

INTRODUCTION
Approximately 30% of patients treated with fluorouracil (FU) develop grade ≥ 3 toxicity.1 Deleterious genetic variations in DPYD, the gene that encodes dihydropyrimidine dehydrogenase (DPD), have been established as predictors of FU-associated toxicity.2-7 The Clinical Pharmacogenetics Implementation Consortium has recently published updated guidelines for FU dosing in carriers of deleterious DPYD variants.8 The guidelines use an activity score for each variant, which is determined by the degree to which a variant impairs DPD activity.8 Despite clear guidance for variants that have been previously characterized and reported, an urgent need remains for providing FU dosing guidance to patients who carry variants of unknown significance and/or complex genotypes.
We present the case of a patient with rectal cancer who experienced severe FU-associated toxicity during neoadjuvant therapy with capecitabine. Genetic testing revealed a compound heterozygous DPYD genotype that included a novel variant of unknown DPD function, p.T132A (NM_000110.3:c.394A>G; NP_000101.2:p.Thr132Ala), and a well-recognized toxicity-associated variant, rs3918290 (NM_000110.3:c.1905+1G>A; DPYD*2A).2,3 We used our recently reported in silico tool (DPYD-Varifier)7 to predict that p.T132A would be deleterious to function, which was subsequently validated using ex vivo and in vitro approaches. These methodologies permitted us to determine an activity score for the patient that was used to calculate a safe adjusted dose of FU for adjuvant therapy.
METHODS
This study was approved by the Mayo Clinic institutional review board. Informed consent to use medical records and patient-derived specimens for research use, and reporting was obtained from the patient. The entire coding region of DPYD was initially sequenced by Mayo Medical Laboratories. DPYD-Varifier was used to predict the function of p.T132A.7 The effect of p.T132A on DPD activity was measured in vitro as previously described.5-7
Total RNA was isolated from the patient’s blood sample using the PAXgene Blood RNA Kit (PreAnalytiX, Hombrechtikon, Switzerland), and cDNA was reverse transcribed using oligo(dT)15 primers (Thermo Fisher Scientific, Waltham, MA). The full-length DPYD open reading frame was amplified (primers: 5′-gtttgtcactggcagactcg-3′, 5′-ttcacagcaactgtttcacaaa-3′) using Q5 High-Fidelity DNA Polymerase (New England Biolabs, Ipswich, MA). The polymerase chain reaction (PCR) product was cloned into the pJET1.2 vector, and 20 cloned constructs were sequenced at the Mayo Clinic Gene Analysis Shared Resource to determine the cis or trans conformation of the variants. Peripheral blood mononuclear cells (PBMCs) were isolated and assessed ex vivo for DPD activity as previously described.9,10
Additional PCR and quantitative PCR reactions were performed with primers specific to canonically (E13/14: 5′-ctcttgataaggacattgtgacaaa-3′, 5′-tttgcagctcttgcgatgc-3′) and alternatively spliced DPYD (E13/15: 5′-ctcttgataagattgtgattgctagc-3′, 5′-tttgcagctcttgcgatgc-3′). GAPDH (primers: 5′-accacagtccatgccatcac-3′, 5′-tccaccaccctgttgctgt-3′) was used as a control gene to normalize expression values.
CASE REPORT
A timeline for this case study is presented in Figure 1. A 32-year-old male with a history of gastroesophageal reflux and hyperlipidemia presented with altered bowel function and bright red blood per rectum. Colonoscopy revealed a polyp in the cecum and a mass approximately 10 cm within the rectosigmoid colon. Biopsy specimens of the colon and perirectal lymph nodes were obtained, and pathology confirmed adenocarcinoma. Carcinoembryonic antigen was noted to be within normal limits. A staging computed tomography scan of the chest, abdomen, and pelvis showed no evidence of distal metastases, which suggested stage III disease.
Fig 1.
