

Abstract
Ion mobility mass spectrometry (IM-MS) allows separation of native protein ions into “conformational families”. Increasing the IM resolving power should allow finer structural information to be obtained and can be achieved by increasing the length of the IM separator. This, however, increases the time that protein ions spend in the gas phase and previous experiments have shown that the initial conformations of small proteins can be lost within tens of milliseconds. Here, we report on investigations of protein ion stability using a multipass traveling wave (TW) cyclic IM (cIM) device. Using this device, minimal structural changes were observed for Cytochrome C after hundreds of milliseconds, while no changes were observed for a larger multimeric complex (Concanavalin A). The geometry of the instrument (Q-cIM-ToF) also enables complex tandem IM experiments to be performed, which were used to obtain more detailed collision-induced unfolding pathways for Cytochrome C. The instrument geometry provides unique capabilities with the potential to expand the field of protein analysis via IM-MS.
The invention of soft-ionization techniques1−3 has allowed the transfer of intact biomolecules and proteins into the gas phase.4−6 Early electrospray mass spectrometry experiments (ESI-MS) showed pronounced differences in charge state distribution depending on the solution conditions, suggesting that solution phase protein structure can be probed using ESI-MS.7 In their landmark work, Clemmer and Jarrold constructed an ESI–ion mobility–mass spectrometer (ESI-IM-MS), which revealed that a single charge state of a protein can be present in a range of conformations.8 This sparked a significant interest in studies of proteins in the gas phase. IM-MS has the advantage of being able to detect multiple and lowly populated conformational ensembles from small sample volumes, which are comparable to those seen in solution when the ions have a low internal energy (Eint).9 Being able to detect these conformational states provides information on protein folding dynamics and requires far lower sample concentrations when compared to other structural techniques such as X-ray crystallography and NMR. Introduction of the first mainstream commercial IM-MS instrumentation (Synapt HDMS, Waters Corporation, 2006)10 accelerated implementation of the technique in protein studies. IM-MS has been used to show the basic dynamic behavior of proteins,11−13 protein domain organization,14 and to identify and investigate the structural dynamics of disordered proteins.15,16 Deliberate activation of ions through increasing the internal energy via collisional heating can cause unfolding,17 which can provide important information on the structural stability of proteins under different conditions or with different states, the effect of modifications or ligands on protein structure and dynamics,16,18,19 and the subunit organization of oligomers.19,20 This can also be applied to protein complexes to investigate oligomerization pathways and subunit organization of complexes.12,21−24
IM-MS functions by introducing analyte ions into a drift-cell containing an inert buffer gas such as helium or nitrogen. Ions drift through the cell under the influence of an electric field and collide with buffer gas molecules. Drift velocity of ions is governed by their mobility which is inversely proportional to their collision cross-section (CCS). Species that are extended (larger CCS) will undergo a greater number of collisions with the buffer gas and have a longer drift time compared to more compact species (smaller CCS). Mass to charge ratios (m/z) are then measured for each ion by a mass spectrometer.
Increasing the resolution of the IM device should, in principle, enable separation of overlapping features, allowing greater understanding of protein structures in the gas phase. The resolution of the IM device depends on the temperature, ion charge, electric field, and the path length.25 There are several examples in the literature where attempts were made to increase the resolution of the IM apparatus by decreasing the temperature,26,27 increasing the electric field,28 and increasing the path length.29,30 Conventionally, the latter is a physical distance that ions travel, and thus, a number of several meters long drift tube (DT) type instruments have been realized.31−33 There are practical limits in this approach related to the physical size of the apparatus and high voltages required. Alternatively, an experimental setup with inverted “frame of reference” can be envisaged, where the ions are trapped in a stream of moving gas by an opposing electric field, as in the case of trapped ion mobility spectrometry technique (TIMS).34 Here, the “effective path length” can be influenced by a separation time scale and gas velocity. Another way of achieving a long separation path is to utilize multipass devices. The cyclotron mobility spectrometer described by Glaskin et al. uses a drift cell made of four curved segments that are joined by ion funnels,35 which refocus ions.29 An electric field applied to subsequent segments/funnels is switched; only the ions with mobilities resonant with the field switching frequency can proceed to the following segments, while others are lost. An IM spectrum is obtained by scanning the field switching frequency. Several developments of high resolution IM instrumentation have been facilitated by traveling wave (T-Wave, TW) technology,36 which relies on a series of voltage pulses that propel ions across the device. T-Wave technology eliminates problems related to high voltages, required for the traditional, linear field DT-IM apparatus. A T-Wave based, multipass cyclic IM (cIM) separator was first introduced by Giles et al.,37 and separation at a path length over 50 m was demonstrated.38 Further advances in a T-Wave technology include structures for lossless ion manipulation (SLIM), notably by Smith et al.39 A multipass SLIM device has allowed IM separations over an extremely long path length (∼1 km).40 Other recent developments in IM technology include tandem methods. A two-stage IM technique was first presented by Koeniger et al.,32,41 where an instrument featuring two drift tubes allowed separation in the first IM stage, mobility selection, activation, and separation of product ions in the second stage. This was further expanded to a three-stage IM method by Merenbloom et al.31 More recently, the multistage IM technology was combined with a multipass cyclic IM separator by Giles et al.,38 allowing for IMSn-type workflows.
Increasing the resolution of IM separation via path length typically increases experiment time scales. Understandably, changes in the nature of analyte ions occurring on the time scale of separation are undesirable. Previous work by Badman et al. showed that Cytochrome C ions underwent structural changes with time.42,43 Utilizing a quadrupole ion trap (QIT)-IM-MS instrument, ions were stored for varying amounts of time in the QIT, prior the IM-MS measurement. The structural changes started occurring at approximately 30 ms and had stabilized after 60 ms, showing that the initial population of +7 to +10 ions contains precursors or conformational intermediates that unfold in the gas phase.42 Similar results were reported for ubiquitin.44 It was suggested that solution specific conformations transform to new gas phase structures in the absence of solvent after a period of 30 ms. More recently, Allen et al. have utilized a SLIM-based tandem IM instrument45 to study the Cytochrome C ions. It was shown that structural changes occurred an order of magnitude later than reported previously. This was attributed primarily to pressure-related differences in effective ion temperatures46 in the QIT and tandem IM systems and also solution-dependent effects, further confirmed by collision-induced unfolding (CIU) experiments.45 Collectively, the previous work suggests that IM separation time scales appropriate for native protein ions is likely to be on the order of tens to hundreds of milliseconds, especially for larger ions.
