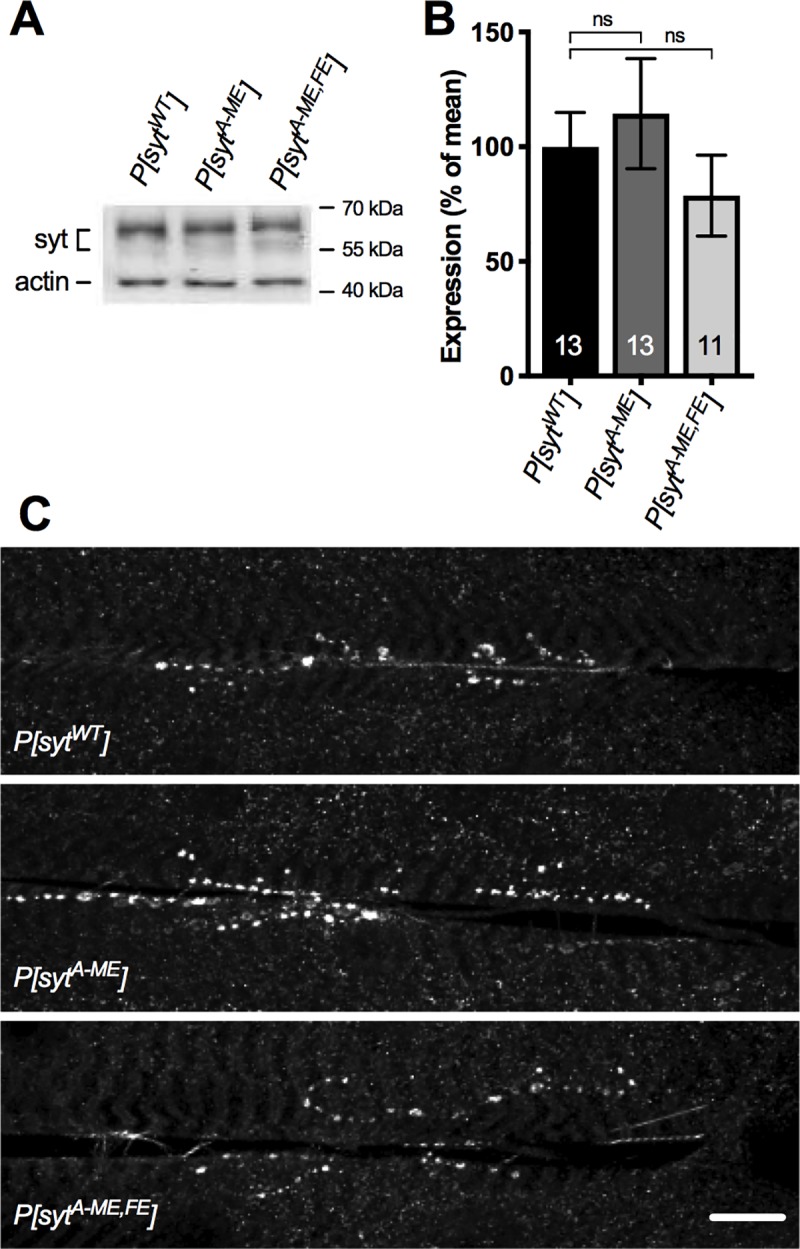
A, Representative western blots of transgenic synaptotagmin expression levels with actin as a loading control. B, Mean protein expression levels ± SEM, normalized to actin (One-way ANOVA, no significant differences). C, Representative confocal images of larval neuromuscular junctions labeled with anti-synaptotagmin antibodies (scale bar = 20 μm). Synaptotagmin is appropriately concentrated at synaptic sites in all three genotypes.
C2A hydrophobic mutations result in decreased release probability
A decrease in evoked transmitter release could result from a decrease in vesicular loading of neurotransmitter which would decrease quantal size and/or a decrease in release probability. The amplitude of spontaneous single vesicle fusion events, mEJPs, at the neuromuscular junction provides a convenient indication of quantal size, assuming there’s no disruption in postsynaptic responsiveness. The amplitude of these single vesicle fusion events was unchanged between the mutants and the control (Fig 4A and 4B; mean ± SEM: P[sytWT] = 1.1 ± 0.06 mV n = 20, P[sytA-ME] = 1.2 ± 0.08 mV n = 16, P[sytA-ME,FE] = 0.9 ± 0.05 mV n = 18; Kruskal-Wallis test H(2) = 7.61, p = 0.0222, Dunn’s multiple comparisons test, P[sytWT] vs P[sytA-ME] p > 0.9999, P[sytWT] vs P[sytA-ME,FE] p = 0.07). The lack of any difference between the control and either mutant is consistent with both vesicular loading and postsynaptic responsiveness being unchanged. Several synaptotagmin mutations result in an increase in the frequency of spontaneous fusion events [5, 32]. Therefore, we analyzed mEJP frequency and found no change between the mutants and the control (Fig 4A and 4C; mean ± SEM: P[sytWT] = 3.9 ± 0.5 Hz n = 20, P[sytA-ME] = 3.7 ± 0.3 Hz n = 16, P[sytA-ME,FE] = 3.9 ± 0.3 Hz n = 18; one-way ANOVA F(2, 51) = 0.1247, p = 0.8806). Thus, neither hydrophobic mutation caused a significant change in either the quantal size or the rate of spontaneous fusion events.
Fig 4. Spontaneous events are unaffected by hydrophobic mutations.