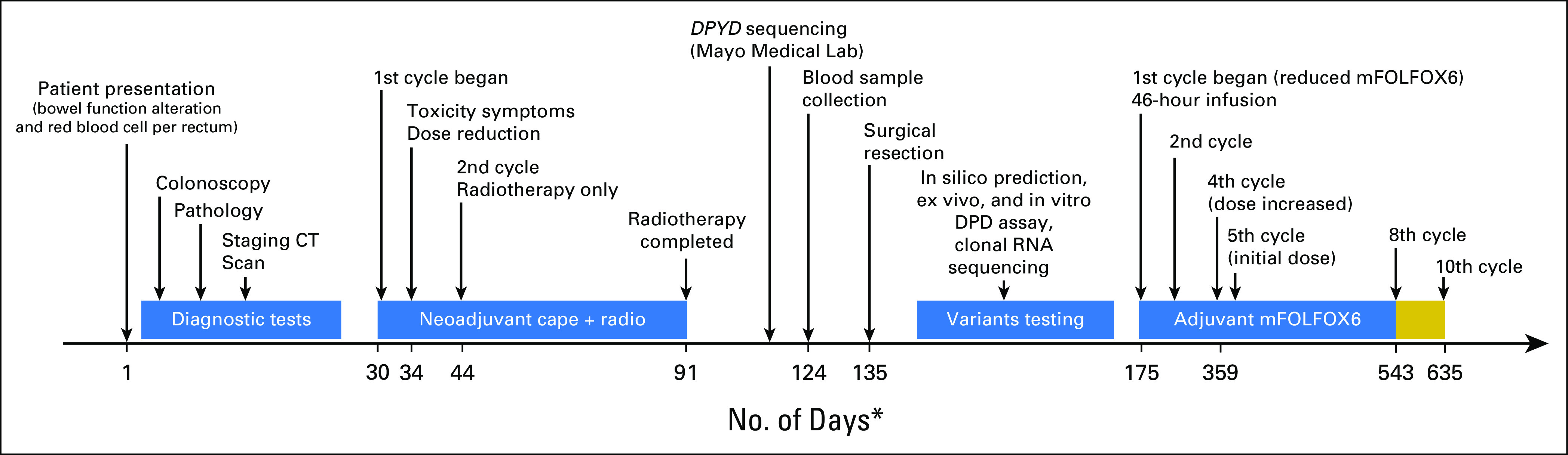
Schematic of the patient’s case report. The chart presents the timeline for the key events in the diagnosis, treatment, and laboratory testing of DPYD variants. The horizontal line represents the number of days from the presentation of symptoms through the 10th cycle of the adjuvant therapy. Gold box represents continued fluorouracil and leucovorin only for an additional two cycles (no oxalipatin). (*) Not drawn to scale. cape, capecitabine; CT, computed tomography; DPD, dihydropyrimidine dehydrogenase; mFOLFOX6, modified infusional fluorouracil, leucovorin, and oxaliplatin; radio, radiotherapy.
The patient was treated shortly after diagnosis with neoadjuvant therapy that consisted of capecitabine (825 mg × m−2 for 5 days each week) and radiotherapy (50.4 Gy in 28 fractions). Five days after starting treatment, the patient reported fatigue, severe diarrhea (10 to 15 bowel movements per day), and skin rash. The capecitabine dose was reduced by approximately 25%, but the patient was hospitalized soon after this change. During hospitalization, absolute neutrophil count decreased to 700 cells/μL (normal reference range, 1,500 to 8,000 cells/μL), which prompted discontinuation of therapy late in the second week of treatment. The patient remained hospitalized for 13 days, during which time he experienced severe neutropenic fever and weight loss (approximately 25 lb). Seven days after discharge from the hospital, the patient’s radiation treatment was resumed and continued for 47 days. Eight weeks after the completion of neoadjuvant therapy, low anterior resection of a 23-cm portion of the rectosigmoid colon was performed. Pathology at the time of resection revealed a microscopic focus (0.5 mm) of residual, moderately differentiated adenocarcinoma identified in a tumor bed measuring 1.3 × 1.0 cm. The surgical resection margins and multiple lymph nodes (n = 16) were negative for tumor. One and a half months after surgery, the patient was scheduled to begin modified infusional FU, leucovorin, and oxaliplatin (mFOLFOX6) adjuvant therapy.