Importantly, Badman et al. utilized a relatively short, linear field DT device, where transit times are on the order of tens of milliseconds, comparable to trapping times in the QIT. Moreover, due to a relatively low velocity of ions in such devices, their effective temperatures are essentially equal to that of the buffer gas. The latter is not necessarily the case in the T-Wave based IM separation. It was reported previously that some ion heating during the T-Wave based IM separation can cause structure perturbation, the effect being especially pronounced for low molecular weight, high mobility species.47,48 The native protein ions typically have relatively low charge and high mass, thus, representing a contrasting case, in principle. Nevertheless, it is of interest and of importance to evaluate the extent of native structure perturbation upon prolonged exposure to T-Waves, particularly in relation to the possibility of high-resolution IM separations in the future.
Here, we utilize a prototype cIM-MS instrument enabling custom experiments designed to further explore the stability of monomeric (Cytochrome C, β-Lactoglobulin) and multimeric (Concanavalin A) proteins, in a T-Wave based separator. Ions were subjected to trapping in a region of the cIM device, and native conformations were found to be stable for hundreds of milliseconds. Additional experiments were also performed to show that extended time within the cIM device under typical separation conditions does not significantly impact protein structure in the gas phase. IMS-CIU-IMS experiments revealed detailed unfolding pathways for Cytochrome C. Our results agree with previous reports suggesting that the native-like conformation or proteins is maintained in the gas phase under extended time scales and, for the first time, show that this is also true within a cIM device. Collectively, our data show that the cIM instrument can be used for studying protein dynamics, stability, and unfolding in the gas phase.
Methods
Sample Preparation
The proteins (equine Cytochrome C (Merck Millipore, U.K.), β-lactoglobulin (Sigma, U.K.), and Concanavalin A (Sigma, U.K.)) were buffer exchanged into 200 mM ammonium acetate solution using 3, 10, or 30 kDa Amicon Ultra 0.5 mL centrifugal spin filters (Merck Millipore, U.K.). The samples were spun a total of three times at 12000 rpm for 15 min at room temperature. The protein was then diluted to 8–10 μM after concentration calculation using a Qubit protein assay (ThermoFisher Scientific, U.K.).
Mass Spectrometry
The samples were introduced into the instrument using a nano-ESI source (Waters Corp., Wilmslow, U.K.). The emitters (manufactured using a P97 Flaming/Brown micropipette puller, coated in gold using a Quorum Q150R S sputter coater) were held at 1.2 kV. The cIM instrument design is discussed in detail elsewhere49,50 and so will only be briefly covered here. The instrument schematic is presented in Figure 1. Ions are transferred from the source through the first vacuum stages using ion guide arrangements (StepWave), which propel ions toward the quadrupole mass filter. The subsequent trap cell is used for accumulating ions prior to IM separation. The resulting ion packets are then transported through an ion guide (IG) and injected into a helium cell. In this work we utilize the injection energy into the He cell to generate some of the collision induced unfolding (CIU) data. The subsequent ion guide (prestore) transports the ions into a multifunction array (Figure 1C) of electrodes forming part of an orthogonal closed loop, the cIM separator (98 cm path length, single pass resolving power (RP) of ∼65 (CCS/ΔCCS) measured using the inverse-sequence peptide pair (SDGRG and GRGDS).50 The cIM chamber is filled directly with nitrogen to a pressure of ∼2.2 mbar (including some contribution from helium gas leaking in from the He cell) and the T-wave height was 40 V. The T-Wave direction in the array can be altered to either match those in the cIM device (i.e., separate) or to inject/eject ions from it. The control software GUI enables creation of custom sequences of functions to facilitate selective ejection of ions from the cyclic IM device and/or activation followed by further separation of product ions.50 The typical sequence of events employed in single/multipass experiments is presented in Figure S1. Post cIM, ions are transported through an ion guide (Post-Store) and on to the ToF via a segmented quadrupole (XS) transfer cell. The transfer cell allows activation of mobility separated ions. The orthogonal acceleration time-of-flight (oa-ToF) features an offset V geometry allowing m/z measurements at resolutions in excess of 60000 fwhm.38
Figure 1.

(A) Instrument schematic showing the Q-cIM-ToF geometry; (B) Cartoon showing the orthogonal arrangement of the cyclic IMS and neighboring optics; (C) Multifunction region.