A, Representative traces of mEJPs for P[sytWT], P[sytA-ME], and P[sytA-ME,FE]. B, Mean mEJP amplitudes ± SEM (Kruskal Wallis test with Dunn’s multiple comparison test, no significance = ns). C, Mean mEJP frequency ± SEM (Kruskal Wallis test with Dunn’s multiple comparison test, no significance = ns).
While changes in quantal size cannot account for the differences in EJP amplitude we observed, another possibility is that the hydrophobic mutations decrease the probability of release. Since the ratio of two paired pulses (paired pulse ratio, PPR) is inversely correlated to release probability [33], we conducted a paired pulse analysis. The PPRs for P[sytA-ME] and P[sytA-ME,FE] were significantly different from control across all interpulse intervals (mean ± SEM. P[sytWT] n = 13: 10 ms = 0.6 ± 0.05, 20 ms = 0.7 ± 0.04, 50 ms = 0.9 ± 0.03, 100 ms = 0.9 ± 0.01. P[sytA-ME] n = 14: 10 ms = 1.1 ± 0.1, 20 ms = 1.0 ± 0.08, 50 ms = 1.1 ± 0.06, 100 ms = 1.1 ± 0.04. P[sytA-ME,FE] n = 12: 10 ms = 2.7 ± 0.3, 20 ms = 1.8 ± 0.2, 50 ms = 1.6 ± 0.1, 100 ms = 1.5 ± 0.2. Two-way repeated measures ANOVA, F (36, 108) = 2.190 p = 0.001; Tukey post hoc–P[sytWT] vs P[sytA-ME]: 10 ms p = 0.0005, 20 ms p = 0.01, 50 ms p = 0.007, 100 ms p = 0.0003. P[sytWT] vs P[sytA-ME,FE]: 10 ms p < 0.0001, 20 ms p = 0.0001, 50 ms p = 0.0008, 100 ms p = 0.04). Furthermore, the PPR of the double mutant was significantly greater than that of the single mutant for every interpulse interval, except 100 ms (Fig 5A and 5B P[sytA-ME] vs P[sytA-ME,FE], Tukey post hoc–P[sytA-ME] vs P[sytA-ME,FE]: 10 ms p = 0.0003, 20 ms p = 0.003, 50 ms p = 0.01, 100 ms p = 0.2). These results indicate that a decreased release probability could account for the decrease in neurotransmitter release, which is further bolstered by the finding that the release probability scales inversely with the severity of the mutation.
Fig 5. Probability of release is decreased by hydrophobic mutations.

Probability of release was determined using a paired pulse protocol with interpulse intervals of 10, 20, 50, and 100ms. A, Representative traces of paired EJPs for P[sytWT], P[sytA-ME], and P[sytA-ME,FE] with a 20ms interpulse interval. B, Mean paired pulse ratios ± SEM for P[sytWT], P[sytA-ME], and P[sytA-ME,FE] (Two-way Repeated Measures ANOVA with Tukey post-hoc, p < 0.05 = *, p < 0.01 = **, p < 0.001 = ***, p < 0.0001 = ****). Indicated differences are between mutants and P[sytWT], though the paired pulse ratio was significantly different (p < 0.05) between P[sytA-ME], and P[sytA-ME,FE], for all interpulse intervals except 100ms.
C2A hydrophobic mutations decrease the apparent Ca2+ affinity of release
Fast, synchronous neurotransmitter release is a Ca2+-dependent, cooperative process [34]. In order to test whether the C2A hydrophobic mutations affect the Ca2+ dependence of synaptotagmin-triggered release, we recorded EJPs at a variety of extracellular Ca2+ concentrations, ranging from 0.05 to 5 mM. In our dose-response curve, the intermediate decrease of EJP amplitude in P[sytA-ME] mutants and the dramatic decrease of EJP amplitude in P[sytA-ME,FE] mutants compared to control are evident at all Ca2+ concentrations (Fig 6A). We used nonlinear regression to fit curves to the data (Fig 6A solid curves). To determine whether these hydrophobic mutations impact the cooperativity of release, we compared the Hill coefficient, calculated from the equations of the curve for each genotype, and found no significant differences (Hill slope: P[sytWT] = 1.8, P[sytA-ME] = 1.8, P[sytA-ME,FE] = 1.8). Thus, mutations of these C2A hydrophobic residues do not impact the cooperativity of Ca2+-dependent neurotransmitter release.
Fig 6. The apparent Ca2+ affinity of release is decreased by hydrophobic mutations.