Because of the severe toxicity during the initial neoadjuvant therapy, genetic testing was ordered. Sequencing of the coding region for DPYD revealed the presence of DPYD*2A and the previously unreported variant p.T132A. With our recently reported in silico prediction algorithm DPYD-Varifier,7 we determined that p.T132A was likely deleterious to function and thus may have contributed to FU-related toxicities during neoadjuvant therapy.
Additional functional studies were performed to determine the phasing of these two variants and the degree to which p.T132A alters DPD activity. Clonal RNA sequencing identified 11 clones that contained only DPYD*2A and nine that contained only p.T132A, which confirmed the trans conformation of the two variants. DPD activity in the patient’s PBMCs was reduced by 64% (92.4 pmol FU × min−1 × mg−1) compared with previously reported reference values obtained from individuals who do not carry a deleterious DPYD variant10 (254.0 pmol FU × min−1 × mg−1; Fig 2A). To determine whether allelic imbalance of DPYD was occurring, allele-specific PCR primers E13/14 and E13/15 were used to amplify canonical and alternatively spliced DPYD (Fig 2B, top). In quantitative studies, efficiencies of both PCR reactions were similar, and both the canonical and the alternatively spliced forms of DPYD were expressed at similar levels (Fig 2C).
Fig 2.
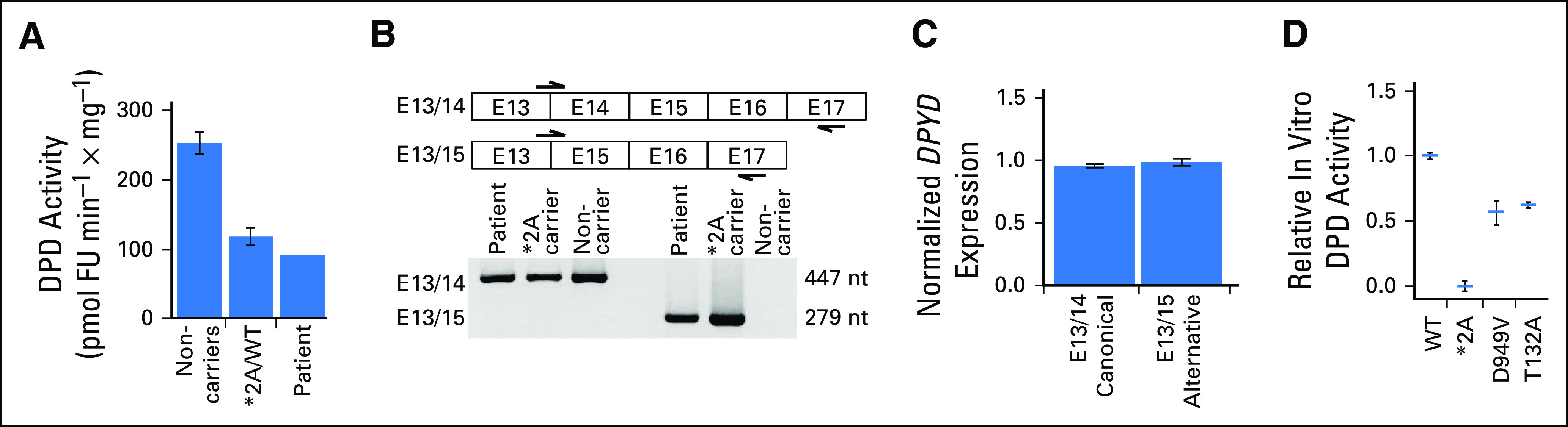
Functional assessment of DPYD variants in the patient. (A) Dihydropyrimidine dehydrogenase (DPD) enzyme activity was measured in peripheral blood mononuclear cells isolated from the patient and was compared with reference values previously published by our laboratory for peripheral blood mononuclear cells from individuals who do not carry known deleterious variants in DPYD (noncarriers) and those heterozygous for *2A (*2A/wild type [WT]).10 (B) Polymerase chain reaction primers were designed as indicated (arrows) to amplify across the exon 13/14 and 13/15 junctions to detect the presence of alternative splicing in the patient and a heterozygous carrier of *2A. (C) Relative expression of canonical and alternatively spliced DPYD was measured using primers described in (B). (D) In vitro DPD activity conferred by recombinant *2A, p.D949V, and p.T132A variants relative to WT DPD activity was compared. For each variant, the mean of two independent experiments is presented as a horizontal bar ± standard deviation. Each independent experiment consisted of three technical replicates. Results for p.D949V were obtained from previously published data.7 nt, nucleotide.