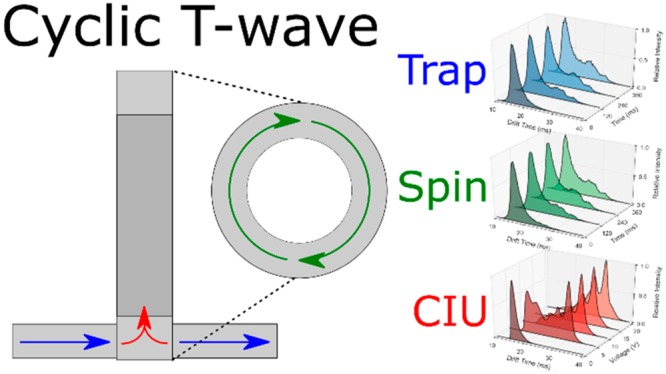
In addition to a typical single/multipass operation (Figure S1), three custom modes of operation were designed to explore protein stability over extended experiment times in the high-pressure cIM device. First, in the “trapping” mode ions are stored in the array for a prolonged period of time, before IM separation takes place (Figure S2). This mode enables the time-resolved assessment of the protein conformation stability in the absence of any (intentional) activation. Second, the “spinning” mode was designed to verify the effect of ion heating during the T-Wave-based IM separation (Figure S3). Normally, the number of passes around the cIM (and, hence, separation time) is limited by so-called wrap-around, a phenomenon where the fastest ions ultimately catch up with the slowest ones.50 To extend the exposure to T-Waves, ions are passed around the cIM device for varying amounts of time (allowing wrap-around) before being recollected in the prestore and subjected to another IM separation before detection. It should be noted that while the ions are being manipulated in the cIM device, the next set of ions are continually being accumulated in the trap region (∼10–2 mbar of N2) of the instrument. Consequently, at the start of the cIM manipulation, the ions will have an average gas-phase lifetime of around half of the cIM experiment time. To minimize activation of ions entering the cIM and retain low-energy conformations, generally the injection voltage into the He cell is kept as low as possible while still maintaining reasonable transmission. Experiments to help elucidate the effect of ion storage in the trap region alone were carried out (see Figures S4 and S5) and indicated no significant activation with time. Lastly, the IMS-CIU-IMS mode of operation is used (Figure S6). Here, a mobility-selected ion population can be ejected and trapped in the prestore, while the remaining ions are removed from the cIM. The selected ions are then reinjected into the array, but with a higher voltage between the two regions to induce activation. This way, we can probe unfolding transitions of selected regions of arrival time space, increasing the specificity of CIU experiments.18,51 In this work we focus on the dual stage method; however, multistage experiments (IMSn) can be performed in an analogous way.
Data Analysis
Data were analyzed using Masslynx v4.1 (Waters Corporation) and Driftscope v2.1 (Waters Corporation). To compare the data, the gross arrival time had the injection time (10 ms) subtracted to give drift-time. In the case of trapping experiments, the time spent in the array (0–360 ms) was also subtracted. For spinning, the extended cyclic motion (0–360 ms) and the reinjection time (45 ms) were subtracted. CIU fingerprint plots were created using Benthesikyme.52 Population fitting was performed using in-house software written in Python 2.7, peak maxima were identified using the second derivative and manually adjusted to ensure the same conformational populations were tracked across the different experiments. During trapping and spinning experiments the data were aligned according to the most intense peak to allow the same centroid value for each Gaussian population for tracking. In the case where a second conformational population became the maximum peak, the two most intense peaks were used for alignment. Each conformational family was approximated by a Gaussian distribution. The sum of all distributions was optimized to produce the best fit to the experimental data or trace. No restraint to the full width half-maximum value of each Gaussian was imposed like in previous work.52 The code is freely available at https://github.com/ThalassinosLab/CIVU.
Results
Cytochrome C
CytC was analyzed, and a very narrow charge state distribution was observed, with high abundance of the +7 charge state (Figure 2A), which was quadrupole selected for experimentation (1765 m/z). Other charge states ranging from +8 to +5 could be seen at low abundance. Minimal activation upon both trapping and spinning experiments was observed for the +7 (Figure 3A–C). Comparing these structural transitions to the CytC +7 CIU plots (Figure 3C), they are equivalent to less than 5 V of activation. The time scales of conformational change are similar to previous studies.45
Figure 2.
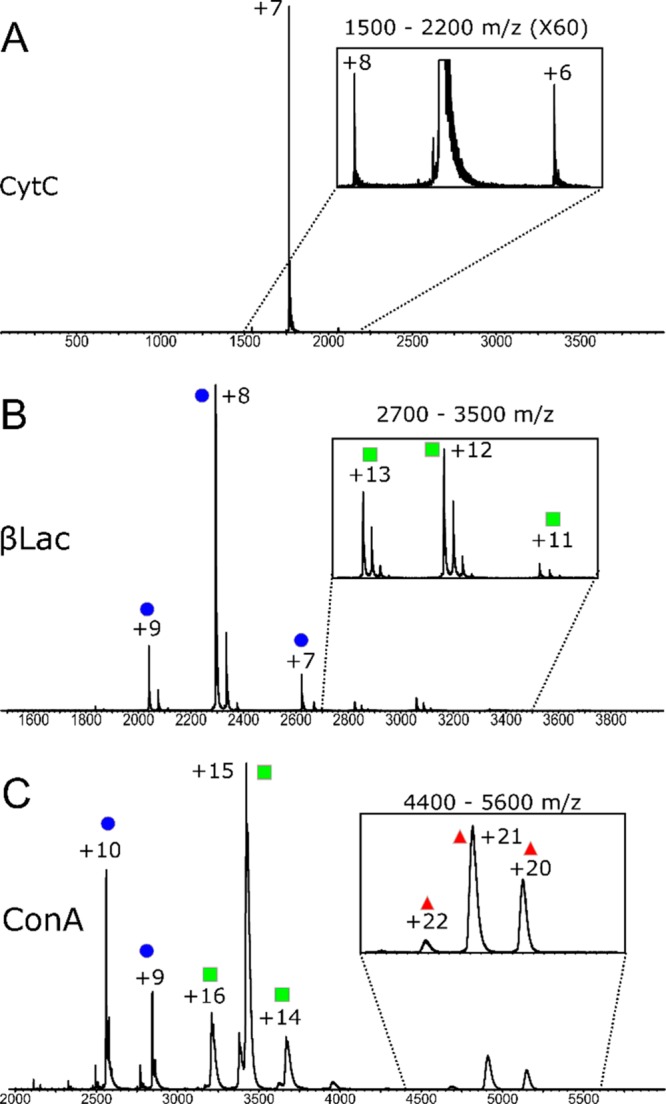
Mass spectra for the proteins (A) Cytochrome C (CytC), (B) β-Lactoglobulin (βLac), and (C) Concanavalin A (ConA). Blue circle = monomer, green square = dimer, red triangle = tetramer.
Figure 3.

Trapping (A, D, G), spinning (B, E, H), and CIU (C, F, I) experiments shown for CytC +7 (A–C, purple), βLac +8 (D–F, blue), ConA + 21 (G–I, red). Each figure is composed of ATD slices, arranged in increasing experimental increment, i.e., trapping time (ms), from dark to light shading. Plotted is the drift time against normalized intensity.