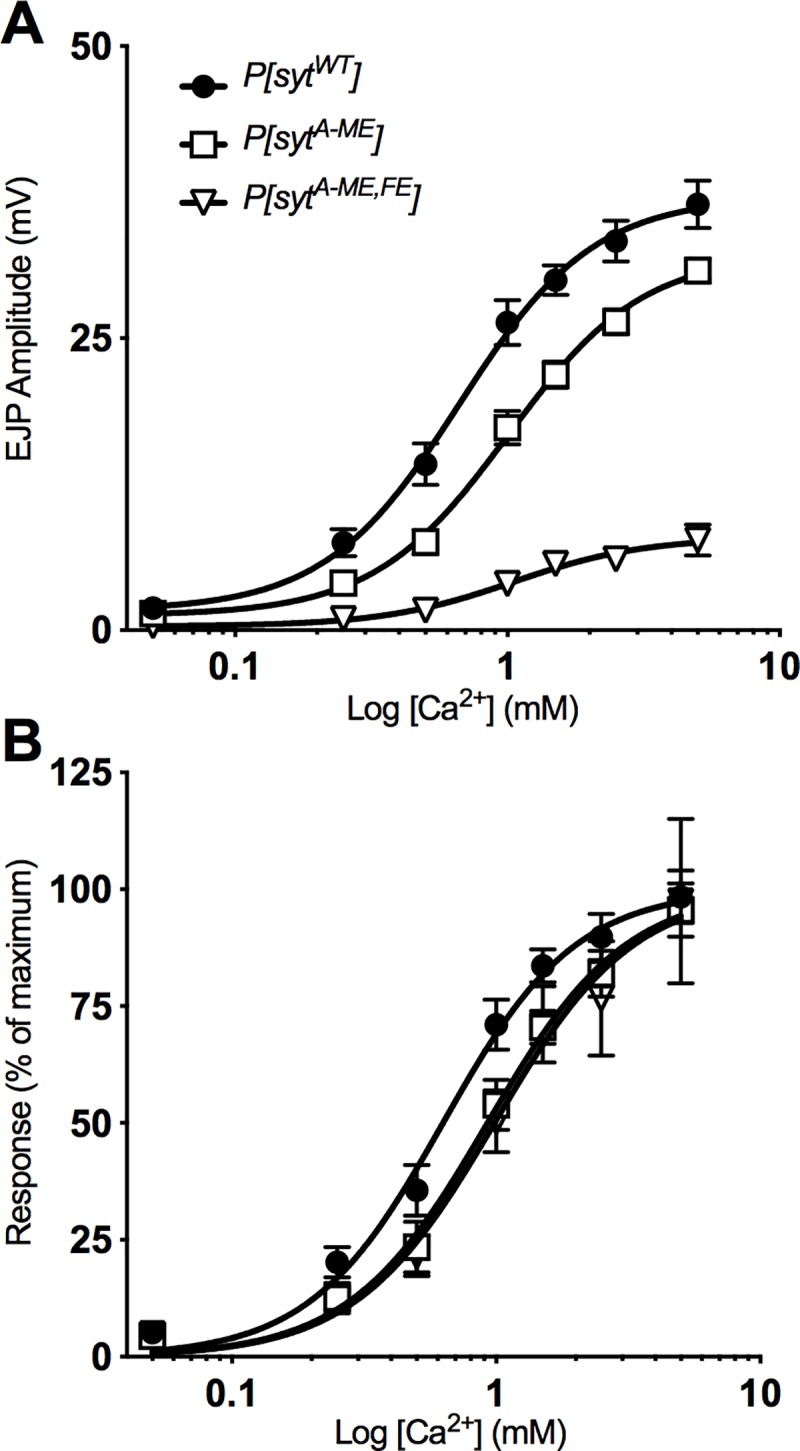
A, Mean EJP amplitude ± SEM across a range of Ca2+ concentrations fit with a nonlinear regression. B, Ca2+ curves normalized to maximum EJP amplitude predicted by the nonlinear regression. The significant rightward shift in the curve (EC50, non overlapping confidence intervals) indicates a decrease in the apparent Ca2+ affinity of neurotransmitter release.
To assess the apparent Ca2+ affinity of release, the response at each Ca2+ level was normalized to the maximal response predicted by the non-linear regression equation for each genotype. Consistent with the decrease in Ca2+ affinity for phospholipid binding mutants reported previously [35], Fig 6B displays a similar shift in the curves for the Ca2+ dependence of neurotransmitter release. (EC50, 95%CI: P[sytWT] = 0.6 mM, 0.5–0.7 mM, n = 13; P[sytA-ME] = 0.9 mM, 0.8–1.0 mM, n = 12; P[sytA-ME,FE] = 1.0 mM, 0.8–1.2 mM, n = 12; non-overlapping confidence intervals compared to control). The rightward shift of the Ca2+ curves demonstrates that both of the C2A hydrophobic mutants caused a decrease in the apparent Ca2+ affinity of release.
Discussion
We investigated the role of two hydrophobic residues in the C2A domain of synaptotagmin in neurotransmitter release. Mutation of the C2A loop 1 hydrophobic residue (sytA-ME) resulted in a 50% reduction of evoked release (Fig 2B), consistent with previous findings for the loop 3 hydrophobic residue (sytA-FE, [19]). Notably, mutation of both of these hydrophobic residues in tandem (sytA-ME,FE) nearly abolished the evoked response (Fig 2B). These deficits could result from the decreased release probability (Fig 5). Analysis of spontaneous release suggests that neither vesicle loading (mEJP amplitude) nor frequency of events (mEJP frequency) played a role in the observed deficits (Fig 4). Evoked responses at varying Ca2+ levels revealed that the apparent Ca2+ affinity of release was decreased by either our single or double hydrophobic residue mutations in C2A, but the Ca2+ cooperativity of release was unaffected (Fig 6). Importantly, the double mutation decreased evoked release significantly more than the complete absence of synaptotagmin 1 (Fig 2D), making it the most severe mutation of the C2A domain of synaptotagmin to date.