To directly quantify the degree to which p.T132A impairs DPD function as a single allele, in vitro analysis of transgenic p.T132A DPD was performed. This amino acid substitution decreased DPD activity by 38% compared with wild-type DPD protein (Fig 2D). This level of reduction is similar to that observed for the well-studied p.D949V variant5,7; thus, the DPD activity score would be expected to be similar. The DPD activity score8 was calculated to be 0.5 for this patient (0.0 for DPYD*2A and 0.5 for p.T132A), which suggested that a 75% reduction in FU dose likely would be safe.
One and a half months after surgery, the patient received the first cycle of dose-adjusted modified infusional FU, leucovorin, and oxaliplatin therapy. No bolus FU was administered, and the 46-hour infusional doses of FU and leucovorin were reduced by 75%. Standard doses of oxaliplatin were administered (85 mg × m−2). The patient experienced grade 1 to 2 diarrhea, dyspepsia, neutropenia, and fatigue during his adjuvant chemotherapy (assessed by the treating physician using Common Terminology Criteria for Adverse Events [version 5.0]). During cycle 4, the infusional dose of FU was increased by 10% to 15%, and an increase in diarrhea and fatigue was observed. Because of this, the subsequent doses were given at the initial dose (75% reduction) for the remainder of therapy. No additional dose interruptions or delays were required. Finally, the decision was made to administer two additional cycles of therapy that did not contain oxaliplatin because of accumulating grade 1 neuropathy (Fig 1, gold box). The patient has completed the 10th cycle of adjuvant therapy and has responded well to the treatment with no severe toxicity and no physical evidence of cancer progression.
DISCUSSION
This report outlines the effective management of toxicity in a patient with rectal cancer treated with an FU-based regimen using a precision oncology approach. To date, genetic testing of DPYD variants is not mandated in the United States, and complete DPD deficiency is only listed as a contraindication to FU treatment.11 Many studies have increasingly performed functional characterizations of DPYD variants,3,5-7,9,10,12,13 and the Clinical Pharmacogenetics Implementation Consortium has published activity-based FU dosing guidelines for many of the reported DPYD variants.8 However, a paucity of information exists with respect to FU dose adjustments in compound heterozygous variant carriers.
In this case report, DPYD genotype and variant function data were used to provide guidance for adjuvant FU dosing in a patient who carries a complex DPYD genotype that includes a novel variant of unknown significance. The 38% reduction in in vitro DPD function attributed to p.T132A also is consistent with the approximately 36% activity measured in the patient’s PBMCs because the p.T132A variant in the patient’s genome is present in a compound complex heterozygous state (trans) with DPYD*2A. Structurally, the p.T132A variant is located in proximity to iron sulfur cluster 1 and 2 (three-dimensional distance < 15 Å). Typically, deleterious variants are in proximity to important domains in the DPD protein.7
Overall, this case study demonstrates that the in silico and in vitro approaches used in this report can be used concurrently with DPYD variant screening to enable informed decisions about FU dose modifications. These approaches are especially valuable with regard to variants with unknown function and/or compound complex genotypes. Overall, this case report illustrates that DPYD genotype-phenotype–guided FU dosing is a promising approach for personalized and precision cancer therapy.
ACKNOWLEDGMENT
We thank Kelly Bouchonville for proofreading and technical editing of the manuscript. We also thank the Mayo Clinic Cancer Center for supporting shared resources used in generating data for this article.
Footnotes
Supported by National Institutes of Health Grant No. CA015083.