β-Lactoglobulin
β-Lactoglobulin (βLac) is a 16 kDa protein, with several disulfide bridges, that exists as a monomer and dimer in solution; with the ratio being dependent on protein concentration and the ionic strength of the solution.53 βLac was detected mainly as monomer (Figure 2B), and the charge states +7 to +9 were quadrupole isolated for further analysis (2663, 2294, and 2940 m/z, respectively). The +7 charge state did not display structural changes upon trapping or spinning (Figures S8A–C and S11); however, the +8 and +9 charges displayed very minimal structural changes over the course of 240 ms (Figures 3D–F, S12, and S13) comparable to 10–20 V of intentional activation (Figure S8I).
Concanavalin A
To investigate the effect of prolonged gas phase exposure on the stability of multimeric complexes we analyzed Concanavalin A (ConA), a protein which exists as a 51 kDa dimer or 102 kDa tetramer. The mass spectrum of ConA contained monomeric, dimeric, and tetrameric species, with the most intense peaks belonging to the dimeric states (Figure 2C). The tetrameric +21 charge state was quadrupole isolated for further analysis (4894 m/z). Here, no structural perturbation upon trapping or spinning (Figure 3H,I, S14) was observed. In addition, no complex dissociation was observed, apart from under deliberate activation conditions (Figure S9). The CIU experiments revealed that minimal conformational changes were observed at up to 50 V activation (Figure 3I).
Increased Resolution
Multipass cIM separation offers increased resolution as a function of the square root of the number of passes, n (√(nz), where z is the ion charge state).37 The above experiments have shown that extended time in both the prestore and, when under T-Wave motion in the cIM, can cause small conformational changes for some protein ions, but not others, and this appears to be strongly related to mass and charge state. The +7 charge state ions of CytC generated from ammonium acetate were subjected to 1–3 passes around the cyclic ion guide (Figure 4).
Figure 4.
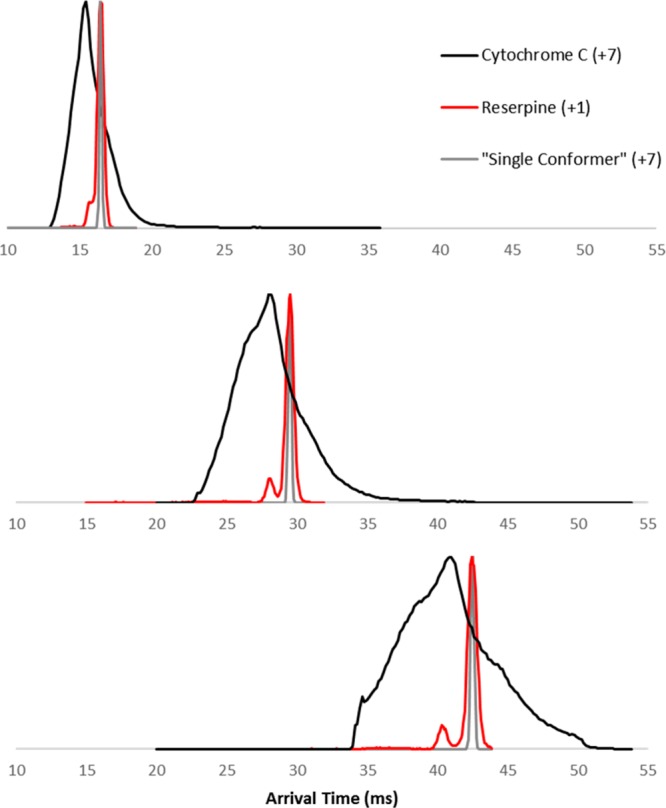
CytC (AmAc) +7 charge state arrival time distribution after multiple passes of the cyclic drift-region 1, 2, and 3 passes in the cIM. The measured ATD of singly charged reserpine (m/z 609) is shown to indicate the expected diffusion-limited peak width along with the derived peak for a +7 charge state species.
Subjecting the +7 ions to higher resolution separation revealed some evidence of new features in the broadened ATD (Figure 4A–C), however, distinct peaks were not seen. This suggests that the initial width of the protein ATD is due to the extremely large variety of highly similar conformational families, unresolvable by the cIM separator operating at a resolving power of ∼300 (CCS/ΔCCS derived for a +7 ion) at 3 passes (the peaks of singly charged reserpine, expected to be diffusion limited, are also shown in Figure 4, for comparison). This, however, is not always the case and is most likely protein, charge state, and solution condition specific. For example, early experiments with bovine insulin ions (generated from denaturing solution) showed that increased number of passes allows greater resolution of previously unresolvable features (Figure S15).
As an alternative approach to probe the presence of structural subpopulations in more detail, we used an IMS-CIU-IMS approach.
IMS-CIU-IMS
An experiment which can be performed on this instrument is multistage IMS (IMSn), where a subset ion population can be selected after IM separation, activated, and subjected to IM separation again. Due to the geometry of the instrument this can theoretically be done a limitless number of times. Here we will focus our attention on phenomena that can be probed in more detail compared to a traditional single stage collision induced unfolding (CIU) analysis. As an example, we use the quadrupole isolated, +7 ion of CytC was activated on injection to the trap (20 V). The initial ATD is presented in Figure 5A. We then use the IMS-CIU-IMS methodology, where the CIU occurs on reinjection to the array from the prestore, to obtain unfolding profiles of 4 subsets of this initial population (B–E). Conformation α, upon activation, directly converts into conformations β, γ, and δ. As longer drift time conformations are selected, populations β to δ are directly accessed. This shows the sequence of unfolding events, however it cannot be confirmed that this is nonreversible. Two conformations, ε (not directly selected) and ζ (not present in initial ATD), appear after the extension of conformation δ, between 60–80 V of activation.
Figure 5.

Slice CIU for an activated +7 CytC ion: (A) arrival time distribution, with the slices which have been removed for further mobility selection bounded by dotted lines; (B–E) CIU fingerprints for slices 16–17, 19–20, 23–24, 26–27 ms. Populations labeled as α, β, γ, δ, ε, and ζ.