Previous in vivo and in culture analyses of C2A and C2B domain function suggested that the C2B domain was essential, while C2A played a secondary role. C2B mutations resulted in dominant negative effects and lethality, while C2A mutations were viable and only decreased release by a maximum of 50–80%. Specifically, mutations disrupting Ca2+ binding resulted in: a dominant-negative effect or lethality in C2B [2, 30], but only a 0–80% decrease in evoked release in C2A [10, 28, 29, 36]. Mutations that altered the polylysine motif resulted in: an ~40–50% decrease in evoked transmitter release in C2B [31, 37–39], yet did not impair evoked release in C2A [40]. Similarly, mutation of a loop 3 positively-charged residue involved in electrostatic interactions with membranes resulted in: a 60–80% decrease in evoked release in C2B [41, 42], see however [39], and only an ~45–55% decrease in C2A [39, 41–43]. Importantly, mutation of the loop 3 hydrophobic residue resulted in: embryonic lethality in C2B, but only a 50% decrease in evoked release in C2A [19]. Taken together, these results led to the understanding that C2B was critical, while C2A only acted in a facilitatory manner.
The current study challenges this longstanding idea regarding the significance of the C2A domain. The lethality caused by mutation of the C2B loop 3 hydrophobic residue is still the most severe synaptotagmin phenotype to date [19] and demonstrates the predominant role of the C2B domain in vivo. Here we report that simultaneous mutation of both the loop 1 and loop 3 hydrophobic tip residues to negatively-charged glutamates in C2A resulted in the most dramatic deficit ever observed for a C2A domain mutation. This could be the result of either removal of hydrophobicity or increased electrostatic repulsion of the negatively charged membrane. However, previous work found that mutation of the loop 3 hydrophobic tip residue in C2A to either a hydrophilic, non-charged tyrosine or a hydrophilic, negatively-charged glutamate had an equal inhibitory impact on evoked transmitter release. Both decreased release by ~50% [19]. Thus, the net charge at the loop 3 hydrophobic site was inconsequential. Rather, the hydrophilic nature of the mutation correlated with the disruption of function [19]. As such, we chose to use glutamate substitutions at both hydrophobic sites in C2A for the current study. While the C2A double glutamate mutant is still viable, this is the first C2A mutation to result in less neurotransmitter release than that observed in sytnull larvae. Thus, the hydrophobic tip residues of both domains are essential for synaptotagmin-triggered vesicle fusion. This demonstrates for the first time that C2A plays more than a facilitatory role; it is absolutely required for synaptotagmin to function as the Ca2+ sensor for fast, synchronous neurotransmitter release.
The mechanism(s) by which these hydrophobic residues exert their effects has been extensively studied in vitro. These include SNARE interactions with, and membrane penetration by, synaptotagmin. The impact of loop 1 and loop 3 hydrophobic residue mutations on SNARE interactions are controversial, with some studies indicating mutation of the hydrophobic residues has an impact on SNARE binding [18, 19] while others indicate that there is no impact on SNARE binding [21]. However, even the studies reporting decreased t-SNARE binding in mutants with decreased hydrophobicity demonstrated that the changes in synaptotagmin’s membrane interactions, and NOT the changes in t-SNARE binding, correlated with the changes in synaptotagmin function in vivo [19]. Furthermore, the experiments that showed decreased Ca2+-dependent interactions between t-SNAREs and hydrophobic synaptotagmin mutants required membrane-embedded t-SNAREs [18, 19]. Thus, the apparent decrease in Ca2+-dependent SNARE interactions may actually be a result of disrupting membrane interactions.
There is, however, ample evidence supporting a role for the hydrophobic tip residues in membrane penetration. The loop 1 and loop 3 hydrophobic residues of both C2 domains insert into membranes following Ca2+ binding, implicating membrane penetration, and resultant membrane bending and lipid disorder, as important downstream effector interactions of Ca2+ binding [15, 20]. In vitro membrane tubulation and liposome fusion assays required Ca2+-dependent synaptotagmin C2 domain insertion into membranes [15, 18]. Mutations that prevented membrane penetration blocked liposome fusion and tubulation [17]. In cultured PC12 cells, these same mutations prevented vesicular cargo release [21]. Mutations that enhanced membrane penetration had the opposite effect [17, 21] and also increased the apparent Ca2+ affinity of neurotransmitter release at hippocampal autapses [44]. These findings indicate that the ability of the loop 1 and 3 hydrophobic residues to insert into the membrane is crucial to synaptotagmin’s role in driving vesicle fusion.
Interestingly, each C2 domain seemed to play equal roles in vitro, with C2B even depending on the presence of C2A to insert into membranes or bind liposomes [16]. Furthermore, in isolated C2A domains, single residue substitutions of the loop 1 or loop 3 hydrophobic residues that blocked membrane penetration had summative effects in decreasing the Ca2+ dependence of liposome binding, while the double mutant effectively prevented liposome binding [35]. Thus, in vitro evidence suggested balanced roles for the C2A and C2B domains with regard to membrane penetration. Our current finding, that the hydrophobic residues in both domains are essential for synaptotagmin-triggered neurotransmitter release, is more consistent with the in vitro studies. However, disruption of C2B function in vivo is still more severe than that of C2A: 1) mutation of the loop 3 hydrophobic residue in C2B results in dominant negative lethality, while mutation of both the loop 1 and loop 3 hydrophobic residues in C2A does not ([19], and this study); and 2) mutations of Ca2+-binding residues in C2B also result in dominant negative phenotypes, while those in C2A do not [2, 10, 29]. The predominance of the C2B domain for synaptotagmin function in vivo could be due to its direct interactions with the SNARE complex [21, 31, 37, 40] or due to its greater ability to induce membrane bending [17, 18, 21].