AUTHOR CONTRIBUTIONS
Conception and design: Shikshya Shrestha, Timothy J. Hobday, Steven M. Offer, Robert B. Diasio
Collection and assembly of data:Shikshya Shrestha, Erin E. Tapper, Colbren (Scout) Trogstad-Isaacson, Timothy J. Hobday, Steven M. Offer
Data analysis and interpretation:Shikshya Shrestha, Timothy J. Hobday, Steven M. Offer
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/po/author-center.
Shikshya Shrestha
No relationship to disclose
Erin E. Tapper
No relationship to disclose
Colbren (Scout) Trogstad-Isaacson
No relationship to disclose
Timothy J. Hobday
Consulting or Advisory Role: Ipsen, AbbVie
Research Funding: Novartis (Inst)
Steven M. Offer
Patents, Royalties, Other Intellectual Property: Royalties for invention of health care–related technology, managed by Mayo Clinic
Robert B. Diasio
No relationship to disclose
REFERENCES
- 1.Lévy E, Piedbois P, Buyse M, et al. Toxicity of fluorouracil in patients with advanced colorectal cancer: Effect of administration schedule and prognostic factors. J Clin Oncol. 1998;16:3537–3541. doi: 10.1200/JCO.1998.16.11.3537. [DOI] [PubMed] [Google Scholar]
- 2.Saif MW, Ezzeldin H, Vance K, et al. DPYD*2A mutation: The most common mutation associated with DPD deficiency. Cancer Chemother Pharmacol. 2007;60:503–507. doi: 10.1007/s00280-006-0392-5. [DOI] [PubMed] [Google Scholar]
- 3.Lee AM, Shi Q, Pavey E, et al. DPYD variants as predictors of 5-fluorouracil toxicity in adjuvant colon cancer treatment (NCCTG N0147) J Natl Cancer Inst. 2014;106:dju298. doi: 10.1093/jnci/dju298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meulendijks D, Henricks LM, Sonke GS, et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015;16:1639–1650. doi: 10.1016/S1470-2045(15)00286-7. [DOI] [PubMed] [Google Scholar]
- 5.Offer SM, Fossum CC, Wegner NJ, et al. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014;74:2545–2554. doi: 10.1158/0008-5472.CAN-13-2482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Offer SM, Wegner NJ, Fossum C, et al. Phenotypic profiling of DPYD variations relevant to 5-fluorouracil sensitivity using real-time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73:1958–1968. doi: 10.1158/0008-5472.CAN-12-3858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shrestha S, Zhang C, Jerde CR, et al. Gene-specific variant classifier (DPYD-Varifier) to identify deleterious alleles of dihydropyrimidine dehydrogenase. Clin Pharmacol Ther. doi: 10.1002/cpt.1020. 10.1002/cpt.1020 [epub ahead of print on January 12, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amstutz U, Henricks LM, Offer SM BJ, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin Pharmacol Ther. 2018;103:210–216. doi: 10.1002/cpt.911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Offer SM, Lee AM, Mattison LK, et al. A DPYD variant (Y186C) in individuals of African ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther. 2013;94:158–166. doi: 10.1038/clpt.2013.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nie Q, Shrestha S, Tapper EE, et al. Quantitative contribution of rs75017182 to dihydropyrimidine dehydrogenase mRNA splicing and enzyme activity. Clin Pharmacol Ther. 2017;102:662–670. doi: 10.1002/cpt.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Er T-K, Bujanda L, Rodrigo M, et al. Pharmacogenomic biomarkers for colorectal cancer treatment. Cancer Treat Commun. 2015;4:121–127. [Google Scholar]
- 12.Elraiyah T, Jerde CR, Shrestha S, et al. Novel deleterious dihydropyrimidine dehydrogenase variants may contribute to 5-fluorouracil sensitivity in an East African population. Clin Pharmacol Ther. 2017;101:382–390. doi: 10.1002/cpt.531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lee AM, Shi Q, Alberts SR, et al. Association between DPYD c.1129-5923 C>G/hapB3 and severe toxicity to 5-fluorouracil-based chemotherapy in stage III colon cancer patients: NCCTG N0147 (Alliance) Pharmacogenet Genomics. 2016;26:133–137. doi: 10.1097/FPC.0000000000000197. [DOI] [PMC free article] [PubMed] [Google Scholar]