Discussion
Our data show that proteins can to a large extent retain their native and multimeric states over the time scales compatible with high resolution IM separations. Importantly, the effect of prolonged exposure to T-waves appears, in most cases, similar to trapping alone, indicating little structural perturbation induced by T-wave based separation itself. In this study we explored time scales up to 360 ms, which would typically exceed the realistic time scales for separation of protein ions. These time scales (and number of passes around the device) are limited by wrap-around in the present setup. The aforementioned CytC work by Allen et al.,45 explored time scales up to 33 s, which at the moment is beyond the scope of our work. The magnitude of change observed over several hundred milliseconds by Allen et al. agrees well with data presented here. This is an order of magnitude longer than reported previously by Badman et al.39 It would seem plausible that this is due to the fact that the quadrupole ion trap (QIT) used to retain the protein ions for increments of time operated at a much lower pressure, approximately 0.0133 mbar42,43 compared to ∼2.2 mbar in the cIM device and the ∼5 mbar used in the SLIM based tandem IM. However, the trapping region of this instrument operates at similar pressures to the QIT and our data shows that increasing the time spent in the trapping cell does not significantly affect ion conformation (Figure S5). We will attempt to explore this in future work.
The loss of native conformation is less apparent for larger proteins. For βLac, the lowest charge state observed (+7) did not undergo any detectable change. Higher charge states (+8, +9) were minimally perturbed, and only after 240 ms. This is in agreement with previous reports showing that low charge states are more reflective of the solution conformation of proteins.54 No loss of initial conformations was observed for ConA. This suggests that the gas phase longevity of the native structures is proportional to the ion mass and, most likely, inversely proportional to its charge. CytC may indeed be particularly sensitive to gas phase studies as previous work showed that while it does not undergo backbone changes, the surface residues are rearranged55−57 and it retains fewer salt bridges when in the gas-phase rather than in solution.58 Gas-phase salt bridges are thought to play a very important role in the retention of native or native-like states, due to the much lower electrostatic permittivity of the vacuum compared to that of aqueous solution.59−61 Interestingly, subjecting native protein ions to an increased number of passes around the cIM device did not resolve overlapping features, suggesting that the ATDs of native ions consist of a very large number or even a continuum of conformers.41 This is perhaps not so surprising if we imagine, for example, a variety of ways in which solvent exposed residues of the protein can be rearranged during desolvation. This observation is somewhat parallel with the previous findings from a study utilizing variable temperature IM instrumentation,26 where only a minimal increase in resolution was observed for native protein ions at cryogenic temperatures. Collectively, this suggests that the native protein ATDs are naturally broad, not due to diffusion or interconversion, but inherent conformational heterogeneity, which is consistent with previous findings in the field.41,62 The above phenomena will be investigated in the future work utilizing the high resolution capabilities of the instrument combined with the IMS-CIU-IMS methodology presented here.
Acknowledgments
C.E. is funded by a BBSRC iCASE Award with Waters BB/L015382/1. J.U., N.T., K.G., and M.M. are all employees of Waters Corporation, which manufactures and sells T-wave IM-MS instruments.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.8b05641.
Detailed instrument information; Stability plots for +7 and +9 βLac; Population tracking for all species; and Mass spectrum for ConA during stability experiments (PDF)
Author Contributions
The cyclic ion mobility device was designed and built by K.G. and J.U. M.M. was involved in scientific discussions around this study. N.T. was involved in some of the preliminary investigations using the cyclic ion mobility device for this study. Experiments were carried out by C.E. and J.U. S.K. wrote the population fitting algorithm and performed data analysis. K.T. designed and supervised experiments. The manuscript was written through contributions of all authors.
The authors declare the following competing financial interest(s): Jakub Ujma, Nick Tomczyk, Kevin Giles and Mike Morris are all employees of Waters Corporation.
Supplementary Material
References
- Macha S. F.; Limbach P. A. Matrix-Assisted Laser Desorption/Ionization (MALDI) Mass Spectrometry of Polymers. Curr. Opin. Solid State Mater. Sci. 2002, 6, 213–220. 10.1016/S1359-0286(02)00036-0. [DOI] [Google Scholar]
- Yamashita M.; Fenn J. B. Electrospray Ion Source. Another Variation on the Free-Jet Theme. J. Phys. Chem. 1984, 88, 4451–4459. 10.1021/j150664a002. [DOI] [Google Scholar]