For both C2A and C2B, mutation of the hydrophobic residues required for Ca2+-dependent membrane penetration resulted in a more severe phenotype than mutation of the Ca2+ binding residues themselves [2, 10, 29]. This combination of findings indicates that: 1) the hydrophobic residues mediate a key effector interaction(s), and 2) the reported mutations of Ca2+ binding residues must not completely block these downstream interactions. For example, the sytA-ME,FE hydrophobic mutation abolished all synaptotagmin-triggered neurotransmitter release (Fig 2D). Yet the mutation of the second of the five Ca2+ binding aspartates to a glutamate (sytA-D2E), which completely blocked Ca2+ binding by C2A in vitro, supported some (20%) synaptotagmin-triggered Ca2+-evoked release in vivo [10]. This combination of effects suggests that in the P[sytA-D2E] mutant, the intact C2B domain may be able to place the C2A pocket close enough to membranes for some hydrophobic interactions to occur, and facilitate a small amount of synaptotagmin-triggered fusion, despite the electrostatic repulsion by C2A. While in the P[sytA-ME,FE] mutant, the required downstream hydrophobic interactions by C2A are completely blocked and no synaptotagmin-triggered release occurs in response to Ca2+ influx. For C2B, the loop 3 hydrophobic mutant resulted in less Ca2+-evoked release than seen in sytnull mutants, no change in spontaneous release, and embryonic lethality ([19] and unpublished observations). On the other hand, mutation of Ca2+ binding aspartates in C2B to neutral asparagines (sytB-DN) resulted in less evoked release than in sytnull mutants, but some could survive to larval or even adult stages, and there was an increase in spontaneous transmitter release [2]. No synaptotagmin-triggered, Ca2+-evoked release was possible in either case. However, the decreased electrostatic repulsion of the presynaptic membrane in the P[sytB-DN] mutants could have permitted membrane interactions by the hydrophobic residues resulting in increased spontaneous neurotransmitter release. Since these mutants were viable to a later stage than the loop 3 hydrophobic mutants, this combination of effects suggests that the increase in spontaneous release in the P[sytB-DN] mutants was beneficial to the organism. Thus, the increased severity of the hydrophobic mutations in both C2 domains, compared to the Ca2+ binding mutations reported to date, suggests that the interactions mediated by the hydrophobic tip residues are absolutely essential for synaptotagmin function, are downstream of Ca2+ binding, and still occur to some degree in Ca2+ binding mutants.
Our results, coupled with previous in vitro and in vivo findings, are consistent with the idea that membrane insertion by both C2 domains is a primary downstream effector interaction in synaptotagmin’s transduction of Ca2+ influx into vesicle fusion. Prior to Ca2+ influx, the negative charge of both Ca2+-binding pockets provides electrostatic repulsion of the negatively-charged presynaptic membrane (Fig 1). After Ca2+ entry, Ca2+ binding results in a net positive charge at the tip of each C2 domain which then attracts the presynaptic membrane–flipping the electrostatic switch [11, 12]. The hydrophobic residues at the tips of each C2 domain are then able to insert into the hydrophobic core of the presynaptic membrane [14, 20, 45]. This insertion has been shown to result in positive curvature of the target membrane [17, 18] and disruption of phospholipid order [46], which could provide the final energy required to drive fusion of the vesicle membrane with the presynaptic membrane. A certain threshold of lipid destabilization could be required for fusion, and inhibiting penetration by C2A may lower membrane destabilization below this threshold.
A recent study [47] suggests a mechanism whereby the membrane disruption resulting from synaptotagmin penetration could directly promote SNARE-mediated vesicle fusion. Synaptotagmin penetration is proposed to disorder lipids near the transmembrane domain of syntaxin allowing the bent linker in the juxtamembrane region to straighten, thereby facilitating the transition from trans- to cis-SNAREs required for driving fusion. Synaptotagmin-triggered membrane curvature [17, 18] could augment this transition. Assuming a certain threshold of lipid disorder is necessary for SNAREs to alter their conformation, partial disruption of the membrane in our P[sytA-ME] mutant or in the previously studied P[sytA-FE] mutant [19] may allow only a subset of SNARE complexes to fully straighten leading to the observed 50% reduction in vesicle fusion. However, our double mutant (P[sytA-ME,FE]), which should prevent any insertion by C2A, would not contribute to the membrane disorder necessary for conformational change in the SNARE complex, thereby preventing fusion.
A remaining question is how do synaptotagmin 1 mutants result in less fast, synchronous fusion than that seen in the absence of the wild type protein (sytnull)? In these cases, the presence of the mutant protein must actively inhibit the residual neurotransmitter release remaining in sytnull mutants (Fig 2D and [2, 19]). The finding that the Ca2+ dependence of evoked release in sytnull mutants is the same as in wild type (no change in EC50 [2]) is consistent with the idea that another isoform of synaptotagmin could be the trigger. However, the only other synaptotagmin isoform expressed at the neuromuscular junction in Drosophila is synaptotagmin IV and it is concentrated in the postsynaptic cell [48]. Whether there is any synaptotagmin IV in the presynaptic terminal or if its expression level is impacted in sytnull mutants is unknown. In short, the Ca2+ sensor that triggers the residual vesicle fusion in sytnull mutants remains to be determined.
Regardless of the mechanism of action, our characterization of the most severe C2A domain mutation to date challenges the predominant model of synaptotagmin function. While all previous in vivo evidence suggested that C2A acted only as a facilitatory domain, disrupting the hydrophobicity of the loop 1 and loop 3 Ca2+-binding pocket residues of C2A (Fig 1) decreased evoked neurotransmitter release to levels less than that in sytnull larvae (Fig 2). These findings are consistent with in vitro evidence indicating important roles for these hydrophobic tip residues in membrane penetration, and potential interactions with membrane embedded SNARE proteins. Our findings now demonstrate that, in contrast to the current view, the C2A domain is absolutely required for synaptotagmin to trigger fast, synchronous vesicle fusion.
Supporting information
(PDF)
Acknowledgments
Special thanks to Dr. Ann Hess, Colorado State University, Dept of Statistics.
Data Availability
All data files associated with this paper are publicly available from the Mountain Scholar database at https://hdl.handle.net/10217/199886 and/or https://doi.org/10.25675/10217/199886.
Funding Statement
NER - IOS-1257363 from the National Science Foundation. www.nsf.gov. NER – CRC from the College of Veterinary Medicine and Biomedical Sciences award. https://vetmedbiosci.colostate.edu. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1.Brose N, Petrenko AG, Südhof TC, Jahn R. Synaptotagmin: a calcium sensor on the synaptic vesicle surface. Science. 1992;256(5059):1021–5. 10.1126/science.1589771 [DOI] [PubMed] [Google Scholar]
- 2.Mackler JM, Drummond JA, Loewen CA, Robinson IM, Reist NE. The C(2)B Ca(2+)-binding motif of synaptotagmin is required for synaptic transmission in vivo. Nature. 2002;418(6895):340–4. Epub 2002/07/12. 10.1038/nature00846 . [DOI] [PubMed] [Google Scholar]
- 3.Broadie K, Bellen HJ, DiAntonio A, Littleton JT, Schwarz TL. Absence of synaptotagmin disrupts excitation-secretion coupling during synaptic transmission. Proc Natl Acad Sci U S A. 1994;91(22):10727–31. 10.1073/pnas.91.22.10727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Geppert M, Goda Y, Hammer RE, Li C, Rosahl TW, Stevens CF, et al. Synaptotagmin I: a major Ca2+ sensor for transmitter release at a central synapse. Cell. 1994;79(4):717–27. 10.1016/0092-8674(94)90556-8 [DOI] [PubMed] [Google Scholar]
- 5.DiAntonio A, Schwarz TL. The effect on synaptic physiology of synaptotagmin mutations in Drosophila. Neuron. 1994;12(4):909–20. Epub 1994/04/01. 10.1016/0896-6273(94)90342-5 . [DOI] [PubMed] [Google Scholar]
- 6.Littleton JT, Stern M, Schulze K, Perin M, Bellen HJ. Mutational analysis of Drosophila synaptotagmin demonstrates its essential role in Ca(2+)-activated neurotransmitter release. Cell. 1993;74(6):1125–34. Epub 1993/09/24. 10.1016/0092-8674(93)90733-7 . [DOI] [PubMed] [Google Scholar]
- 7.Perin MS, Johnston PA, Ozcelik T, Jahn R, Francke U, Sudhof TC. Structural and functional conservation of synaptotagmin (p65) in Drosophila and humans. The Journal of biological chemistry. 1991;266(1):615–22. Epub 1991/01/05. . [PubMed] [Google Scholar]
- 8.Fernandez I, Arac D, Ubach J, Gerber SH, Shin O, Gao Y, et al. Three-dimensional structure of the synaptotagmin 1 C2B-domain: synaptotagmin 1 as a phospholipid binding machine. Neuron. 2001;32(6):1057–69. Epub 2002/01/05. 10.1016/s0896-6273(01)00548-7 . [DOI] [PubMed] [Google Scholar]
- 9.Sutton RB, Ernst JA, Brunger AT. Crystal structure of the cytosolic C2A-C2B domains of synaptotagmin III. Implications for Ca(+2)-independent snare complex interaction. The Journal of cell biology. 1999;147(3):589–98. Epub 1999/11/05. 10.1083/jcb.147.3.589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Striegel AR, Biela LM, Evans CS, Wang Z, Delehoy JB, Sutton RB, et al. Calcium binding by synaptotagmin's C2A domain is an essential element of the electrostatic switch that triggers synchronous synaptic transmission. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2012;32(4):1253–60. Epub 2012/01/27. 10.1523/jneurosci.4652-11.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Davletov B, Perisic O, Williams RL. Calcium-dependent membrane penetration is a hallmark of the C2 domain of cytosolic phospholipase A2 whereas the C2A domain of synaptotagmin binds membranes electrostatically. The Journal of biological chemistry. 1998;273(30):19093–6. Epub 1998/07/21. 10.1074/jbc.273.30.19093 . [DOI] [PubMed] [Google Scholar]
- 12.Ubach J, Zhang X, Shao X, Sudhof TC, Rizo J. Ca2+ binding to synaptotagmin: how many Ca2+ ions bind to the tip of a C2-domain? The EMBO journal. 1998;17(14):3921–30. Epub 1998/07/22. 10.1093/emboj/17.14.3921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murray D, Honig B. Electrostatic control of the membrane targeting of C2 domains. Mol Cell. 2002;9(1):145–54. 10.1016/s1097-2765(01)00426-9 . [DOI] [PubMed] [Google Scholar]