- Fenn J. B. Electrospray Ionization Mass Spectrometry: How It All Began. J. Biomol. Technol. 2002, 13, 101–118. 10.1126/science.2675315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siuzdak G.; Bothner B.; Yeager M.; Brugidou C.; Fauquet C. M.; Hoey K.; Change C. M. Mass Spectrometry and Viral Analysis. Chem. Biol. 1996, 3, 45–48. 10.1016/S1074-5521(96)90083-6. [DOI] [PubMed] [Google Scholar]
- Ruotolo B. T.; Giles K.; Campuzano I.; Sandercock A. M.; Bateman R. H.; Robinson C. V. Biochemistry: Evidence for Macromolecular Protein Rings in the Absence of Bulk Water. Science (Washington, DC, U. S.) 2005, 310, 1658–1661. 10.1126/science.1120177. [DOI] [PubMed] [Google Scholar]
- Dole M.; Hines R. L.; Mack L. L.; Mobley R. C.; Ferguson L. D.; Alice M. B. Gas Phase Macroions. Macromolecules 1968, 1, 96–97. 10.1021/ma60001a017. [DOI] [Google Scholar]
- Chowdhury S. K.; Katta V.; Chait B. T. Probing Conformational Changes in Proteins by Mass Spectrometry. J. Am. Chem. Soc. 1990, 112, 9012–9013. 10.1021/ja00180a074. [DOI] [Google Scholar]
- Clemmer D. E.; Hudgins R. R.; Jarrold M. F. Naked Protein Conformations: Cytochrome c in the Gas Phase. J. Am. Chem. Soc. 1995, 117, 10141–10142. 10.1021/ja00145a037. [DOI] [Google Scholar]
- Chen S. H.; Russell D. H. How Closely Related Are Conformations of Protein Ions Sampled by IM-MS to Native Solution Structures?. J. Am. Soc. Mass Spectrom. 2015, 26, 1433–1443. 10.1007/s13361-015-1191-1. [DOI] [PubMed] [Google Scholar]
- Pringle S. D.; Giles K.; Wildgoose J. L.; Williams J. P.; Slade S. E.; Thalassinos K.; Bateman R. H.; Bowers M. T.; Scrivens J. H. An Investigation of the Mobility Separation of Some Peptide and Protein Ions Using a New Hybrid Quadrupole/Travelling Wave IMS/Oa-ToF Instrument. Int. J. Mass Spectrom. 2007, 261, 1–12. 10.1016/j.ijms.2006.07.021. [DOI] [Google Scholar]
- Shelimov K. B.; Clemmer D. E.; Hudgins R. R.; Jarrold M. F. Protein Structure in Vacuo: Gas-Phase Conformations of BPTI and Cytochrome C. J. Am. Chem. Soc. 1997, 119, 2240–2248. 10.1021/ja9619059. [DOI] [Google Scholar]
- Wojnowska M.; Yan J.; Sivalingam G. N.; Cryar A.; Gor J.; Thalassinos K.; Djordjevic S. Autophosphorylation Activity of a Soluble Hexameric Histidine Kinase Correlates with the Shift in Protein Conformational Equilibrium. Chem. Biol. 2013, 20, 1411–1420. 10.1016/j.chembiol.2013.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacholarz K. J.; Barran P. E. Distinguishing Loss of Structure from Subunit Dissociation for Protein Complexes with Variable Temperature Ion Mobility Mass Spectrometry. Anal. Chem. 2015, 87, 6271–6279. 10.1021/acs.analchem.5b01063. [DOI] [PubMed] [Google Scholar]
- Zhong Y.; Han L.; Ruotolo B. T. Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angew. Chem., Int. Ed. 2014, 53, 9209–9212. 10.1002/anie.201403784. [DOI] [PubMed] [Google Scholar]
- Beveridge R.; Covill S.; Pacholarz K. J.; Kalapothakis J. M. D.; Macphee C. E.; Barran P. E. A Mass-Spectrometry-Based Framework to Define the Extent of Disorder in Proteins. Anal. Chem. 2014, 86, 10979–10991. 10.1021/ac5027435. [DOI] [PubMed] [Google Scholar]
- Dickinson E. R.; Jurneczko E.; Pacholarz K. J.; Clarke D. J.; Reeves M.; Ball K. L.; Hupp T.; Campopiano D.; Nikolova P. V.; Barran P. E. Insights into the Conformations of Three Structurally Diverse Proteins: Cytochrome c, P53, and MDM2, Provided by Variable-Temperature Ion Mobility Mass Spectrometry. Anal. Chem. 2015, 87, 3231–3238. 10.1021/ac503720v. [DOI] [PubMed] [Google Scholar]
- Clemmer D. E.; Jarrold M. F. Ion Mobility Measurements and Their Applications to Clusters and Biomolecules. J. Mass Spectrom. 1997, 32, 577–592. . [DOI] [Google Scholar]
- Nyon M. P.; Prentice T.; Day J.; Kirkpatrick J.; Sivalingam G. N.; Levy G.; Haq I.; Irving J. A.; Lomas D. A.; Christodoulou J.; et al. An Integrative Approach Combining Ion Mobility Mass Spectrometry, X-Ray Crystallography, and Nuclear Magnetic Resonance Spectroscopy to Study the Conformational Dynamics of 1-Antitrypsin upon Ligand Binding. Protein Sci. 2015, 24, 1301–1312. 10.1002/pro.2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landreh M.; Marklund E. G.; Uzdavinys P.; Degiacomi M. T.; Coincon M.; Gault J.; Gupta K.; Liko I.; Benesch J. L. P.; Drew D.; et al. Integrating Mass Spectrometry with MD Simulations Reveals the Role of Lipids in Na+/H+ Antiporters. Nat. Commun. 2017, 8, 13993. 10.1038/ncomms13993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyung S. J.; Robinson C. V.; Ruotolo B. T. Gas-Phase Unfolding and Disassembly Reveals Stability Differences in Ligand-Bound Multiprotein Complexes. Chem. Biol. 2009, 16, 382–390. 10.1016/j.chembiol.2009.02.008. [DOI] [PubMed] [Google Scholar]