- 14.Vermaas JV, Tajkhorshid E. Differential Membrane Binding Mechanics of Synaptotagmin Isoforms Observed in Atomic Detail. Biochemistry. 2017;56(1):281–93. Epub 2016/12/21. 10.1021/acs.biochem.6b00468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Herrick DZ, Sterbling S, Rasch KA, Hinderliter A, Cafiso DS. Position of synaptotagmin I at the membrane interface: cooperative interactions of tandem C2 domains. Biochemistry. 2006;45(32):9668–74. Epub 2006/08/09. 10.1021/bi060874j . [DOI] [PubMed] [Google Scholar]
- 16.Bai J, Wang P, Chapman ER. C2A activates a cryptic Ca(2+)-triggered membrane penetration activity within the C2B domain of synaptotagmin I. Proceedings of the National Academy of Sciences of the United States of America. 2002;99(3):1665–70. Epub 2002/01/24. 10.1073/pnas.032541099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martens S, Kozlov MM, McMahon HT. How synaptotagmin promotes membrane fusion. Science (New York, NY). 2007;316(5828):1205–8. Epub 2007/05/05. 10.1126/science.1142614 . [DOI] [PubMed] [Google Scholar]
- 18.Hui E, Johnson CP, Yao J, Dunning FM, Chapman ER. Synaptotagmin-mediated bending of the target membrane is a critical step in Ca(2+)-regulated fusion. Cell. 2009;138(4):709–21. Epub 2009/08/26. 10.1016/j.cell.2009.05.049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Paddock BE, Wang Z, Biela LM, Chen K, Getzy MD, Striegel A, et al. Membrane penetration by synaptotagmin is required for coupling calcium binding to vesicle fusion in vivo. J Neurosci. 2011;31(6):2248–57. 10.1523/JNEUROSCI.3153-09.2011 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chapman ER, Davis AF. Direct interaction of a Ca2+-binding loop of synaptotagmin with lipid bilayers. The Journal of biological chemistry. 1998;273(22):13995–4001. Epub 1998/06/05. 10.1074/jbc.273.22.13995 . [DOI] [PubMed] [Google Scholar]
- 21.Lynch KL, Gerona RR, Kielar DM, Martens S, McMahon HT, Martin TF. Synaptotagmin-1 utilizes membrane bending and SNARE binding to drive fusion pore expansion. Molecular biology of the cell. 2008;19(12):5093–103. Epub 2008/09/19. 10.1091/mbc.E08-03-0235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Loewen CA, Mackler JM, Reist NE. Drosophila synaptotagmin I null mutants survive to early adulthood. Genesis (New York, NY: 2000). 2001;31(1):30–6. Epub 2001/10/23. 10.1002/gene.10002 . [DOI] [PubMed] [Google Scholar]
- 23.DiAntonio A, Parfitt KD, Schwarz TL. Synaptic transmission persists in synaptotagmin mutants of Drosophila. Cell. 1993;73(7):1281–90. Epub 1993/07/02. 10.1016/0092-8674(93)90356-u . [DOI] [PubMed] [Google Scholar]
- 24.Bischof J, Maeda RK, Hediger M, Karch F, Basler K. An optimized transgenesis system for Drosophila using germ-line-specific phiC31 integrases. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(9):3312–7. Epub 2007/03/16. 10.1073/pnas.0611511104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brand AH, Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development (Cambridge, England). 1993;118(2):401–15. Epub 1993/06/01. . [DOI] [PubMed] [Google Scholar]
- 26.Yao KM, White K. Neural specificity of elav expression: defining a Drosophila promoter for directing expression to the nervous system. Journal of neurochemistry. 1994;63(1):41–51. Epub 1994/07/01. 10.1046/j.1471-4159.1994.63010041.x . [DOI] [PubMed] [Google Scholar]
- 27.Feng Y, Ueda A, Wu CF. A modified minimal hemolymph-like solution, HL3.1, for physiological recordings at the neuromuscular junctions of normal and mutant Drosophila larvae. Journal of neurogenetics. 2004;18(2):377–402. Epub 2005/03/15. 10.1080/01677060490894522 . [DOI] [PubMed] [Google Scholar]
- 28.Stevens CF, Sullivan JM. The synaptotagmin C2A domain is part of the calcium sensor controlling fast synaptic transmission. Neuron. 2003;39(2):299–308. Epub 2003/07/23. 10.1016/s0896-6273(03)00432-x . [DOI] [PubMed] [Google Scholar]
- 29.Robinson IM, Ranjan R, Schwarz TL. Synaptotagmins I and IV promote transmitter release independently of Ca(2+) binding in the C(2)A domain. Nature. 2002;418(6895):336–40. Epub 2002/07/12. 10.1038/nature00915 . [DOI] [PubMed] [Google Scholar]
- 30.Nishiki T, Augustine GJ. Dual roles of the C2B domain of synaptotagmin I in synchronizing Ca2+-dependent neurotransmitter release. J Neurosci. 2004;24(39):8542–50. 10.1523/JNEUROSCI.2545-04.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Loewen CA, Lee SM, Shin YK, Reist NE. C2B polylysine motif of synaptotagmin facilitates a Ca2+-independent stage of synaptic vesicle priming in vivo. Molecular biology of the cell. 2006;17(12):5211–26. Epub 2006/09/22. 10.1091/mbc.E06-07-0622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liu H, Dean C, Arthur CP, Dong M, Chapman ER. Autapses and networks of hippocampal neurons exhibit distinct synaptic transmission phenotypes in the absence of synaptotagmin I. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2009;29(23):7395–403. Epub 2009/06/12. 10.1523/jneurosci.1341-09.2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annual review of physiology. 2002;64:355–405. Epub 2002/02/05. 10.1146/annurev.physiol.64.092501.114547 . [DOI] [PubMed] [Google Scholar]
- 34.Dodge FA Jr., Rahamimoff R. Co-operative action a calcium ions in transmitter release at the neuromuscular junction. The Journal of physiology. 1967;193(2):419–32. Epub 1967/11/01. 10.1113/jphysiol.1967.sp008367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gerber SH, Rizo J, Sudhof TC. Role of electrostatic and hydrophobic interactions in Ca(2+)-dependent phospholipid binding by the C(2)A-domain from synaptotagmin I. Diabetes. 2002;51 Suppl 1:S12–8. Epub 2002/01/30. 10.2337/diabetes.51.2007.s12 . [DOI] [PubMed] [Google Scholar]
- 36.Fernandez-Chacon R, Shin OH, Konigstorfer A, Matos MF, Meyer AC, Garcia J, et al. Structure/function analysis of Ca2+ binding to the C2A domain of synaptotagmin 1. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2002;22(19):8438–46. Epub 2002/09/28. 10.1523/JNEUROSCI.22-19-08438.2002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mackler JM, Reist NE. Mutations in the second C2 domain of synaptotagmin disrupt synaptic transmission at Drosophila neuromuscular junctions. The Journal of comparative neurology. 2001;436(1):4–16. Epub 2001/06/20. . [PubMed] [Google Scholar]
- 38.Borden CR, Stevens CF, Sullivan JM, Zhu Y. Synaptotagmin mutants Y311N and K326/327A alter the calcium dependence of neurotransmission. Molecular and cellular neurosciences. 2005;29(3):462–70. Epub 2005/05/12. 10.1016/j.mcn.2005.03.015 . [DOI] [PubMed] [Google Scholar]
- 39.Li L, Shin OH, Rhee JS, Arac D, Rah JC, Rizo J, et al. Phosphatidylinositol phosphates as co-activators of Ca2+ binding to C2 domains of synaptotagmin 1. The Journal of biological chemistry. 2006;281(23):15845–52. Epub 2006/04/06. 10.1074/jbc.M600888200 . [DOI] [PubMed] [Google Scholar]
- 40.Mace KE, Biela LM, Sares AG, Reist NE. Synaptotagmin I stabilizes synaptic vesicles via its C(2)A polylysine motif. Genesis (New York, NY: 2000). 2009;47(5):337–45. Epub 2009/04/10. 10.1002/dvg.20502 . [DOI] [PubMed] [Google Scholar]
- 41.Paddock BE, Striegel AR, Hui E, Chapman ER, Reist NE. Ca2+-dependent, phospholipid-binding residues of synaptotagmin are critical for excitation-secretion coupling in vivo. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2008;28(30):7458–66. Epub 2008/07/25. 10.1523/jneurosci.0197-08.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wang P, Wang CT, Bai J, Jackson MB, Chapman ER. Mutations in the effector binding loops in the C2A and C2B domains of synaptotagmin I disrupt exocytosis in a nonadditive manner. The Journal of biological chemistry. 2003;278(47):47030–7. Epub 2003/09/10. 10.1074/jbc.M306728200 . [DOI] [PubMed] [Google Scholar]
- 43.Fernandez-Chacon R, Konigstorfer A, Gerber SH, Garcia J, Matos MF, Stevens CF, et al. Synaptotagmin I functions as a calcium regulator of release probability. Nature. 2001;410(6824):41–9. Epub 2001/03/10. 10.1038/35065004 . [DOI] [PubMed] [Google Scholar]
- 44.Rhee JS, Li LY, Shin OH, Rah JC, Rizo J, Sudhof TC, et al. Augmenting neurotransmitter release by enhancing the apparent Ca2+ affinity of synaptotagmin 1. Proceedings of the National Academy of Sciences of the United States of America. 2005;102(51):18664–9. Epub 2005/12/15. 10.1073/pnas.0509153102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bai J, Tucker WC, Chapman ER. PIP2 increases the speed of response of synaptotagmin and steers its membrane-penetration activity toward the plasma membrane. Nature structural & molecular biology. 2004;11(1):36–44. Epub 2004/01/14. 10.1038/nsmb709 . [DOI] [PubMed] [Google Scholar]
- 46.Lai AL, Tamm LK, Ellena JF, Cafiso DS. Synaptotagmin 1 modulates lipid acyl chain order in lipid bilayers by demixing phosphatidylserine. The Journal of biological chemistry. 2011;286(28):25291–300. Epub 2011/05/26. 10.1074/jbc.M111.258848 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kiessling V, Kreutzberger AJB, Liang B, Nyenhuis SB, Seelheim P, Castle JD, et al. A molecular mechanism for calcium-mediated synaptotagmin-triggered exocytosis. Nature structural & molecular biology. 2018;25(10):911–7. Epub 2018/10/07. 10.1038/s41594-018-0130-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Adolfsen B, Saraswati S, Yoshihara M, Littleton JT. Synaptotagmins are trafficked to distinct subcellular domains including the postsynaptic compartment. The Journal of cell biology. 2004;166(2):249–60. 10.1083/jcb.200312054 [DOI] [PMC free article] [PubMed] [Google Scholar]