- Benesch J. L. P. Collisional Activation of Protein Complexes: Picking Up the Pieces. J. Am. Soc. Mass Spectrom. 2009, 20, 341–348. 10.1016/j.jasms.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Bernstein S. L.; Dupuis N. F.; Lazo N. D.; Wyttenbach T.; Condron M. M.; Bitan G.; Teplow D. B.; Shea J. E.; Ruotolo B. T.; Robinson C. V.; et al. Amyloid-β 2 Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem. 2009, 1, 326–331. 10.1038/nchem.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bleiholder C.; Dupuis N. F.; Wyttenbach T.; Bowers M. T. Ion Mobilityg-Mass Spectrometry Reveals a Conformational Conversion from Random Assembly to β-Sheet in Amyloid Fibril Formation. Nat. Chem. 2011, 3, 172–177. 10.1038/nchem.945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eschweiler J. D.; Rabuck-Gibbons J. N.; Tian Y.; Ruotolo B. T. CIUSuite: A Quantitative Analysis Package for Collision Induced Unfolding Measurements of Gas-Phase Protein Ions. Anal. Chem. 2015, 87, 11516–11522. 10.1021/acs.analchem.5b03292. [DOI] [PubMed] [Google Scholar]
- St. Louis R. H.; Hill H. H.; Eiceman G. A. Ion Mobility Spectrometry in Analytical Chemistry. Crit. Rev. Anal. Chem. 1990, 21, 321–355. 10.1080/10408349008050848. [DOI] [Google Scholar]
- Ujma J.; Giles K.; Morris M.; Barran P. E. New High Resolution Ion Mobility Mass Spectrometer Capable of Measurements of Collision Cross Sections from 150 to 520 K. Anal. Chem. 2016, 88, 9469–9478. 10.1021/acs.analchem.6b01812. [DOI] [PubMed] [Google Scholar]
- May J. C.; Russell D. H. A Mass-Selective Variable-Temperature Drift Tube Ion Mobility-Mass Spectrometer for Temperature Dependent Ion Mobility Studies. J. Am. Soc. Mass Spectrom. 2011, 22, 1134–1145. 10.1007/s13361-011-0148-2. [DOI] [PubMed] [Google Scholar]
- Dugourd P.; Hudgins R. R.; Clemmer D. E.; Jarrold M. F. High-Resolution Ion Mobility Measurements. Rev. Sci. Instrum. 1997, 68, 1122–1129. 10.1063/1.1147873. [DOI] [Google Scholar]
- Glaskin R. S.; Ewing M. A.; Clemmer D. E. Ion Trapping for Ion Mobility Spectrometry Measurements in a Cyclical Drift Tube. Anal. Chem. 2013, 85, 7003–7008. 10.1021/ac4015066. [DOI] [PubMed] [Google Scholar]
- Merenbloom S. I.; Glaskin R. S.; Henson Z. B.; Clemmer D. E. High-Resolution Ion Cyclotron Mobility Spectrometry. Anal. Chem. 2009, 81, 1482–1487. 10.1021/ac801880a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Merenbloom S. I.; Koeniger S. L.; Valentine S. J.; Plasencia M. D.; Clemmer D. E. IMS-IMS and IMS-IMS-IMS/MS for Separating Peptide and Protein Fragment Ions. Anal. Chem. 2006, 78, 2802–2809. 10.1021/ac052208e. [DOI] [PubMed] [Google Scholar]
- Koeniger S. L.; Merenbloom S. I.; Valentine S. J.; Jarrold M. F.; Udseth H. R.; Smith R. D.; Clemmer D. E. An IMS-IMS Analogue of MS-MS. Anal. Chem. 2006, 78, 4161–4174. 10.1021/ac051060w. [DOI] [PubMed] [Google Scholar]
- Kemper P. R.; Dupuis N. F.; Bowers M. T. A New, Higher Resolution, Ion Mobility Mass Spectrometer. Int. J. Mass Spectrom. 2009, 287, 46–57. 10.1016/j.ijms.2009.01.012. [DOI] [Google Scholar]
- Silveira J. A.; Michelmann K.; Ridgeway M. E.; Park M. A. Fundamentals of Trapped Ion Mobility Spectrometry Part II: Fluid Dynamics. J. Am. Soc. Mass Spectrom. 2016, 27, 585–595. 10.1007/s13361-015-1310-z. [DOI] [PubMed] [Google Scholar]
- Shaffer S. A.; Prior D. C.; Anderson G. A.; Udseth H. R.; Smith R. D. An Ion Funnel Interface for Improved Ion Focusing and Sensitivity Using Electrospray Ionization Mass Spectrometry. Anal. Chem. 1998, 70, 4111–4119. 10.1021/ac9802170. [DOI] [PubMed] [Google Scholar]
- Giles K.; Pringle S. D.; Worthington K. R.; Little D.; Wildgoose J. L.; Bateman R. H. Applications of a Travelling Wave-Based Radio-Frequency-Only Stacked Ring Ion Guide. Rapid Commun. Mass Spectrom. 2004, 18, 2401–2414. 10.1002/rcm.1641. [DOI] [PubMed] [Google Scholar]
- Giles K.; Wildgoose J.; Pringle S.; Garside J.; Carney P.; Nixon P.; Langridge D.. 62nd ASMS Conference on Mass Spectrometry and Allied Topics; June 15–19, 2014, ASMS, 2014.
- Giles K.; Ujma J.; Wildgoose J.; Green M. R.; Richardson K.; Langridge D.; Tomczyk N.. Design and Performance of a Second-Generation Cyclic Ion Mobility Enabled Q-ToF. 65th ASMS Conference on Mass Spectrometry and Allied Topics; ASMS, 2017
- Webb I. K.; Garimella S. V. B.; Tolmachev A. V.; Chen T. C.; Zhang X.; Norheim R. V.; Prost S. A.; LaMarche B.; Anderson G. A.; Ibrahim Y. M.; et al. Experimental Evaluation and Optimization of Structures for Lossless Ion Manipulations for Ion Mobility Spectrometry with Time-of-Flight Mass Spectrometry. Anal. Chem. 2014, 86, 9169–9176. 10.1021/ac502055e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng L.; Webb I. K.; Garimella S. V. B.; Hamid A. M.; Zheng X.; Norheim R. V.; Prost S. A.; Anderson G. A.; Sandoval J. A.; Baker E. S.; et al. Serpentine Ultralong Path with Extended Routing (SUPER) High Resolution Traveling Wave Ion Mobility-MS Using Structures for Lossless Ion Manipulations. Anal. Chem. 2017, 89, 4628–4634. 10.1021/acs.analchem.7b00185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koeniger S. L.; Clemmer D. E. Resolution and Structural Transitions of Elongated States of Ubiquitin. J. Am. Soc. Mass Spectrom. 2007, 18, 322–331. 10.1016/j.jasms.2006.09.025. [DOI] [PubMed] [Google Scholar]
- Badman E. R.; Hoaglund-Hyzer C. S.; Clemmer D. E. Monitoring Structural Changes of Proteins in an Ion Trap over ∼ 10–200 Ms: Unfolding Transitions in Cytochrome c Ions. Anal. Chem. 2001, 73, 6000–6007. 10.1021/ac010744a. [DOI] [PubMed] [Google Scholar]
- Badman E. R.; Myung S.; Clemmer D. E. Evidence for Unfolding and Refolding of Gas-Phase Cytochrome c Ions in a Paul Trap. J. Am. Soc. Mass Spectrom. 2005, 16, 1493–1497. 10.1016/j.jasms.2005.04.013. [DOI] [PubMed] [Google Scholar]
- Myung S.; Badman E. R.; Lee Y. J.; Clemmer D. E. Structural Transitions of Electrosprayed Ubiquitin Ions Stored in an Ion Trap over < 10 Ms to 30 S. J. Phys. Chem. A 2002, 106, 9976–9982. 10.1021/jp0206368. [DOI] [Google Scholar]
- Allen S. J.; Eaton R. M.; Bush M. F. Structural Dynamics of Native-Like Ions in the Gas Phase: Results from Tandem Ion Mobility of Cytochrome C. Anal. Chem. 2017, 89, 7527–7534. 10.1021/acs.analchem.7b01234. [DOI] [PubMed] [Google Scholar]
- Viehland L. A.; Mason E. A. Gaseous Ion Mobility and Diffusion in Electric Fields of Arbitrary Strength. Ann. Phys. (Amsterdam, Neth.) 1978, 110, 287–328. 10.1016/0003-4916(78)90034-9. [DOI] [Google Scholar]
- Morsa D.; Gabelica V.; De Pauw E. Effective Temperature of Ions in Traveling Wave Ion Mobility Spectrometry. Anal. Chem. 2011, 83, 5775–5782. 10.1021/ac201509p. [DOI] [PubMed] [Google Scholar]
- Merenbloom S. I.; Flick T. G.; Williams E. R. How Hot Are Your Ions in TWAVE Ion Mobility Spectrometry?. J. Am. Soc. Mass Spectrom. 2012, 23, 553–562. 10.1007/s13361-011-0313-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ujma J.; Ropartz D.; Giles K.; Richardson K.; Langridge D.; Wildgoose J.; Green M.; Pringle S. Cyclic Ion Mobility Mass Spectrometry Distinguishes Anomers and Open-Ring Forms of Pentasaccharides. J. Am. Soc. Mass Spectrom. 2019, 30, 1028. 10.1007/s13361-019-02168-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giles K.; Ujma J.; Wildgoose J.; Pringle S.; Richardson K.; Langridge D.; Green M. A Cyclic Ion Mobility-Mass Spectrometry System. Anal. Chem. 2019, na. 10.1021/acs.analchem.9b01838. [DOI] [PubMed] [Google Scholar]
- Tian Y.; Han L.; Buckner A. C.; Ruotolo B. T. Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Anal. Chem. 2015, 87, 11509–11515. 10.1021/acs.analchem.5b03291. [DOI] [PubMed] [Google Scholar]
- Sivalingam G. N.; Cryar A.; Williams M. A.; Gooptu B.; Thalassinos K. Deconvolution of Ion Mobility Mass Spectrometry Arrival Time Distributions Using a Genetic Algorithm Approach: Application to 1-Antitrypsin Peptide Binding. Int. J. Mass Spectrom. 2018, 426, 29–37. 10.1016/j.ijms.2018.01.008. [DOI] [Google Scholar]
- Sakurai K.; Oobatake M.; Goto Y. Salt-Dependent Monomer-Dimer Equilibrium of Bovine β-Lactoglobulin at PH 3. Protein Sci. 2001, 10, 2325–2335. 10.1110/ps.17001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarff C. A.; Thalassinos K.; Hilton G. R.; Scrivens J. H. Travelling Wave Ion Mobility Mass Spectrometry Studies of Protein Structure: Biological Significance and Comparison with X-Ray Crystallography and Nuclear Magnetic Resonance Spectroscopy Measurements. Rapid Commun. Mass Spectrom. 2008, 22, 3297–3304. 10.1002/rcm.3737. [DOI] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. Native Electron Capture Dissociation for the Structural Characterization of Noncovalent Interactions in Native Cytochrome C. Angew. Chem., Int. Ed. 2003, 42, 4900–4904. 10.1002/anie.200351705. [DOI] [PubMed] [Google Scholar]
- Breuker K.; McLafferty F. W. The Thermal Unfolding of Native Cytochrome c in the Transition from Solution to Gas Phase Probed by Native Electron Capture Dissociation. Angew. Chem., Int. Ed. 2005, 44, 4911–4914. 10.1002/anie.200500668. [DOI] [PubMed] [Google Scholar]
- Steinberg M. Z.; Elber R.; McLafferty F. W.; Gerber R. B.; Breuker K. Early Structural Evolution of Native Cytochrome c after Solvent Removal. ChemBioChem 2008, 9, 2417–2423. 10.1002/cbic.200800167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z.; Browne S. J.; Vachet R. W. Exploring Salt Bridge Structures of Gas-Phase Protein Ions Using Multiple Stages of Electron Transfer and Collision Induced Dissociation. J. Am. Soc. Mass Spectrom. 2014, 25, 604–613. 10.1007/s13361-013-0821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonner J.; Lyon Y. A.; Nellessen C.; Julian R. R. Photoelectron Transfer Dissociation Reveals Surprising Favorability of Zwitterionic States in Large Gaseous Peptides and Proteins. J. Am. Chem. Soc. 2017, 139, 10286–10293. 10.1021/jacs.7b02428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konermann L. Addressing a Common Misconception: Ammonium Acetate as Neutral PH “Buffer” for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom. 2017, 28, 1827–1835. 10.1007/s13361-017-1739-3. [DOI] [PubMed] [Google Scholar]
- Porrini M.; Rosu F.; Rabin C.; Darré L.; Gómez H.; Orozco M.; Gabelica V. Compaction of Duplex Nucleic Acids upon Native Electrospray Mass Spectrometry. ACS Cent. Sci. 2017, 3, 454–461. 10.1021/acscentsci.7b00084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen S. J.; Giles K.; Gilbert T.; Bush M. F. Ion Mobility Mass Spectrometry of Peptide, Protein, and Protein Complex Ions Using a Radio-Frequency Confining Drift Cell. Analyst 2016, 141, 884–891. 10.1039/C5AN02107C. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.